

Abstract
Chimeric MS/MS spectra contain fragments from multiple precursor ions and therefore hinder compound identification in metabolomics. Historically, deconvolution of these chimeric spectra has been challenging and relied upon specific experimental methods that introduce variation in the ratios of precursor ions between multiple tandem mass spectrometry (MS/MS) scans. DecoID provides a complementary, method-independent approach where database spectra are computationally mixed to match an experimentally acquired spectrum by using LASSO regression. We validated that DecoID increases the number of identified metabolites in MS/MS datasets from both data-independent and data-dependent acquisition without increasing the false discovery rate. We applied DecoID to publicly available data from the MetaboLights repository and to data from human plasma, where DecoID increased the number of identified metabolites from data-dependent acquisition data by over 30% compared to direct spectral matching. DecoID is compatible with any user-defined MS/MS database and provides automated searching for some of the largest MS/MS databases currently available.
Introduction
Compound identification is generally recognized as the major bottleneck when performing untargeted metabolomics with liquid chromatography/mass spectrometry (LC/MS)1,2. An important step in the identification process is matching MS/MS data from a feature (defined by a unique combination of retention time and m/z values) in the research sample to a reference MS/MS spectrum in metabolomic databases. Even small variations in an MS/MS spectrum can indicate structural differences in the precursor ions (Supplementary Figure 1)3,4. This necessitates exact matches of both m/z and intensity for all fragments in the research and reference MS/MS spectra. However, reference MS/MS spectra are typically derived from pure chemical standards, while research MS/MS spectra are obtained by analyzing much more complex sample matrices that contain many compounds. When more than one of these compounds is simultaneously fragmented in the same MS/MS experiment, the resulting spectra are said to be “chimeric” (Figure 1a). By definition, chimeric spectra do not match reference data from pure chemical standards and therefore hinder compound identification when not deconvolved.
Figure 1. Deconvolution with DecoID to identify metabolites with chimeric MS/MS spectra.

(a) Schematic of a chimeric (bottom) and non-chimeric (top) MS/MS spectrum. Each color represents a unique precursor ion. Smaller circles indicate fragments. When searched in MS/MS databases, chimeric spectra do not lead to identifications (or, even worse, they lead to incorrect identifications). (b) Histogram showing the percentage contamination of MS/MS spectra from the analysis of NIST SRM 1950 human plasma with DDA and a 1 m/z isolation window. Despite using a narrow isolation window, greater than 50% of the acquired spectra have more than 10% contamination. (c) MS/MS spectra that were not able to be identified with spectral matching to mzCloud had significantly higher levels of MS/MS contamination (two-sided two-sample Kolmogorov–Smirnov test) than those spectra that were able to be identified. Horizontal lines on top and bottom of violin plot represent the maximum and minimum values. (d) Diagram of the DecoID search algorithm. A library of reference MS/MS spectra is assembled from metabolomic databases, predicted isotopologue spectra, and pure unknowns. This library of reference MS/MS spectra is filtered on the basis of MS1 information, the size of the MS/MS isolation window, and retention time (if available) for each experimentally observed MS/MS pattern in the user’s data. The experimentally observed MS/MS spectrum is then reconstructed by using the filtered library spectra and non-negative LASSO regression. All components used in the deconvolution are scored against the library spectra on the basis of accurate mass and spectral similarity. Lastly, all reference library spectra used for the deconvolution undergo a redundancy check to determine whether an equally good deconvolution could have been achieved by using a different set of library spectra. Potentially redundant components are flagged in the report provided to the user.
Historically, deconvolution of chimeric spectra in metabolomics has relied upon experimental variation in the ratios of precursor ions between MS/MS scans. When the ratio of two precursor ions varies between MS/MS scans, it leads to proportionate changes in the intensity of each precursor’s product ions. The variation in precursor ions can occur due to differences in the precursors’ chromatographic elution profiles, differences in precursor concentrations between samples, or by shifting the location of the MS/MS isolation window. There are several algorithms that make use of these principles to deconvolve chimeric spectra in specific experimental workflows for metabolomics3,5–8 and proteomics9–14. Notably, however, deconvolution of metabolomic MS/MS spectra on the basis of these experimental factors is not always successful3,5. While it is possible to analyze samples with different combinations of experimental conditions to improve success, this is low throughput and requires designing sample-specific methods. Moreover, it is not compatible with analysis of most chimeric data in public repositories such as MetaboLights15 and the Metabolomics Workbench16. Thus, we sought to develop a complementary strategy to deconvolve metabolomic MS/MS spectra independent of how the spectra were acquired.
When performing metabolomics with data-independent acquisition (DIA), wide MS/MS isolation windows are applied (e.g., >20 m/z). The majority of the fragmentation data obtained are chimeric and require deconvolution3,17. In contrast, metabolomic workflows using data-dependent acquisition (DDA) typically apply a narrow MS/MS isolation window (e.g., 1–3 m/z)18. Although DDA workflows aim to only fragment a single precursor in each MS/MS experiment, previous studies have suggested that chimeric data are still prevalent3,19. We applied a simple metric inspired by Lawson et al.19 to estimate the percentage of the MS/MS signal that arises from analytes other than the targeted precursor (Methods and Supplementary Figure 2) in a DDA analysis of the NIST 1950 reference plasma. We find that, even with a 1 m/z isolation window, more than half of the acquired MS/MS spectra have some degree of contamination (Figure 1b). Further, precursors whose MS/MS spectra did not return any high-scoring hits after searching mzCloud had significantly higher levels of contamination (Figure 1c), suggesting that deconvolution of DDA spectra may help improve metabolite identification rates (the number of identified features divided by the number of features detected).
To fill the need for acquisition-independent deconvolution of any MS/MS spectrum in metabolomics, we have developed DecoID, which builds upon a proteomic method20 to perform a database-assisted deconvolution. DecoID is a Python library with a graphical-user interface to facilitate data processing and visualization (Supplementary Figure 3). DecoID accepts raw data from any vendor file format that is compatible with MSConvert21 and utilizes user-defined MS/MS databases as well as built-in support and automated searching of some of the largest MS/MS databases currently available: Human Metabolome Database (HMDB, hmdb.ca)22, Mass Bank of North America (MoNA, https://mona.fiehnlab.ucdavis.edu/)23, and mzCloud (www.mzcloud.org).
Results
DecoID uses non-negative LASSO regression24 to deconvolve any MS/MS spectrum into a linear combination of database spectra. Once deconvolved, the purified spectrum corresponding to the precursor of interest can be extracted and scored against the reference spectrum to identify the compound and check for redundancies (that is other metabolites in the database with highly similar MS/MS patterns, Methods). The workflow of DecoID is depicted in Figure 1d and a detailed description of the algorithm is provided in the Methods. The goal of DecoID is to be universally applicable to any experimental workflow for both MS/MS acquisition (DDA and DIA) and sample introduction (liquid chromatography, flow injection, and direct infusion). DecoID can operate on a single spectrum or it can leverage an entire dataset through full-scan MS1 information and grouping of MS/MS spectra if peak information is provided (Methods). In addition, DecoID can also use retention time information from user-provided databases to gain further specificity in metabolite identifications and more accurate deconvolutions (Supplementary Figure 4).
Although MS/MS databases are rapidly growing25, there will still be spectra contaminated with precursors that are not contained in any MS/MS database. One notable source of contamination is what we refer to here as orphan isotopologues. We say that an MS/MS spectrum is contaminated by an orphan isotopologue when the MS/MS spectrum contains fragments from an M+1 isotopologue of a contaminating compound (but not the parent M+0 of this same contaminating compound, Figure 2a). These orphan M+1 isotopologues primarily arise from naturally occurring carbon-13 and are challenging to deconvolve because only MS/MS data for M+0 isotopologues are typically included in MS/MS databases. In our evaluation of human plasma by DDA with 1 m/z isolation windows, we estimate that approximately 10–15% of the acquired spectra will be contaminated by orphan isotopologues (Methods). To remove this contamination, spectra for orphan carbon-13 isotopologues are computationally predicted from the M+0 database spectrum by using an approach similar to that of isoMETLIN26 (Figure 2b–c, Supplementary Figure 5, and Methods) and applied to deconvolve spectra contaminated with orphan isotopologues. Additionally, when a non-chimeric MS/MS spectrum is acquired on a precursor whose reference spectrum is not contained in any of the MS/MS databases searched, DecoID uses this pure spectrum for potential deconvolution. To this end, DecoID creates an “on-the-fly” unknown spectral library containing these pure MS/MS data. This enables deconvolution of MS/MS spectra within the same dataset that are contaminated by unknown compounds, pending their inclusion in the on-the-fly library (Supplementary Figure 6).
Figure 2. Orphan isotopologue contamination and MS/MS spectrum prediction.

(a) An orphan carbon-13 isotopologue can cause MS/MS contamination if the M+0 parent of a contaminating compound (red) is excluded from the isolation window (gray) but the M+1 carbon-13 peak is not. Such chimeric MS/MS spectra cannot be deconvolved by considering only database spectra and will severely impact the ability to identify the targeted precursor (blue). (b-c) Schematic of predicting the MS/MS spectrum of an M+1 isotopologue arising from naturally occurring carbon-13. (b) First, based on the chemical formula of the precursor ion, all possible subformulas are enumerated, and the m/z of each subformula is computed. Then, for each observed fragment, the possible subformulas are assigned and the mean number of carbon atoms for all possible subformulas of each fragment are computed. (c) Using the computed number of carbons in each fragment, the intensity of each fragment in the M+1 spectrum is computed by distributing the intensity of the M+0 fragment ions according to the number of carbons in each fragment. All carbon atoms are equally likely to be carbon-13, thus for a seven-carbon fragment and an eight-carbon precursor, there is a 7/8 probability that the carbon-13 is retained in the fragment and a 1/8 probability that it is removed as a neutral loss. Carbon atoms are represented by gray and red circles for carbon-12 and carbon-13, respectively.
To test the effectiveness of the unknown library, we applied DecoID to a DDA dataset of 526 metabolite standards from the Mass Spectrometry Metabolite Library (IROA Technologies) in a single mixture (the IROA standard mixture, Methods). MS/MS data were acquired with a 1 m/z isolation window. The dataset was deconvolved with DecoID and two versions of our in-house database: the full in-house database and a partial in-house database in which 50% of the entries were arbitrarily removed to simulate incomplete database coverage of a sample. When an unknown library was created on-the-fly and then used for deconvolution, database similarity scores from the partial database were restored to nearly the same level as those from the full database (Supplementary Figure 7). Further details are described in the Methods and an example is available in Supplementary Figure 6. We note that the on-the-fly deconvolution capability of DecoID benefits from MS/MS methods where multiple spectra are acquired on each precursor. As such, performance of the on-the-fly deconvolution capability will vary depending on the specific experimental workflow used.
Null Evaluations
To verify that DecoID performs faithful deconvolutions, three null evaluations were performed. One of our objectives was to ensure that DecoID does not falsely deconvolve noisy, but non-chimeric, MS/MS spectra into several components by using simulated DDA and DIA spectra. The simulated spectra were composed of database spectra from MoNA with random noise added (Methods). In the first null evaluation, DecoID was used to deconvolve the noisy, but non-chimeric, spectra by searching the spectra back against the unmodified MoNA database using various LASSO parameter values, which is the primary parameter for DecoID’s deconvolution algorithm. For all tested non-zero parameter values, DecoID did not find more than a single component, meaning no unfaithful deconvolutions were performed (Supplementary Figure 8).
As a second independent null evaluation, the same simulated spectra were again deconvolved with DecoID. This time, however, the reference spectra for the compounds in the simulated spectra were removed from the MoNA database. Using this partial database, DecoID did not falsely combine database spectra to match the non-chimeric spectra that were absent from the database. In both of the first two null evaluations, the only case where unfaithful deconvolutions were performed was when the LASSO parameter was set to zero. A LASSO parameter of zero amounts to a non-negative linear regression, which has been successfully utilized to deconvolve proteomic MS/MS spectra20. However, our results show that a comparable approach cannot be applied to metabolomic MS/MS spectra without the LASSO penalty term (Supplementary Figure 8).
After optimizing the DecoID LASSO parameter (Methods and Supplementary Figure 9) to maximize metabolite identification accuracy, which found an optimal value of 5.0 for both DIA and DDA datasets, we sought to verify that the optimal parameters enabled faithful deconvolutions on chimeric data. To do this, we performed a third null evaluation where we analyzed synthetic DDA and DIA datasets formed of reference spectra for the compounds used to optimize the DecoID parameters. These reference spectra were mixed according to the retention times of each compound to create a chimeric dataset with a known absolute ground-truth. With DecoID, we then deconvolved these datasets using mzCloud and a subset of mzCloud where the spectra of the mixture compounds were removed from the database, creating a partial database. For the DDA dataset, a LASSO parameter of 5.0 was sufficient to produce no significant difference between the database similarity scores when the decoy database was used with and without DecoID deconvolution. For the DIA dataset, a LASSO parameter of 5.0 was insufficient to produce a non-significant result, indicating that DecoID was unfaithfully increasing database similarity scores. Using a LASSO parameter of 50.0, on the other hand, was sufficient and the performance in metabolite identification accuracy was negligibly different to when a parameter value of 5.0 was used (Supplementary Figure 9 and Supplementary Figure 10).
Validation with IROA standard mixture
To verify the accuracy and performance of DecoID, we again applied it to the IROA mixture dataset of DDA and DIA spectra. For the DDA experiments, isolation windows of 1, 3, and 5 m/z were used to create a gradient of contamination in the datasets (Supplementary Figure 11). To validate the performance of DecoID across diverse MS/MS databases, the DDA standard mixture datasets were searched with and without deconvolution using MoNA, HMDB, mzCloud, and our in-house database. Each database covers various portions of the IROA standards (Supplementary Table 1). With all databases, similarity scores for correctly identified metabolites did not decrease as the degree of contamination increased when using DecoID (Figure 3a, Supplementary Figure 12, and Supplementary Figure 13). To verify that DecoID actually improves metabolite identification and does not just falsely increase database scores, receiver operating characteristic (ROC) curves were drawn from the DDA results for all databases using the top three hits of each feature (Methods and Supplementary Figure 14). DecoID gives a higher area under the ROC curve (auROC) compared to direct database searching when using the in-house database or mzCloud. When using HMDB or MoNA, DecoID gives a slightly worse auROC that we speculate is due to the diversity of spectra in those databases (QTOF, Orbitrap, and QqQ) compared to the in-house database and mzCloud, which are entirely composed of Orbitrap spectra (the experimental spectra used in this study are from an Orbitrap instrument). When aggregating the results across all databases and examining the false discovery rate (FDR) and true positive rate (TPR) for the top hit of each feature, however, DecoID shows the same FDR and a higher TPR relative to searching the acquired spectra against the database for nearly all dot product thresholds (Figure 3b–c). Further, when using a dot product threshold of 80, DecoID significantly increased the TPR without increasing the FDR, thereby improving metabolite identification in DDA spectra (Figure 3d).
Figure 3. DecoID improves metabolite identification in the DDA and DIA IROA datasets.
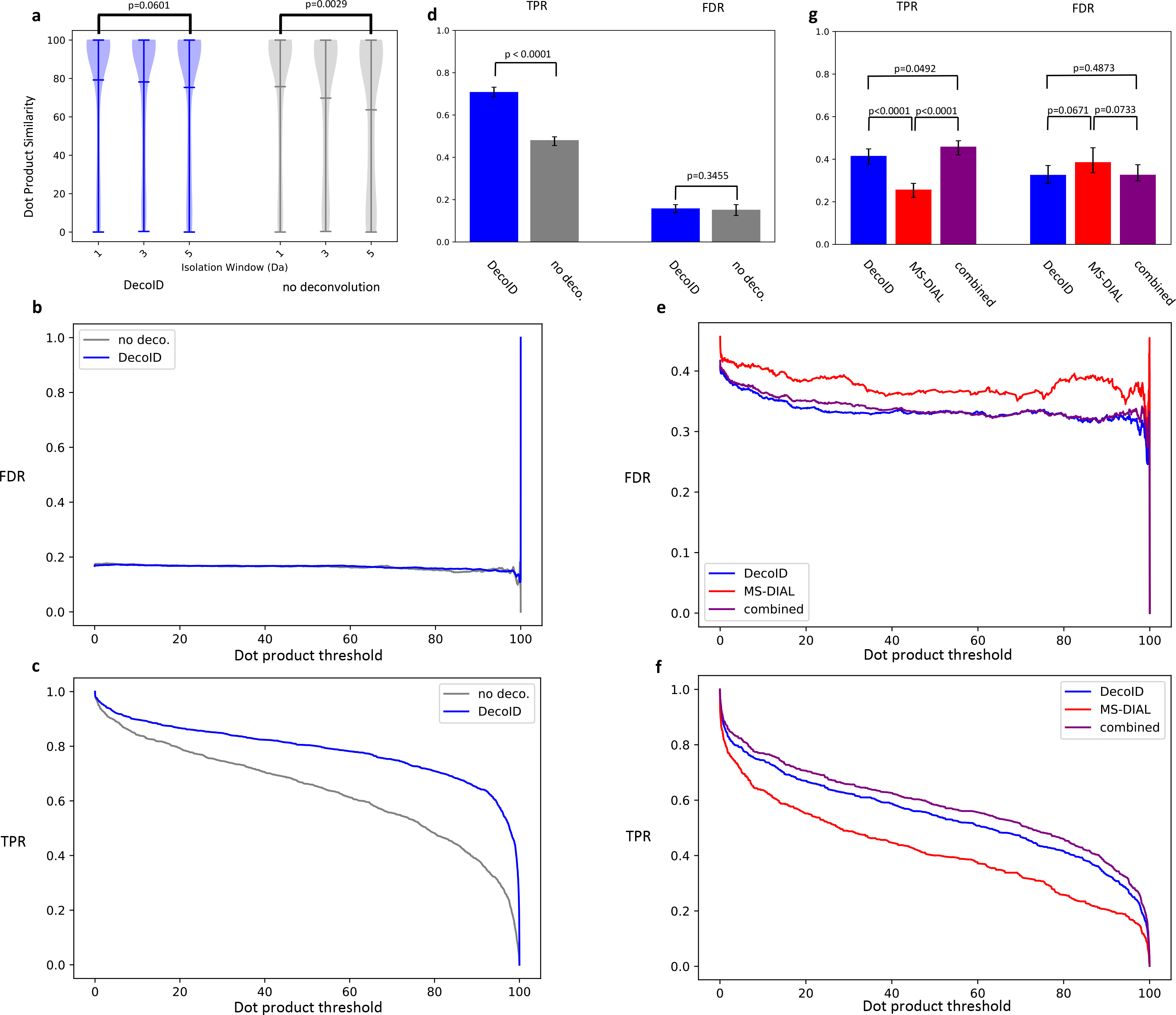
(a) Without deconvolution, dot product similarity to a database spectrum decreased as the MS/MS isolation window increased (producing more contaminated spectra) in the negative mode IROA DDA dataset. After deconvolving with DecoID, no decrease in similarity occurred. The horizontal lines on the violin plots represent the mean, maximum, and minimum similarity values. (b-c) The FDR (b) and TPR (C) are plotted as a function of dot product threshold for DecoID and directly searching the acquired spectra (no deconvolution) from the IROA DDA dataset. At all thresholds, DecoID had nearly the same FDR as when no deconvolution was performed and a higher TPR. (d) Using DecoID and a dot product threshold of 80, there was no significant increase in FDR, but there was a significant increase in TPR. (e-f) When DecoID was used alone or when the combined approach was used, the FDR (e) was intrinsically lower and the TPR (f) was higher than when MS-DIAL was used alone on the IROA DIA dataset. (g) DecoID and the combined approach significantly increased the TPR relative to MS-DIAL when using a dot product threshold of 80. Results shown in b-g are from the top MS/MS match for each metabolite in the positive-mode and negative-mode data. Results are aggregated from the in-house database, mzCloud, HMDB, and MoNA. Data shown in (d) and (g) represent mean FDR/TPR +/− 95% empirical confidence interval derived from bootstrap resampling (n=10,000) the IROA DDA and DIA dataset and calculating the FDR and TPR on each independently resampled dataset (see Methods). Statistical significance in (a) was assessed using the two-sided two-sample Kolmogorov–Smirnov test. Statistical significance in (d) and (g) was assessed through 2-sided comparison of the bootstrapped FDR and TPR distributions (Methods).
To validate the utility of DecoID to DIA spectra and to compare its performance to the widely used DIA deconvolution software MS-DIAL5, we collected SWATH27 DIA spectra (Methods) on the IROA mixture and deconvolved the acquired spectra with both DecoID and MS-DIAL. We compared the performance of the software in three scenarios: (1) DecoID used alone, (2) MS-DIAL used alone, (3) or both software used in parallel (Methods and Supplementary Figure 15) by computing the auROC in each case. To promote a fair comparison, the MS-DIAL software was optimized to give the highest auROC (Methods and Supplementary Figure 16). When using MoNA and the in-house database, a higher auROC was achieved with DecoID compared to MS-DIAL. With HMDB and mzCloud, in contrast, the reverse occurred. When using the combined approach, the highest auROC was achieved in all databases except HMDB, thereby demonstrating the complementary performance of both software (Supplementary Figure 17). MS-DIAL is unable to deconvolve chimeric spectra when the precursors have highly similar chromatographic peak shapes, even though the precursors may have different m/z values. This limitation allows DecoID to be more successful in some cases because DecoID does not use chromatographic information to deconvolve MS/MS spectra. We expect the difference in performance to be particularly pronounced in shorter LC methods where peaks are not well separated. Supplementary Figure 18 shows an example of a spectrum that DecoID was able to successfully deconvolve that MS-DIAL could not. We also compared the FDR and TPR as functions of dot product threshold for both DecoID, MS-DIAL, and the combined approach when the top hit was considered for each feature. We found that using either DecoID alone or using the combined approach resulted in a lower FDR and a higher TPR at all thresholds compared to when MS-DIAL was used alone (Figure 3e–f). Importantly, when using a dot product cutoff of 80, DecoID was able to significantly improve the TPR while decreasing the FDR compared to MS-DIAL. When using the combined approach, an even more pronounced increase in the TPR was achieved (Figure 3g). This striking result prompted us to develop a semi-automated workflow to use the two algorithms in parallel (Supplementary Figure 15). In this workflow, DecoID can read the output of an MS-DIAL deconvolution and automatically combine the results from MS-DIAL and DecoID. An example script is available on the DecoID GitHub page (https://github.com/e-stan/DecoID/).
Validation in different sample matrices
In addition to the strong performance of DecoID on the IROA standard mixture datasets, we also sought to verify that DecoID can improve metabolite identification in various sample matrices. To accomplish this, we spiked metabolite standards into E. coli, human plasma, and P. pastoris metabolite extracts (Methods). We then compared how well DecoID identified the spiked-in metabolites compared to directly searching the DDA spectra against the databases without deconvolution. We also compared the success of identifying the spiked-in metabolites when using a DIA workflow with DecoID alone, MS-DIAL alone, or DecoID with MS-DIAL. The auROC was computed for DDA spectra (Supplementary Figure 11, Supplementary Figure 19, Supplementary Figure 20, and Supplementary Figure 21) and DIA spectra (Supplementary Figure 22, Supplementary Figure 23, and Supplementary Figure 24). In eleven of the twelve database/sample matrix pairs, DecoID increased the auROC on DDA spectra. In all database/sample matrix pairs, the combined usage of DecoID and MS-DIAL outperformed MS-DIAL used individually, and, in eight of the twelve cases, it outperformed DecoID used individually.
Application to NIST SRM 1950
To demonstrate that DecoID leads to more identifications compared to conventional MS/MS database searching of DDA spectra when analyzing a biological sample, DDA HILIC/MS/MS spectra were acquired for the NIST SRM 1950 plasma sample and processed with DecoID (Methods). Consistent with a recent LC/MS/MS analysis of human plasma28, when searched against HMDB, MoNA, mzCloud, and our in-house database without deconvolution, 164 metabolites were identified on the basis of accurate mass and matching of experimental MS/MS data to reference spectra in metabolite databases (Level 2a29, Methods)30. After deconvolution with DecoID, 215 features were identified. This represents a greater than 30% increase in identification rate compared to applying no deconvolution (Figure 4a). As an example, after deconvolution, creatinine was correctly identified (Figure 4b). The identification was confirmed with a pure standard and a retention time match. The breakdown of which features were able to be identified in which databases, along with the individual increases in identification rate for each database, is given in Figure 4c–g. Given the complementary nature of MS/MS databases, we suggest using multiple databases to give the best identification rate. DecoID facilitates this approach by allowing databases (e.g., HMDB, MoNA, and mzCloud) to be easily switched in the user interface (see Supplementary Figure 3). The complete table of identifications made with DecoID is available in Supplementary Table 2.
Figure 4. DecoID improves identification rates in NIST SRM 1950.
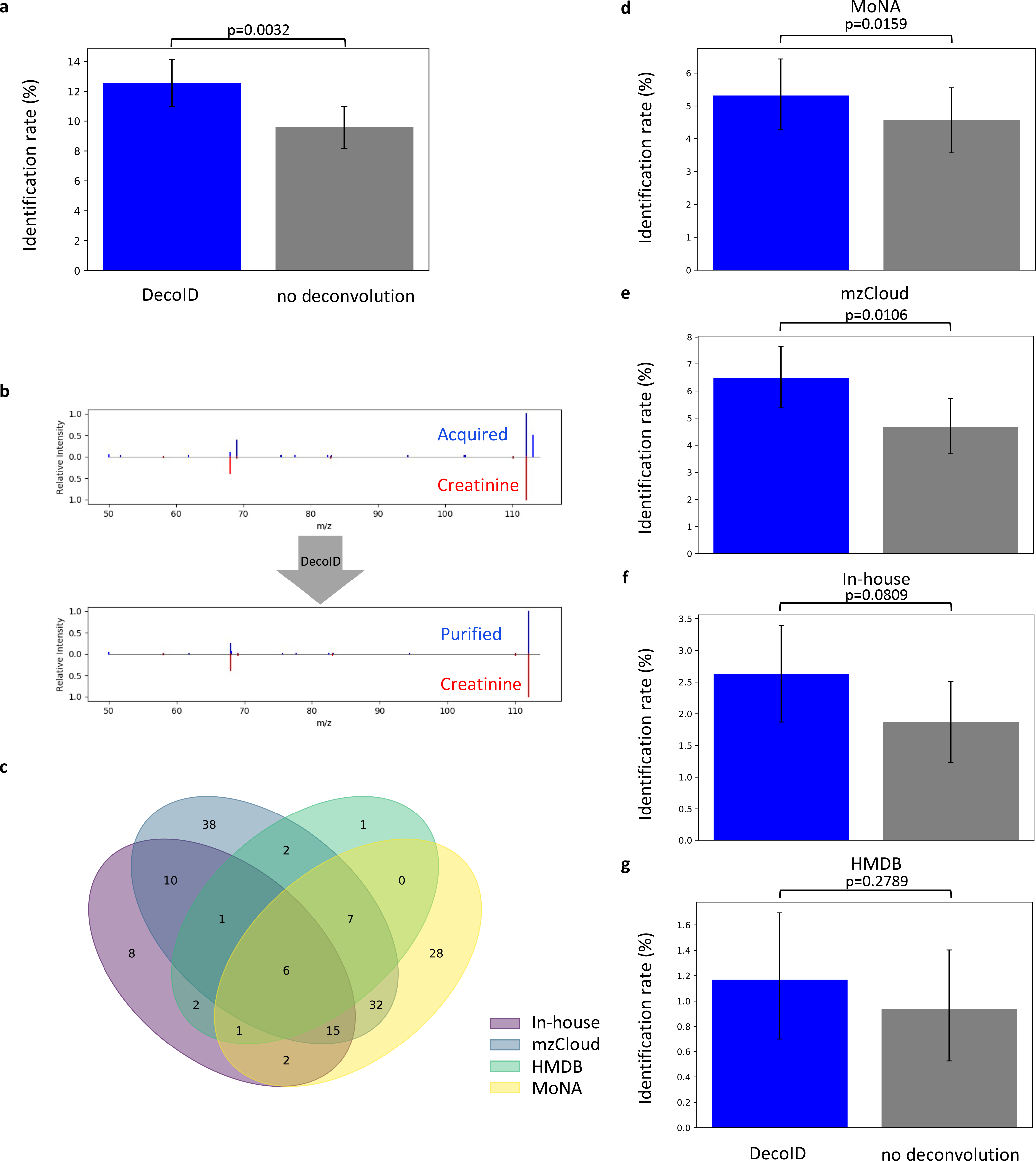
(a) Analysis of NIST SRM 1950 human plasma in a DDA experiment with a 1 m/z isolation window led to a greater than 30% increase in identification rate when using DecoID and aggregating results from HMDB, MoNA, mzCloud, and our in-house database. (b) Example identification from the NIST SRM 1950 plasma dataset would not have been possible without DecoID. The MS/MS similarity to the reference spectrum increased after deconvolution with DecoID compared to no deconvolution. Identification was confirmed with a retention-time match. (c) Venn diagram showing which features were able to be identified when the different databases were used. The breakdown shows that MS/MS databases can offer complementary identifications that boost the identification rate. (d-g) When using the four databases MoNA (d) mzCloud (e), our in-house database (f), and HMDB (g), DecoID increases the identification rate when compared to no deconvolution. Data shown in in (a) and (d-g) represent mean identification rate +/− 95% empirical confidence interval found from bootstrap resampling (n=10,000) the NIST SRM 1950 dataset and calculating the identification rate on each independently resampled dataset (Methods). Statistical significance in (a) and (d-g) was assessed through 1-sided comparison of the bootstrapped identification rate distributions (Methods).
Application of DecoID to a Human Plasma DIA Dataset
To verify that DecoID can also improve the number of identified metabolites in a biological DIA dataset, a human plasma DIA dataset was deconvolved by using DecoID alone, MS-DIAL alone, or the two software packages together. We identified 235 features (level 2a29) with MS-DIAL and 183 features with DecoID. When used in parallel, however, 339 features (40% more than just using MS-DIAL alone) were able to be identified, highlighting the complementary nature of the two deconvolution algorithms and the benefit of using both approaches in parallel (Figure 5a–b). As an example, cyclic AMP was able to be identified using DecoID (Figure 5c), but not by using MS-DIAL (Figure 5d). The breakdown of the three approaches for individual databases shown in Figure 5e–i. The complete list of identifications using the combined approach can be found in Supplementary Table 3.
Figure 5. DecoID increases the identification rate in a human plasma DIA dataset.
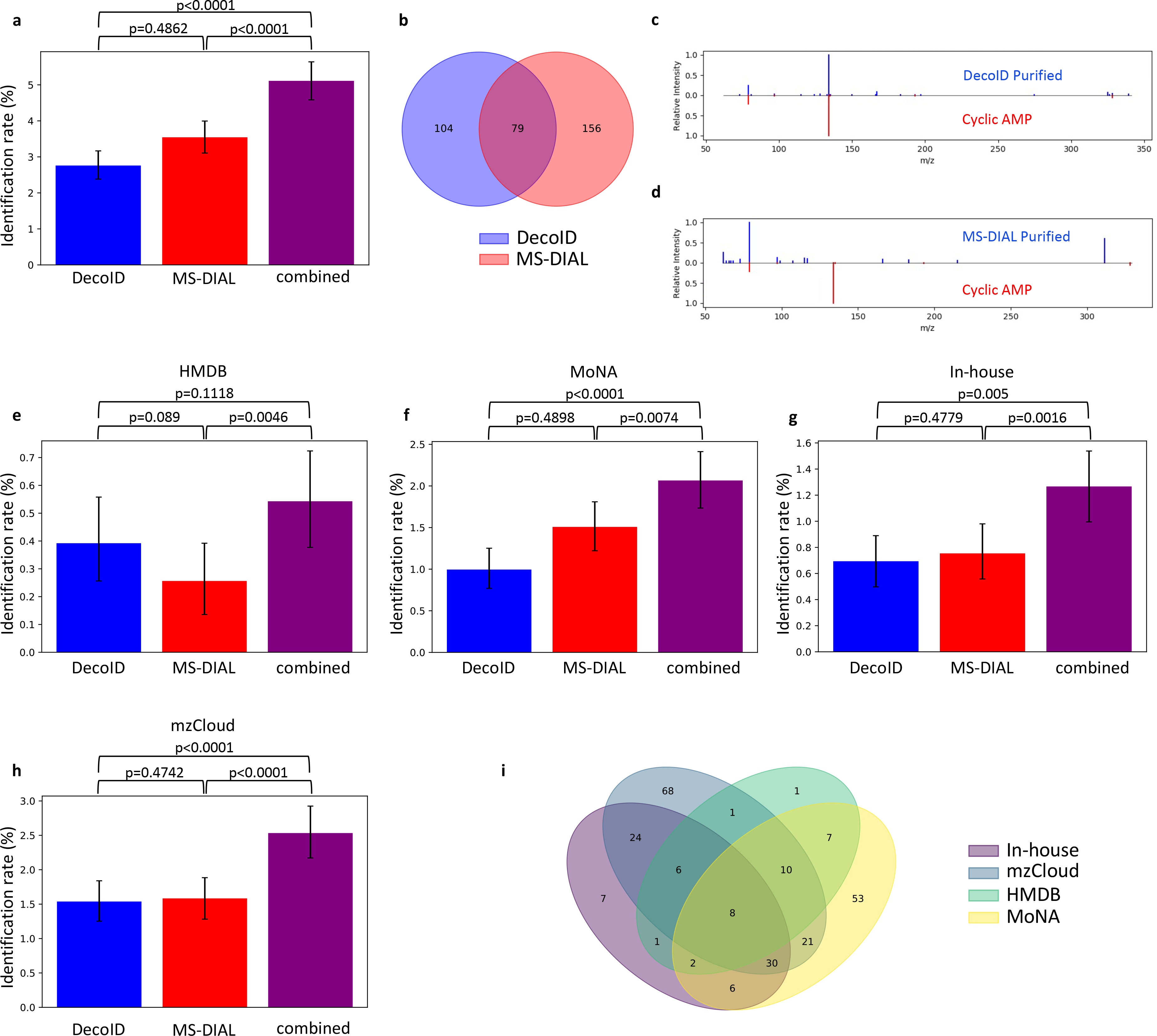
DecoID and MS-DIAL were applied to a plasma DIA dataset. All features detected by MS-DIAL were deconvolved with both DecoID and MS-DIAL. The deconvolved spectra were searched against HMDB, MoNA, the in-house database, and mzCloud. The results from MS-DIAL and DecoID were combined by taking the best hit for each feature amongst the results from MS-DIAL and DecoID (combined). (a) when combining the results for all databases, this parallel approach yielded greater identification rates compared to using either method alone. (b) Venn diagram showing the overlap in features that are identified using either DecoID or MS-DIAL. (c-d) Example identification from the negative mode DIA dataset that was found after deconvolution with DecoID (c) but that would not have been possible when using MS-DIAL (d). Identification was confirmed with a retention time match. (e-h) Identification rates of DecoID, MS-DIAL, and the combined approach when applied to the plasma DIA dataset with HMDB (e), MoNA (f), our in-house database (g), and mzCloud (h). (i) Venn diagram showing which features were able to be identified from which database after combining the results of MS-DIAL and DecoID. Data shown in (a) and (e-h) represent mean identification rate +/− 95% empirical confidence interval derived from bootstrap resampling (n=10,000) the plasma DIA dataset and calculating the identification rate on each independently resampled dataset (Methods). Statistical significance in (a) and (e-h) was assessed through 1-sided comparison of the bootstrapped identification rate distributions (Methods).
Improved Identification in MetaboLights Dataset
The acquisition-independent workflow of DecoID enables deconvolution of publicly available datasets to be processed with DecoID to improve metabolite identification and gain new insights from prior studies. We note that most of the metabolomic datasets in public repositories were not acquired with methods that are amenable to experimental deconvolution by existing strategies such as MS-DIAL, highlighting a benefit of DecoID. As an example, a reversed-phase liquid chromatography (RPLC)/MS/MS DDA study investigating the effect of a ketogenic diet on mouse xenograft tumor models31 found on the MetaboLights15 online repository was analyzed with DecoID (Methods). After deconvolution with DecoID, 71 additional features, including citrate, were able to be identified (Level 2a29) by searching MoNA, HMDB, mzCloud, and our in-house database, representing a greater than 20% improvement in identification rate compared to searching the acquired spectra directly (Figure 6a–b). By examining the breakdown of which features were able to be identified using which databases, we again see the complimentary nature of the reference databases (Figure 6c). The performance with the individual databases is given in Figure 6d–g. The complete list of identifications from all databases is available in Supplementary Table 4. A detailed summary of the important statistics, metrics, and relevant figures for all the datasets analyzed in this study is provided in Supplementary Table 5.
Figure 6. DecoID increases the identification rate of metabolites from a publicly available mouse xenograft RPLC/MS/MS dataset.
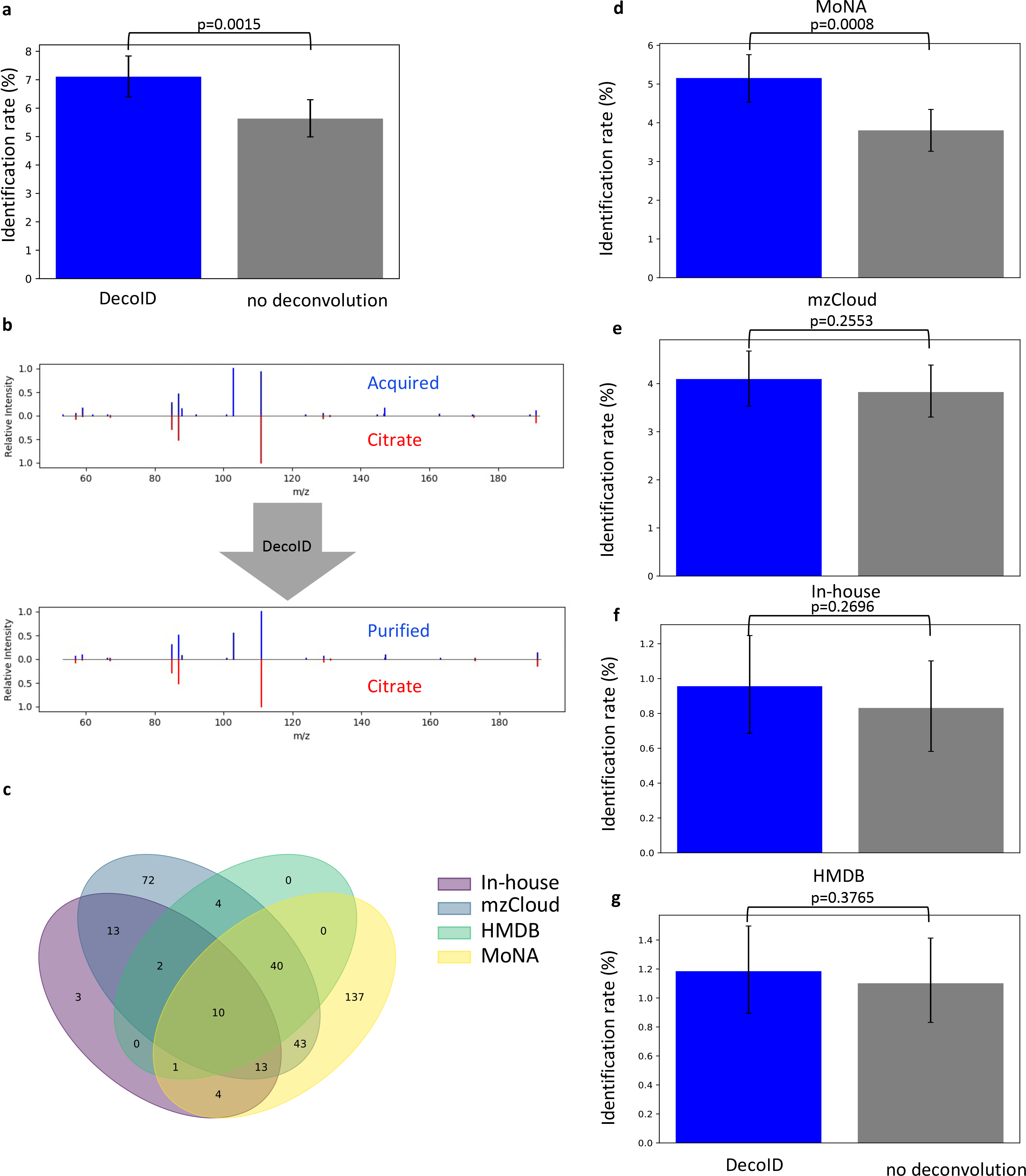
DecoID was applied to a published RPLC/MS/MS dataset31 uploaded to the MetaboLights repository. (a) DecoID increased the number of identifications made when aggregating the results from MoNA, mzCloud, the our in-house database, and HMDB. (b) Example deconvolution from the mzCloud database that led to the identification of citrate (bottom) that was not possible without deconvolution (top). (c) Using multiple databases provide complementary identifications. The Venn diagram shows which features were able to be identified from which databases after deconvolution. (d-g) Identification rates when using DecoID and no deconvolution to identify metabolites in the RPLC/MS/MS dataset with MoNA (d), mzCloud (e), in-house databse (f), and HMDB (g). With all databases, DecoID increased the identification rate compared to no deconvolution. Data shown in (a) and (d-g) represent the mean identification rate +/− 95% empirical confidence interval derived from bootstrap resampling (n=10,000) the RPLC/MS/MS dataset and calculating the identification rate on each independently resampled dataset (Methods). Statistical significance in (a) and (d-g) was assessed through 1-sided comparison of the bootstrapped identification rate distributions (Methods).
Discussion
DecoID is an acquisition-independent method to deconvolve metabolomic MS/MS spectra. We have shown that DecoID successfully and faithfully deconvolves DDA and DIA spectra from various sample matrices and increases the number of identified features in both workflows. The complementary nature of DecoID to the commonly used deconvolution algorithm MS-DIAL enables large improvements in metabolite identification in DIA studies when both algorithms are employed in parallel. Further, DecoID is backwards compatible with all MS/MS data that have been deposited in public repositories such as MetaboLights and the Metabolomics Workbench, whereas other existing deconvolution software tools such as MS-DIAL are not. DecoID is open source and freely available on the Patti Lab website (http://pattilab.wustl.edu/software/DecoID).
Methods
Standards, chemicals and samples
Acetonitrile, methanol, and water (all LC/MS grade) were purchased from Fisher Scientific or Millipore Sigma. Ammonium bicarbonate, ammonium hydroxide, and methylenediphosphonic (medronic) acid were ordered as eluent additives for LC/MS from Millipore Sigma. The Mass Spectrometry Metabolite Library (IROA Technologies) was purchased from Sigma-Aldrich (St. Louis, MO). The metabolite standards of plates 1–6 were reconstituted according to the protocol from IROA. An aliquot of each well was taken to prepare a pool. After drying in a vacuum concentrator, the standard mixture was reconstituted in 50% acetonitrile, 50% water to yield a final concentration of approximately 5–10 μM (dependent on the molecular weight). The list of IROA metabolites detected in both polarities, along with their retention times, is available in Supplementary Table 6. The NIST SRM 1950 (frozen human plasma) was ordered from the National Institute of Standards and Technology. It was extracted with 80% ethanol (1:10 dilution), kept at −20 °C for 1 h, and centrifuged (14,000 g, 10 min, 4 °C). The supernatant was directly used for LC/MS analysis.
An additional standard mix containing 81 metabolites was prepared for the spike-in experiments (Supplementary Table 7). Dried unlabeled metabolite yeast extract from P. pastoris was purchased from Cambridge Isotope Laboratories. It was reconstituted in 1 mL water and diluted at a ratio of 1:20 in 50% acetonitrile. Dried unlabeled E. coli extract was obtained as part of the credentialed E. coli kit from CIL. It was reconstituted in 100 μL of 50% acetonitrile. Pooled human plasma was purchased from Innovative Research, Inc (Novi, MI, USA), extracted with 80% methanol, and incubated at −20 °C for one hour. All three extracts were then centrifuged (14,000 g, 10 min, 4 °C) and spiked with the metabolite standard mix to yield a final concentration of 10 μM (1:10 dilution) before to analysis. Only the portion of the spiked-in standards not designated for parameter optimization were used to evaluate performance in the spike-in datasets.
Liquid chromatography/mass spectrometry
Liquid chromatography was performed with a SeQuant® ZIC®-pHILIC column (100 × 2.1 mm, 5 μm, polymer, including a guard column 20×2.1 mm, 5 μm, polymer, Merck-Millipore). Mobile phase A was 95% water, 5% acetonitrile with 20 mM ammonium bicarbonate, 0.1% ammonium hydroxide (25% ammonia in water) and 2.5 μM medronic acid. Mobile phase B was 95% acetonitrile, 5% water (vol/vol) with 2.5 μM medronic acid. Medronic acid and/or phosphate alone has been shown to improve peak shapes32. A Vanquish Horizon UHPLC system (Thermo Fisher Scientific) was used at a flow rate of 0.250 mL min−1 and 40 °C. The following linear gradient was applied: 0–1 min 90% B, 1–14 min decrease to 25% B, 14–14.5 min 25% B, and 90% B for re-equilibration until 22 min. The flow rate was increased to 0.400 mL min−1 from 15.5–20 min. The samples were kept at 6 °C in the autosampler, and the injection volume was 2 μL. The LC system was coupled to an Orbitrap ID-X Tribrid mass spectrometer (Thermo Fisher Scientific) via electrospray ionization in positive and negative mode with a spray voltage of 3.5 and 2.8 kV, respectively. The RF lens value was 60%. Data were acquired in data-dependent acquisition (DDA) mode and data-independent acquisition (DIA) mode with a mass range of 67–900 m/z. For DDA, an inclusion list with the m/z values ([M+H]+ for positive mode, [M-H]- for negative mode) for the standard mixture compounds was used. MS1 scans were acquired at a resolution of 120K with an automatic gain control (AGC) target of 2e5 and a maximum injection time of 200 ms. Different isolation windows of 1, 3, and 5 m/z were used. A normalized collision energy of 40% was used. Data were acquired with a resolution of 15,000, an AGC target of 2.5e4, and a maximum injection time of 50 ms. For DIA, full scans with 60K resolution, an AGC target of 2e5, and a maximum injection time of 100 ms were acquired. The isolation window for fragmentation was 20 m/z, the normalized collision energy 40%, the resolution was 15,000, the AGC target 4e5, and the maximum injection time was 22 ms. All isolation windows used for the datasets analyzed can be found in Supplementary Table 5.
MS/MS Database Preparation
Three publicly available MS/MS databases were tested with DecoID: Mass Bank of North America (MoNA), Human Metabolome Database (HMDB), and mzCloud. MoNA experimental spectra were downloaded from the MoNA web interface (https://mona.fiehnlab.ucdavis.edu/downloads) as NIST libraries (.MSP format). MSP formatted files are directly compatible with DecoID. DecoID processes the MSP file in a spectrum-by-spectrum fashion, keeping those that have entries for the “ExactMass” or “PrecursorMZ” field as well as “Ion_mode” and “DB”. If the InChIKey33 is provided in the MSP file, that is used as the compound identifier. If not, the compound name is used as the compound identifier. After this filtration, ~125,000 experimental spectra were loaded in DecoID for use with deconvolution.
HMDB does not have a direct MSP download format available on the HMDB website. To allow for DecoID compatibility, the downloaded XML (Extensible Markup Language) file for all experimental MS/MS spectra (https://hmdb.ca/downloads) was parsed and converted to MSP. The Jupyter notebook “DecoID/housekeeping/HMDB_xml_to_MSP.ipynb” handles this conversion. The same requirements as with MoNA were applied (non-empty “database-id”, “ionization-mode”, “spectra-type”, and “instrument-type”). Metabolite information (name, InChIKey, mass, and formula) were extracted from the All Metabolites XML file available for download at HMDB. After processing, ~3,000 experimental spectra were available for deconvolution. After processing the MSP files downloaded for MoNA and built for HMDB, DecoID predicts all carbon-13 M+1 isotopologue spectra and adds them to the database before writing binary versions of these databases for faster loading on future usages. Additionally, for MSP formatted database files, if the “retention time:” field exists for a spectrum, it will be recorded for optional retention time constrained deconvolutions. We note that, in general, retention times in reference databases are not applicable unless the exact same analytical conditions are used. However, researchers may have predicted retention times or have in-house databases with retention times that can be used to improve metabolite identification accuracy and confidence. This can be especially helpful in distinguishing between isomeric compounds. In the case of the IROA compounds, about 60% of the compounds with an isomer in the IROA library were separated by more than 30 seconds in retention time.
Interfacing with mzCloud was completed with usage of the proprietary mzCloud application programming interface. Support for the application programming interface is included with DecoID, however, usage requires an access key granted by Thermo Fisher Scientific. Recalibrated MS/MS spectra from the reference database were used for deconvolution.
Generating Reference Data for Our In-House Database
In addition to the publicly available MS/MS databases, we wished to obtain MS/MS data and retention times for each metabolite in our IROA standard mixture when using the same chromatography as applied above to the research samples. To establish ground truth, we created non-isobaric mixtures containing approximately twenty metabolite standards each. The non-isobaric mixtures were then individually evaluated by HILIC/MS to determine reference retention times. Retention time bounds were calculated and manually inspected by using Skyline (v20.0.1). Reference MS/MS spectra for the IROA metabolites were obtained by using flow-injection analysis to evaluate each individual standard in a separate experiment. The reference retention times and the MS/MS spectra from flow injection analysis were then combined for all metabolites into a single MSP file that can be read by DecoID.
DecoID Algorithm
Input:
The first step of the DecoID workflow is MS/MS data import. DecoID accepts vendor formatted files that are compatible with MS-Convert21. DecoID uses MS-Convert21 to automatically perform vendor data centroiding and conversion of raw data (both MS1 and MS/MS) into mzML. It is also possible to directly provide a .mzML file to DecoID that has already been centroided. The performance of DecoID on profile data has not been evaluated. The user can supply an optional intensity threshold where all detected MS/MS fragments below this absolute intensity will be removed from downstream analysis. This can prevent overfitting to low intensity fragments that are likely noise. The user can also import a peak table that provides the m/z values and retention time bounds for unknown features of interest. These peak definitions can be found through many means such as XCMS34 or any other peak detection platform such as Compound Discoverer (Thermo-fisher Scientific) or MassHunter Profinder (Agilent Technologies). If peak information is provided, only those MS/MS spectra corresponding to one of the input peaks is deconvolved. Peak information is required for DIA MS/MS data. For DDA, if no peak information is provided, each MS/MS spectrum is treated as a unique feature for identification.
Candidate spectra selection
To deconvolve an MS/MS spectrum, DecoID assumes a linear model of the fragmentation process where an observed MS/MS spectrum (a vector), y, can be thought of as the product of a matrix of MS/MS spectra, X, of individual precursors multiplied by a vector of precursor abundances, β (Equation 1). An MS/MS spectrum can easily be converted into a real valued vector by binning MS/MS fragments based on their m/z values. DecoID uses a bin size of 0.01 m/z and a maximum m/z of 5,000 to bin the spectra, serving as a balance between resolution and computational cost. The matrix, X, is formed by concatenating the column vectors of database MS/MS spectra into a matrix.
| (1) |
Before X can be formed, the spectra to consider for deconvolution must be selected. DecoID makes this selection based on the isolation window size used. If the M +/− H adduct of a database compound with an MS/MS spectrum in the relevant polarity has an m/z value that falls within the isolation window, it is considered for use in the deconvolution. The distribution of the number of candidate compounds considered to deconvolve DIA and DDA spectra acquired from a human plasma sample can be seen in Supplementary Figure 25. In larger spectral databases, there may be several spectra for a single compound. In this case, DecoID compares the similarity of the query MS/MS spectrum to each spectrum for the compound in the database and selects the spectrum with the greatest similarity. For all spectral similarity assessments, DecoID uses the normalized dot product. Several other metrics exist for the scoring of similarity35, but for its ease of interpretation and robust performance in many applications, we chose the normalized dot product (Equation 2). DecoID can be reconfigured to use any other simple scoring metric. The deconvolution operates independently from the similarity metric, which is only used for selecting spectra before the deconvolution and scoring the MS/MS hits after deconvolution.
| (2) |
If desired, the MS/MS database can be expanded by predicting the MS/MS spectrum for the carbon-13 M+1 isotopologue for each compound in the database. This enables removal of contamination arising from orphan isotopologues (Orphan isotopologue spectrum prediction). Further, pure spectra collected during acquisition can be used to deconvolve chimeric spectra contained in the same dataset (On-the-fly unknown library). To prevent spurious deconvolution, candidate spectra were only used for deconvolution if, in the nearest MS1 scan, there was a peak of less than a ppm tolerance, Δppm, away from the database compound’s m/z. Δppm values should be set based on the mass accuracy of the instrument. For carbon-13 M+1 isotopologues, there must be a peak for the M+0 and the M+1 ions. If no full scan data are contained in the raw data file, this filtration step is skipped. Full scan data are required to use the M+1 isotopologue prediction and the on-the-fly unknown library. If retention times are available for database spectra and retention time filtration is selected, candidate spectra are further filtered based on the database retention times and the retention time of the MS/MS spectrum.
Deconvolution
Once the matrix of database spectra, X, is assembled, a non-negative LASSO24 regression problem is formed (Equation 3) that enables determination of the contribution of each precursor spectra of X to the observed spectrum, y. For DDA MS/MS data, λ = 5.0 is used. For DIA MS/MS data, λ = 50.0 is used. λ is a hyperparameter for LASSO regression that regularizes the system to favor a sparse solution36. See Parameter optimization for details on how λ is set.
| (3) |
The non-negative LASSO problem is solved using coordinate descent with the Scikit-learn37 Python package.
Feature Identification
To average the output of the deconvolution for all acquired spectra of a particular feature, the spectra, yi, and abundance vector, βi, for all acquired spectra within the retention time bounds of the feature are collected. The averaged reconstructed spectrum is computed by taking the vector sum of each reconstructed spectrum. The averaged observed spectrum is computed by taking the vector sum of all acquired spectra for a feature.
The residual noise, ϵ, that remains after the deconvolution is given by subtracting the averaged reconstructed spectrum from the averaged observed spectrum and dividing by the number of non-zero abundance elements, n, in the summed abundance vector, ∑βi (Equation 4).
| (4) |
Each purified component, wj, used to reconstruct the acquired spectrum is defined by adding the residual noise to the library spectrum (column j of the matrix X) multiplied by its coefficient (element j of the vector β) as given in Equation 5. Because it is possible that, after this summation, an element of wj may be negative, all negative elements are set to zero before scoring similarity.
| (5) |
Each component found for a feature that had a precursor m/z value that is more than Δppm away from the feature of interest’s m/z value is discarded. Additionally, the database spectra that constituted X are also discarded if their precursor’s m/z value is more than Δppm away from the feature of interest’s m/z, value regardless of their coefficient in the LASSO regression. The remaining database spectra and pure components are then subjected to a redundancy check (details are given below). The score for each database hit is taken to be the maximum similarity it has amongst the remaining components, the original averaged spectrum, and the residual noise. An identification is made based on the accurate mass and MS/MS similarity amounting to a Level 2a29 identification. The coefficients themselves cannot directly be used for feature identification due to the non-uniqueness of MS/MS spectra to a particular metabolite. For example, isomeric compounds often have highly similar MS/MS spectra (Supplementary Figure 1). As such, these coefficients should not be used as a measure of the relative abundance of the precursors in the MS/MS spectrum.
Output
After deconvolution and feature identification, three files are generated to summarize the results. The first of these files gives all hits for each feature in the dataset. This includes the mass error and dot product similarity along with the compound name, formula, database ID, and the spectrum ID for the exact spectral match. The component of the deconvolution where the match occurred is also listed by the compound ID of the spectrum used in the deconvolution. The information on each component that contributed to the deconvolution, along with the purified spectrum for that component, can be found in the second output file. Lastly, a binary DecoID is generated that contains details on each hit that enables the visualization of features through the user interface (Supplementary Figure 3). The user interface was implemented with the Python tkinter package. In addition to showing the metabolite hits, the reconstructed MS/MS spectrum is also shown using the solution to Equation 3. Further, the MS1 spectrum is also reconstructed by taking the precursor m/z values of each component found in the deconvolution and summing the regression coefficients at each m/z value to give an MS1 spectrum.
Orphan Isotopologue Spectrum Prediction
To predict the carbon-13 M+1 isotopologue MS/MS spectrum, the formula of the precursor ion and the database M+0 spectrum are used. The first step of the prediction is to enumerate all possible subformula of the precursor ion. For example, for the precursor molecule C8H8NO4, there are (8+1)x(8+1)x(1+1)x(4+1) = 810 possible subformulas, given no constraints on the chemical composition of the subformulas. Next, the theoretical m/z value of each subformula is computed and candidate formulas are assigned to the M+0 database spectrum’s fragments based on the computed m/z value of the subformula and the user-defined mass tolerance, which should be set based on the mass accuracy within the database spectra. For the M+1 spectra predicted in this study, a mass tolerance of 15 ppm was used. We note that, in some cases, multiple subformulas are possible for a single fragment. However, this will only affect the intensities of the fragment ions in the predicted spectra, not their m/z values. Moreover, only the number of carbons in the fragment is important for the prediction, and we found fragments with high variance in the number of possible carbon atoms to be infrequent. After fragments are assigned subformulas, the mean number of carbon atoms in each fragment is computed. If no subformula matches an observed fragment’s m/z value, then that fragment is removed from further consideration as it is most likely a noise peak or a fragment ion from a contaminating precursor. After the mean number of carbons is computed for each fragment, the intensities of the M+0 spectrum’s fragments are distributed according to the number of carbons in the precursor and the number of carbons in the fragment. For example, for an eight-carbon precursor and a seven-carbon fragment, the intensity of the fragment will be distributed with 7/8 of the original fragment’s intensity going to a new fragment shifted +1.003 m/z and the original fragment being 1/8 as intense as it was in the M+0 spectrum. This process is depicted in Figure 2.
On-the-Fly Unknown Library
Over the course of a DDA MS/MS experiment, it is possible that two isobaric features will partially co-elute. If, during this co-elution, an MS/MS spectrum is acquired, a chimeric spectrum will be produced. However, it is possible that a non-chimeric spectrum of one of the precursors might be acquired earlier or later. If this is the case, even if this compound is not in the MS/MS database, it can still be used to deconvolve this second chimeric spectrum (Supplementary Figure 6). DecoID implements this framework by first identifying non-contaminated MS/MS spectra in the input data and running them through the DecoID deconvolution and identification workflow. Spectra are classified as non-contaminated if their contamination is less than 10% and the total ion current is greater than 1e4 (Quantifying MS/MS contamination). These parameters can be modified by the user to better fit specific instrumentation. If no identification is returned with a similarity greater than 80, the acquired MS/MS spectrum is used to deconvolve other spectra that were acquired within the retention time bounds of the feature. This workflow is only applicable to DDA MS/MS data.
Redundancy Check
MS/MS spectra are not unique to a single metabolite. In the case of isomeric compounds, approximately 25% of spectra for compounds with an isomer have a similarity greater than 90 to at least one isomeric spectrum. In the case of isobaric (but not isomeric) compounds, it is much less (Supplementary Figure 1). However, approximately 40% of all compounds in HMDB have an isomeric compound in the database. This necessitates that the uniqueness of the DecoID deconvolution be assessed by a redundancy check, which searches each purified component against all database spectra (filtered by the exact mass of the component). If another database spectrum has a dot product similarity between the database spectrum and the component that is greater than 90% (user-defined threshold) of the dot product similarity between the database spectrum for the component and the component itself, then the component fails the redundancy check and any match to that component will be reported as redundant. We note that a failed redundancy check does not affect the quality of the deconvolution, merely its uniqueness. This check is meant to flag cases where an inconclusive identification is likely.
Parameter Optimization
The primary parameter for the DecoID deconvolution is the LASSO penalty term. To fit this parameter, we tested five different values on two separate metabolite standard mixture datasets consisting of 81 metabolite standards (one DIA and one DDA) acquired in both positive and negative mode. These 81 metabolites were the same as what were spiked into the biological matrices. About 50% of the metabolites that are detected in each polarity were used for parameter optimization, and the other 50% were used for performance evaluation in the spike-in datasets. Supplementary Table 7 annotates which compounds were used for optimization and evaluation. Receiver operating characteristic (ROC) curves were constructed and the area under the ROC (auROC) curve was computed for each parameter value (Supplementary Figure 9) using the top three hits for each of the optimization metabolites in the mixture. After combining the results across the four tested databases, a LASSO parameter of 5.0 was found to be optimal for both DIA and DDA. For the DIA dataset, this parameter was too weak to prevent unfaithful deconvolutions in the null evaluations, so a LASSO parameter of 50.0 was used for DIA data. To optimize MS-DIAL, the “sigma” deconvolution parameter was tuned by trying ten values within the MS-DIAL recommended range (0.1–1.0) on the same DIA dataset used to optimize the DecoID LASSO parameter. The auROC was computed at each parameter value and a parameter value of 0.9 was selected as optimal (Supplementary Figure 16).
Simulated Spectra for Null Evaluations
The simulated DDA (1 m/z isolation window) and DIA (20 m/z isolation window) spectra were generated by randomly selecting 500 positive mode spectra. Then, for each spectrum, the number of noise fragments to add was selected by sampling from a uniform distribution ranging between ten and one hundred. The m/z values of the fragments were determined by sampling from the empirical distribution of fragment masses within MoNA. The noise peaks were scaled to represent 0%, 25%, 50%, 75%, or 100% of the signal in the simulated spectra.
Quantifying MS/MS Contamination
MS/MS contamination is quantified in a method similar to MSPurity19. The nearest MS1 scan to the MS/MS spectrum of interest was selected, and the fraction of the signal coming from co-isolated analytes other than the targeted precursor was calculated (Supplementary Figure 2). The frequency with which orphan isotopologues contribute to MS/MS contamination was determined through analysis of the NIST SRM 1950 plasma sample. We predicted orphan isotopologue contamination of an MS/MS spectrum based on the assumption that each feature found on a de-isotoped peak list (for details on the peak list see below) will produce an M+1 ion. We then cross referenced our predicted M+1 peak list against the retention time and isolation window of each acquired MS/MS spectrum to estimate the frequency of contamination.
DecoID Performance Evaluation
To evaluate the performance of DecoID compared to directly searching the acquired DDA MS/MS spectra, the DecoID workflow was applied identically except without the LASSO regression (the returned abundance vector is the zero vector). This ensures all scoring and spectral processing remains exactly the same. Manually inspected peak boundaries were provided for the standard mixture metabolites that were detected in at least one polarity. These peak boundaries were given as input to DecoID and used to establish the ground-truth identifications for the standard mixtures. For each feature, the maximum scoring hit using a Δppm of 10 (used for all analyses) was considered. If the maximum hit had a spectral similarity of greater than 80 (as assessed with the dot product similarity), it was considered identified. The true positive rate (TPR) was calculated by taking the number of correct identifications divided by the sum of the true positive identifications and the false negative identifications. Correct identifications were assigned on the basis of InChIKey33 for HMDB and MoNA. Correct identifications were assigned by using compound ID for mzCloud. The false discovery rate (FDR) was calculated by taking the number of false positives and dividing by the sum of the false positives and true positives. Confidence intervals for the FDR and TPR were calculated via a bootstrapping procedure in which the complete dataset was resampled with replacement 10,000 times and the FDR and TPR were calculated at each iteration. Empirical p-values for the probability of identical FDRs and TPRs were calculated based on the frequency of resampled datasets that resulted in higher or lower values between two methods (DecoID, no deconvolution, MS-DIAL, and combined). The 2.5 and 97.5 percentiles of the FDR and TPR were used as the empirical 95% confidence interval. Features for which MS/MS data were collected, but for which no hits were returned, were not used in the calculation of the FDR and TPR. Receiver operating characteristic (ROC) curves were drawn by considering the top three hits for each feature to create balanced positive and negative classes. The maximum ID rates (the fraction of compounds in the dataset with reference spectra in a particular database) for all ground-truth datasets are available in Supplementary Table 5. ROC thresholds and the resulting false positive rates and true positive rates were computed with the Python package Scikit-learn37. The area under the ROC curve (auROC) was computed using numerical integration. All comparisons between DecoID and directly searching the acquired spectra for DDA only used features that DecoID separated into more than one component (the remaining features will have the exact same results as direct database searching). Comparisons to MS-DIAL were only made on features detected by MS-DIAL.
MS-DIAL Usage
Comparison to MS-DIAL5 was performed by first converting the DIA .raw files to .mzML files with MS-Convert21 using vendor peak picking to centroid the data. Files were then converted to .abf with Reifycs ABF converter (http://www.reifycs.com/AbfConverter/index.html) for compatibility with MS-DIAL. MS-DIAL version 4.12 was used to deconvolve the DIA spectra for both positive and negative mode. Mass accuracy was set to 0.005 Da for “MS1 tolerance” and 0.01 for “MS2 tolerance”. Peak detection used a minimum peak height of 1000 and a mass slice width of 0.1. The “sigma” parameter for the “MS2Dec” was set to 0.9 after optimization (Parameter optimization and Supplementary Figure 16). All other parameters were left at their default values. After deconvolution, the deconvoluted spectra were exported to a .txt file. The spectra in this file were fed into DecoID for identification, with deconvolution disabled by using the MS-DIAL linkage built into DecoID. We note that it would be possible to enable deconvolution with DecoID of the already deconvolved spectra exported from MS-DIAL. However, the effectiveness of this joint approach has not been evaluated.
Comparing Performance on Biological Datasets
The NIST SRM 1950 plasma sample was searched against mzCloud without deconvolution as described above. The peak lists for the plasma sample were extracted from the AcquireX38 inclusion lists (Thermo Fisher Scientific). Inclusion lists were formed on the basis of peak detection, de-isotoping, annotation of M +/− H ions, and annotation of background signals (peaks that are not at least three times more intense than in the extraction blank). Peaks that were unable to be identified with the thresholds outlined above, where MS/MS data were acquired, had significantly more contamination than those that were able to be identified based on a Kolmogorov–Smirnov two-sample, two-sided test.
The publicly available RPLC/MS/MS dataset31 of a mouse xenograft was downloaded from the MetaboLights15 repository as .mzML files. Files were centroided with MS-Convert and peak picking was performed by using the centWave39 algorithm within XCMS34. Peak correspondence was also performance within XCMS. A feature was considered identified if an MS/MS match with a dot product similarity greater than 80 was found. Deconvolution and identification were carried out with the experimental MoNA database, HMDB, mzCloud, and the in-house database.
The DIA plasma dataset was the same dataset used for the spike-in evaluation. However, the peak list consisted of the features detected by MS-DIAL, not just the spiked-in metabolites. The peak boundaries used were also from MS-DIAL. The MS-DIAL deconvoluted spectra were searched against MoNA, HMDB, mzCloud, and the in-house database using the DecoID linkage for MS-DIAL. A dot-product similarity of greater than 80 was considered an identification for both the MS-DIAL and DecoID results.
The identification rate is the number of features that were able to be identified divided by the number of features detected. After deconvolution and identification with DecoID, the improvement in identification rate was assessed by using a bootstrapping procedure where the identification/no identification status of each feature was resampled 10,000 times and the identification rate was calculated at each iteration. The 2.5th and 97.5th percentiles of the identification rate were used as the empirical 95% confidence interval. Empirical p-values for the probability of identical identification rates were calculated based on the frequency of resampled datasets that resulted in lower identification rates between two methods (DecoID, no deconvolution, MS-DIAL, and combined).
Data availability
All MS/MS data used in the evaluation of DecoID has been uploaded to the MetaboLights repository as study MTBLS2207 and is also available on the DecoID GitHub release (https://github.com/e-stan/DecoID/releases/). The publicly available dataset analyzed is available on MetaboLights as study MTBLS1066 (all reversed-phase negative mode datafiles were used). The MS/MS databases applied can be obtained at the curators’ websites (https://mona.fiehnlab.ucdavis.edu, https://www.mzcloud.org, and https://hmdb.ca). The in-house IROA metabolite database is available within the DecoID release on GitHub (https://github.com/e-stan/DecoID/releases/), and the reference spectra have been uploaded to MoNA (submitter: Ethan Stancliffe, origin file: IROA_DB_for_mona_filtered_exported_addedInfo.msp).
Code availability
Source code is available on Zenodo40 and GitHub (https://github.com/e-stan/DecoID). Included is an example dataset along with documentation for both the DecoID Python package and user interface. A standalone executable built for Windows can alternatively be downloaded from the Patti Lab website (http://pattilab.wustl.edu/software/DecoID).
Reporting Summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institutes of Health grants U01 CA235482 (G.J.P.), R35 ES028365 (G.J.P.), and R24 OD024624 (G.J.P.).
Footnotes
Competing Interests Statement
G.J.P. is a scientific advisory board member for Cambridge Isotope Laboratories and has a research collaboration agreement with Thermo Fisher Scientific.
References
- 1.Blaženović I, Kind T, Ji J & Fiehn O Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 8, 31 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baker ES & Patti GJ Perspectives on Data Analysis in Metabolomics: Points of Agreement and Disagreement from the 2018 ASMS Fall Workshop. J. Am. Soc. Mass Spectrom (2019) doi: 10.1007/s13361-019-02295-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nikolskiy I, Mahieu NG, Chen Y-J, Tautenhahn R & Patti GJ An Untargeted Metabolomic Workflow to Improve Structural Characterization of Metabolites. Anal. Chem 85, 7713–7719 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nash WJ & Dunn WB From mass to metabolite in human untargeted metabolomics: Recent advances in annotation of metabolites applying liquid chromatography-mass spectrometry data. TrAC Trends Anal. Chem 120, 115324 (2019). [Google Scholar]
- 5.Tsugawa H et al. MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat. Methods 12, 523–526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Samanipour S, Reid MJ, Bæk K & Thomas KV Combining a Deconvolution and a Universal Library Search Algorithm for the Nontarget Analysis of Data-Independent Acquisition Mode Liquid Chromatography−High-Resolution Mass Spectrometry Results. Environ. Sci. Technol 52, 4694–4701 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Li H, Cai Y, Guo Y, Chen F & Zhu Z-J MetDIA: Targeted Metabolite Extraction of Multiplexed MS/MS Spectra Generated by Data-Independent Acquisition. Anal. Chem 88, 8757–8764 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Yin Y, Wang R, Cai Y, Wang Z & Zhu Z-J DecoMetDIA: Deconvolution of Multiplexed MS/MS Spectra for Metabolite Identification in SWATH-MS-Based Untargeted Metabolomics. Anal. Chem 91, 11897–11904 (2019). [DOI] [PubMed] [Google Scholar]
- 9.Ting YS et al. PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat. Methods 14, 903–908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsou C-C et al. DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat. Methods 12, 258–264 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J et al. MSPLIT-DIA: sensitive peptide identification for data-independent acquisition. Nat. Methods 12, 1106–1108 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, Pirmoradian M, Chernobrovkin A & Zubarev RA DeMix Workflow for Efficient Identification of Cofragmented Peptides in High Resolution Data-dependent Tandem Mass Spectrometry. Mol. Cell. Proteomics 13, 3211–3223 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dorfer V, Maltsev S, Winkler S & Mechtler K CharmeRT: Boosting Peptide Identifications by Chimeric Spectra Identification and Retention Time Prediction. J. Proteome Res 17, 2581–2589 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houel S et al. Quantifying the Impact of Chimera MS/MS Spectra on Peptide Identification in Large-Scale Proteomics Studies. J. Proteome Res 9, 4152–4160 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haug K et al. MetaboLights: a resource evolving in response to the needs of its scientific community. Nucleic Acids Res 48, D440–D444 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sud M et al. Metabolomics Workbench: An international repository for metabolomics data and metadata, metabolite standards, protocols, tutorials and training, and analysis tools. Nucleic Acids Res. 44, D463–D470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kind T et al. Identification of small molecules using accurate mass MS/MS search. Mass Spectrom. Rev 37, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhu X, Chen Y & Subramanian R Comparison of Information-Dependent Acquisition, SWATH, and MSAll Techniques in Metabolite Identification Study Employing Ultrahigh-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry. Anal. Chem 86, 1202–1209 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Lawson TN et al. msPurity: Automated Evaluation of Precursor Ion Purity for Mass Spectrometry-Based Fragmentation in Metabolomics. Anal. Chem 89, 2432–2439 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Peckner R et al. Specter: linear deconvolution for targeted analysis of data-independent acquisition mass spectrometry proteomics. Nat. Methods 15, 371–378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kessner D, Chambers M, Burke R, Agus D & Mallick P ProteoWizard: open source software for rapid proteomics tools development. Bioinformatics 24, 2534–2536 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wishart DS et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horai H et al. MassBank: a public repository for sharing mass spectral data for life sciences. J. Mass Spectrom 45, 703–714 (2010). [DOI] [PubMed] [Google Scholar]
- 24.Tibshirani R Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B Methodol 58, 267–288 (1996). [Google Scholar]
- 25.Vinaixa M et al. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. TrAC Trends Anal. Chem 78, 23–35 (2016). [Google Scholar]
- 26.Cho K et al. isoMETLIN: A Database for Isotope-Based Metabolomics. Anal. Chem 86, 9358–9361 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonner R & Hopfgartner G SWATH data independent acquisition mass spectrometry for metabolomics. TrAC Trends Anal. Chem 115278 (2018) doi: 10.1016/j.trac.2018.10.014. [DOI] [Google Scholar]
- 28.Telu KH, Yan X, Wallace WE, Stein SE & Simón‐Manso Y Analysis of human plasma metabolites across different liquid chromatography/mass spectrometry platforms: Cross-platform transferable chemical signatures. Rapid Commun. Mass Spectrom 30, 581–593 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schymanski EL et al. Identifying Small Molecules via High Resolution Mass Spectrometry: Communicating Confidence. Environ. Sci. Technol 48, 2097–2098 (2014). [DOI] [PubMed] [Google Scholar]
- 30.Fiehn O et al. The metabolomics standards initiative (MSI). Metabolomics 3, 175–178 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Licha D et al. Untargeted Metabolomics Reveals Molecular Effects of Ketogenic Diet on Healthy and Tumor Xenograft Mouse Models. Int. J. Mol. Sci 20, 3873 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 32.Spalding JL, Naser FJ, Mahieu NG, Johnson SL & Patti GJ Trace Phosphate Improves ZIC-pHILIC Peak Shape, Sensitivity, and Coverage for Untargeted Metabolomics. J. Proteome Res 17, 3537–3546 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heller S, McNaught A, Stein S, Tchekhovskoi D & Pletnev I InChI - the worldwide chemical structure identifier standard. J. Cheminformatics 5, 7 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification | Analytical Chemistry. 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 35.Stein SE & Scott DR Optimization and testing of mass spectral library search algorithms for compound identification. J. Am. Soc. Mass Spectrom 5, 859–866 (1994). [DOI] [PubMed] [Google Scholar]
- 36.Chen Y & Wang M Hardness of approximation for sparse optimization with L0 norm. (2016).
- 37.Pedregosa F et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830 (2011). [Google Scholar]
- 38.Cho K et al. Targeting unique biological signals on the fly to improve MS/MS coverage and identification efficiency in metabolomics. Anal. Chim. Acta 1149, 338210 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tautenhahn R, Böttcher C & Neumann S Highly sensitive feature detection for high resolution LC/MS. BMC Bioinformatics 9, 504 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stancliffe Ethan. e-stan/DecoID: DecoID. (Zenodo, 2021). doi: 10.5281/zenodo.4783380. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All MS/MS data used in the evaluation of DecoID has been uploaded to the MetaboLights repository as study MTBLS2207 and is also available on the DecoID GitHub release (https://github.com/e-stan/DecoID/releases/). The publicly available dataset analyzed is available on MetaboLights as study MTBLS1066 (all reversed-phase negative mode datafiles were used). The MS/MS databases applied can be obtained at the curators’ websites (https://mona.fiehnlab.ucdavis.edu, https://www.mzcloud.org, and https://hmdb.ca). The in-house IROA metabolite database is available within the DecoID release on GitHub (https://github.com/e-stan/DecoID/releases/), and the reference spectra have been uploaded to MoNA (submitter: Ethan Stancliffe, origin file: IROA_DB_for_mona_filtered_exported_addedInfo.msp).