

Abstract
Background and purpose
Variants in the glucocerebrosidase (GBA) gene are recognized as a common and important genetic risk factor for Parkinson disease (PD). However, the impact of variant severity on the clinical phenotype of PD in the Chinese population remains unclear. Thus, the present study aimed to determine the frequency of GBA‐related PD (GBA‐PD) and the relationship of GBA variant severity with clinical characteristics in a large Chinese cohort.
Methods
Long‐range polymerase chain reaction and next generation sequencing were performed for the entire GBA gene. GBA variant severity was classified into five classes: mild, severe, risk, complex, and unknown.
Results
Among the total 737 PD patients, 47 GBA variants were detected in 79 (10.72%) patients, and the most common GBA variants were R163Q, L444P, and R120W. Complete demographic and clinical data were obtained for 673 patients, which revealed that 18.50% of early onset PD patients had GBA variants. Compared with patients without GBA variants, GBA‐PD patients experienced PD onset an average of 4 years earlier and had more severe motor and nonmotor symptoms. Patients carrying severe and complex variants had a higher burden of nonmotor symptoms, especially depression, and more mood/cognitive and gastrointestinal symptoms than patients carrying mild variants.
Conclusions
GBA‐PD is highly prevalent in the Chinese population. The severity of GBA variants underlies distinct phenotypic spectrums, with PD patients carrying severe and complex variants seeming to have similar phenotypes. PD patient stratification by GBA variant severity should become a prerequisite for selecting specific treatments.
Keywords: GBA, genotype–phenotype correlations, Parkinson disease, prevalence
The severity of GBA variants underlies distinct phenotypic spectrums, with Parkinson disease (PD) patients carrying severe and complex variants seeming to have similar phenotypes. PD patient stratification by GBA variant severity should become a prerequisite for selecting specific treatments.
![]()
INTRODUCTION
Parkinson disease (PD) is a chronic progressive neurodegenerative disorder defined by the loss of dopaminergic neurons in the substantia nigra and the presence of alpha‐synuclein (α‐syn) protein aggregation [1, 2]. It is well known that genetic factors influence PD susceptibility, especially variants in the glucocerebrosidase (GBA) gene [3, 4]. Heterozygous variants in the GBA gene, which encodes the lysosomal enzyme β‐glucocerebrosidase (GCase) that hydrolyzes glucocerebroside to glucose and ceramide, are recognized as the most common and important genetic risk factor for sporadic PD, increasing the risk of developing PD by 5%–30% [5, 6, 7, 8].
It is estimated that 7%–15% of PD patients worldwide harbor a heterozygous GBA variant. However, due to ethnic heterogeneity in GBA variants, the frequency of GBA variants varies greatly between populations, ranging from 10%–31% in the Ashkenazi Jewish (AJ) population to 3%–12% in non‐AJ North Americans and 2.1%–8.7% in the Chinese population [9, 10, 11, 12]. Notably, the presence of a highly homologous pseudogene (GBAP1), which is located 16 kb downstream of the functional GBA gene with 96% of the shared exon sequence, makes GBA sequencing challenging [13, 14]. As a result, most studies are limited to screening the most common GBA variants, such as L444P and N370S [15, 16, 17, 18], and the prevalence may thus be underestimated.
Overall, GBA‐related PD (GBA‐PD) patients have earlier onset, higher prevalence of the postural instability gait difficulty (PIGD) phenotype, worse motor symptoms, more frequent nonmotor symptoms (NMSs; especially cognitive impairment), more rapid progression, and reduced survival compared with non‐GBA‐mutated PD (NM‐PD) patients [17, 19, 20, 21, 22]. A recent study explored the impact of GBA status on the clinical phenotype of PD in the Italian population, and found that different variant types exhibited distinct phenotypic characteristics [23]. To the best of our knowledge, studies linking the severity of different GBA variants to clinical features in the Chinese PD population have not been performed previously, so the genotype–phenotype correlations of GBA‐PD patients cannot be fully elucidated.
In the present study, the entire GBA gene was screened in a large cohort of Chinese PD patients. We then conducted a comprehensive comparison of demographic, motor, and nonmotor characteristics in PD patients with and without GBA variants, as well as in patients with GBA variants of varying severity in the GBA‐PD group.
METHODS
Participants
From January 2012 to June 2020, a total of 737 unrelated Chinese PD patients were recruited at the Department of Neurology of the Affiliated Brain Hospital of Nanjing Medical University. All patients participating in this study were evaluated by a movement disorder specialist and diagnosed with PD based on the UK Parkinson's Disease Society Brain Bank clinical diagnostic criteria [24]. The exclusion criteria for this study were as follows: (i) atypical or secondary parkinsonism; (ii) severe chronic diseases such as heart failure, kidney failure, etc.; and (iii) clinically significant lesions revealed by brain magnetic resonance imaging. This study was approved by the Medical Ethics Committee of the Affiliated Brain Hospital of Nanjing Medical University (2011‐KY003, 2015‐KY030, and 2019‐KY019‐01) and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all participants before starting the experiment.
Clinical assessment
We successfully collected demographic, motor, and nonmotor characteristics for 673 of the 737 patients. The demographic data included age, gender, years of formal education, age at onset, frequency of early onset PD (EOPD), family history for PD, years of disease duration, and levodopa equivalent daily dose (LEDD). The age at onset in PD patients was defined as the age when motor symptoms first appeared. Patients with an age at onset of ≤50 years were classified as EOPD [25]. Information about dopaminergic drug use was collected, and the LEDD was calculated [26]. The evaluated motor features included the Unified Parkinson's Disease Rating Scale (UPDRS) part II, UPDRS part Ⅲ, and modified Hoehn‐Yahr (H‐Y) stage, which reflect activities of daily living (ADL), motor disability, and severity of disease, respectively. According to the formula proposed by Jankovic et al. [27], PD patients were divided into tremor‐dominant (TD), indeterminate, and PIGD subtypes. The assessed nonmotor features included global cognition, mood, and sleep. General cognition was assessed by the Mini‐Mental State Examination and Montreal Cognitive Assessment (MoCA) [28]. To correct for the education effect, if the education period of PD patients was ≤12 years, the MoCA score (if < 30) was increased by 1 point. Depression and anxiety were measured using the Hamilton Depression Scale (HAMD) and Hamilton Anxiety Scale, respectively. The Parkinson Disease Sleep Scale (PDSS) was used to evaluate sleep. The Non‐Motor Symptoms Questionnaire (NMSQuest) for PD was used to evaluate patients' NMSs and divided into nine domains, namely, cardiovascular, sleep, mood/cognitive, perception/hallucinations, attention/memory, gastrointestinal, urinary, sexual function, and miscellaneous [29, 30].
Molecular analysis of GBA variants
To avoid sequencing the nearby GBAP1, the long‐range polymerase chain reaction (LR‐PCR) protocol was implemented with GBA gene‐specific primers (F: AGGTCCCTGAGACAGATACTGG; R: CAATGAGACTTGAGGAAGGGCTC) and the TaKaRa LA Taq DNA Polymerase Hot‐Start Version (Takara Bio) to amplify the entire GBA gene. An amplicon with a length of 11,246 base pairs (bp) was obtained. The cycling conditions for amplification were as follows: initial denaturation at 94°C for 60 s, 30 cycles of denaturation at 98°C for 10 s, annealing at 68°C for 12 min, and extension at 72°C for 20 min. Lastly, samples were held at 4°C. After LR‐PCR, the PCR products were fragmented using Hieff Smearase (YEASEN) to an average size of 150–250 bp before sequencing. Library preparation was performed using the Hieff NGS OnePot DNA Library Prep Kit for Illumina (YEASEN). Next generation sequencing (paired‐end 150 bp) was then performed using a HiSeq 4000 sequencer (Illumina).
Based on whether they carried GBA variants, PD patients were divided into GBA‐PD and NM‐PD groups. Based on the criteria proposed by Petrucci et al. [23], the severity of GBA variants in the GBA‐PD group was further classified into five classes: mild (causing nonneuropathic subtypes of Gaucher disease [GD] type 1, such as N370S), severe (known to cause neuropathic phenotypes of GD type 2 or 3, such as L444P), risk (related to risk factors of PD but not meaningful for GD, such as E326K and T369M), complex (two or more GBA variants, such as L444P‐A456P‐V460V), and unknown.
Statistical analysis
Descriptive statistics were calculated for demographic and clinical characteristics; continuous variables were reported as the mean and standard deviation, whereas categorical variables were reported as frequencies and proportions. For comparisons between the NM‐PD and GBA‐PD groups, linear regression analysis was used for continuous variables and logistic regression analysis was used for binary variables. Multinomial logistic regression with TD subtype as the comparator was performed to compare motor subtypes between the two groups. Probability values were two‐tailed and calculated after adjusting for potential confounding factors, as listed in the tables. Furthermore, GBA‐PD patients were divided into subgroups according to the severity of the GBA variant. Comparisons of demographic and clinical features among the mild, severe, and complex subgroups were performed using one‐way analysis of variance, Kruskal–Wallis H‐test, or Fisher exact test followed by post hoc analysis with Bonferroni adjustment. Results were deemed to be statistically significant if the p‐value was <0.05. All statistical analyses were performed using IBM SPSS (v25.0).
RESULTS
Identified GBA variants
GBA gene sequencing was performed on 737 Chinese PD patients, 79 (GBA‐PD, 10.72%) of whom carried 47 distinct GBA variants. The variant severity levels were eight (10.13%) mild, 28 (35.44%) severe, one (1.27%) risk, seven (8.86%) complex, and 35 (44.30%) unknown (Figure S1). Surprisingly, R163Q was the most common variant, with a cumulative frequency of 15.19% (12 cases, including 10 isolated cases and two cases as part of a recombinant allele). The second and third most common variants were L444P and R120W, with cumulative frequencies of 12.66% (10 cases, including seven isolated cases and three cases as part of a recombinant allele) and 7.59% (six isolated cases), respectively (Table S1).
Demographic and clinical features between GBA‐PD and NM‐PD groups
Of the 737 PD patients whose GBA gene was sequenced, demographic and clinical data were obtained for 673 patients. According to whether they carried GBA variants, these 673 PD patients were divided into GBA‐PD (n = 79) and NM‐PD (n = 594) groups. Of note, 32 of the 173 EOPD patients (18.50%) carried GBA variants.
The demographic, motor, and nonmotor characteristics of the GBA‐PD and NM‐PD groups are compared in Table 1. In terms of demographic data, there were no statistically significant differences in gender, formal education, disease duration, or LEDD between the two groups. However, patients with GBA‐PD were younger (mean age = 57.2 years, SD = 9.7) compared with the NM‐PD group (mean age = 61.2 years, SD = 9.6), had a significantly earlier age at onset (mean = 53.3 years, SD = 9.7) compared with the NM‐PD group (mean = 57.1 years, SD = 10.1), and had higher prevalence of EOPD (40.5%) compared with the NM‐PD group (23.9%), when adjusting for gender and disease duration (p = 0.001). In addition, patients with GBA‐PD had a higher frequency of family history of PD (13.9%) compared with the NM‐PD group (5.7%), when adjusting for age, gender, and disease duration (p = 0.014). With respect to motor characteristics, the prevalence of the PIGD subtype, which reflects more axial symptoms and less tremor, was not significantly different between the NM‐PD and GBA‐PD groups; this subtype was the most common phenotype in both groups. However, patients with GBA‐PD had more severe UPDRS ADL scores, UPDRS motor scores, and modified H‐Y stages compared with the NM‐PD group. Regarding the NMSs, there were no significant group differences in cognitive impairment. However, depression, anxiety, and sleep impairments were more prevalent in the GBA‐PD group than the NM‐PD group.
TABLE 1.
Comparison of demographic and clinical characteristics between GBA‐PD and NM‐PD patients
| Variable | GBA‐PD, n = 79 | NM‐PD, n = 594 | p a |
|---|---|---|---|
| Age, years | 57.2 ± 9.7 | 61.2 ± 9.6 | 0.001 b * |
| Gender, male, n (%) | 41 (51.9) | 330 (55.6) | 0.471 c |
| Formal education, years | 9.3 ± 4.1 | 10.2 ± 4.1 | 0.073 |
| Age at onset, years | 53.3 ± 9.7 | 57.1 ± 10.1 | 0.001 b * |
| EOPD, n (%) | 32 (40.5) | 142 (23.9) | 0.001 b * |
| Family history of PD, n (%) | 11 (13.9) | 34 (5.7) | 0.014* |
| Disease duration, years | 3.9 ± 3.1 | 4.2 ± 3.9 | 0.728 d |
| LEDD, mg/day | 405.7 ± 309.4 | 375.2 ± 330.3 | 0.077 |
| UPDRS ADL score | 11.8 ± 6.6 | 10.3 ± 6.0 | 0.001* |
| UPDRS motor score | 30.0 ± 16.5 | 24.9 ± 14.2 | <0.001 e * |
| H‐Y stage | 2.5 ± 1.2 | 1.9 ± 0.8 | <0.001* |
| Motor subtype, n (%) | |||
| TD | 20 (25.3) | 138 (23.2) | |
| PIGD | 44 (55.7) | 355 (59.8) | 0.712 |
| Indeterminate | 15 (19.0) | 101 (17.0) | 0.889 |
| MMSE score | 26.4 ± 3.7 | 26.7 ± 3.7 | 0.402 f |
| MoCA score | 22.6 ± 4.9 | 22.6 ± 4.9 | 0.893 f |
| HAMD score | 12.8 ± 9.1 | 10.3 ± 7.4 | 0.004* |
| HAMA score | 10.2 ± 8.1 | 7.5 ± 7.2 | 0.001* |
| PDSS score | 103.5 ± 32.4 | 116.3 ± 23.0 | <0.001* |
| NMSQuest score | 11.3 ± 6.3 | 9.0 ± 4.9 | <0.001* |
Data are reported as mean ± SD or n (%). Probability values were calculated using linear regression or logistic regression.
Abbreviations: ADL, activities of daily living; EOPD, early onset PD; GBA‐PD, GBA‐related PD; HAMA, Hamilton Anxiety Scale; HAMD, Hamilton Depression Scale; H‐Y, Hoehn‐Yahr; LEDD, levodopa equivalent daily dose; MMSE, Mini‐Mental State Examination; MoCA, Montreal Cognitive Assessment; NM‐PD, non‐GBA‐mutated PD; NMSQuest, Non‐Motor Symptoms Questionnaire; PD, Parkinson disease; PDSS, Parkinson Disease Sleep Scale; PIGD, postural instability gait difficulty; TD, tremor‐dominant; UPDRS, Unified Parkinson's Disease Rating Scale.
*p < 0.05.
Adjusted for age, gender, and disease duration (unless otherwise indicated).
Adjusted for gender and disease duration.
Adjusted for age and disease duration.
Adjusted for age and gender.
Adjusted for age, gender, disease duration, and LEDD.
Adjusted for age, gender, disease duration, and years of formal education.
Demographic and clinical features among GBA‐PD subgroups
Considering there was only one GBA‐PD patient in the risk group, we compared demographic and clinical data among the mild, severe, and complex subgroups (Table 2). Among the demographic and motor features, the only significant subgroup difference emerged for UPDRS motor scores. In the post hoc analysis, GBA‐PD patients in the severe subgroup had more severe UPDRS motor scores than those in the mild subgroup. Regarding scores linked to NMSs, HAMD, PDSS, and NMSQuest scores were significantly different among the three subgroups. Post hoc analysis revealed that GBA‐PD patients in the complex and severe subgroups had more severe HAMD and NMSQuest scores than those in the mild subgroup. In addition, GBA‐PD patients in the complex subgroup had higher PDSS scores than those in the mild subgroup.
TABLE 2.
Comparison of demographic and clinical characteristics among GBA‐related PD patients grouped by GBA variant classes
| Variable | Mild, n = 8 | Severe, n = 28 | Complex, n = 7 | p | Post hoc |
|---|---|---|---|---|---|
| Age, years | 58.9 ± 9.8 | 55.5 ± 10.2 | 51.0 ± 6.2 | 0.299 | |
| Gender, male, n (%) | 4 (50.0) | 18 (64.3) | 3 (42.9) | 0.540 | |
| Formal education, years | 9.0 ± 2.4 | 10.0 ± 3.8 | 10.6 ± 1.8 | 0.589 | |
| Age at onset, years | 55.3 ± 8.8 | 51.0 ± 10.1 | 47.7 ± 7.1 | 0.308 | |
| EOPD, n (%) | 3 (37.5) | 15 (53.6) | 5 (71.4) | 0.545 | |
| Family history of PD, n (%) | 1 (12.5) | 3 (10.7) | 2 (28.6) | 0.527 | |
| Disease duration, years | 3.6 ± 2.7 | 4.5 ± 3.5 | 3.3 ± 2.1 | 0.574 | |
| LEDD, mg/day | 407.1 ± 274.6 | 469.9 ± 349.4 | 439.3 ± 379.9 | 0.895 | |
| UPDRS ADL score | 8.5 ± 4.2 | 12.6 ± 6.6 | 12.1 ± 8.2 | 0.307 | |
| UPDRS motor score | 17.6 ± 6.1 | 34.5 ± 15.7 | 31.4 ± 22.0 | 0.036* | 0.032 a * |
| H‐Y stage | 1.8 ± 0.6 | 2.8 ± 1.3 | 2.6 ± 1.7 | 0.104 | |
| MMSE score | 26.8 ± 3.3 | 27.2 ± 2.1 | 27.4 ± 1.5 | 0.993 | |
| MoCA score | 22.8 ± 4.6 | 23.7 ± 3.6 | 22.7 ± 3.9 | 0.725 | |
| HAMD score | 6.1 ± 5.7 | 15.0 ± 9.3 | 17.7 ± 8.1 | 0.022* | 0.042 a *, 0.038 b * |
| HAMA score | 8.9 ± 4.5 | 11.0 ± 8.2 | 12.7 ± 8.8 | 0.724 | |
| PDSS score | 121.3 ± 21.1 | 102.4 ± 31.4 | 76.9 ± 48.3 | 0.044* | 0.039 b * |
| NMSQuest score | 6.3 ± 5.2 | 12.0 ± 5.8 | 14.3 ± 5.1 | 0.018* | 0.045*, 0.025 b * |
Data are reported as mean ± SD or n (%). Group comparisons were calculated using analysis of variance, Kruskal–Wallis H‐test, or Fisher exact test followed by post hoc analysis with Bonferroni adjustment.
Abbreviations: ADL, activities of daily living; EOPD, early onset PD; HAMA, Hamilton Anxiety Scale; HAMD, Hamilton Depression Scale; H‐Y, Hoehn‐Yahr; LEDD, levodopa equivalent daily dose; MMSE, Mini‐Mental State Examination; MoCA, Montreal Cognitive Assessment; NMSQuest, Non‐Motor Symptoms Questionnaire; PD, Parkinson disease; PDSS, Parkinson Disease Sleep Scale; UPDRS, Unified Parkinson's Disease Rating Scale.
*p < 0.05.
Statistically significant difference between the mild and severe subgroups.
Statistically significant difference between the mild and complex subgroups.
NMSs based on NMSQuest
Because there were significant differences in NMSQuest scores between the GBA‐PD and NM‐PD groups, and among the mild, severe, and complex subgroups, the frequency of the nine domains in NMSQuest were further compared (Figure 1). Patients with GBA‐PD had a higher prevalence of sleep, mood/cognitive, perception/hallucinations, gastrointestinal, urinary, sexual function, and miscellaneous symptoms compared to the NM‐PD group, adjusting for age, gender, and disease duration (Table S2). Among the subgroups, GBA‐PD patients in the complex and severe subgroups had a higher prevalence of mood/cognitive and gastrointestinal symptoms compared with the mild subgroup. Additionally, GBA‐PD patients carrying severe variants had a higher prevalence of miscellaneous domain symptoms than GBA‐PD patients carrying mild variants (Table S3).
FIGURE 1.
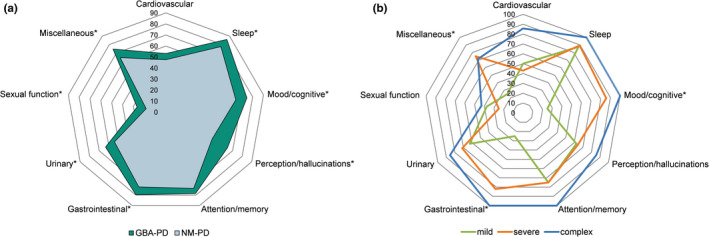
Comparison of frequency of NMSs classified by domain among different patient groups. (a) Frequency (%) of the nine domains of NMSs in GBA‐PD versus NM‐PD groups. (b) Frequency (%) of the nine domains of NMSs among GBA‐PD subgroups carrying mild, severe, and complex variants. *p < 0.05. Statistical comparisons are shown in Tables S2 and S3. Abbreviations: GBA‐PD, GBA‐related PD; NM‐PD, non‐GBA‐mutated PD; NMSs, nonmotor symptoms; PD, Parkinson disease [Color figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
To our knowledge, this is the first comprehensive analysis of the entire GBA gene in a large cohort of Chinese PD patients. We detected GBA variants in 79 (10.72%) patients. For the results of the genotype–phenotype correlations, GBA‐PD patients had an approximately 4 year earlier age at onset and more severe motor and nonmotor characteristics than NM‐PD patients. Furthermore, among the three GBA‐PD subgroups, patients carrying severe and complex variants had a higher burden of NMSs, especially depression, and more prevalent mood/cognitive and gastrointestinal symptoms than patients carrying mild variants. Therefore, the effect of GBA variant status on phenotypic profile seems to depend on the severity, and complex variants and severe variants may underlie similar phenotypes.
In our cohort, 10.72% of PD patients carried one or more variants in the GBA gene. This is much higher than the GBA variant rate (3.64%) in the Chinese population estimated in a previous meta‐analysis [31], which may be attributed to previous studies in the Chinese population focusing on detecting relatively common GBA variants, such as L444P and R120W [32, 33]. Notably, the detected frequency is slightly higher than the GBA variant rate (8.7%) in 187 Chinese PD patients with sequencing of the entire GBA gene [12], which may be due to the large difference in sample size. Surprisingly, R163Q was the most common GBA variant (1.63%) among the 737 patients with PD in this study. Because R163Q is a relatively common variant (0.3%) in healthy Asian populations according to the Genome Aggregation Database, it is currently difficult to correctly classify the severity of the R163Q variant in GBA‐PD patients. However, carrying the R163Q variant may still be a risk factor for PD. Therefore, it is necessary to increase knowledge about GBA variants that are common in healthy Asian populations and to accumulate information about GBA variants other than L444P and R120W. After R163Q, L444P was the most common (1.36%) pathogenic GBA variant identified in PD patients in this study, which is consistent with the results of a previous study in China [31]. Thus, this finding supports the view of L444P as a common pan‐racial variant.
The prevalence of GBA variants in EOPD patients (age at onset ≤ 50 years) rose to 18.50% in the present study, which is in line with previous reports [23, 34, 35]. Recently, several studies have performed whole‐exome sequencing of EOPD patients in the Chinese population to better understand the clinical and genetic correlations of GBA, SNCA, and LRRK2 in EOPD [11, 36], indicating that GBA screening in Chinese EOPD patients is important for diagnosis.
Compared with NM‐PD cases, GBA‐PD patients in the present study were approximately 4 years younger at the onset of symptoms. This finding is similar to results reported by studies from North America and the United Kingdom [19, 37] as well as a previous meta‐analysis of the Chinese population [38]. However, a study from Germany reported that the age at onset of GBA‐PD could be as much as 6 years earlier compared with NM‐PD [17].
We confirmed the significant association of GBA variants with more severe motor symptoms and NMSs (specifically depression, anxiety, and sleep disorders), which was mainly due to the correlation between the extent of Lewy body pathology and clinical symptoms. Autopsy pathology has revealed that GBA‐PD patients tend to have more diffuse and widespread neocortical Lewy body‐type pathology compared to NM‐PD patients [9]. In addition, experimental data have shown that, mechanistically, GCase and α‐syn form a bidirectional pathogenic loop [39]: (i) the functional loss of GCase caused by GBA variant compromises the degradation of lysosomal α‐syn, leading to accumulation of α‐syn; and (ii) aggregated α‐syn itself inhibits the lysosomal activity of GCase. Consequently, GBA‐PD patients satisfy the two conditions of this bidirectional pathogenic loop in parallel, thereby forming an autoreinforcement mechanism. However, the relationship between the severity of GBA variant and the level of GCase activity has not been clearly elucidated due to the limited number of GBA‐PD patients carrying variants of varying severity [23, 40]. Therefore, clinical data of patients with different severity of GBA variants are still needed for detailed comparison.
This study demonstrated that GBA‐PD patients with complex and severe classes had more severe NMSs compared with patients with mild variants, which is mostly consistent with the results of previous studies [15, 23]. Importantly, this study provides the first evidence that the existence of depression in PD is variant‐specific. A recent study within the GBA‐PD subgroups focused on psychiatric symptoms, including hallucinations, delusions, and impulsive–compulsive behavior, but ignored depression [23]. Lastly, considering the nine domains of NMSs, the frequencies of mood/cognitive and gastrointestinal domain symptoms in the complex and severe subgroups were higher than those in the mild subgroup. Therefore, the clinical features of NMSs in GBA‐PD patients seem to be influenced by the severity of GBA variants. Importantly, our clinical data support the view that complex variants are similar to severe variants, which is consistent with a recent review [41].
The strengths of the present study are as follows: (i) the establishment of a relatively large GBA‐PD cohort in the Chinese population for meaningful epidemiological comparisons; (ii) screening of the entire GBA gene and avoidance of GBAP1 interference, which makes the variant‐specific analysis more accurate; and (iii) the support of complex variants and severe variants to share similar phenotypic profiles, according to clinical data.
The present study is also subject to several limitations. (i) In GBA‐PD patients, the limited number of subjects carrying variants of varying severity may mask additional significant differences. (ii) Considering the heterogeneity of the cohort, we recommend that these findings are explored in de novo GBA‐PD cohorts and, ideally, in cohorts of GBA nonmanifesting carriers. (iii) All patients with idiopathic PD in this study underwent GBA gene testing, but this does not rule out the possibility that some PD patients have common PD pathogenic genes, such as SNCA and LRRK2, which may have impacted the results. (iv) We unfortunately did not test the onset and longitudinal evolution of motor complications (motor fluctuations and dyskinesia) and key NMSs (such as cognitive impairment and hallucinations), which will be important in future research.
In conclusion, the prevalence of GBA‐PD patients in the Chinese population is very high, which contributes to the large proportion of EOPD cases. Our study has expanded the spectrum of nonmotor characteristics in GBA‐PD patients and indicates that variant severity may underlie different phenotypic features. Notably, the severe and complex groups seem to have similar phenotypes. In this context, stratifying PD patients based on the severity of GBA variants should become a necessary prerequisite for selecting specific treatments.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to disclose.
AUTHOR CONTRIBUTIONS
Jingru Ren: Conceptualization (lead), data curation (equal), formal analysis (lead), investigation (equal), methodology (equal), validation (equal), writing–original draft (lead), writing–review & editing (equal). Ronggui Zhang: Conceptualization (equal), formal analysis (equal), investigation (equal), validation (equal), visualization (equal). Chenxi Pan: Methodology (equal), supervision (equal), validation (equal), visualization (equal), writing–review & editing (equal). Jianxia Xu: Data curation (equal), formal analysis (equal), methodology (equal), supervision (equal), validation (equal). Haochen Sun: Data curation (equal), formal analysis (equal), supervision (equal), writing–review & editing (equal). Ping Hua: Supervision (equal), validation (equal), visualization (equal), writing–review & editing (equal). Li Zhang: Supervision (equal), validation (equal), visualization (equal), writing–review & editing (equal). Wenbin Zhang: Formal analysis (equal), methodology (equal), validation (equal), visualization (equal), writing–review & editing (equal). Pingyi Xu: Supervision (equal), validation (equal), visualization (equal), writing–review & editing (equal). Changyan Ma: Methodology (equal), supervision (equal), validation (equal), writing–review & editing (equal). Weiguo Liu: Conceptualization (equal), funding acquisition (lead), investigation (equal), methodology (equal), project administration (equal), supervision (lead), validation (lead), writing–review & editing (equal).
Supporting information
Supplementary material
ACKNOWLEDGMENTS
The authors gratefully acknowledge the active participation of the patients, the cooperation of the families, and the assistance of Shanghai WeHealth BioMedical Technology Co. (Shanghai, China).
Ren J, Zhang R, Pan C, et al. Prevalence and genotype–phenotype correlations of GBA‐related Parkinson disease in a large Chinese cohort. Eur J Neurol.2022;29:1017–1024. doi: 10.1111/ene.15230
Funding information
This work was supported by the National Key Research and Development Program of China (2017YFC1310302, and 2016YFC1306600), National Natural Science Foundation of China (81571348), and Science and Technology Program of Jiangsu Province (BE2019611)
DATA AVAILABILITY STATEMENT
The original data for this study can be obtained from the corresponding author via email upon reasonable request.
REFERENCES
- 1. Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. 2021;397(10291):2284‐2303. [DOI] [PubMed] [Google Scholar]
- 2. Obeso JA, Stamelou M, Goetz CG, et al. Past, present, and future of Parkinson's disease: a special essay on the 200th Anniversary of the Shaking Palsy. Mov Disord. 2017;32(9):1264‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vollstedt EJ, Kasten M, Klein C. Using global team science to identify genetic parkinson's disease worldwide. Ann Neurol. 2019;86(2):153‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Do J, McKinney C, Sharma P, Sidransky E. Glucocerebrosidase and its relevance to Parkinson disease. Mol Neurodegener. 2019;14(1):36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aharon‐Peretz J, Rosenbaum H, Gershoni‐Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med. 2004;351(19):1972‐1977. [DOI] [PubMed] [Google Scholar]
- 6. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med. 2009;361(17):1651‐1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Anheim M, Elbaz A, Lesage S, et al. Penetrance of Parkinson disease in glucocerebrosidase gene mutation carriers. Neurology. 2012;78(6):417‐420. [DOI] [PubMed] [Google Scholar]
- 8. Beavan M, McNeill A, Proukakis C, Hughes DA, Mehta A, Schapira AH. Evolution of prodromal clinical markers of Parkinson disease in a GBA mutation‐positive cohort. JAMA Neurol. 2015;72(2):201‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Neumann J, Bras J, Deas E, et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson's disease. Brain. 2009;132(Pt 7):1783‐1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gan‐Or Z, Amshalom I, Kilarski LL, et al. Differential effects of severe vs mild GBA mutations on Parkinson disease. Neurology. 2015;84(9):880‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li N, Wang L, Zhang J, et al. Whole‐exome sequencing in early‐onset Parkinson's disease among ethnic Chinese. Neurobiol Aging. 2020;90:150. e155‐150. e111. [DOI] [PubMed] [Google Scholar]
- 12. Yu Z, Wang T, Xu J, et al. Mutations in the glucocerebrosidase gene are responsible for Chinese patients with Parkinson's disease. J Hum Genet. 2015;60(2):85‐90. [DOI] [PubMed] [Google Scholar]
- 13. Horowitz M, Wilder S, Horowitz Z, Reiner O, Gelbart T, Beutler E. The human glucocerebrosidase gene and pseudogene: structure and evolution. Genomics. 1989;4(1):87‐96. [DOI] [PubMed] [Google Scholar]
- 14. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567‐583. [DOI] [PubMed] [Google Scholar]
- 15. Thaler A, Bregman N, Gurevich T, et al. Parkinson's disease phenotype is influenced by the severity of the mutations in the GBA gene. Parkinsonism Relat Disord. 2018;55:45‐49. [DOI] [PubMed] [Google Scholar]
- 16. Thaler A, Gurevich T, Bar Shira A, et al. A "dose" effect of mutations in the GBA gene on Parkinson's disease phenotype. Parkinsonism Relat Disord. 2017;36:47‐51. [DOI] [PubMed] [Google Scholar]
- 17. Brockmann K, Srulijes K, Pflederer S, et al. GBA‐associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord. 2015;30(3):407‐411. [DOI] [PubMed] [Google Scholar]
- 18. Lerche S, Schulte C, Srulijes K, et al. Cognitive impairment in Glucocerebrosidase (GBA)‐associated PD: Not primarily associated with cerebrospinal fluid Abeta and Tau profiles. Mov Disord. 2017;32(12):1780‐1783. [DOI] [PubMed] [Google Scholar]
- 19. Malek N, Weil RS, Bresner C, et al. Features of GBA‐associated Parkinson's disease at presentation in the UK Tracking Parkinson's study. J Neurol Neurosurg Psychiatry. 2018;89(7):702‐709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cilia R, Tunesi S, Marotta G, et al. Survival and dementia in GBA‐associated Parkinson's disease: the mutation matters. Ann Neurol. 2016;80(5):662‐673. [DOI] [PubMed] [Google Scholar]
- 21. Maple‐Grødem J, Dalen I, Tysnes OB, et al. Association of GBA genotype with motor and functional decline in patients with newly diagnosed Parkinson disease. Neurology. 2021;96(7):e1036‐e1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stoker TB, Camacho M, Winder‐Rhodes S, et al. Impact of GBA1 variants on long‐term clinical progression and mortality in incident Parkinson's disease. J Neurol Neurosurg Psychiatry. 2020;91(7):695‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petrucci S, Ginevrino M, Trezzi I, et al. GBA‐related Parkinson's disease: dissection of genotype‐phenotype correlates in a large Italian cohort. Mov Disord. 2020;35(11):2106‐2111. [DOI] [PubMed] [Google Scholar]
- 24. Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry. 1988;51(6):745‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Butterfield PG, Valanis BG, Spencer PS, Lindeman CA, Nutt JG. Environmental antecedents of young‐onset Parkinson's disease. Neurology. 1993;43(6):1150‐1158. [DOI] [PubMed] [Google Scholar]
- 26. Tomlinson CL, Stowe R, Patel S, Rick C, Gray R, Clarke CE. Systematic review of levodopa dose equivalency reporting in Parkinson's disease. Mov Disord. 2010;25(15):2649‐2653. [DOI] [PubMed] [Google Scholar]
- 27. Jankovic J, McDermott M, Carter J, et al. Variable expression of Parkinson's disease: a base‐line analysis of the DATATOP cohort. The Parkinson Study Group. Neurology. 1990;40(10):1529‐1534. [DOI] [PubMed] [Google Scholar]
- 28. Nasreddine ZS, Phillips NA, Bédirian V, et al. The Montreal Cognitive Assessment, MoCA: a brief screening tool for mild cognitive impairment. J Am Geriatr Soc. 2005;53(4):695‐699. [DOI] [PubMed] [Google Scholar]
- 29. Chaudhuri KR, Martinez‐Martin P, Schapira AHV, et al. International multicenter pilot study of the first comprehensive self‐completed nonmotor symptoms questionnaire for Parkinson's disease: the NMSQuest study. Mov Disord. 2006;21(7):916‐923. [DOI] [PubMed] [Google Scholar]
- 30. Chaudhuri KR, Martinez‐Martin P, Brown RG, et al. The metric properties of a novel non‐motor symptoms scale for Parkinson's disease: results from an international pilot study. Mov Disord. 2007;22(13):1901‐1911. [DOI] [PubMed] [Google Scholar]
- 31. Chen J, Li W, Zhang T, Wang YJ, Jiang XJ, Xu ZQ. Glucocerebrosidase gene mutations associated with Parkinson's disease: a meta‐analysis in a Chinese population. PLoS One. 2014;9(12):e115747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Huang CL, Wu‐Chou YH, Lai SC, et al. Contribution of glucocerebrosidase mutation in a large cohort of sporadic Parkinson's disease in Taiwan. Eur J Neurol. 2011;18(10):1227‐1232. [DOI] [PubMed] [Google Scholar]
- 33. Wu YR, Chen CM, Chao CY, et al. Glucocerebrosidase gene mutation is a risk factor for early onset of Parkinson disease among Taiwanese. J Neurol Neurosurg Psychiatry. 2007;78(9):977‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early‐onset Parkinson disease. Neurology. 2007;69(12):1270‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Duran R, Mencacci NE, Angeli AV, et al. The glucocerobrosidase E326K variant predisposes to Parkinson's disease, but does not cause Gaucher's disease. Mov Disord. 2013;28(2):232‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chen Y, Gu X, Ou R, et al. Evaluating the role of SNCA, LRRK2, and GBA in Chinese patients with early‐onset Parkinson's disease. Mov Disord. 2020;35(11):2046‐2055. [DOI] [PubMed] [Google Scholar]
- 37. Mata IF, Leverenz JB, Weintraub D, et al. GBA Variants are associated with a distinct pattern of cognitive deficits in Parkinson's disease. Mov Disord. 2016;31(1):95‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zhang Y, Sun QY, Zhao YW, et al. Effect of GBA mutations on phenotype of parkinson's disease: a study on chinese population and a meta‐analysis. Parkinsons Dis. 2015;2015:916971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and α‐synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37‐52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lerche S, Schulte C, Wurster I, et al. The mutation matters: CSF profiles of GCase, sphingolipids, α‐synuclein in PD(GBA). Mov Disord. 2021;36(5):1216‐1228. [DOI] [PubMed] [Google Scholar]
- 41. Menozzi E, Schapira AHV. Exploring the genotype‐phenotype correlation in GBA‐parkinson disease: clinical aspects, biomarkers, and potential modifiers. Front Neurol. 2021;12:694764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Data Availability Statement
The original data for this study can be obtained from the corresponding author via email upon reasonable request.