

ABSTRACT
Sclerostin is a negative regulator of the Wnt/β‐catenin signaling and is, therefore, an important inhibitor of bone formation and turnover. Because ectopic vascular calcification develops in a similar way to bone formation, one might reasonably attribute a role to sclerostin in this pathological process. Ectopic calcification, especially vascular calcification, importantly contributes to mortality in elderly and patients with diabetes, osteoporosis, chronic kidney disease (CKD), and hypertension. The central players in this ectopic calcification process are the vascular smooth muscle cells that undergo dedifferentiation and thereby acquire characteristics of bonelike cells. Therefore, we hypothesize that depletion/deactivation of the Wnt/β‐catenin signaling inhibitor sclerostin may promote the development of ectopic calcifications through stimulation of bone‐anabolic effects at the level of the arteries. We investigated the role of sclerostin (encoded by the Sost gene) during vascular calcification by using either Sost −/− mice or anti‐sclerostin antibody. Sost −/− and wild‐type (WT) mice (C57BL/6J background) were administered an adenine‐containing diet to promote the development of CKD‐induced vascular calcification. Calcifications developed more extensively in the cardiac vessels of adenine‐exposed Sost −/− mice, compared to adenine‐exposed WT mice. This could be concluded from the cardiac calcium content as well as from cardiac tissue sections on which calcifications were visualized histochemically. In a second experiment, DBA/2J mice were administered a warfarin‐containing diet to induce vascular calcifications in the absence of CKD. Here, warfarin exposure led to significantly increased aortic and renal tissue calcium content. Calcifications, which were present in the aortic medial layer and renal vessels, were significantly more pronounced when warfarin treatment was combined with anti‐sclerostin antibody treatment. This study demonstrates a protective effect of sclerostin during vascular calcification. © 2022 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: MATRIX MINERALIZATION, BONE HISTOMORPHOMETRY, SYSTEMS BIOLOGY – BONE INTERACTORS, Wnt/BETA‐CATENIN
Sclerostin, a well‐known inhibitor of physiological bone formation, protects against vascular calcification development in mice. A significantly increased calcium content was observed in: (i) cardiac vessels of Sost −/− mice versus WT mice, after induction of renal failure and (ii) aorta and renal vessels of anti‐sclerostin antibody‐treated versus vehicle‐treated mice.
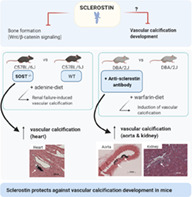
Introduction
Sclerostin is a protein that is encoded by the SOST gene, and functions as an important inhibitor of Wnt/β‐catenin signaling to regulate bone formation.( 1 , 2 ) In the bone, this 22‐kilodalton (kDa) protein binds to the low‐density lipoprotein receptor‐related protein 4/5/6 (LRP4/5/6) co‐receptors, thereby preventing the Wnt ligands from binding to their receptors. Genetic defects of the SOST gene can lead to the development of sclerosteosis and Van Buchem disease. Both diseases are characterized by, respectively, undetectable or very low serum sclerostin levels which result in massive bone overgrowth.( 3 ) To gain a better understanding of the effects of sclerostin deficiency, Sost −/− mice were generated. Similar to the phenotype observed in sclerosteosis and van Buchem disease patients, these mice had an increased bone mineral density, bone formation, bone volume, and bone strength.( 4 ) Given its function as an inhibitor of canonical Wnt signaling, and thus bone formation, antibodies were developed against sclerostin to serve as a therapeutic to stimulate bone formation, mainly in the context of osteoporosis.( 5 )
Because pathological (ectopic) calcification of the vessels or vascular calcification in many aspects resembles the physiological process of bone formation, it has been hypothesized that sclerostin also in this process may have an important role. Vascular calcification importantly contributes to mortality in elderly and patients with diabetes, osteoporosis, chronic kidney disease (CKD), and hypertension.( 6 ) In these patient groups, vascular calcification often is present as calcification in the medial arterial layer, which is also called arteriosclerosis or Mönckeberg's sclerosis. The presence of these calcifications in the vasculature leads to increased arterial stiffness and reduced vessel compliance, resulting in increased pulse pressure, cardiac hypertrophy, and heart failure.( 7 ) The pathological process of vascular calcification is actively regulated by vascular smooth muscle cells (VSMCs). During the vascular calcification process, as a result of pathological stimuli, VSMCs undergo dedifferentiation; ie, the cells lose their smooth muscle cell markers and obtain characteristics of other mesenchymal‐derived cells with bone‐forming capacities such as osteoblasts and chondrocytes.( 8 ) As a result, Wnt/β‐catenin signaling, a signaling pathway exerting anabolic effects on the bone, is likely to also affect the process of vascular media calcification.( 9 , 10 ) Despite its well‐known role in bone turnover, studies investigating the role of sclerostin in vascular calcification come to conflicting conclusions.( 11 , 12 ) Although some studies found a negative association( 12 , 13 , 14 , 15 , 16 ) between serum sclerostin levels and mortality, suggesting a protective role of sclerostin through inhibiting vascular calcification, others reported a positive( 17 ) or no association.( 18 , 19 , 20 , 21 ) In CKD patients, serum sclerostin levels are increased and seem to be higher in the presence of vascular calcification.( 22 , 23 , 24 ) As Ceijka and colleagues( 25 ) showed, the increase in serum sclerostin levels not to be due to renal retention, but rather results from an increased production of sclerostin CKD. Recently, we showed that sclerostin production by the osteocytes remained stable during the development of vascular calcification while it was distinctly induced in calcified arteries.( 26 ) This supports the hypothesis that sclerostin, which is locally produced in the calcified vessels, may spill over into the circulation, thereby contributing to increased serum sclerostin levels.
To determine the exact function of sclerostin in vascular calcification in general, we investigated calcification development in mice with genetic depletion of sclerostin (Sost −/− mice) and in mice treated with an anti‐sclerostin antibody, using vascular calcification models induced by either renal failure (adenine model) or inhibition of matrix Gla protein (warfarin model). This research significantly contributes to the existing knowledge regarding the role of sclerostin in ectopic vascular calcification.
Subjects and Methods
Statement of ethics
All experimental procedures were conducted in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals 85‐23 (1996) and approved by the University of Antwerp Ethical Committee for Animal Experiments (ECD‐code 2014‐97, 2016‐55).
Animals
In the first experiment, 33 Sost −/− mice (B6‐SOSTtm1Dgen mice( 27 , 28 )) and 37 wild‐type (WT) controls (Charles River, Saint‐Germain‐Nuelles, France) on a C57BL/6J background, were included. In the second experiment, 64 WT DBA/2J (Charles River) mice were included. The origin of animals is summarized in supplemental table 1. All animals were 14 weeks old and housed at a maximum capacity of eight animals per cage. Cages were kept at constant temperature and humidity and were exposed to a 12‐hour light/dark cycle. The mice had free access to tap water and their assigned diet. Male mice were used because compared to female animals they are known to be more vulnerable to the development of CKD and vascular calcification.( 29 , 30 )
Vascular calcification models
Two mice models of vascular calcification were used to investigate the role of sclerostin in this process. The first model is a model of adenine‐induced CKD, in the C57BL/6J mice. Although in this model no calcifications are detected in large arteries (aorta), which is in line with the fact that C57BL/6J mice are highly resistant against vascular calcification, calcifications of smaller arteries in the heart are clearly observed. DBA/2J mice, on the other hand, are less resistant to vascular calcification development.( 31 ) Therefore, these mice were used in a second study in which large artery (aortic) calcifications were induced through dietary exposure to warfarin.
Because Sost −/− mice are on a C57BL/6J genetic background, they were given adenine to investigate the effect of sclerostin absence on vascular calcification in cardiac vessels. To be able to investigate a possible role for sclerostin in large artery (aortic) calcification, warfarin‐administered DBA/2J mice were treated with anti‐sclerostin‐antibodies.
Next to the heart and large elastic arteries, medium‐sized and smaller vessels in the renal tissue are also known to be prone to the development of ectopic calcifications.( 32 ) So calcification of renal tissue was also investigated in both models. This, together with the use of two models; ie, a CKD model (adenine) as well as a model with normal renal function (warfarin), allowed us to investigate the role of sclerostin on general vascular calcification and interpret our data in a broader perspective.
Study design experiment 1: Adenine‐administration to Sost −/− and WT mice to investigate the effect of sclerostin absence on vascular calcification development in cardiac vessels
This model was developed based on our experience with dietary adenine‐administration to rats( 33 ) and on an adenine‐induced renal failure mice model as described by Jia and colleagues.( 34 ) Under normal circumstances, adenine levels in the blood and urine are kept low through efficient metabolization of adenine by the adenine phosphoribosyltransferase enzyme.( 35 ) However, when adenine is administered in large amounts, the activity of this enzyme is saturated and adenine is oxidized by xanthine dehydrogenase to 2.8 dihydroxyadenine.( 36 ) Due to the very low solubility of this metabolite, 2.8 dihydroxyadenine crystals precipitate in the nephrons where they cause severe kidney damage.( 37 ) The diet administered to the mice (21 Sost −/− mice and 21 WT mice) was changed every 10 and 14 days. During the 10‐day period, the diet contained 0.30% adenine and 1.2% phosphate; during the 14‐day period the adenine concentration was halved (0.15%) while the phosphate concentration remained the same (SSNIFF Spezialdiäten GmbH, Soest, Germany). In total, the mice were subjected to three periods of a 0.30% adenine concentration and two periods of 0.15% adenine concentration. The two control groups, consisting of 12 Sost −/− mice and 16 WT mice, received a control diet and were euthanized at the same age as the adenine‐exposed mice. Sost −/− and WT mice were randomly assigned to the treatment and control groups.
Study design experiment 2: Effect of anti‐sclerostin antibody treatment on warfarin‐induced vascular calcification development in large arteries
Vascular calcification was induced by administration of a warfarin‐containing diet (3 mg warfarin/g diet and 1.5 mg vitamin K1/g diet( 31 ); SSNIFF Spezialdiäten GmbH). Exposure to warfarin prevents the production of biologically active matrix γ‐carboxyglutamic acid protein (MGP), which is a local inhibitor of vascular calcification.( 31 , 38 ) In more detail, warfarin prevents the recycling of vitamin K, which is a crucial cofactor for γ‐glutamyl carboxylation of MGP.( 39 ) This posttranslational modification is needed for MGP to become biologically active. Both the warfarin‐containing and control were administered during the full duration of the study (9 weeks). Four weeks after the start of the experiment, mice were subcutaneously injected twice weekly with the anti‐sclerostin antibody (recombinant anti‐SOST therapeutic antibody MOB‐012LC; Creative Biolabs, Shirley, NY, USA) or vehicle (PBS). The antibody was administrated at a dose of 25 mg/kg, as reported earlier for similar anti‐sclerostin antibodies.( 40 , 41 ) Animals were randomly assigned to the different groups (control diet + vehicle, n = 16; warfarin diet + vehicle, n = 24; and warfarin diet + anti‐sclerostin antibody, n = 24).
Serum markers of mineral metabolism and renal function
At euthanasia, mice were exsanguinated via the retro‐orbital plexus after anesthesia with 60 mg/kg ketamine/16 mg/kg xylazine, diluted in sterile saline and administered via intraperitoneal injection. Serum levels of creatinine (Crystal Chem Inc., Elk Grove Village, IL, USA; Cat. 80350), phosphorus (Ecoline®S Phosphate kit; Diasys, Holzheim, Germany), fibroblast growth factor 23 (FGF23) (Kianos Laboratories, Tokyo, Japan; Cat. CY‐4000), and intact parathyroid hormone (PTH) (Immutopics, San Clemente, CA, USA; Cat. 60‐2305) were measured.
Evaluation of vascular calcification
At euthanasia, the aorta, heart, and kidneys were carefully isolated and the aorta was stripped from adherent tissue. For histological analyses, the abdominal aorta was fixed in neutral buffered formalin for 90 minutes and cut into 2‐mm‐thick to 3‐mm‐thick rings. These rings were then embedded upright in paraffin of which 4‐μm sections were cut and stained for calcification with von Kossa and counterstained with hematoxylin and eosin (H&E).
To determine the calcium content, the thoracic aorta, distal part of the heart, and right kidney were weighed on a precision balance. The tissue samples were then digested overnight in 65% HNO3 at 60°C, followed by dilution in 0.1% La(NO3)3 to eliminate chemical interference during atomic absorption spectrometric analysis (Perkin‐Elmer, Waltham, MA, USA). Results were expressed as mg calcium/g wet tissue.
Evaluation of gene expression
Total mRNA was extracted from the distal part of the abdominal aorta using the Isolate II RNA mini kit (Bioline Meridian Bioscience, Memphis, TN, USA). Complementary DNA was generated with the High‐Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA, USA). Real‐time PCR amplification was performed based on the TaqMan fluorescence methodology (QuantStudio 3; Thermo Fisher Scientific, Waltham, MA, USA). TaqMan probes and primers for glyceraldehyde 3‐phosphate dehydrogenase (Gapdh: Mm99999915_g1), Wnt/β‐catenin pathway genes, ie, low‐density lipoprotein receptor‐related protein 5 and 6 (Lrp5: Mm01227476_m1 and Lrp6: Mm00999795_m1), canonical Wnt‐signaling target genes, ie, alkaline phosphatase (Alp: Mm00475834_m1), Cyclin D1 (CycD1: Mm00432359_m1), T‐cell factor 4 (Tcf4: Mm00443210_m1), and Axin 2 (Mm00443610_m1), and osteochondrogenic marker, ie, SRY‐box transcription factor 9 (Sox9: Mm00448840_m1) and bone‐morphogenetic protein 2 (Bmp2: Mm01340178_m1), were purchased from Thermo Fisher Scientific as a TaqMan® gene expression assay on demand. For each sample, the expression of the tested transcripts was analyzed in triplicate and normalized to the expression of Gapdh, the housekeeping gene. The comparative threshold cycle (CT) method, using tissues of control mice as calibrator samples, was applied to calculate the gene expression levels.
Evaluation of bone metabolism
Because sclerostin’s primary role is as a bone anabolic protein and the close link between ectopic, vascular calcification, and physiological bone formation, the left tibia was isolated at euthanasia and fixed overnight in 70% ethanol. After dehydration and embedding in 100% methyl methacrylate (Merck, Hohenbrunn, Germany), 5‐μm‐thick sections were stained by Goldner’s method for quantitative histology to determine static bone parameters using Axiovision image analysis software (release 4.5; Carl Zeiss, Oberkoch, Germany). Bone analysis was performed on the trabecular bone tissue, in the area directly next to the proximal growth plate and the cortical bone (magnification ×200). Total bone area, mineralized bone area, osteoid width and area, osteoblast perimeter, eroded perimeter, osteoclast perimeter, and trabecular number were measured using this software platform.
To obtain quick proof for functional sclerostin blocking by the anti‐sclerostin antibody, the left femur of all animals of experiment 2 was evaluated by ex vivo micro–computed tomography (μCT) using a SkyScan 1076 μCT scanner (Bruker, Kontich, Belgium). After isolation, the femur was placed in a radio translucent sample holder filled with 70% ethanol. μCT scanning was performed with a resolution of 8.86 mm (at tube energy of 80 kV with an intensity of 110 μA). Images were reconstructed using NRecon (SkyScan software; Bruker) and 1 mm of trabecular bone in the distal metaphysis of the femur, at a distance of 1.95 mm from the beginning of the bone, was analyzed using CtAnalyser (SkyScan software; Bruker). A fixed threshold (same for all groups: lower gray threshold 80, upper gray threshold 255) was used for segmentation of trabecular bone and bone marrow on the μCT images.
All results were determined using blinded measurements.
Statistical analysis
All statistical analyses were performed using PRISM software, version 6.0 (GraphPad, San Diego, CA, USA). Data are presented as the mean ± SD. Statistical differences between groups were investigated with the Kruskal‐Wallis test, followed by the Mann–Whitney U test (two‐tailed) when significant. Bonferroni correction was applied when multiple group post hoc testing was done. Values of p <0.05 were considered statistically significant.
Results
Effect of sclerostin‐absence on CKD‐induced vascular calcification
Body weight and evaluation of mineral metabolism
As described,( 34 ) dietary adenine supplementation results in significant body weight loss (p < 0.0001). However, no differences were observed between Sost −/− and WT mice receiving either the adenine‐containing diet or the control diet (Table 1).
Table 1.
Body Weight
| Group | Body weight (g) mean ± SD |
|---|---|
| Sost −/− + adenine | 20.2 ± 2.0 |
| WT + adenine | 20.0 ± 0.9 |
| Sost −/− + control diet | 32.3 ± 3.5 |
| WT + control diet | 29.8 ± 2.2 |
Administration of adenine led to CKD development, which could be concluded from the significant increases in serum creatinine (Fig 1. A), phosphate (Fig 1. B), FGF23 (Fig 1. C), and parathyroid (PTH) (Fig 1. D) levels in the adenine‐treated animals as compared to those receiving a control diet. No differences were observed for these parameters between Sost −/− mice and WT mice exposed to either the adenine diet or the control diet.
Fig. 1.
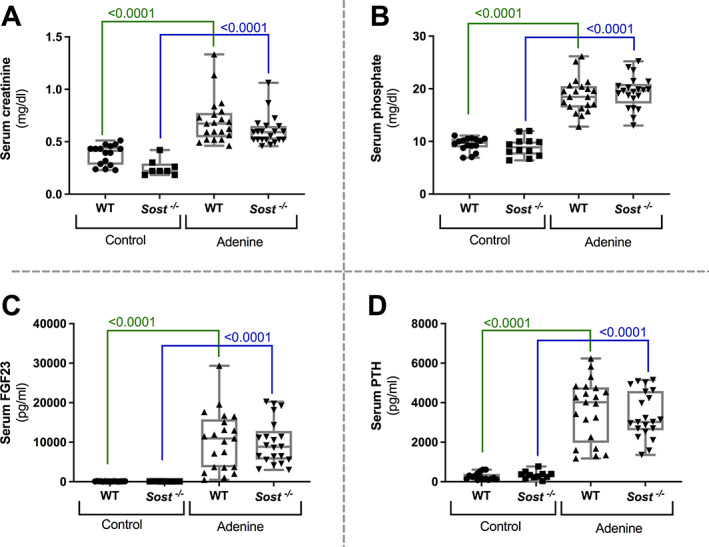
Development of renal failure due to adenine‐exposure. Biochemical evaluation of renal function: serum (A) creatinine, (B) phosphate, (C) FGF23, and (D) PTH levels in Sost −/− and control mice, exposed to adenine or control diet.
Evaluation of vascular calcification development
Neither WT mice, nor Sost −/− mice receiving a control diet developed calcification in either the aorta, kidney, or heart. In WT mice, no aorta calcification developed as a result of adenine‐exposure, because the calcium content of the (thoracic) aorta of adenine‐exposed mice was not different from that of control mice (Fig. 2A ). This was further confirmed by the negative von Kossa staining of (abdominal) aorta tissue sections. In adenine‐exposed Sost −/− mice, the calcium content was slightly, however significantly, elevated compared to Sost −/− mice receiving control diet (Fig. 2A ). In the kidney (Fig. 2B ) adenine exposure led to a similar, significantly increased calcium content in both Sost −/− and WT mice. Von Kossa staining of renal sections, however, made clear that renal areas of calcification were primarily observed in the renal tubules and thus could not be categorized as vascular calcifications. Sost −/− mice exposed to adenine had a significantly higher calcium content in the heart compared to WT mice exposed to adenine as well as compared to the Sost −/− mice receiving a control diet. The cardiac calcium content of the latter was not significantly different from that of control WT mice without renal failure (Fig. 2C ). Cardiac calcifications were located in the vasculature (Fig. 2C ). A histological overview of cardiac calcification is presented in Supplementary Fig. 1.
Fig. 2.
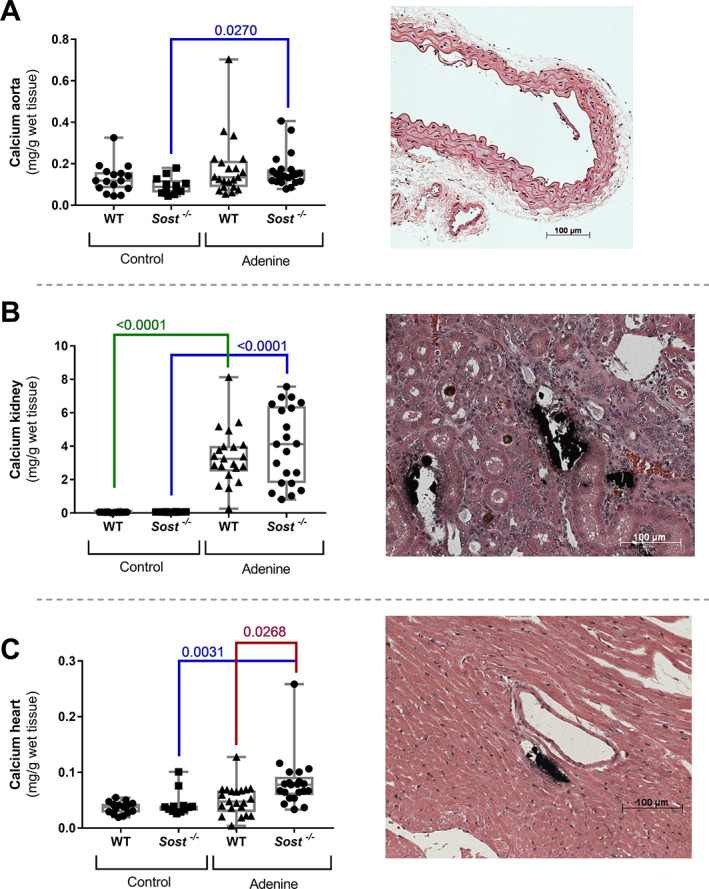
Development of cardiac vascular calcifications in Sost −/− mice with renal failure. Calcium content of (A) the aorta, (B) kidney, and (C) heart of Sost −/− and WT mice exposed to adenine or control diet, accompanied by representative images of von Kossa‐stained sections of the aorta, kidney and heart of Sost −/− mice (counterstained with H&E). (A) von Kossa staining was not able to reveal calcifications in the aortic tissue sections, (B) areas of calcification are present in the renal tubules, (C) cardiac calcifications are located in the vasculature.
CKD and Sost −/− induced effects on the bone
The bone phenotype of Sost −/− mice is characterized by a significantly increased bone and mineralized area and trabecular thickness and number, as well as a significantly decreased osteoid area, compared to WT mice (Supplementary Fig. 2).
Adenine‐induced CKD in Sost −/− and WT mice led to a significant increase in eroded perimeter (Fig. 3A ), as well as a significant decrease in bone area (Fig. 3B ), mineralized area (Fig. 3C ), and trabecular number (Fig. 3D ) compared to the respective control mice (either Sost −/− or WT). Adenine‐exposed Sost −/− mice also had a significantly higher osteoid area (Fig. 3E ), osteoblast perimeter (Fig. 3F ), and osteoid width (Fig. 3G ), and a significantly decreased trabecular thickness (Fig. 3H ), compared to control Sost −/− mice.
Fig. 3.
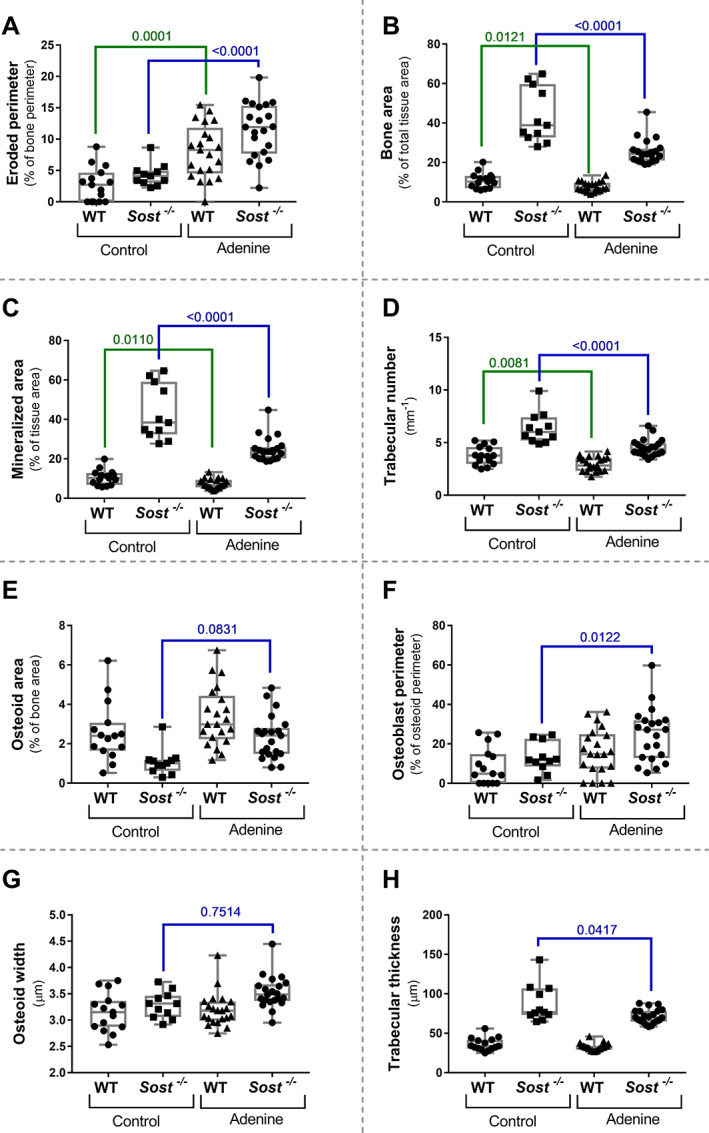
Influence of CKD on bone metabolism in WT and Sost −/− mice. (A) Eroded perimeter, (B) bone area, (C) mineralized area (% of tissue area), (D) trabecular number, (E) osteoid area, (F) osteoblast perimeter (relative to the osteoid perimeter), (G) osteoid width, (H) trabecular thickness.
Effect of anti‐sclerostin antibody treatment on warfarin‐induced vascular calcification
Body weight and evaluation of mineral metabolism
Warfarin‐exposure and/or anti‐sclerostin antibody treatment did not result in chances in body weight (Table 2).
Table 2.
Body Weight
| Group | Body weight (g) mean ± SD |
|---|---|
| Anti‐Scl Ab + warfarin | 27.5 ± 2.4 |
| Vehicle + warfarin | 28.2 ± 2.4 |
| Vehicle + control diet | 28.6 ± 1.7 |
Ab = antibody.
Serum levels of creatinine, phosphate, PTH, and FGF23 in vehicle‐treated warfarin‐exposed animals were similar to those of control mice (Fig. 4A–D ). Serum phosphate levels of warfarin‐exposed mice treated with the anti‐sclerostin antibody, however, were significantly higher, compared to control animals (Fig. 4B ). Furthermore, serum FGF23 levels in this group were significantly lower compared to vehicle‐treated, warfarin‐exposed mice and control mice. No differences in serum creatinine and serum PTH levels were observed.
Fig. 4.
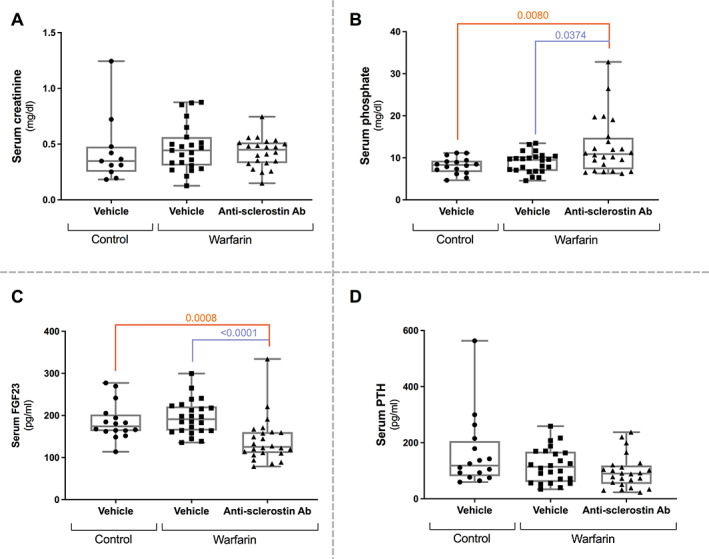
Anti‐sclerostin antibody‐treatment leads to increased serum phosphate levels and decreased serum FGF23 levels. Serum creatinine (A), phosphate (B), FGF23 (C), and PTH (D) levels in mice treated with anti‐sclerostin antibody or vehicle, exposed to a warfarin‐containing or control diet. FGF23 = fibroblast growth factor 23, PTH = parathyroid hormone.
Evaluation of vascular calcification development
Warfarin exposure led to a significant increase in calcium content in the aorta (Fig. 5A ) and the kidney (Fig. 5B ). Mice exposed to warfarin and treated with the anti‐sclerostin antibody had a significantly higher calcium content in the aorta and the kidney compared to warfarin‐exposed mice treated with vehicle. Calcifications in the aorta were located in the medial layer, following the elastic lamellae. Quantification of the von Kossa–positive area showed a significant increase in calcified area in aortas of anti‐sclerostin antibody‐treated mice versus vehicle‐treated mice (Fig. 5A ). In the kidney, areas of calcification were confined to the vasculature (Fig. 5B ).
Fig. 5.
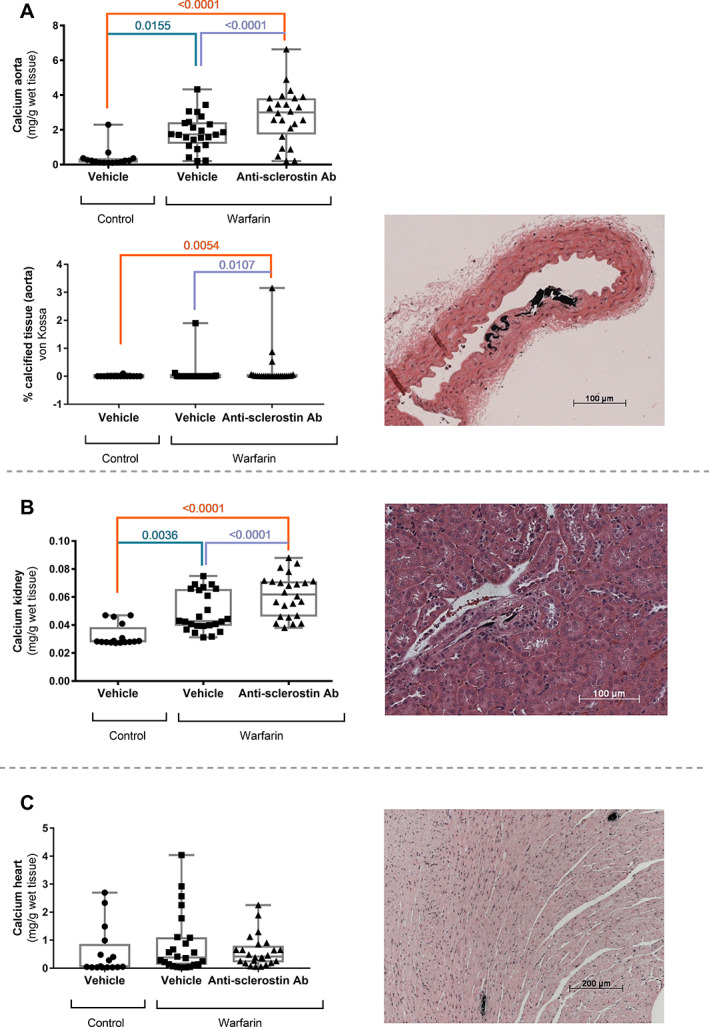
Anti‐sclerostin antibody treatment leads to increased development of vascular calcifications. Calcium content of (A, upper panel) the aorta, (B) kidney, and (C) heart of mice treated with anti‐sclerostin antibody or vehicle after exposure to warfarin‐containing or control diet, accompanied by representative images of von Kossa‐stained tissue sections of anti‐sclerostin antibody‐treated mice (counterstained with H&E). (A, lower panel) Quantification of the calcified area as measured on von Kossa–stained tissues sections. These calcifications are located in the medial layer of the aorta, following the elastic lamellae. (B) Areas of calcification in the kidney were confined to the vasculature. (C) Spontaneous calcifications developed in the myocardium of DBA/2 mice.
Cardiac calcium content of DBA/2J mice (Fig. 5C ) is known to be relatively high because of spontaneous calcification development (20 times as high as those in the C57BL/6J WT mice at the same age). No differences in these spontaneous calcifications were seen between the different groups.
Gene expression analysis
In order to investigate the involvement of the canonical Wnt signaling during vascular calcification development, we determined the expression of (i) genes encoding proteins that are involved in canonical Wnt signaling (Lrp 5 and 6) and (ii) Wnt/β‐catenin target genes (Axin2, Alp, CycD1, and Tcf4).
Lrp5 expression was significantly upregulated in the calcified vessels of warfarin‐exposed animals, compared to mice that received control diet (Fig. 6A ). There was no effect of anti‐sclerostin antibody treatment on Lrp5 mRNA expression. No differences were observed in Lrp6 mRNA (Fig. 6B ) expression between the different treatment groups.
Fig. 6.
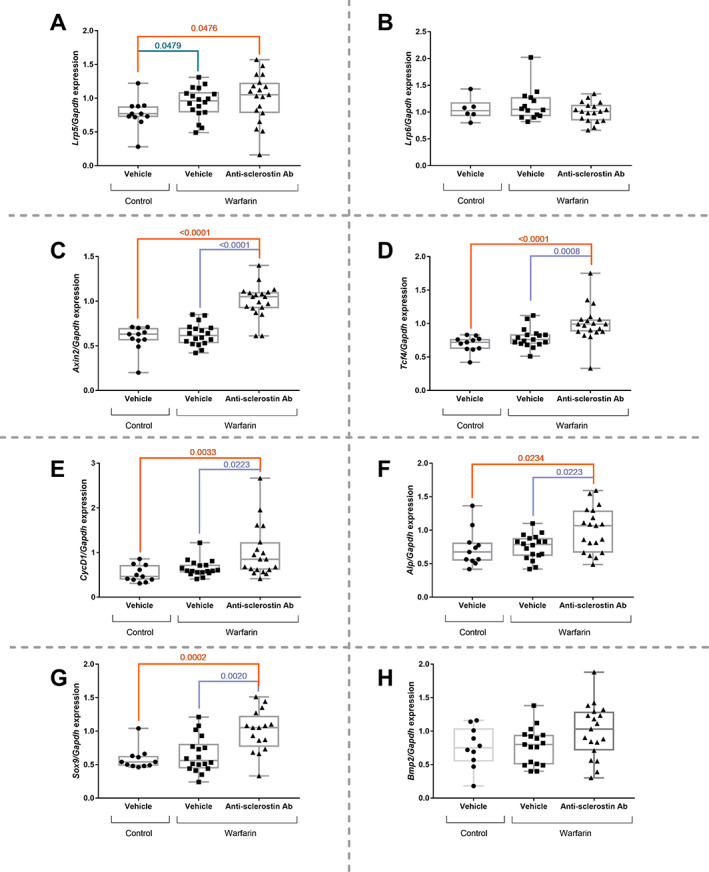
Anti‐sclerostin antibody treatment leads to upregulated aortic mRNA expression of Wnt target genes. (A) Lrp5, (B) Lrp6, (C) Axin2, (D) Tcf4, (E) CycD1, (F) Alp, (G) Sox9, and (H) Bmp2 mRNA expression profile in the abdominal aorta of mice treated with anti‐sclerostin antibody or vehicle after exposure to warfarin‐containing or control diet. Alp = alkaline phosphatase; Bmp2 = bone‐morphogenetic protein 2; CycD1 = cyclin D1; GAPDH = glyceraldehyde 3‐phosphate dehydrogenase; Lrp5 = low‐density lipoprotein receptor‐related protein 5; Lrp6 = low‐density lipoprotein receptor‐related protein 6; Sox9 = SRY‐box transcription factor 9; Tcf4 = transcription factor 4; WT = wild type.
Gene expression of Wnt/β‐catenin target genes, Axin2 (Fig. 6C ), Tcf4 (Fig. 6D ), CycD1 (Fig. 6E ), and Alp (Fig. 6F ), was significantly increased in anti‐sclerostin antibody‐treated mice versus vehicle treated‐mice (exposed to warfarin).
Last, in order to investigate whether, in addition to Alp, other osteochondrogenic markers were upregulated, the expression of Sox9 (Fig. 6G ) and Bmp2 (Fig. 6H ) was determined. Similar expression patterns were found as for Alp: gene expression was increased in anti‐sclerostin antibody‐treated mice compared to vehicle‐treated mice; however, without reaching the significance level for Bmp2.
Functional sclerostin blocking
Preparing bone tissue for quantitative histomorphometric analysis is a time‐consuming and labor‐intensive procedure, not possible to be performed in a period shorter than 8 weeks. To get relatively fast proof for anti‐sclerostin antibody‐induced functional sclerostin blocking, μCT analysis was performed in advance of further bone histomorphometric analysis. μCT analysis (overview in Fig. 7E) showed a significantly increased bone volume (Fig. 7A) and trabecular thickness (Fig. 7C) and number (Fig. 7D), in combination with a decreased porosity (Fig. 7B) when mice were treated with the anti‐sclerostin antibody compared to vehicle‐treated mice.
Fig. 7.
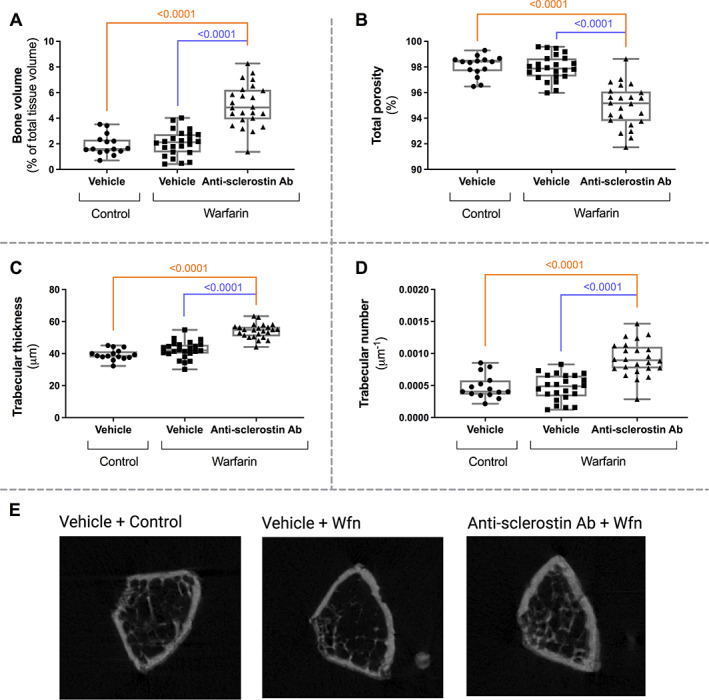
Increased bone formation in anti‐sclerostin antibody‐treated mice as evaluated by μCT analysis. Evaluation of (A) bone volume, (B) total porosity, (C) trabecular thickness, and (D) trabecular number in warfarin‐exposed mice treated with anti‐sclerostin antibody or vehicle and mice receiving the control diet. (E) μCT image of a cross section of the bone for each group.
Bone histomorphometric data confirmed μCT analysis: significantly increased bone area (Fig. 8A) and trabecular thickness (Fig. 8B) when treated with the anti‐sclerostin antibody. No other bone histomorphometric data differed significantly between the three groups.
Fig. 8.
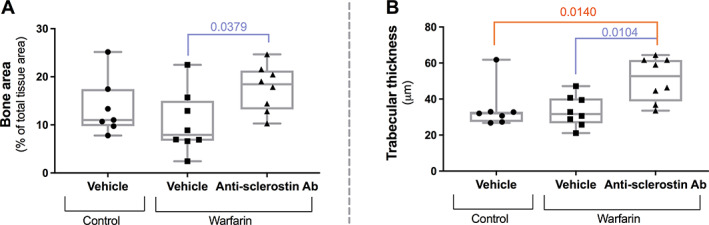
Increased bone formation in anti‐sclerostin antibody‐treated mice as evaluated by bone histomorphometric analysis. Evaluation of (A) bone area and (B) trabecular thickness in mice treated with the anti‐sclerostin antibody versus vehicle‐treated mice.
Discussion
This study confirmed the known catabolic effects of sclerostin on bone turnover,( 42 ) because absence of sclerostin, either by knocking out the Sost gene or by treating mice with an anti‐sclerostin antibody, resulted in significantly higher bone formation/mineralization, which was evidenced by a series of static bone parameters. However, there might be another side of the coin, because it has been suggested that these beneficial effects on bone possibly might be accompanied by an increased risk for vascular calcifications.( 43 )
In this study, we investigated whether absence of sclerostin or its functional inhibition through the use of an anti‐sclerostin antibody indeed promoted vascular calcifications in the aorta, heart, and kidney. To fully understand the possible role of sclerostin in vascular calcification, two different but complementary experiments were set up.
In the first experiment, the effect of sclerostin absence was investigated by studying Sost −/− mice (on C57BL/6J background). Adenine‐induced renal failure led to the development of vascular calcifications in cardiac vessels, and this was significantly more pronounced in Sost −/− mice compared to WT mice. Furthermore, Sost −/− mice exposed to adenine had a significantly higher aortic calcium content compared to Sost −/− mice with normal renal function, which was not the case for the WT animals. This observation argues for the fact that CKD‐induction was able to increase the aortic calcium content of Sost −/− mice but not in WT mice. This significant increase may also be related to the numerically lower aortic calcium levels of control Sost −/− versus control WT mice. The latter can, possibly, be ascribed to the more pronounced buffering capacity of the bone, hence better incorporation of calcium and phosphate in the bone compartment in Sost −/− mice versus WT mice (see next paragraph).
Because Sost −/− mice have a condition which is similar to that of sclerosteosis and Van Buchem disease patients, and compensate for the loss of sclerostin by upregulating the expression of other Wnt inhibitors such as Dickkopf‐related protein 1 (DKK1),( 44 ) the role of sclerostin as an inhibitor of vascular calcification might even be more important than what could be deduced from the data of this experiment. One should also not overlook the fact that there is a close link between the mineralization of the bone and the vasculature. The bone has an important homeostatic function because it serves as a mineral reservoir, in particular for calcium and phosphate. Compared to WT mice, Sost −/− mice have an increased bone formation and thus increased bone buffering capacity.( 4 , 28 ) It is remarkable that despite this increased bone buffering capacity, the Sost −/− mice developed significantly more calcifications in cardiac vessels compared to WT mice.
In the second experiment, DBA/2J mice received a warfarin‐containing diet in order to induce vascular calcifications (in absence of renal failure). DBA/2J mice are known to have reduced levels of pyrophosphate due to a hypomorphic mutation in the Abcc6 gene( 45 ) and are, compared to C57BL/6J mice, more vulnerable to develop ectopic calcifications.( 31 ) In the DBA/2J mice on a warfarin diet, calcifications developed in the aorta and renal arteries, which were significantly more pronounced when mice were also treated with an anti‐sclerostin antibody.
The results of both experiments are consistent in that more calcifications develop in the absence of (functional) sclerostin. This observation is in line with the reported suggestion that sclerostin can act as an inhibitor of calcification in the vasculature (aorta and vasculature of kidney and heart). Several findings support this hypothesis: (i) mRNA and protein expression of sclerostin were demonstrated in calcified vessels of mice, rats and humans( 26 , 46 ) and (ii) negative associations were found between serum sclerostin levels and mortality in CKD patients.( 12 , 15 ) These findings suggest that sclerostin is locally produced in the calcified tissue to act as a negative feedback mechanism to slow down the ectopic calcification process. Furthermore, the Active‐Controlled Fracture Study in Postmenopausal Women With Osteoporosis at High Risk (ARCH) study, a recent clinical trial comparing anti‐sclerostin antibody (romosozumab) treatment for 12 months followed by alendronate treatment (12 months) versus alendronate treatment alone (24 months), revealed an increased risk of serious adverse (cardio)vascular events in postmenopausal women during the first year treatment with anti‐sclerostin antibody.( 47 ) These findings, together with the results obtained in this study, point to a protective role for sclerostin during vascular calcification.
Several mechanisms might be involved in the way sclerostin inhibits the vascular calcification process. First, similar to its function in the bone, sclerostin by binding to LRP5 (significantly increased Lrp5 expression is warfarin mice), might inhibit Wnt signaling, and thereby directly act on VSMCs to prevent ectopic calcification, which is in agreement with the significantly higher aortic expression of Wnt‐target genes and as a result osteochondrogenic markers in warfarin animals treated with anti‐sclerostin antibody. The fact that warfarin‐induced calcifications can be intensified with the Wnt signaling agonist lithium chloride( 9 ) is also in accordance with this. Second, sclerostin might indirectly stimulate FGF23, via inhibition of phosphate‐regulating neutral endopeptidase, X‐linked (PHEX).( 48 ) FGF23 then stimulates urinary phosphate excretion, thereby lowering the concentration of this well‐known calcification inducer.( 49 ) In the present study, sclerostin inhibition in the non‐CKD model (warfarin model) resulted in significantly decreased serum FGF23 and increased serum phosphate levels, which is in agreement with the fact that sclerostin might stimulate FGF23 production. However, in the presence of renal failure, the absence of sclerostin in Sost −/− mice was not able to overcome the expected rise in serum phosphate and FGF23 levels due to loss of renal function.
Anti‐sclerostin antibodies have proven to be an adequate treatment for osteoporosis patients and evidence for reducing the risk of bone fractures has been provided.( 50 ) The (significant) increase in severe cardiovascular events due to anti‐sclerostin treatment (2.5% versus 1.9% in the control group receiving alendronate) was relatively small, which is in line with the rather mild effect of (functional) sclerostin absence on the induction of vascular calcification in our animal models. Overall, the advantages of a beneficial effect of anti‐sclerostin treatment on bone health and the significant improvement of an individual's quality of life going along herewith, probably outweigh the risk of adverse cardiovascular events. Nevertheless, continuous alertness is recommended in patients on long‐term treatment with anti‐sclerostin antibody, in particular when its use is also being considered in CKD patients.
To conclude, this study presents evidence for a protective role of sclerostin during the development of vascular calcification, and thereby importantly contributes to the existing knowledge on the similarities between the systemic bone and vascular (patho)physiological mechanisms.( 51 ) Our findings again point to the importance of considering the bone‐vascular axis when developing new therapeutics to either treat impaired bone metabolism or vascular calcification.
Author Contributions
Annelies De Maré: Conceptualization; data curation; formal analysis; funding acquisition; investigation; writing – original draft. Britt Opdebeeck: Conceptualization; investigation; methodology. Ellen Neven: Conceptualization; project administration; supervision. Patrick C D'Haese: Project administration; supervision; writing – review and editing. Anja Verhulst: Conceptualization; funding acquisition; methodology; project administration; supervision; writing – review and editing.
Conflict of Interest
ADM, BO, EV, PCD and AV have nothing to disclose.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4503.
Supporting information
Supplementary Fig. 1 Histological overview of cardiac vascular calcifications in Sost −/− mice with renal failure. Images of von Kossa‐stained sections of the heart of Sost −/− mice (counterstained with H&E), from left to right: increasing magnifications.
Supplementary Fig. 2 Bone phenotype of Sost −/− mice. (A) Bone area, (B) mineralized area, (C) trabecular thickness, (D) trabecular number and (E) osteoid area. WT = wild type.
Supplementary Table 1 Animals Supplemental Table.
Acknowledgments
This work was supported by the Research Foundation Flanders (1182417N and 1182419N to ADM). We especially thank Simonne Dauwe and Geert Dams for their excellent technical assistance. We thank Novartis Pharma (Basel Switzerland), for the supply of Sost −/− mice. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Data Availability Statement
Data available on request from the authors
References
- 1. van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte‐expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199(6):805‐814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ukita M, Yamaguchi T, Ohata N, Tamura M. Sclerostin enhances adipocyte differentiation in 3T3‐L1 cells. J Cell Biochem. 2016;117(6):1419‐1428. [DOI] [PubMed] [Google Scholar]
- 3. van Lierop AH, Hamdy NAT, van Egmond ME, Bakker E, Dikkers FG, Papapoulos SE. Van Buchem disease: clinical, biochemical, and densitometric features of patients and disease carriers. J Bone Miner Res. 2013;28(4):848‐854. [DOI] [PubMed] [Google Scholar]
- 4. Li XD, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860‐869. [DOI] [PubMed] [Google Scholar]
- 5. McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412‐420. [DOI] [PubMed] [Google Scholar]
- 6. Tolle M, Reshetnik A, Schuchardt M, Hohne M, van der Giet M. Arteriosclerosis and vascular calcification: causes, clinical assessment and therapy. Eur J Clin Invest. 2015;45(9):976‐985. [DOI] [PubMed] [Google Scholar]
- 7. Van den Bergh G, Opdebeeck B, D'Haese PC, Verhulst A. The vicious cycle of arterial stiffness and arterial media calcification. Trends Mol Med. 2019;25(12):1133‐1146. [DOI] [PubMed] [Google Scholar]
- 8. Neven E, De Schutter TM, De Broe ME, D'Haese PC. Cell biological and physicochemical aspects of arterial calcification. Kidney Int. 2011;79(11):1166‐1177. [DOI] [PubMed] [Google Scholar]
- 9. Nie B, Zhang SY, Guan SM, Zhou SQ, Fang X. Role of Wnt/beta‐catenin pathway in the arterial medial calcification and its effect on the OPG/RANKL system. Curr Med Sci. 2019;39(1):28‐36. [DOI] [PubMed] [Google Scholar]
- 10. Freise C, Kretzschmar N, Querfeld U. Wnt signaling contributes to vascular calcification by induction of matrix metalloproteinases. BMC Cardiovasc Disord. 2016;16:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Viaene L, Behets GJ, Claes K, et al. Sclerostin: another bone‐related protein related to all‐cause mortality in haemodialysis? Nephrol Dial Transpl. 2013;28(12):3024‐3030. [DOI] [PubMed] [Google Scholar]
- 12. Lips L, van Zuijdewijn CLMD, ter Wee PM, et al. Serum sclerostin: relation with mortality and impact of hemodiafiltration. Nephrol Dial Transpl. 2017;32(7):1217‐1223. [DOI] [PubMed] [Google Scholar]
- 13. Drechsler C, Evenepoel P, Vervloet MG, et al. High levels of circulating sclerostin are associated with better cardiovascular survival in incident dialysis patients: results from the NECOSAD study. Nephrol Dial Transpl. 2015;30(2):288‐293. [DOI] [PubMed] [Google Scholar]
- 14. Yang CY, Chang ZF, Chau YP, et al. Circulating Wnt/beta‐catenin signalling inhibitors and uraemic vascular calcifications. Nephrol Dial Transpl. 2015;30(8):1356‐1363. [DOI] [PubMed] [Google Scholar]
- 15. Jean G, Chazot C, Bresson E, Zaoui E, Cavalier E. High serum sclerostin levels are associated with a better outcome in haemodialysis patients. Nephron. 2016;132(3):181‐190. [DOI] [PubMed] [Google Scholar]
- 16. Wang XR, Yuan LA, Zhang JJ, Hao L, Wang DG. Serum sclerostin values are associated with abdominal aortic calcification and predict cardiovascular events in patients with chronic kidney disease stages 3–5D. Nephrol Ther. 2017;22(4):286‐292. [DOI] [PubMed] [Google Scholar]
- 17. Goncalves FLC, Elias RM, dos Reis LM, et al. Serum sclerostin is an independent predictor of mortality in hemodialysis patients. BMC Nephrol. 2014;15:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jorgensen HS, Winther S, Dupont L, et al. Sclerostin is not associated with cardiovascular event or fracture in kidney transplantation candidates. Clin Nephrol. 2018;90(1):18‐26. [DOI] [PubMed] [Google Scholar]
- 19. Sato M, Hanafusa N, Kawaguchi H, Tsuchiya K, Nitta K. A prospective cohort study showing no association between serum sclerostin level and mortality in maintenance hemodialysis patients. Kidney Blood Press Res. 2018;43(3):1023‐1033. [DOI] [PubMed] [Google Scholar]
- 20. Nowak A, Artunc F, Serra AL, et al. Sclerostin quo vadis? – is this a useful long‐term mortality parameter in prevalent hemodialysis patients? Kidney Blood Press Res. 2015;40(3):266‐276. [DOI] [PubMed] [Google Scholar]
- 21. Delanaye P, Krzesinski JM, Warling X, et al. Clinical and biological determinants of sclerostin plasma concentration in hemodialysis patients. Nephron Clin Pract. 2014;128(1‐2):127‐134. [DOI] [PubMed] [Google Scholar]
- 22. Koos R, Brandenburg V, Mahnken AH, et al. Sclerostin as a potential novel biomarker for aortic valve. J Heart Valve Dis. 2013;22(3):317‐325. [PubMed] [Google Scholar]
- 23. Brandenburg VM, Kramann R, Koos R, et al. Relationship between sclerostin and cardiovascular calcification in hemodialysis patients: a cross‐sectional study. BMC Nephrol. 2013;14:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Claes KJ, Viaene L, Heye S, Meijers B, d'Haese P, Evenepoel P. Sclerostin: another vascular calcification inhibitor? J Clin Endocrinol Metab. 2013;98(8):3221‐3228. [DOI] [PubMed] [Google Scholar]
- 25. Cejka D, Marculescu R, Kozakowski N, et al. Renal elimination of sclerostin increases with declining kidney function. J Clin Endocr Metab. 2014;99(1):248‐255. [DOI] [PubMed] [Google Scholar]
- 26. De Mare A, Maudsley S, Azmi A, et al. Sclerostin as regulatory molecule in vascular media calcification and the bone‐vascular axis. Toxins. 2019;11(7):428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kramer I, Loots GG, Studer A, Keller H, Kneissel M. Parathyroid hormone (PTH)‐induced bone gain is blunted in SOST overexpressing and deficient mice. J Bone Miner Res. 2010;25(2):178‐189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kaesler N, Verhulst A, De Mare A, et al. Sclerostin deficiency modifies the development of CKD‐MBD in mice. Bone. 2018;107:115‐123. [DOI] [PubMed] [Google Scholar]
- 29. Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. 2004;24(7):1161‐1170. [DOI] [PubMed] [Google Scholar]
- 30. Silbiger SR, Neugarten J. The impact of gender on the progression of chronic renal disease. Am J Kidney Dis. 1995;25(4):515‐533. [DOI] [PubMed] [Google Scholar]
- 31. Kruger T, Oelenberg S, Kaesler N, et al. Warfarin induces cardiovascular damage in mice. Arterioscler Thromb Vasc Biol. 2013;33(11):2618‐2624. [DOI] [PubMed] [Google Scholar]
- 32. C‐m Qin, Wei X, Gong CP, et al. [Expression of BMP2/Smad1/Runx2 signal pathway in renal artery of rat with vascular calcification]. Sichuan Da Xue Xue Bao Yi Xue Ban 2016;47(2):180‐3. Chinese. [PubMed] [Google Scholar]
- 33. Neven E, Vervaet B, Brand K, et al. Metformin prevents the development of severe chronic kidney disease and its associated mineral and bone disorder. Kidney Int. 2018;94(1):102‐113. [DOI] [PubMed] [Google Scholar]
- 34. Jia T, Olauson H, Lindberg K, et al. A novel model of adenine‐induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013;14:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kelley WN, Levy RI, Rosenbloom FM, Henderson JF, Seegmiller JE. Adenine phosphoribosyltransferase deficiency – a previously undescribed genetic defect in man. J Clin Invest. 1968;47(10):2281‐2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wyngaarden JB, Dunn JT. 8‐Hydroxyadenine as the intermediate in the oxidation of adenine to 2,8‐dihydroxyadenine by xanthine oxidase. Arch Biochem Biophys. 1957;70(1):150‐156. [DOI] [PubMed] [Google Scholar]
- 37. Fye KH, Sahota A, Hancock DC, et al. Adenine phosphoribosyltransferase deficiency with renal deposition of 2,8‐dihydroxyadenine leading to nephrolithiasis and chronic‐renal‐failure. Arch Intern Med. 1993;153(6):767‐770. [PubMed] [Google Scholar]
- 38. Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol. 1998;18(9):1400‐1407. [DOI] [PubMed] [Google Scholar]
- 39. Schurgers LJ, Cranenburg ECM, Vermeer C. Matrix Gla‐protein: the calcification inhibitor in need of vitamin K. Thromb Haemost. 2008;100(4):593‐603. [PubMed] [Google Scholar]
- 40. Li XD, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24(4):578‐588. [DOI] [PubMed] [Google Scholar]
- 41. Fulzele K, Dedic C, Lai F, et al. Loss of Gs alpha in osteocytes leads to osteopenia due to sclerostin induced suppression of osteoblast activity. Bone. 2018;117:138‐148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Poole KES, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19(10):1842‐1844. [DOI] [PubMed] [Google Scholar]
- 43. McClung MR, Grauer A. Romosozumab in postmenopausal women with osteopenia. Reply. N Engl J Med. 2014;370(17):1664‐1665. [DOI] [PubMed] [Google Scholar]
- 44. van Lierop AH, Moester MJC, Hamdy NAT, Papapoulos SE. Serum Dickkopf 1 levels in sclerostin deficiency. J Clin Endocr Metab. 2014;99(2):E252‐E256. [DOI] [PubMed] [Google Scholar]
- 45. Babler A, Schmitz C, Buescher A, et al. Microvasculopathy and soft tissue calcification in mice are governed by fetuin‐A, magnesium and pyrophosphate. PLoS One. 2020;15(2):e0228938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhu DX, Mackenzie NCW, Millan JL, Farquharson C, MacRae VE. The appearance and modulation of osteocyte marker expression during calcification of vascular smooth muscle cells. PLoS One. 2011;6(5):e19595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Saag KG, Petersen J, Brandi ML, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377(15):1417‐1427. [DOI] [PubMed] [Google Scholar]
- 48. Ryan ZC, Ketha H, McNulty MS, et al. Sclerostin alters serum vitamin D metabolite and fibroblast growth factor 23 concentrations and the urinary excretion of calcium. Proc Natl Acad Sci U S A. 2013;110(15):6199‐6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Giachelli CM. The emerging role of phosphate in vascular calcification. Kidney Int. 2009;75(9):890‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375(16):1532‐1543. [DOI] [PubMed] [Google Scholar]
- 51. Towler DA. Commonalities between vasculature and bone an osseocentric view of arteriosclerosis. Circulation. 2017;135(4):320‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1 Histological overview of cardiac vascular calcifications in Sost −/− mice with renal failure. Images of von Kossa‐stained sections of the heart of Sost −/− mice (counterstained with H&E), from left to right: increasing magnifications.
Supplementary Fig. 2 Bone phenotype of Sost −/− mice. (A) Bone area, (B) mineralized area, (C) trabecular thickness, (D) trabecular number and (E) osteoid area. WT = wild type.
Supplementary Table 1 Animals Supplemental Table.
Data Availability Statement
Data available on request from the authors