

Abstract
Aims
Chronic inflammation is a risk factor for cardiovascular disease (CVD). IL‐6 signalling perturbation through IL‐6 or IL‐6R blockade may have potential benefit on cardiovascular risk. It is unknown whether targeting either IL‐6 or IL‐6 receptor may result in similar effects on CVD and adverse events. We compared the anticipated effects of targeting IL‐6 and IL‐6 receptor on cardiometabolic risk and potential side effects.
Methods
We constructed four instruments: two main instruments with genetic variants in the IL6 and IL6R loci weighted for their association with CRP, and two after firstly filtering variants for their association with IL‐6 or IL‐6R expression. Analyses were performed for coronary artery disease (CAD), ischemic stroke, atrial fibrillation (AF), heart failure, type 2 diabetes (T2D), rheumatoid arthritis (RA), infection endpoints, and quantitative haematological, metabolic and anthropometric parameters.
Results
A 1 mg/L lower CRP by the IL6 instrument was associated with lower CAD (odds ratio [OR] 0.86, 95% confidence interval [CI] 0.77;0.96), AF and T2D risk. A 1 mg/L lower CRP by the IL6R instrument was associated with lower CAD (OR 0.90, 95% CI 0.86;0.95), any stroke and ischemic stroke, AF, RA risk and higher pneumonia risk. The eQTL‐filtered results were in concordance with the main results, but with wider confidence intervals.
Conclusions
IL‐6 signalling perturbation by either IL6 or IL6R genetic instruments is associated with a similar risk reduction for multiple cardiometabolic diseases, suggesting that both IL‐6 and IL‐6R are potential therapeutic targets to lower CVD. Moreover, IL‐6 rather than IL‐6R inhibition might have a more favourable pneumonia risk.
Keywords: cardiovascular disease, classical signalling, IL‐6, trans‐signalling
What is already known about this subject
The residual risk for CVD is at least partly attributed to chronic inflammation.
MR studies have robustly shown the effect of IL‐6 receptor genetic variants on CVD.
Some IL6R variants are likely to upregulate IL‐6 trans‐signalling, while IL6 variants likely inhibit both the classical and trans‐signalling pathway.
What this study adds
Inhibition of the IL‐6 signalling pathway, by targeting either IL‐6 or IL‐6R, is likely to result in beneficial effects on the risk for CAD, stroke, AF and type 2 diabetes.
IL‐6 receptor perturbation but not IL‐6 perturbation was associated with increased pneumonia risk, which suggests a differential effect.
1. INTRODUCTION
The burden of cardiovascular disease (CVD) on morbidity and mortality remains high, despite major advances in the (early) diagnosis and treatment. The residual risk for CVD is at least partly attributed to chronic inflammation. 1 Inflammatory markers, including interleukin‐6 (IL‐6) and C‐reactive protein (CRP), have been consistently associated with adverse cardiovascular outcomes both in subjects with and without a history of CVD. 1 , 2 , 3 , 4 However, dissecting the causative nature of the different markers of inflammation has proven troublesome often due to confounding and reverse causation. 5
Finding the culprit factor in the inflammatory process is pivotal to curb residual risk. The Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS) showed that subcutaneously administered canakinumab, a monoclonal antibody directed against IL‐1 beta, reduced both CRP levels by a median of 54.1% (interquartile range [IQR]: −74.4, −16.5%) from baseline and the incidence of cardiovascular disease by 15% (odds ratio [OR] 0.85, 95% confidence interval [CI] 0.74–0.98). 6 A post hoc analysis revealed that CVD treatment benefit was most pronounced in patients who achieved interleukin‐6 (IL‐6) levels below 1.65 ng/L (OR for MACE: 0.68, 95% CI 0.56–0.82, compared to an OR of 0.90 for participants with IL‐6 levels above the median of 1.65 ng/L [95% CI 0.76–1.07]). 7 While conditioning on post‐randomization events may induce selection bias, 8 baseline IL‐6 concentrations and future CVD event rates have been correlated independently in epidemiological studies. 9 , 10 However, no cardiovascular outcome trial addressing the potential beneficial role of direct lowering of IL‐6 signalling has been conducted to date.
Mendelian randomization (MR) studies can be of help in estimating the potential effects in clinical intervention trials. 11 MR studies have robustly shown the causal effect of IL‐6R perturbation on coronary heart disease (CAD) and related phenotypes such as ischaemic stroke, aortic aneurysm, atrial fibrillation and carotid plaque. 12 , 13 , 14 , 15 The effect of IL‐6 perturbation has, however, not been studied with similar vigour.
Differentiating between IL‐6 and IL‐6R perturbation is important because IL‐6 signalling is activated by binding of the circulating IL‐6 ligand to either the soluble or membrane‐bound IL‐6R. 16 Signal transduction via the soluble IL‐6R (trans‐signalling) is generally considered to be pro‐inflammatory, while transduction through membrane‐bound IL‐6R (classical signalling) is considered anti‐inflammatory. 16 Paradoxically, IL6R variants associated with reduced risk of CAD are associated with increased soluble IL‐6R. 12 , 15 , 17 Blockade of the IL‐6R by tocilizumab and inhibition of IL‐6 by siltuximab results in inhibition of both the classical and trans‐signalling pathways, and the observed increase of soluble IL‐6R by IL6R variants complicate the translation of results from IL6R MR studies to pharmacological effects. Moreover, multiple other ligands, including CNTF and IL‐30, have also shown to bind to IL‐6R. 18
Downregulation of IL‐6 signalling through IL6 variants will likely lead to both reduced trans‐ and classical signalling, without incorporating effects of any other ligands binding to IL‐6R. It is unknown whether perturbation of the IL‐6 ligand results in similar effects compared to perturbation of IL‐6R. Here, we performed a two‐sample MR study to evaluate and compare the phenotypic consequences of IL‐6 signalling perturbation through IL‐6R and IL‐6.
2. METHODS
In this MR study, we used the naturally occurring variation within and around the IL6 and IL6R gene to estimate the effect of inhibition of IL‐6 and IL‐6R on various clinical biomarkers and outcome. These models allow exploration of the directionality of a therapeutic compound on clinical outcome in trials and exploration of unanticipated on‐target side effects. 19 , 20 MR for drug–target discovery and validation and its assumptions have been reviewed elsewhere. 11 We used publicly available data, and the original studies were all approved by the relevant ethical committees.
2.1. Construction of genetic instruments
We constructed two distinct genetic instruments for IL‐6 and IL‐6R, by selecting genetic variants from within a 100 kb window around the genes encoding IL‐6 (IL6, ENSG00000136244) and IL‐6R (IL6R, ENSG00000160712). Instruments were extracted from a UK Biobank genome‐wide association study (GWAS), based on their associations (P‐value < 5 × 10−4) with CRP (http://www.nealelab.is/uk-biobank/). 21 We selected CRP levels as our exposure variable since, unlike IL‐6 levels, CRP levels are available for 361 194 participants. This results in more potential genetic variants compared to smaller IL‐6 level GWAS and additionally protects against weak instrument bias and bias through measurement error. 22 To strengthen our results and to support the assumption that our main genetic instruments indeed affect the targeted gene, we additionally created two genetic instruments following an eQTL criterion, first removing variants from the initial pool of potential CRP‐associated variants within 100 kb around the genes that did not associate with mRNA expression of IL6 or IL6R, using data on expression levels from eQTLGen and GTEx portal release V8 (results accessed on 6 May 2020) 23 , 24 (P‐value < 5 × 10−4; Figure 1). From the eQTL‐filtered variants we identified the final set of variants based on their CRP association (P‐value < 5 × 10−4). While this approach results in fewer variants available for analysis and thus less power, it ensures selected variants have an impact on the targeted gene. MR analyses were subsequently performed on both the main set of instruments (the IL6 and IL6R instruments without eQTL filter), and the eQTL‐filtered set. For each instrument, only variants with a minor allele frequency (MAF) of >0.01 and in low linkage disequilibrium (LD) (R 2 < 0.1, based on a 1000 Genomes European reference sample set 25 ) with the other variants in the genetic instrument eligible for inclusion. Clumping of the genetic instruments was performed using ieugwasr. 26 Due to differences in array coverage of the various GWAS, proxy variants (R 2 > 0.9, 1000 Genomes European reference sample set) were used to substitute unavailable variants where necessary. Variants for which no proxy was available were omitted from the analysis for that specific outcome.
FIGURE 1.
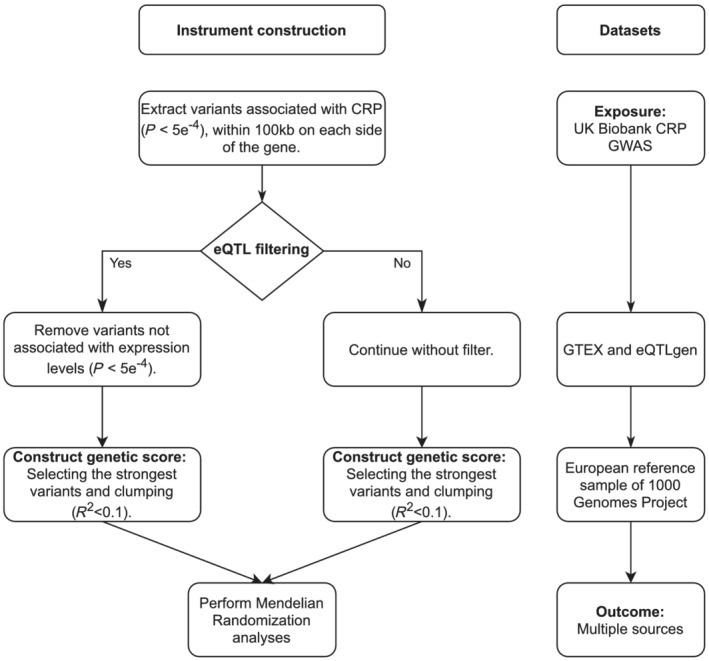
Genetic instrument construction. Diagram showing the protocol for selecting the genetic instruments used in this study. CRP, C‐reactive protein; eQTL, expression quantitative trait loci; GWAS, genome‐wide association study
2.2. Data and contributing studies
We sought to validate our genetic instruments by testing for their associations with IL‐6 levels and IL‐6 receptor levels, available from a GWAS employing Olink protein assays. 27 , 28 We tested our genetic instruments for associations with the following clinical outcome parameters of interest for future cardiovascular outcome trials: coronary artery disease (CAD), 29 any stroke and ischaemic stroke, 30 heart failure (HF), 31 type 2 diabetes (T2D) 32 and atrial fibrillation (AF). 33 Because therapeutic agents targeting IL‐6R are available for rheumatoid arthritis (RA), we also included RA in our analysis as a positive control. 34 To assess possible adverse impact of IL‐6 signalling therapy, we tested the associations of our genetic instruments with immunity biomarkers including white blood cell counts and differentiation (WBC), retrieved from studies including the UK Biobank and blood cell consortium. 35 , 36 Since tocilizumab treatment in RA patients results in increased LDL‐C levels, we extracted lipid and lipoprotein levels from a GWAS from the MAGNETIC consortium. 37 A previous study observed a suggestive effect of IL‐6 signalling on the risk for T2D. 13 Therefore, we included GWAS on glucose, HbA1c and body mass index (BMI) in our analysis. 37 , 38 , 39 As infections will be the main safety cause of concern in cardiovascular outcome trials, we assessed the possible adverse impact of IL‐6‐targeted therapy by including ICD10 summary data on any infection, and pneumonia from participants of the FinnGen study. 40 A full list of the datasets used for this analysis is provided in Table S1 in the Supporting Information.
2.3. Statistical analyses
The effects of on‐target IL‐6 or IL‐6R perturbation were estimated using Mendelian randomization, specifically using the inverse‐variance weighed (IVW) and the more robust MR‐Egger estimators. 41 , 42 MR‐Egger provides valid estimates even in extreme settings were 100% of the variants show a horizontal pleiotropy effect; however, this comes at the cost of comparatively low power. Hence, we subsequently applied the Rücker model selection framework to decide between both estimates. 42 First, we calculated both the IVW and Egger models. If the difference in Q – Q' between the IVW and the Egger model is significant, we considered the Egger model a better fit. Using Cochran's Q and Rucker's Q' statistic in assessment of pleiotropy improves prediction by penalizing outlying variants with a random effects model. Thus, based on the chosen model's Q, we then chose a fixed or random‐effects model. We present the results of the model with the best fit in the main text, and the fixed‐ or random‐effect results of all methods in Tables S3–S6 in the Supporting Information. In further sensitivity analyses, we also used the weighted median method. The potential for horizontal pleiotropy bias was further limited by selecting variants based on CRP from small cis‐regions known to encode IL‐6 or IL‐6R, and through the development of two additional genetic scores by firstly filtering based on the association with IL‐6 and IL‐6R mRNA and afterwards on CRP. All effects are standardized to a 1 mg/L reduction in CRP levels, and presented as mean difference (MD) or odds ratio (OR) with 95% confidence interval (CI). We provide estimates using a 95% confidence interval, and indicate which outcomes achieve a multiple testing threshold (defined as 0.05/28 outcomes/2 genes = 8.92 × 10−4) with a ‘#’. Furthermore, we employed the Kolmogorov–Smirnov test on the instrument‐specific 28 P‐values to assess if our results are due to multiple testing. 43 Finally, we provide precision of our results by calculating the squared standard error (Table S3–S6 in the Supporting Information), where a lower value indicates a higher precision, compared to the other analyses for that outcome. All analyses were performed using R version 3.6.1. 44 The plots were made using ggplot2. 45
2.4. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 46
3. RESULTS
3.1. Characteristics of genetic instruments for IL6‐signalling
Depending on the variant coverage of the outcome dataset available for analysis, up to eight variants were used in our IL6 instrument, and up to three variants using the eQTL filtering approach (Figures S1 and S2 in the Supporting Information). The combined F‐statistic for the main IL6 instrument was 164, and for the instrument with eQTL filtering 66. For IL6R, up to 19 variants were available for our instrument, while up to six variants were available for the eQTL‐filtered instrument. The combined F‐statistic for the main IL6R instrument was 1221, and for the instrument with eQTL filtering 770 (Figures S3–S4 in the Supporting Information). All variants included in the genetic instruments are listed in Table S2 in the Supporting Information and the number of variants available per analysis is listed in Table S3–S6 in the Supporting Information.
3.2. Effect of genetic instruments on IL‐6 levels
Genetically predicted lower CRP by the main instrument for IL6 was indeed associated with reduced IL‐6 levels (−0.17 SD units, 95% CI −0.30;−0.04; Figure 2) and genetically predicted lower CRP by the main IL6R instrument was associated with increased IL‐6 receptor levels (5.16 SD units, 95% CI: 3.86;6.45) and increased IL‐6 levels (0.43 SD units, 95% CI: 0.26;0.60; Figure 2). The associations of the main IL6R instrument with IL‐6 and IL‐6R levels and the eQTL‐filtered IL6R instrument with IL‐6R levels reached the multiple testing threshold. The results for the eQTL‐filtered instruments were consistent with the conventionally selected instruments (Tables S3–S6 in the Supporting Information).
FIGURE 2.
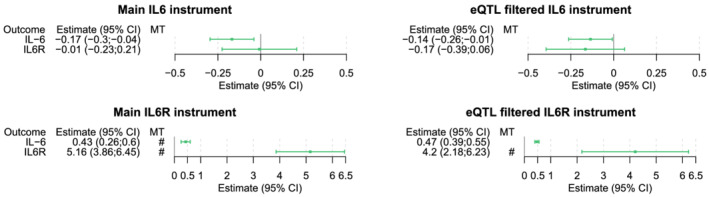
Effects of the genetic instruments on IL‐6 and IL‐6R levels. Forest plots representing the change in IL‐6 and IL‐6R levels for all genetic instruments, scaled to a 1 mg/L reduction in CRP levels. The bracket marks an association of the genetic instrument with IL‐6 or IL‐6R that meets the multiple testing threshold (8.92 × 10−4). CRP, C‐reactive protein; IL‐6, Interleukin‐6
3.3. Effect of inhibition of interleukin 6 signalling on clinical outcomes
Genetically predicted CRP reduction by the IL6 instrument is associated with lower odds for CAD (OR 0.86, 95% CI 0.77;0.96), AF (OR 0.86, 95% CI 0.77;0.95) and T2D (OR 0.83, 95% CI 0.75;0.92; Figure 3). The eQTL‐filtered analysis showed an association between CRP reduction by the IL6 instrument and HF (OR 0.77, 95% CI 0.63;0.94), and similar effect estimates with remaining outcomes compared to the IL6 instrument, albeit with wider confidence intervals. Only the association of the IL6 instrument with diabetes reached the multiple testing threshold.
FIGURE 3.
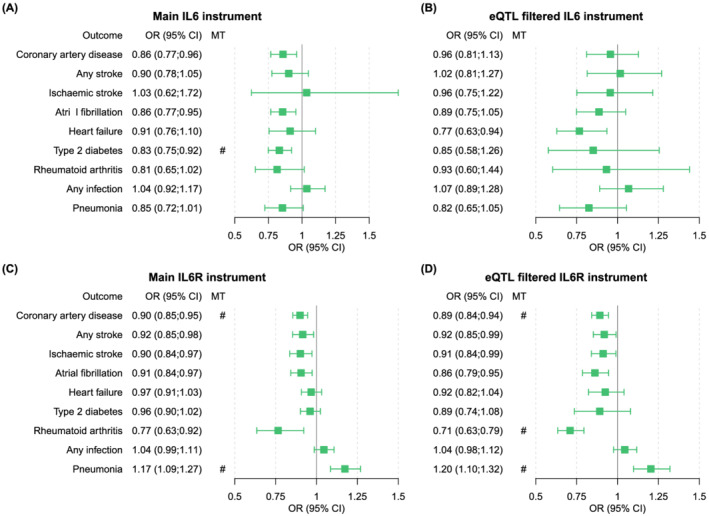
Drug target MR effects of IL‐6 and IL‐6R on clinical outcome, per 1 mg/L reduction in CRP. Forest plots representing the risk of clinical outcome parameters for all genetic instruments, scaled to a 1 mg/L reduction in CRP levels. The hash mark indicates an association of the genetic instrument with the clinical outcome that meets the multiple testing threshold (8.92 × 10−4)
The Mendelian randomization analysis of IL6R suggested that 1 mg/L lower CRP levels due to the IL6R instrument are associated with a lower risk for CAD (OR 0.90, 95% CI 0.86;0.95), any stroke (OR 0.92, 95% CI 0.85;0.98) and any ischaemic stroke (OR 0.90, 95% CI 0.84;0.97), AF (OR 0.91, 95% CI 0.84;0.97) and RA (OR 0.77, 95% CI 0.64;0.92; Figure 3). The IL6R instrument was also associated with an increased risk for pneumonia (OR 1.17, 95% CI 1.09;1.27). The eQTL‐filtered IL6R analysis confirmed effects on CAD (OR 0.89, 95% CI 0.84;0.95), any stroke (OR 0.92, 95% CI 0.85;0.99), any ischaemic stroke (OR 0.91, 95% CI 0.84;0.99), AF (OR 0.86, 95% CI 0.79;0.95), RA (OR 0.71, 95% CI 0.63;0.79) and pneumonia (OR 1.20, 95% CI 1.10;1.32). The associations of the IL6R main instrument with CAD and with pneumonia reached the multiple testing threshold, and the associations of the eQTL‐filtered IL6R instrument with CAD, RA and pneumonia also reached the multiple testing threshold. The estimates of the weighted median method were very similar to the effects of the IVW method (Tables S3–S6 and Figures S5–S12 in the Supporting Information).
3.4. Effect of inhibition of interleukin 6 signalling on safety biomarkers
The IL6 model was associated with reduced platelet counts (−0.100, 95% CI −0.16; −0.04), fibrinogen levels (−0.115, 95% CI −0.204; −0.026), HDL‐C (−0.865, 95% CI −1.335; −0.395), ApoA1 (−0.763, 95% CI −1.259; −0.266) and BMI (−0.068, 95% CI −0.107; −0.029), and with increased basophil counts (0.198, 95% CI 0.091;0.304). The eQTL‐filtered analysis showed associations between the IL6 instrument and increased white blood cell (0.817, 95% CI 0.439;1.195), neutrophil (0.464, 95% CI 0.07;0.859), basophil (0.468, 95% CI 0.044;0.891) and eosinophil counts (1.38, 95% CI 0.97;1.79), with reduced platelet counts (−0.100, 95% CI −0.188; −0.013) and with lower BMI (−0.052, 95% CI −0.103; −0.001). The associations of the main IL6 instrument with basophils, HDL‐C and BMI and the associations of the eQTL‐filtered IL6 instrument with white blood cell count and eosinophils reached the multiple testing threshold. The IL6R instrument was associated with increased monocyte (0.07, 95% CI 0.05;0.091), platelet counts (0.032, 95% CI 0.006;0.058) and with BMI (0.076, 95% CI 0.007;0.145), and was not associated with an effect on other leukocytes. The IL6R eQTL‐filtered analysis confirmed associations with increased monocytes (0.075, 95% CI 0.059;0.09) and showed associations with increased eosinophils (0.032, 95% CI 0.004;0.06), and with reduced lymphocyte counts (−0.072, 95% CI −0.121; −0.023) (Figure 4, and point estimates provided in Tables 3–6 in the Supporting Information). The associations of the main and eQTL‐filtered IL6R instrument with monocytes reached the multiple testing threshold. The estimates of the weighted median method for the safety biomarkers were also similar to the effects of the IVW method (Tables S3–S6 and Figures S5–S12 in the Supporting Information).
FIGURE 4.
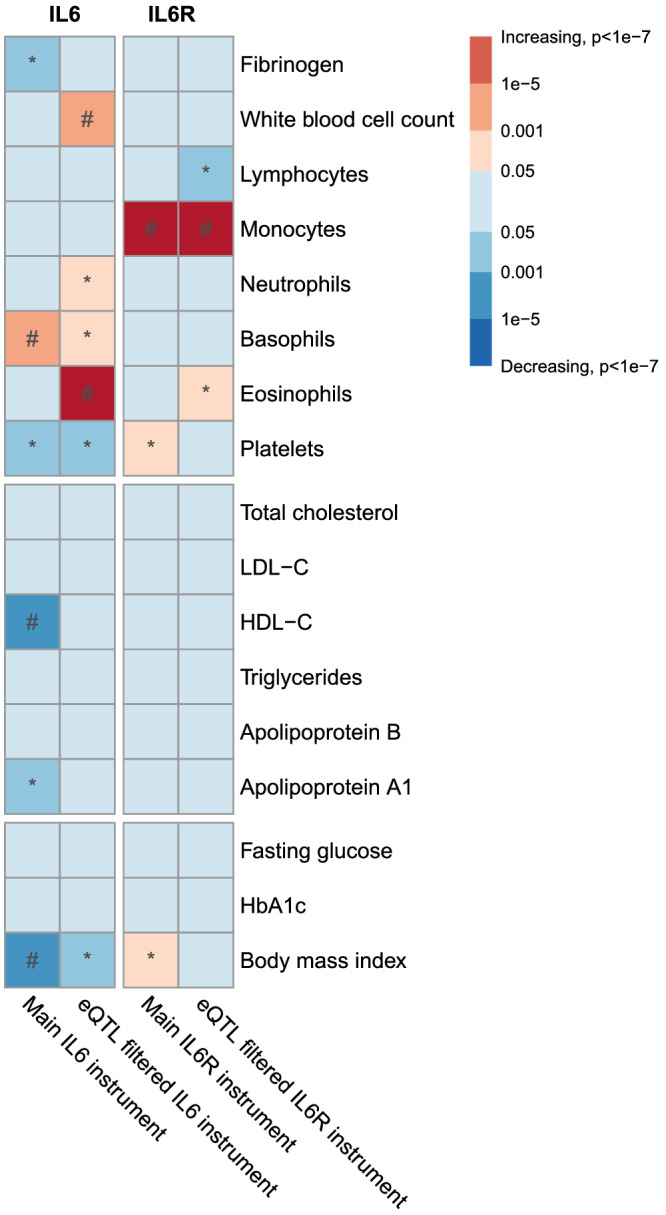
Drug target MR effects of IL‐6 and IL‐6R on biomarkers. The strength of the association for each genetic instrument with the outcome is depicted by the P‐value times the direction of the effect, per 1 mg/L CRP reduction. The asterisks mark an association of the genetic instrument with the trait of P < .05. The hash mark indicates an association of the genetic instrument with the trait meeting the multiple testing threshold (8.92 × 10−4). eQTL, expression quantitative trait loci
The Kolmogorov–Smirnov test was 1.0 × 10−3 and 9.8 × 10−3 for the main and eQTL‐filtered IL6 instrument, respectively, and 4.2 × 10−4 and 3.0 × 10−3 for the main and eQTL‐filtered IL6R instrument, respectively, indicating that our results are unlikely to be driven by false‐positive results (Figure S13 in the Supporting Information).
4. DISCUSSION
In this study, we used Mendelian randomization to predict the clinical effects of reducing IL‐6 signalling by pharmacologic inhibition of either IL‐6 ligand or IL‐6 receptor. We show that inhibition of IL‐6 ligand, mimicked through IL6, is nominally associated with risk reductions of CAD, AF and T2D, and observed a similar effects profile of IL‐6R inhibition, mimicked by IL6R, which was nominally associated with reduced risk for CAD, (ischaemic) stroke, AF and RA, at the potential cost of increased pneumonia risk. The association of the IL6 instrument with T2D and IL6R instrument with CAD and pneumonia reached the multiple testing threshold. Targeting IL‐6 signalling through pharmacologic inhibition of IL‐6 or IL‐6R will likely elicit similar favourable effects on clinical CVD outcomes. Unlike previous MR studies of these targets, we created additional genetic scores using variants associated with both CRP levels and mRNA expression of IL6 or IL6R. The results from these analyses generally resulted in wider confidence intervals, but were largely directional concordant, and any discordant results were within the 95% CI of each other. The directional concordance of the two eQTL‐filtered instruments with the main instruments provide further support for the causal effects of IL‐6‐signalling perturbation through either IL‐6 or IL‐6R on cardiometabolic disease.
Our observations that IL‐6 signalling inhibition through IL6 variants reduces the risk for CAD and atrial fibrillation is in accordance with the literature on IL6R variants and provides evidence for the protective benefits of perturbing IL‐6 signalling through inhibition of either IL‐6 or IL‐6R in CAD. 12 , 13 , 14 , 15 This is of importance as IL6 genetic variants likely inhibit both the classical and trans‐signalling pathway, unlike IL6R variants, from which some are described to be likely to upregulate IL‐6 trans‐signalling in certain tissues. 17 , 47 , 48 For example, the Asp358Ala variant in IL6R was shown to increase soluble IL‐6R and is associated with increased risk for asthma, atopic dermatitis and faster disease progression in ALS patients, and it was suggested that this is due to upregulation of the trans‐signalling pathway. 47 , 49 , 50 Based on our observations that both IL6 and IL6R instruments are associated with reduced CAD risk, we hypothesize that it is probably the classical‐signalling pathway that is involved in cardiovascular disease. Inhibition of the IL‐6 ligand may be pharmacologically preferential over targeting IL‐6R, as IL‐6R monoclonal antibodies have been shown to inhibit the IL‐6R ligands ciliary neurotrophic factor and IL‐30. 51 It is unlikely that a similar effect will be observed in patients treated with a monoclonal antibody directed against the IL‐6 ligand. Moreover, targeting the IL‐6 ligand instead of the IL‐6 receptor may also be preferential based on the potentially required therapeutic range of monoclonal antibodies, as levels of the IL‐6 ligand are normally in the range of 1 pg/mL but can rise dramatically in periods of acute inflammation, compared to a relatively stable range of 50–75 ng/mL for IL‐6R. 52 It is possible that the effective therapeutic range of IL‐6 monoclonal antibodies for CAD prevention will be lower than that for IL‐6R antibodies, potentially resulting in less influence of IL‐6 antibodies on acute phase reactions (such as infections), compared to IL‐6R antibodies.
We show that inhibition of IL‐6 signalling by our IL6 instrument is associated with a protective effect on the risk for type 2 diabetes, even after correction for multiple testing. A large epidemiological study previously showed that IL‐6 levels and CRP levels are associated with the risk for T2D, 53 and genetic studies also observed a directionally consistent association between variation in the IL6R locus and T2D, with IL6R variant rs7529229 showing a trend towards a lower risk (OR 0.97, 95% CI 0.94;1.00). 13 , 54 Although IL‐6 has been shown to reduce hepatic insulin sensitivity, the exact role of IL‐6 in glucose metabolism has not been elucidated, as some studies have also shown a beneficial role of IL‐6 on peripheral insulin sensitivity. 55 It is of note that we did not observe an effect on either glucose or HbA1c. The observed beneficial effect of tocilizumab on insulin sensitivity and HbA1c levels (in both T2D and non‐T2D subjects) supports the notion that inhibition of the IL‐6 signalling pathway may have beneficial effects on glucose metabolism, 56 , 57 a finding that was subsequently confirmed in clinical trials by showing improved fasting blood glucose and HbA1c levels in RA patients with T2D randomized to the IL‐6R monoclonal antibody sarilumab. 58 In contrast, a number of MR studies have shown that LDL‐C lowering by statins and proprotein convertase subtilisin–kexin type 9 inhibition confers a low, but consistent increased risk for T2D. 19 , 20 , 59 , 60
In anticipation on any adverse effects of interest for future clinical trials, we observed that IL‐6 signalling inhibition by IL6R variants was associated with increased risk for pneumonia. In contrast, IL6 variants showed a trend towards a lower risk for pneumonia. These results might indicate that inhibition of IL‐6 might have a more favourable infection risk profile than inhibition of IL‐6R. The Asp358Ala variant in IL6R was associated with increased soluble IL‐6R in lung tissue and was shown to act pro‐inflammatorily in lung cells. 47 The variant was associated with atopic asthma, but not with COPD. 47 A recent MR study showed that IL6R variants were associated with increased pneumonia. 61 This is in line with findings for tocilizumab, which is associated with increased risk for opportunistic and serious bacterial infections in a dose‐dependent manner, with pneumonia, urinary tract infections and cellulitis most frequently mentioned. 62 Future research is warranted to investigate the discrepancy between IL‐6 inhibition through IL6 or IL6R variants observed in this study.
This is the first MR study attempting to directly compare IL‐6 signalling inhibition through either IL6 or IL6R genetic variants on various clinical outcomes and biomarkers. The IL6 locus is a relatively well‐preserved locus with few genetic variants strongly affecting IL6, limiting the power of these analyses unlike for IL6R. It is therefore reassuring that the effect direction of inhibition of IL‐6 signalling inhibition through either IL6 or IL6R for the expected clinical outcomes (e.g., CAD, RA) was similar. However, this could imply that a number of non‐significant concordant associations are simply due to a lack of power. In addition, we have also observed some surprising associations and discrepancies between IL6 and IL6R, and between the main and eQTL‐filtered instruments (e.g., the effect on monocytes, platelets, BMI). This is also the first of a number of limitations in our study that warrant further discussion. First, we selected our IL6 and IL6R variants based on the downstream biomarker CRP, which is an indirect estimation of their function. However, IL‐6 and IL‐6R GWAS are small compared to the CRP GWAS we used and would preclude us from comparing the effects of both targets based on a standardized measurement. Our model framework using an eQTL filter provides an additional layer of evidence, but at the cost of power, since these instruments contain many fewer variants. Second, we included a proxy variant when a variant from our preferred genetic instrument was not available for a specific trait. However, since proxy variants were not always available, the number of variants in some of the analyses was limited. We guarded against weak instrument bias by selecting variants with an F‐statistic above 10. Third, we did not correct for multiple testing, since this article was meant to be hypothesis‐generating to inform the ongoing trial effort. Especially regarding adverse outcomes, we consider it important to report any association we find, as these results can be important for safety analyses in clinical trials. Fourth, while the cis focus of our analysis severely limited the potential for horizontal pleiotropy, some variant known to encode IL6 or IL6R were nevertheless in LD with variants affecting neighbouring genes. None of these neighbouring genes for which multiple associations exist with our included variants were, however, related to the considered phenotypes (see Tables S7 and S8 in the Supporting Information), limiting the potential for any LD‐based horizontal pleiotropy. Moreover, we have implemented several steps to guard against any inadvertent horizontal pleiotropy bias, hence should any bias remain this is likely minimal. We selected the 100 kb region because approximately 92% of all lead cis‐eQTL variants are anticipated to reside within 100 kb of the gene. 23 Fifth, our analyses make use of data derived mainly from European cohorts. Caution is warranted in translating our results to other ethnicities. Last, our model is based on the effect of modest genetic disturbances over the course of a lifetime. Caution is warranted in directly translating these effects towards an anticipated effect of a therapeutic agent that will have a potentially larger effect but over a shorter period of time.
In summary, in this study we show that inhibition of the IL‐6 signalling pathway, either by targeting IL‐6 or IL‐6R is likely to result in beneficial effects on the risk for CAD, stroke, AF and type 2 diabetes, but the observed association of IL6R with pneumonia risk warrants caution and should be evaluated in clinical trials.
COMPETING INTERESTS
A.J.C. has nothing to disclose. F.W.A. and A.F.S. have received Servier funding for unrelated work. P.N. has received research support from Apple, Amgen and Boston Scientific, personal fees from Apple, Genentech, Novartis and Blackstone Life Sciences, and spousal employment at Vertex, all unrelated to the present work. P.M.R. has served as a consultant to Flame, Agepha, Corvidia, Inflammazome, Novartis, Amgen, Merck and Civi Bio and is listed as co‐inventor on patents related to the use of inflammatory biomarkers in CVD and diabetes that are no longer active. G.K.H. has received funding from Regeneron, Aegerion, Amgen, AstraZeneca, Eli Lilly, Genzyme, Kowa, Pfizer, Regeneron Pharmaceuticals, Roche, Sanofi, The Medicines Company, Ionis and personal fees from Novo Nordisk.
CONTRIBUTORS
A.J.C., F.W.A., G.K.H. and A.F.S. designed the study. A.J.C. and A.F.S. performed the analyses. A.J.C., F.W.A., P.N., P.M.R., G.K.H. and A.F.S. contributed to the interpretation of the data and the drafting of the manuscript. A.J.C. and A.F.S. had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Supporting information
Table S1 List of outcomes
Table S2 List of instruments
Table S3 Results from main IL6 instrument
Table S4 Results from eQTL filtered IL6 instrument
Table S5 Results from main IL6R instrument
Table S6 Results from eQTL filtered IL6R instrument
Table S7 eQTL associations for IL6 variants
Table S8 eQTL associations for IL6R variants
Figure S1 Locus plot for IL6 showing the genomic region around the IL6 gene
Figure S2 CRP effect estimates for each included genetic variant in the IL6 scores (in mg/L). The first mentioned allele is the CRP‐ reducing effect allele. Red are the eQTL‐filtered variants, blue are the regularly selected variants
Figure S3 Locus plot for IL6R showing the genomic region around the IL6R gene
Figure S4 CRP effect estimates for each included genetic variant in the IL6R scores (in mg/L). The first mentioned allele is the CRP‐reducing effect allele. Red are the eQTL‐filtered variants, blue are the regularly selected variants.
Figure S5 Forest plot for main IL6 instrument and biomarkers
Figure S6 Forest plot for main IL6 instrument and clinical outcome
Figure S7 Forest plot for eQTL‐filtered IL6 instrument and biomarkers
Figure S8 Forest plot for eQTL‐filtered IL6 instrument and clinical outcome
Figure S9 Forest plot for main IL6R instrument and biomarkers
Figure S10 Forest plot for main IL6R instrument and clinical outcome
Figure S11 Forest plot for eQTL‐filtered IL6R instrument and biomarkers
Figure S12 Forest plot for eQTL‐filtered IL6R instrument and clinical outcome
Figure S13 Distribution of P‐values for each instrument, compared to the uniform distribution
Genetic variant candidates and R markdown script
ACKNOWLEDGEMENTS
A.J.C. is supported by grants from the Atheros fund and the AMC Young Talent Fund. F.W.A. is supported by UCL Hospitals NIHR Biomedical Research Centre. P.N. is supported by grants from the National Heart, Lung, and Blood Institute (R01HL142711, R01HL148565, R01HL148050, R01HL151283), Fondation Leducq (TNE‐18CVD04), and Hassenfeld Scholar Award from the Massachusetts General Hospital. P.M.R. has received investigator‐initiated research grants from Kowa, Novartis, Pfizer, AstraZeneca, NHLBI and NCI. G.K.H. is supported by grants from Netherlands Organization for Scientific Research, grants from Klinkerpad fonds, and grants from the European Union. A.F.S. is supported by BHF grant PG/18/5033837 and the UCL BHF Research Accelerator AA/18/6/34223.
Cupido AJ, Asselbergs FW, Natarajan P, et al. Dissecting the IL‐6 pathway in cardiometabolic disease: A Mendelian randomization study on both IL6 and IL6R . Br J Clin Pharmacol. 2022;88(6):2875-2884. doi: 10.1111/bcp.15191
DATA AVAILABILITY STATEMENT
The datasets were derived from sources in the public domain and analysed using in‐house scripts. A list of the data sources and R code is enclosed in the supplemental materials.
REFERENCES
- 1. Ridker PM. From C‐reactive protein to interleukin‐6 to interleukin‐1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118(1):145‐156. doi: 10.1161/CIRCRESAHA.115.306656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ridker PM, Cushman M, Stampfer MJ, Tracy RP, Hennekens CH. Inflammation, aspirin, and the risk of cardiovascular disease in apparently healthy men. N Engl J Med. 1997;336(14):973‐979. doi: 10.1056/NEJM199704033361401 [DOI] [PubMed] [Google Scholar]
- 3. Kaptoge S, Di Angelantonio E, Lowe G, et al. C‐reactive protein concentration and risk of coronary heart disease, stroke, and mortality: an individual participant meta‐analysis. The Lancet. 2010;375(9709):132‐140. doi: 10.1016/S0140-6736(09)61717-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ridker PM. Anticytokine agents: targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ Res. 2019;124(3):437‐450. doi: 10.1161/CIRCRESAHA.118.313129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eiriksdottir G, Harris TB, Launer LJ, et al. Association between C reactive protein and coronary heart disease: Mendelian randomisation analysis based on individual participant data. BMJ. 2011;342(7794):425‐432. doi: 10.1136/bmj.d548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377(12):1119‐1131. doi: 10.1056/NEJMoa1707914 [DOI] [PubMed] [Google Scholar]
- 7. Ridker PM, Libby P, MacFadyen JG, et al. Modulation of the interleukin‐6 signalling pathway and incidence rates of atherosclerotic events and all‐cause mortality: analyses from the Canakinumab Anti‐Inflammatory Thrombosis Outcomes Study (CANTOS). Eur Heart J. 2018;39(38):3499‐3507. doi: 10.1093/eurheartj/ehy310 [DOI] [PubMed] [Google Scholar]
- 8. Desai M, Pieper KS, Mahaffey K. Challenges and solutions to pre‐ and post‐randomization subgroup analyses. Curr Cardiol Rep. 2014;16(10):1‐8. doi: 10.1007/s11886-014-0531-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Danesh J, Kaptoge S, Mann AG, et al. Long‐term interleukin‐6 levels and subsequent risk of coronary heart disease: two new prospective studies and a systematic review. PLoS Med. 2008;5(4):600‐610. doi: 10.1371/journal.pmed.0050078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cainzos‐Achirica M, Enjuanes C, Greenland P, et al. The prognostic value of interleukin 6 in multiple chronic diseases and all‐cause death: the Multi‐Ethnic Study of Atherosclerosis (MESA). Atherosclerosis. 2018;278:217‐225. doi: 10.1016/j.atherosclerosis.2018.09.034 [DOI] [PubMed] [Google Scholar]
- 11. Schmidt AF, Finan C, Gordillo‐Marañón M, et al. Genetic drug target validation using Mendelian randomisation. Nat Commun. 2020;11(1):3255. doi: 10.1038/s41467-020-16969-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sarwar N, Butterworth AS, Freitag DF, et al. Interleukin‐6 receptor pathways in coronary heart disease: a collaborative meta‐analysis of 82 studies. The Lancet. 2012;379(9822):1205‐1213. doi: 10.1016/S0140-6736(11)61931-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Swerdlow DI, Holmes MV, Kuchenbaecker KB, et al. The interleukin‐6 receptor as a target for prevention of coronary heart disease: a Mendelian randomisation analysis. The Lancet. 2012;379(9822):1214‐1224. doi: 10.1016/S0140-6736(12)60110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rosa M, Chignon A, Li Z, et al. A Mendelian randomization study of IL6 signaling in cardiovascular diseases, immune‐related disorders and longevity. NPJ Genom Med. 2019;4:23. doi: 10.1038/s41525-019-0097-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Georgakis MK, Malik R, Gill D, Franceschini N, Sudlow CLM, Dichgans M. Interleukin‐6 signaling effects on ischemic stroke and other cardiovascular outcomes: a Mendelian randomization study. Circ Genom Precis Med. 2020;13(3):2872. doi: 10.1161/CIRCGEN.119.002872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Garbers C, Heink S, Korn T, Rose‐John S. Interleukin‐6: designing specific therapeutics for a complex cytokine. Nat Rev Drug Discov. 2018;17(6):395‐412. doi: 10.1038/nrd.2018.45 [DOI] [PubMed] [Google Scholar]
- 17. Van Dongen J, Jansen R, Smit D, et al. The contribution of the functional IL6R polymorphism rs2228145, eQTLs and other genome‐wide SNPs to the heritability of plasma sIL‐6R levels. Behav Genet. 2014;44(4):368‐382. doi: 10.1007/s10519-014-9656-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Garbers C, Aparicio‐Siegmund S, Rose‐John S. The IL‐6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75‐82. doi: 10.1016/j.coi.2015.02.008 [DOI] [PubMed] [Google Scholar]
- 19. Schmidt AF, Swerdlow DI, Holmes MV, et al. PCSK9 genetic variants and risk of type 2 diabetes: a Mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5(2):97‐105. doi: 10.1016/S2213-8587(16)30396-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ference BA, Robinson JG, Brook RD, et al. Variation in PCSK9 and HMGCR and risk of cardiovascular disease and diabetes. N Engl J Med. 2016;375(22):2144‐2153. doi: 10.1056/NEJMoa1604304 [DOI] [PubMed] [Google Scholar]
- 21. Palmer TM, Sterne JAC, Harbord RM, et al. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol. 2011;173(12):1392‐1403. doi: 10.1093/aje/kwr026 [DOI] [PubMed] [Google Scholar]
- 22. Bowden J, Fabiola Del Greco M, Minelli C, Smith GD, Sheehan NA, Thompson JR. Assessing the suitability of summary data for two‐sample Mendelian randomization analyses using MR‐Egger regression: the role of the I2 statistic. Int J Epidemiol. 2016;45(6):1961‐1974. doi: 10.1093/ije/dyw220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Võsa U, Claringbould A, Westra HJ, et al. Unraveling the polygenic architecture of complex traits using blood eQTL metaanalysis. bioRxiv. 2018;18:447367. doi: 10.1101/447367 [DOI] [Google Scholar]
- 24. Aguet F, Barbeira AN, Bonazzola R, et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369(6509):1318‐1330. doi: 10.1126/SCIENCE.AAZ1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. doi: 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hemani G, Zheng J, Elsworth B, et al. The MR‐Base platform supports systematic causal inference across the human phenome. eLife. 2018;7. doi: 10.7554/eLife.34408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ahluwalia TS, Prins BP, Abdollahi M, et al. Genome‐wide association study of circulating interleukin 6 levels identifies novel loci. Hum Mol Genet. 2021;30(5):393‐409. doi: 10.1093/HMG/DDAB023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Folkersen L, Gustafsson S, Wang Q, et al. Genomic and drug target evaluation of 90 cardiovascular proteins in 30,931 individuals. Nat Metab. 2020;2(10):1135‐1148. doi: 10.1038/s42255-020-00287-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. 2018;122(3):433‐443. doi: 10.1161/CIRCRESAHA.117.312086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Malik R, Chauhan G, Traylor M, et al. Multiancestry genome‐wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50(4):524‐537. doi: 10.1038/s41588-018-0058-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shah S, Henry A, Roselli C, et al. Genome‐wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. 2020;11:163. doi: 10.1038/s41467-019-13690-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mahajan A, Taliun D, Thurner M, et al. Fine‐mapping type 2 diabetes loci to single‐variant resolution using high‐density imputation and islet‐specific epigenome maps. Nat Genet. 2018;50(11):1505‐1513. doi: 10.1038/s41588-018-0241-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nielsen JB, Thorolfsdottir RB, Fritsche LG, et al. Biobank‐driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50(9):1234‐1239. doi: 10.1038/s41588-018-0171-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014;506(7488):376‐381. doi: 10.1038/nature12873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Astle WJ, Elding H, Jiang T, et al. The allelic landscape of human blood cell trait variation and links to common complex disease. Cell. 2016;167(5):1415‐1429.e19. doi: 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vuckovic D, Bao EL, Akbari P, et al. The polygenic and monogenic basis of blood traits and diseases. Cell. 2020;182(5):1214‐1231.e11. doi: 10.1016/J.CELL.2020.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kettunen J, Demirkan A, Würtz P, et al. Genome‐wide study for circulating metabolites identifies 62 loci and reveals novel systemic effects of LPA. Nat Commun. 2016;7:11122. doi: 10.1038/ncomms11122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prins BP, Kuchenbaecker KB, Bao Y, et al. Genome‐wide analysis of health‐related biomarkers in the UK Household Longitudinal Study reveals novel associations. Sci Rep. 2017;7:11008. doi: 10.1038/s41598-017-10812-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yengo L, Sidorenko J, Kemper KE, et al. Meta‐analysis of genome‐wide association studies for height and body mass index in ∼700000 individuals of European ancestry. Hum Mol Genet. 2018;27(20):3641‐3649. doi: 10.1093/HMG/DDY271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. FinnGen FinnGen Documentation of R4 release. https://finngen.gitbook.io/documentation/. Published 2020. Accessed September 9, 2020.
- 41. Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol. 2013;37(7):658‐665. doi: 10.1002/gepi.21758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bowden J, Spiller W, Del Greco FM, et al. Improving the visualization, interpretation and analysis of two‐sample summary data Mendelian randomization via the Radial plot and Radial regression. Int J Epidemiol. 2018;47(4):1264‐1278. doi: 10.1093/ije/dyy101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Storey JD. A direct approach to false discovery rates. J R Stat Soc Series B Stat Methodology. 2002;64(3):479‐498. doi: 10.1111/1467-9868.00346 [DOI] [Google Scholar]
- 44. R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing. 2019. https://www.r-project.org/. Published 2019. Accessed December 30, 2021.
- 45. Wickham H. Ggplot2: Elegant Graphics for Data Analysis. New York: Springer; 2016. [Google Scholar]
- 46. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Catalytic receptors. Br J Pharmacol. 2019;176(S1):S247‐S296. doi: 10.1111/BPH.14751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Farahi N, Paige E, Balla J, et al. Neutrophil‐mediated IL‐6 receptor trans‐signaling and the risk of chronic obstructive pulmonary disease and asthma. Hum Mol Genet. 2017;26(8):1584‐1596. doi: 10.1093/hmg/ddx053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mahajan A, Sim X, Ng HJ, et al. Identification and functional characterization of G6PC2 coding variants influencing glycemic traits define an effector transcript at the G6PC2‐ABCB11 locus. PLoS Genet. 2015;11(1):1‐25. doi: 10.1371/journal.pgen.1004876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ferreira MAR, Matheson MC, Duffy DL, et al. Identification of IL6R and chromosome 11q13.5 as risk loci for asthma. The Lancet. 2011;378(9795):1006‐1014. doi: 10.1016/S0140-6736(11)60874-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wosiski‐Kuhn M, Robinson M, Strupe J, et al. IL6 receptor 358 Ala variant and trans‐signaling are disease modifiers in amyotrophic lateral sclerosis. Neurol Neuroimmunol Neuroinflamm. 2019;6(6):e631. doi: 10.1212/NXI.0000000000000631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nitz R, Lokau J, Aparicio‐Siegmund S, Scheller J, Garbers C. Modular organization of interleukin‐6 and interleukin‐11 α‐receptors. Biochimie. 2015;119:175‐182. doi: 10.1016/j.biochi.2015.11.005 [DOI] [PubMed] [Google Scholar]
- 52. Schaper F, Rose‐John S. Interleukin‐6: biology, signaling and strategies of blockade. Cytokine Growth Factor Rev. 2015;26(5):475‐487. doi: 10.1016/j.cytogfr.2015.07.004 [DOI] [PubMed] [Google Scholar]
- 53. Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM. C‐reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286(3):327‐334. doi: 10.1001/jama.286.3.327 [DOI] [PubMed] [Google Scholar]
- 54. Aparicio‐Siegmund S, Garbers Y, Flynn CM, et al. The IL‐6‐neutralizing sIL‐6R‐sgp130 buffer system is disturbed in patients with type 2 diabetes. Am J Physiol Endocrinol Metab. 2019;317(2):E411‐E420. doi: 10.1152/ajpendo.00166.2019 [DOI] [PubMed] [Google Scholar]
- 55. Akbari M, Hassan‐Zadeh V. IL‐6 signalling pathways and the development of type 2 diabetes. Inflammopharmacology. 2018;26(3):685‐698. doi: 10.1007/s10787-018-0458-0 [DOI] [PubMed] [Google Scholar]
- 56. Ogata A, Morishima A, Hirano T, et al. Improvement of HbA1c during treatment with humanised anti‐interleukin 6 receptor antibody, tocilizumab. Ann Rheum Dis. 2011;70(6):1164‐1165. doi: 10.1136/ard.2010.132845 [DOI] [PubMed] [Google Scholar]
- 57. Otsuka Y, Kiyohara C, Kashiwado Y, et al. Effects of tumor necrosis factor inhibitors and tocilizumab on the glycosylated hemoglobin levels in patients with rheumatoid arthritis; an observational study. PLoS One. 13(4):e0196368. doi: 10.1371/journal.pone.0196368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Genovese MC, Fleischmann R & Hagino O et al. The effect of sarilumab in combination with Dmards on fasting glucose and glycosylated hemoglobin in patients with rheumatoid arthritis with and without Diabetes. 2017 American College of Rheumatology Annual Meeting.
- 59. Swerdlow DI, Preiss D, Kuchenbaecker KB, et al. HMG‐coenzyme A reductase inhibition, type 2 diabetes, and bodyweight: evidence from genetic analysis and randomised trials. The Lancet. 2015;385(9965):351‐361. doi: 10.1016/S0140-6736(14)61183-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. White J, Swerdlow DI, Preiss D, et al. Association of lipid fractions with risks for coronary artery disease and diabetes. JAMA Cardiol. 2016;1(6):692‐699. doi: 10.1001/jamacardio.2016.1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Larsson SC, Burgess S, Gill D. Genetically proxied interleukin‐6 receptor inhibition: opposing associations with COVID‐19 and pneumonia. Eur Respir J. 2020;57(1):2003545. doi: 10.1183/13993003.03545-2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Winthrop KL, Mariette X, Silva JT, et al. ESCMID Study Group for Infections in Compromised Hosts (ESGICH) Consensus Document on the safety of targeted and biological therapies: an infectious diseases perspective (Soluble immune effector molecules [II]: agents targeting interleukins, immunoglobulins and complement factors). Clin Microbiol Infect. 2018;24:S21‐S40. doi: 10.1016/j.cmi.2018.02.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 List of outcomes
Table S2 List of instruments
Table S3 Results from main IL6 instrument
Table S4 Results from eQTL filtered IL6 instrument
Table S5 Results from main IL6R instrument
Table S6 Results from eQTL filtered IL6R instrument
Table S7 eQTL associations for IL6 variants
Table S8 eQTL associations for IL6R variants
Figure S1 Locus plot for IL6 showing the genomic region around the IL6 gene
Figure S2 CRP effect estimates for each included genetic variant in the IL6 scores (in mg/L). The first mentioned allele is the CRP‐ reducing effect allele. Red are the eQTL‐filtered variants, blue are the regularly selected variants
Figure S3 Locus plot for IL6R showing the genomic region around the IL6R gene
Figure S4 CRP effect estimates for each included genetic variant in the IL6R scores (in mg/L). The first mentioned allele is the CRP‐reducing effect allele. Red are the eQTL‐filtered variants, blue are the regularly selected variants.
Figure S5 Forest plot for main IL6 instrument and biomarkers
Figure S6 Forest plot for main IL6 instrument and clinical outcome
Figure S7 Forest plot for eQTL‐filtered IL6 instrument and biomarkers
Figure S8 Forest plot for eQTL‐filtered IL6 instrument and clinical outcome
Figure S9 Forest plot for main IL6R instrument and biomarkers
Figure S10 Forest plot for main IL6R instrument and clinical outcome
Figure S11 Forest plot for eQTL‐filtered IL6R instrument and biomarkers
Figure S12 Forest plot for eQTL‐filtered IL6R instrument and clinical outcome
Figure S13 Distribution of P‐values for each instrument, compared to the uniform distribution
Genetic variant candidates and R markdown script
Data Availability Statement
The datasets were derived from sources in the public domain and analysed using in‐house scripts. A list of the data sources and R code is enclosed in the supplemental materials.