

Abstract
The ribose 2′-hydroxyl is the key chemical difference between RNA and DNA and primary source of their divergent structural and functional characteristics. Macromolecular X-ray diffraction experiments typically do not reveal the positions of hydrogen atoms. Thus, standard crystallography cannot determine 2′-OH orientation (H2′-C2′-O2′-HO2′ torsion angle) and its potential roles in sculpting the RNA backbone and the expansive fold space. Here, we report the first neutron crystal structure of an RNA, the Escherichia coli rRNA Sarcin-Ricin Loop (SRL). 2′-OD orientations were established for all 27 residues and revealed O-D bonds pointing toward backbone (O3′, 13 observations), nucleobase (11) or sugar (3). Most riboses in the SRL stem region show a 2′-OD backbone-orientation. GAGA-tetraloop riboses display a 2′-OD base-orientation. An atypical C2′-endo sugar pucker is strictly correlated with a 2′-OD sugar-orientation. Neutrons reveal the strong preference of the 2′-OH to donate in H-bonds and that 2′-OH orientation affects both backbone geometry and ribose pucker. We discuss 2′-OH and water molecule orientations in the SRL neutron structure and compare with results from a solution phase 10 μs MD simulation. We demonstrate that joint cryo-neutron/X-ray crystallography offers an all-in-one approach to determine the complete structural properties of RNA, i.e. geometry, conformation, protonation state and hydration structure.
INTRODUCTION
The ribose 2′-hydroxyl group (2′-OH) plays key roles in RNA chemistry, biology, structure, function and stability. At the nucleotide level, the 2′-OH shifts the conformational equilibrium of the five-membered sugar ring toward the C3′-endo pucker (1–3). At the oligomeric level, this alters the sugar-phosphate backbone conformation relative to B-DNA to the stubbier A-form, with 2′-OH groups jutting into the shallow minor groove. There, they serve as bridgeheads for water tandems that cross the groove (4). The hydroxyl group can establish H-bonds to multiple water molecules, thereby contributing to the increased thermodynamic stability of RNA relative to DNA duplexes that is enthalpy-based. The H-bond acceptor and donor capabilities of the 2′-OH group also underlie its important role in RNA tertiary structure and inter-strand contacts, including the so-called ribose zipper (5–9). Crucially, the ribose hydroxyl serves as the nucleophile in spliceosomal nuclear pre-mRNA splicing (10,11), group II intron self-splicing (12–14), and the catalytic cleavage of the phosphodiester bond by various ribozymes (14–16).
Recently a hypothesis based on QM calculations was put forth in regard to the potential of the 2′-OH group to increase the propensity for non-canonical RNA structures (17). Accordingly, 2′-OH directed toward the base stabilizes the canonical duplex, but orientations of 2′-OH toward the backbone allow sampling of a wider range of conformations. Thus, the latter ‘O3′ orientation’ significantly alters the intrinsic flexibility of the RNA backbone. The 2′-OH group could thus act as a switch, stabilizing the duplex in the ‘base orientation’ (Figure 1A ‘b’) and enabling a wealth of tertiary structures when flipped to the ‘O3′ orientation’ (Figure 1A ‘p’). Moreover, using QM, QM/MM calculations and MD simulations, it was proposed that the orientation of the 2′-OH group can affect the ribose pucker and that a ‘sugar orientation’ (Figure 1A, ‘s’) flips the sugar to the uncommon DNA-like C2′-endo conformation (18). The 2′-OH sugar orientation was also predicted for a C2′-endo puckered ribose based on an early MD simulation of a tRNAAsp 17mer that sought to establish rules governing the orientation of the hydroxyl group (19).
Figure 1.
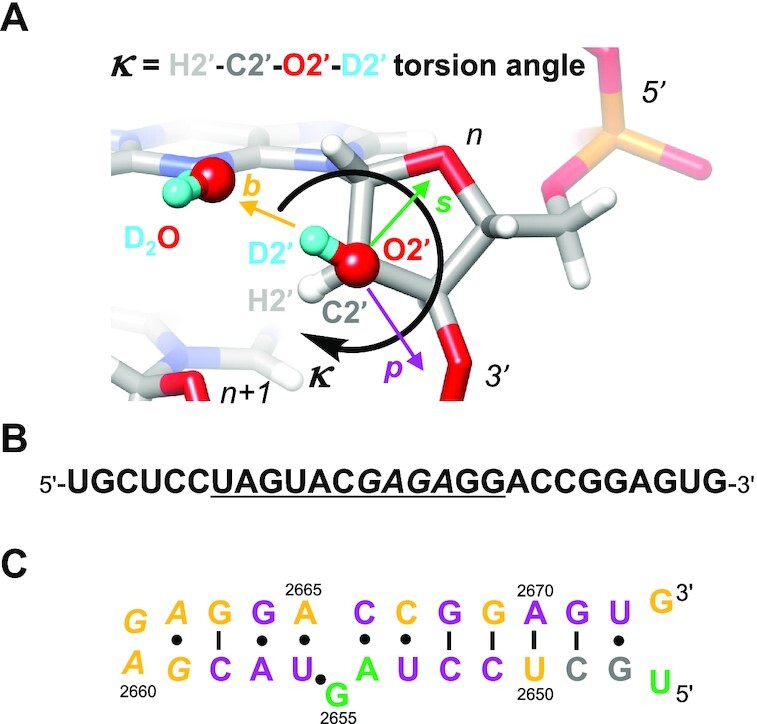
(A) The κ angle defines the orientation of the 2′-hydroxyl group, with HO2′ directed toward base (b, goldenrod), phosphate (p, purple) or sugar (s, green). Please see also similar torsion angle definitions put forth by others (17–19). (B) Sequence of SRL RNA. The underlined 12mer is universally conserved and the tetraloop is highlighted in italicized font. (C) Secondary structure of SRL RNA. Residue numbers correspond to E. coli 23S rRNA and residues are colored according to the orientation of their 2′-OH groups as defined in panel A and established by neutron crystallography. Throughout the text and in the PDB coordinate file, SRL nucleotides are numbered (U)47 to (G)73.
Efforts based on solution NMR to gain insight into the positions of the 2′-OH H atoms (HO2′) were reported for a range of RNA fragments (20,21). Fohrer et al. concluded that the O-H bond orientations alternate between the O3′ region and the base and that the latter orientation is preferred in canonical duplexes based on correlations with exchange rates. However, reliably establishing the orientations of individual 2′-OH groups requires isotope labeling and has not been demonstrated for RNAs beyond simple stem-loop constructs. Consequently, H-bond donor and acceptor patterns involving 2′-OH moieties have remained largely hidden in 3D structures of RNAs large and small (22). X-ray crystal structures of macromolecules including oligonucleotide-size DNA and RNA fragments even at a resolution of 1 Å do not reveal the positions of hydrogen atoms (23), e.g. those of HO2′ atoms. Thus, crucial roles attributed to the particular orientation of the 2′-OH group by theoretical means, i.e. those underlying RNA’s expanded fold space relative to DNA (17) and actual control of the sugar conformation (18), have escaped experimental scrutiny. Moreover, neither solution NMR nor X-ray crystallography at high resolution (24,25) can pinpoint the orientations of first-shell water molecules associated with H-bond donor and acceptor functions of DNA and RNA nucleobases and the sugar-phosphate backbone.
RNA HO2′ atoms can be exchanged for deuterium (D, 2H) in crystals grown from D2O and their positions determined by neutron diffraction as the coherent scattering length of D, b = 6.671 fm, is very similar to that of C, b = 6.646 fm (for scattering lengths, see https://www.ncnr.nist.gov/resources/n-lengths/). Remarkably, no neutron single crystal structure for RNA has been reported to date. To examine the hypothesis that the O–H direction of 2′-hydroxyl groups influences RNA backbone conformation and that orientations that sample backbone atoms are correlated with non-canonical conformations, we selected the Escherichia coli 23S rRNA sarcin/ricin loop (SRL) 27mer of sequence 5′-UGC UCC UAG UAC GAG AGG ACC GGA GUG-3′ (Figure 1B) (26,27). This fragment contains a nearly canonical duplex (bold font nucleotides) and extensive non-canonical structure, including pyrimidine-pyrimidine pairs, base triplets, cross-strand stacking and a GAGA tetraloop (underlined) (Figure 1C). Crystals of this RNA diffract X-rays to atomic resolution [1.1 Å (28–30)]. We used cryo-neutron crystallography [CNC (31–33)] with the goal to establish the orientations of all 2′-OD groups, and to reveal the orientations of first-shell and as many second-shell water molecules as possible (34,35). We also conducted a 10 μs solution phase MD simulation of SRL RNA with no deuterium atoms to compare and contrast solution phase conformations with structural details observed in the low-temperature diffraction experiments. These comparisons help us to identify any structural features that may be influenced by low temperature and/or crystal packing effects.
We demonstrate that riboses with the C2′-endo conformation (three observations in the SRL) are exclusively associated with a sugar orientation of the hydroxyl group. Both, 2′-OH groups directed toward the backbone and those directed toward the base can stabilize non-canonical backbone conformations. Surprisingly, riboses of residues in the canonical stem region of the SRL do not commonly show a base-oriented 2′-OH group. Conversely, the 2′-OH groups of all nucleotides in the tetraloop are directed toward the nucleobase. Therefore, the particular orientation of the ribose hydroxyl group is subject to several external factors that include solvent and packing interactions. The neutron crystal structure reveals the detailed water structure around SRL RNA and a potentially conserved pattern of hydration for the G:U mismatch that involves the exocyclic amino group of guanine and the 2′-OH of uridine. CNC thus offers a unique approach to pinpointing the diverse H-bond acceptor and donor roles of the ribose hydroxyl group in sculpting RNA backbone conformation, tertiary structure and packing as well as organizing the surrounding water with implications for stability and function.
MATERIALS AND METHODS
RNA synthesis and purification
The 27mer SRL RNA was synthesized using standard solid phase phosphoramidite chemistry and deprotection protocols and purified by anion-exchange high-performance liquid chromatography (IEX-HPLC) as previously described (30). The RNA contains a 1:1 mixture of RP- and SP-phosphorothioate moieties at G61 that stabilizes the SRL fold significantly relative to the parent phosphate (30).
Crystallization
The SRL RNA oligomer was dissolved in a buffered D2O solution composed of 1 mM sodium EDTA (pH 8.0) and 10 mM Tris–HCl (pH 8.0; not titrated with DCl) to a concentration of 350 μM. Annealing was performed by heating this solution to 65°C for 2 min followed by slow cooling to room temperature and storage overnight at 4°C. For crystallization 4 μl of the RNA solution were mixed with 2 μl of a crystallization buffer composed of 3.0 M perdeuterated ammonium sulfate (ND4)2SO4, 10 mM magnesium chloride (MgCl2), 10 mM manganese chloride (MnCl2), and 50 mM potassium 2-(N-morpholino)ethanesulfonic acid-d13 (MES-d13, pH 7.0) at 18°C. Crystals appeared in ∼1−2 weeks. They were cryoprotected in a reservoir solution containing 3.4 M (ND4)2SO4, 10 mM MgCl2, 10 mM MnCl2, 50 mM potassium MES-d13 (pH 7.0) and 15% (v/v) perdeuterated glycerol-d8 and then flash-frozen in liquid nitrogen.
Neutron diffraction data collection and processing
Neutron diffraction data were collected for a crystal with a size <0.1 mm3 (0.5 × 0.5 × 0.3 mm) using the macromolecular neutron diffractometer (MaNDi) instrument (36,37) on beamline 11B of the Spallation Neutron Source (SNS) at Oak Ridge National Laboratory (ORNL, Oak Ridge, TN). This instrument is a neutron time-of-flight, wavelength-resolved Laue diffractometer that uses a Crystal Logic (Los Angeles, CA) custom goniometer with a built-in Oxford Cryosystems Cobra nitrogen cryostream. The cryostream at MaNDi/SNS is positioned above the vertically mounted goniostat and rotation axis such that the sample mount makes an angle close to 180° with the cryostream. The instrument permits use of standard cryoloops and pins and thus allows for facile transfer of crystals from the neutron to an X-ray source in shipping dewars for joint data collections and subsequent refinement. In the data collection mode, the crystal is surrounded by a spherical array of 46 custom-made Anger camera detectors. A total of 15 diffraction images were collected at 100 K with an exposure time of 12 h each. The crystal was held static during each diffraction image and was rotated 10° between images. Data collection was completed in 7.5 days, yielding a dataset of good completeness to a resolution of 2.2 Å (Supplementary Table S1). These data were reduced with the Mantid program (38) using newly developed 3D profile fitting for time-of-flight diffraction data (39). Integrated intensities were then scaled using Lauenorm from the Lauegen suite (40,41). The reduced and corrected data statistics are shown in Supplementary Table S1.
X-ray diffraction data collection and processing
X-ray data for the same SRL RNA crystal were collected to 1.0 Å resolution using the D8 Venture system (Bruker AXS, Madison, WI) in the Biomolecular Crystallography Facility at the Vanderbilt University Center for Structural Biology. The setup includes an Excillum D2 + MetalJet X-ray source with Helios MX optics providing Ga Kα radiation at a wavelength of 1.3418 Å. The crystal was mounted on a kappa axis goniometer and maintained at 100 K using an Oxford Cryosystems Cryostream 800 cryostat. The detector was a PHOTON III charge-integrating pixel array detector (CPAD). Data collection was performed in shutterless mode. Diffraction data were reduced using Proteum3 software (Bruker AXS). Data collection statistics are summarized in Supplementary Table S2.
Joint X-ray/neutron refinement
Initial phases were obtained by molecular replacement, using the SRL RNA search model based on the X-ray structure of crystals grown under identical conditions (PDB ID 7JJD) (30). All crystallographic refinements were carried out with the program PHENIX (42,43) combining the two data sets. Refinement targets and libraries were maximum likelihood and the standard libraries incorporated in Phenix. Model building and manual fitting was performed using the COOT molecular graphics program (44). Non-exchangeable hydrogens were set to full occupancy except for C8 in purines. D2O molecules were automatically oriented using the Python script ‘Neutron Water Orientation’ [NWO; https://github.com/OCald/NeutronWaterOrientation (45)]. Selected crystal data, data collection and refinement statistics are summarized in Table 1.
Table 1.
Selected crystal data, diffraction data collection and X-ray/neutron joint refinement statistics
| Diffraction method | SRL Cryo X-ray data | SRL Cryo neutron data |
|---|---|---|
| Wavelength [Å] | 1.342 | 2.0–4.0 |
| Resolution range [Å] | 16.05–0.998 (1.033- 0.998) | 12.34–2.25 (2.33–2.25) |
| Space group | P43 | |
| Unit cell: a= b, c [Å] | 29.495, 76.52 | |
| Total reflections | 99 309 | 9850 (771) |
| Unique reflections | 35 326 (3,461) | 2887 (280) |
| T min/Tmax | 0.5940/0.7488 | |
| Multiplicity | 13.68 (10.96) | 3.40 (2.73) |
| Completeness [%] | 99.67 (98.30) | 92.32 (92.41) |
| Mean I/sigma(I) | 21.66 (4.45) | 9.8 (2.8) |
| Wilson B-factor [Å2] | 8.30 | 25.34 |
| R-merge | 0.055 (0.412) | 0.212 (0.299) |
| R-meas | 0.029 (0.404) | 0.247 (0.357) |
| R-pim | 0.020 (0.186) | 0.121 (0.189) |
| CC1/2 | 0.999 (0.849) | 0.886 (0.300) |
| Reflections in refinement | 35 272 (3,456) | 2886 (280) |
| Reflections for R-free | 1760 (174) | 143 (13) |
| R-work | 0.1785 (0.2452) | 0.2447 (0.3310) |
| R-free | 0.1965 (0.2838) | 0.3079 (0.3171) |
| Number of non-H atoms | 709 | |
| RNA | 624 | |
| ligands | 5 | |
| solvent | 240 | |
| RMSD bonds [Å] | 0.014 | 0.090 |
| RMSD angles [°] | 1.78 | 1.61 |
| Clashscore | 2.11 | 42.39 |
| Average B-factor [Å2] | 17.2 | |
| ligands | 61.1 | |
| solvent | 31.3 | |
| PDB ID code | 7UCR | |
Statistics for the highest-resolution shell are shown in parentheses.
Joint refinement by combining X-ray and neutron data sets collected using the same crystal results in much improved neutron Rwork and Rfree as well as nuclear density maps, allowing for more precise positioning of deuterium positions ((43,46) and data not shown). Neutron data alone tend to be of lower completeness and the orders of magnitude difference between the cross-section of interaction of neutrons and X-rays with matter in the crystal results in lower intensities of reflections for neutron diffraction even with vastly extended exposure times. The advantages of joint refinement may result from a number of causes, including an increase in observation to parameter ratio when combining neutron data with typically higher resolution X-ray data. Partially deuterated crystals, as in the present structure, may result in cancellation effects using neutron data alone when the negative scattering length of hydrogen offsets scattering from the atom to which it is bound. Refinements using neutron data alone may result in over-fitting due to the treatment of hydrogen/deuterium atom position as independent atoms rather than modeled as riding positions. For these and perhaps many other reasons, joint X-ray and neutron refinement is becoming the standard.
MD simulation
Classical MD simulations were performed using the PMEMD AMBER module (47) (https://ambermd.org/). The AMBER ROC force field (48) (i.e. AMBER nucleic acid parameters with ‘Rochester’ torsion angle modifications) was used together with the TIP3P water model (49) and Joung/Cheatham parameters for monovalent ions (50) for all calculations. The SRL RNA was solvated in a truncated octahedron box with a 14 Å buffer zone between any RNA atom and the closest box wall. The resultant unit cell contained 1 SRL molecule, 26 sodium cations to impose net charge neutrality to facilitate periodic boundary calculations, and 9926 water molecules, yielding a solution of ∼5 mM RNA and ∼130 mM sodium cations. The starting conformation was subjected to a three-step minimization procedure. First, RNA atoms were relaxed for 10 000 steps of conjugate gradient minimization while water molecules and counterions were restrained at starting positions. Next, all solvent and counterions were relaxed for 10 000 steps while RNA atoms were restrained. Finally, all restraints were removed and the entire system was minimized for 10 000 additional steps. The minimized complex was then heated gradually from 0 to 300 K during a 200 ns canonical ensemble (NVT) MD simulation, followed by a 300 ns NPT ensemble simulation. Production MD simulations were then run with a 1.5 fs timestep for 10 μs. Energy and force calculations were performed using minimal image periodic boundary conditions, a 12Å nonbonded cutoff for real space interactions, a homogeneity assumption to approximate the contributions of long-range Lennard-Jones forces to the virial tensor, and staggered particle-mesh Ewald for long-range electrostatics correction (51). A Langevin thermostat with collision frequency 3 ps–1 was used to maintain the system temperature (52). All bonds containing hydrogen atoms were constrained using the SHAKE algorithm (53) and the SETTLE method was used to maintain rigid water geometry (54). Final analysis of all MD trajectories was performed using the cpptraj package (55).
Illustrations
Graphical analyses of the MD trajectories and all figure generations (with the exception of Figure 2) were performed using UCSF Chimera (56). Figure 2 panels were made using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
Figure 2.

Examples of the quality of the final nuclear and electron densities, shown as meshworks contoured at 1.5 σ and colored in blue and red, respectively. (A) Pairing between A57 and G64. (B) A70 with 2′-OH in the phosphate orientation. (C) C67 with associated D2O molecules. (D) G61 with 2′-OH in the base orientation. (E) A54 with 2′-OH in the sugar orientation. (F) G69 with associated D2O molecules. Carbon, oxygen, nitrogen, phosphorus, hydrogen and deuterium atoms are colored in green, red, blue, orange, white and black, respectively, and residues and water molecules are labeled. Figure panels were made using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC).
RESULTS
SRL RNA neutron structure: overview
We report here the first example of the structure of an RNA molecule analyzed by a combination of X-ray and neutron crystallography. The SRL is an iconic motif since it contains a universally conserved 12mer sequence, including the tetraloop that is also the target of two ribosome-inactivating cytotoxins, α-sarcin and ricin (Figure 1B). The former cleaves the phosphodiester bond between G2661 and A2662 (E. coli numbering) in the SRL and the latter depurinates A2660 (Figure 1C) (27,57). The SRL features complex tertiary structural motifs in an oligonucleotide-size fragment and produces crystals that diffract X-rays to atomic resolution. We collected cryo neutron diffraction data for an SRL crystal of volume <0.1 mm3 on the MaNDi/SNS instrument at Oak Ridge National Laboratory to 2.2 Å resolution. This is the smallest sample for which viable data were acquired on this instrument to date, with data collection lasting just over a week. The crystal was taken off the MaNDi goniostat and then transported in a dryshipper to Vanderbilt University, where an X-ray diffraction data set to 1 Å resolution was collected on the in-house Bruker MetalJet instrument. The combination of cryogenic X-ray and neutron diffraction data for joint refinement affords a more reliable water structure around the nucleic acid molecule compared to traditional neutron data collected at room temperature (35). Together with the 27 ribonucleotides, the SRL joint X-ray/neutron crystal structure revealed 83 D2O molecules of which 75 belong to the first hydration shell, and a sulfate ion. However, we did not observe cations, e.g. ND4+, in the final density maps. Accurate determination of the water structure around the RNA molecule is imperative for a meaningful interpretation of the orientations of 2′-OH moieties and potential roles of O2′-H direction in controlling backbone geometry (18), sculpting tertiary structure (17), and enhancing stability by electrostatics and H-bonding (58,59). A summary of crystallographic data collection and refinement parameters is listed in Table 1 and examples of the quality of the final electron and nuclear densities are depicted in Figure 2.
An overall view of the SRL RNA anatomy is depicted in Figure 3A (see also Figure 1C for a secondary structure diagram). The base of the SRL is made up of a hexamer stem that includes two G•U pairs, U47•G73 and G48•U72. However, the terminal pair is disrupted, with U47 looping away and G73 stacking underneath G48. The three G:C pairs, a single A:U pair and the terminal G•U wobble assume a more or less canonical A-form conformation, although both G48 and C49 (highlighted with gray carbon atoms in Figure 3A) exhibit twofold orientation disorder (Supplemental Figure S1). The stem continues with the U53•C67 pair that is stabilized by a bifurcated H-bond between O2 (U) and N4(D)2 (C). The nucleobases of the adjacent ‘pair’, A54 and C66, are nearly coplanar, but the base moieties do not engage in H-bonding. However, the adenine N6(D)2 exocylic amino group forms H-bonds with the phosphate moiety (OP2) of C66. The next layer of the irregular SRL stem is composed of a base triple contributed by G55, U56 and A65, whereby G55 adopts a syn orientation (the only nucleoside to do so in the SRL) and stacks onto G64 from the opposite strand (Figures 3 and 4; ‘cross-strand stack’). Both N1D and N2(D)2 of G55 are H-bonded to the phosphate of A65 and the exocyclic amino group also forms an H-bond to O4 of U56. The uracil engages in two H-bonds to A65, O4…N6(D)2 and N3D…N7, and a third H-bond is potentially formed between O2 (U) and C8H (A) (Figure 4; there is no evidence that H8 of adenine has been partially exchanged for deuterium). Whereas five of the nine guanines exhibit partial H-D exchange at the C8 position (between 7 and 42%; average 28%), adenines do not. The G•U•A base triple stacks against the sheared A57•G64 purine-purine pair that is stabilized by N6(D)2…N3 and N7…N2(D)2 H-bonds (Figure 4, background). Finally, the C58:G63 Watson-Crick pair seals the GAGA loop that features the hallmarks of a GNRA-type RNA tetraloop. These include the sheared G59•A62 pair, a three-layer stack involving A60, G61 and A62, and phosphates of G61and A62 that stack onto and H-bond with the base moiety of G59, respectively (30 and cited references).
Figure 3.

Tertiary structure and hydration of SRL RNA. (A) Overall view of the SRL crystal structure based on joint X-ray/neutron refinement. The color code of individual nucleotide carbon atoms is identical to that in Figure 1 and denotes the orientation of the 2′-OH group. Phosphate groups, oxygen, nitrogen and deuterium atoms are colored in black, red, blue and cyan, respectively, nucleotides are labeled, and pairing and tertiary structural motifs are indicated. Nucleotides G48 and C49 exhibit dual conformational disorder and their carbon atoms are colored in gray. Please see Supplementary Figure S1 for a close-up view and Table 3 for details of torsion angles. (B) Content of the crystallographic asymmetric unit (a.u.): one SRL molecule (gray with 2′-OD O and D atoms colored in red and cyan, respectively) surrounded by D2O molecules (O blue, D light blue) and a single sulfate ion (bottom right). Water molecules and 2′-OD moieties are shown in ball-and-stick mode.
Figure 4.
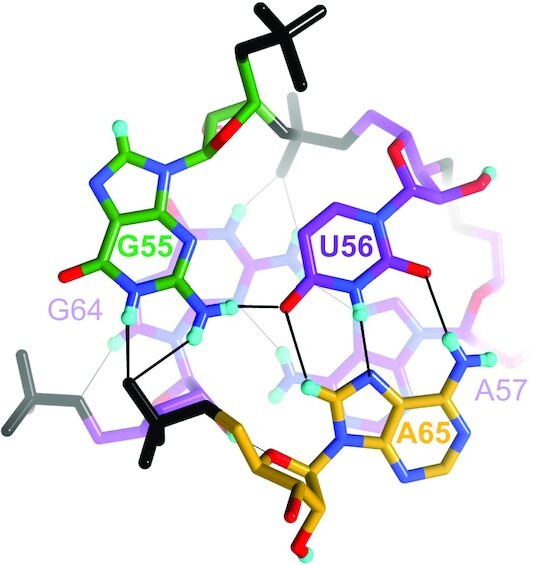
Pairing motifs in two adjacent layers of the SRL RNA stem. Foreground: The G55•U56•A65 base triple. Background: The sheared A57•G64 pair. G55 and G64 engage in a cross-strand stack. The hydrogen atoms on their respective C8 atoms have been partially exchanged for deuterium. The color code is identical to that in Figure 3A, H-bonds are drawn with thin solid lines and residues are labeled. Note the H-bonding interactions between N1D and N2(D)2 of G64 and the phosphate of U56 (top) and 2′-OD of G64 and O4′ of A65 (bottom, slightly obscured).
MD simulation: overview
The MD simulation yields structures that are generally in excellent agreement with the diffraction results. The time-averaged SRL RNA structure exhibits an RMSD of ∼1.8 Å relative to the crystal structure, if the terminal U47•G73 ‘base pair’ is excluded (Figure 5). The overall RMSD agreement rises to ∼2.6 Å if we include the terminal bases in this analysis, as they are both extremely mobile and display many disordered structures similar to that observed in the crystal structure. While the ensemble-averaged SRL structure agrees well with the crystal structure, we do observe some large structural fluctuations during the simulation. The most dramatic fluctuation is a conformation that evolves around 100 ns and involves a dramatic swing of A54 into the major groove, where it stacks directly above G55, as shown in Figure 6. The G55•U56•A65 base triple below A54 is well maintained, consistent with the crystal structure, and base stacking and base pairs above A54 are also well maintained. This distorted structure persists for only 2–3 ns, and reverts fully to a structure consistent with the ensemble average and the crystal structure within 5 ns. Numerous other less dramatic structural fluctuations are also observed during the simulation, and all are quite transient. These occasional large structural fluctuations do suggest that the simulation has sampled significant conformational space, so the good agreement between the crystal structures and the ensemble-averaged MD structure is not due to limited conformational sampling.
Figure 5.
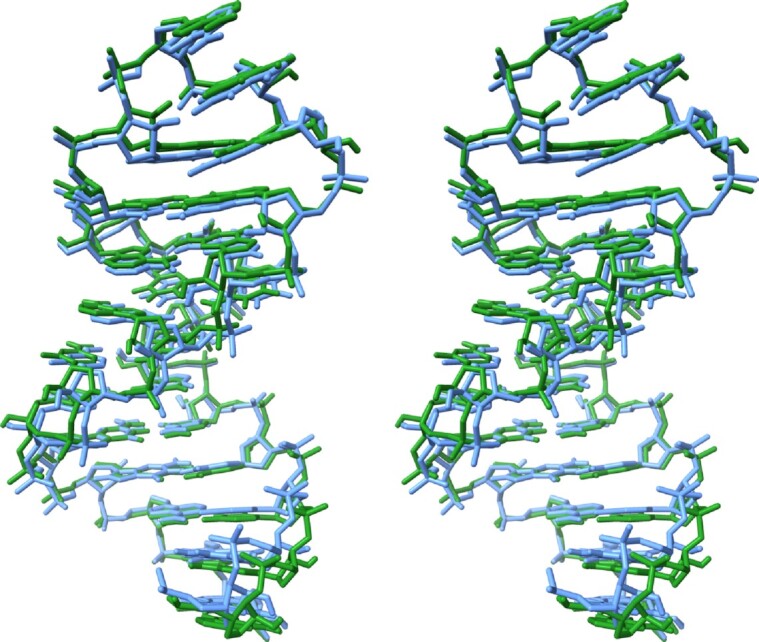
Stereo plot displaying a superposition of the SRL crystal structure (light blue), residues G48 through U72, with the ensemble-averaged simulation structure over the full 10 μs trajectory (green). The terminal U47•G73 ‘base pair’ is omitted in this overlay, as these terminal bases are highly mobile and disordered during most of the trajectory. The G48•U72 pair is at the bottom and the GAGA tetraloop is at the top.
Figure 6.
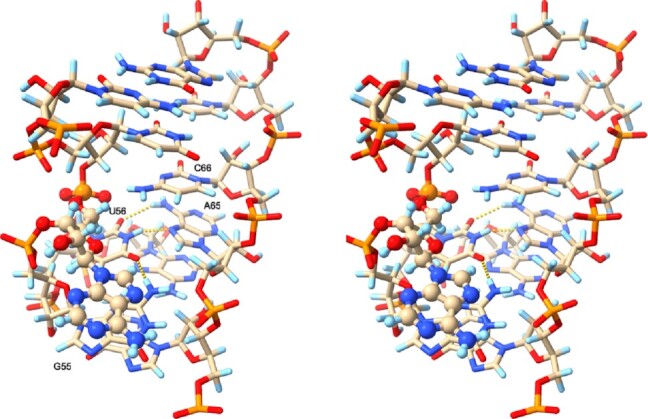
Stereo plot of a transient SRL conformation where A54 (ball-and-stick rendering) swings into the major groove, stacking above G55. TheG55•U56•A65 base triple below A54 is well maintained and depicted with dashed yellow lines, while base stacking and pairs above A54 are also well maintained. Carbon atoms are colored beige, nitrogen atoms blue, oxygen atoms red, phosphorous atoms orange and hydrogen atoms light blue.
H-D exchange at 2′-hydroxyl groups
The incentive for using neutron diffraction to determine the orientations of 2′-hydroxyl groups is that the HO2′ hydrogen is replaced by deuterium upon dissolving RNA in D2O. H-D exchange is not uniform and is sometimes not complete even with a fast exchanging species such as the ribose 2′-OH or the terminal 5′- and 3′-OH groups. Crystallographic refinement then provides a direct measure of the occupancy of exchangeable deuterium atoms in the crystal (from 0 to 100%). For SRL RNA 2′-OH groups the average H-D exchange was 79% (min. 23%, max. 100%). Therefore, the occupancy of these deuterium atoms is sufficient to reliably determine their positions and thus the orientations of 2′-OD moieties. At positions G48 and C49, nucleotides were found to assume two orientations but, in both cases, these could be resolved in the electron and nuclear densities and refined with individual occupancies (Figure 3B). The RNA backbone is extensively hydrated and a thorough analysis of the ribose environments and 2′-OD orientations requires knowledge of the water structure. We previously demonstrated the benefits of low temperature neutron data collection to do so (35). Here, we have applied this approach to establish the positions and orientations of 83 unique D2O molecules in the asymmetric unit of the SRL crystal structure, many of which serve either as donor or acceptor in an H-bond to a 2′-OH(D) moiety (Figure 3B).
2′-hydroxyl orientation: overview
Of the 27 riboses in the SRL, 13 exhibit a 2′-OD directed toward the backbone as defined by κ torsion angles (Figure 1A) that fall into the sc+ and ac+ ranges (ca. 20°…130°). Somewhat fewer, 11, have their 2′-OD directed toward the base, characterized by κ torsion angles between ca. 0° and −70° (sp- and sc- ranges). And the κ angles of three riboses fall into the ap+ range (150°, 171°, 172°), indicative of a 2′-OD moiety with a sugar orientation. Table 2 provides an overview of κ angles, sugar puckers and 2′-OD environments, and Figures 1C and 3A depict SRL secondary and tertiary structural diagrams, respectively, with residues colored according to their 2′-OD orientations (see Figure 1A for the color code). In the case of residue C49 that exhibits dual occupancy, riboses exhibit deviating 2′-OD orientations. The ribose in one orientation (66% occupancy) features a 2′-OD with backbone orientation (κ = +129°). The 2′-OD of the nucleotide in the alternative orientation (34% occupancy) exhibits a base orientation (κ = -64°).
Table 2.
κ angles, sugar puckers and 2′-OH(D) environments in the SRL RNA crystal structure*
| Residue | κ [°] | κ sector | Pucker, P [°] | 2′-OD environment; distances are between DO2′ and O/N |
|---|---|---|---|---|
| U47 | +171 | s (p!) | C2′-endo, 154 | Intra-nucleotide H-bond to O3′ (2.3 Å), but quite far from O2 (3.4 Å). |
|
G48 a
b |
– – |
– – (p!) |
C3′-endo, 1 C3′-endo, 1 |
Two residue orientations; no clear interaction with other atoms. |
|
C49 a
b |
+129 −64 |
p (p!) b |
C3′-endo, 7 C3′-endo, 7 |
Two residue orientations. a, Intra-nucleotide H-bond to O3′ (2.4 Å); b, O2′ donates in long H-bond to water (2.7 Å). |
| U50 | +7 | b (p,b) | C3′-endo, 16 | Two water molecules link D2′ to O2 of uracil. |
| C51 | +98 | p (p,b) | C3′-endo, 17 | Intra-nucleotide H-bond to O3′ (2.2 Å) and engaged in ribose O2′…O2′ zipper with adjacent molecule (C58#, 1.9 Å). |
| C52 | +46 | p (p,b) | C3′-endo, 14 | Intra-nucleotide H-bond to O3′ (2.6 Å) and long H-bond to water (2.7 Å). |
| U53 | +21 | p (p) | C3′-endo, 18 | Donates in H-bonds to two waters, one being linked to phosphate via a second water and the other being linked to N4 of C67 via another water. Both waters H-bonded to 2′-OD are also donating in H-bonds to the sulfate ion. |
| A54 | +150 | s (p) | C2′-endo, 163 | Tandem water bridge to N3 of its own base (adenine) and intra-nucleotide H-bond to O3′ (2.1 Å). |
| G55 | +172 | s (s) | C2′-endo, 163 | H-bonded to O6 of G64 and in-line orientation O2′-P-O5′ with its 3′-phosphate. |
| U56 | +102 | p (p.b) | C3′-endo, 12 | Intra-nucleotide H-bond to O3′ (2.2 Å). |
| A57 | +110 | p (p,b) | C3′-endo, 18 | Intra-nucleotide H-bond to O3′ (2.2 Å). |
| C58 | +121 | p (p,b) | C3′-endo, 17 | Intra-nucleotide H-bond to O3′ (2.3 Å) and ribose zipper with O2′ from neighboring molecule (C51#, 1.8 Å). |
| G59 | −28 | b (b,p) | C3′-endo, 19 | H-bond to N7 of G61 in loop (1.8 Å). O6 of the same residue is farther removed (2.8 Å). |
| A60 | +6 | b (p) | C3′-endo, 8 | H-bonds to O6 from G73# (donor) and water molecule (acceptor), 1.8 and 1.9 Å, resp. |
| G61 | −52 | b (p!) | C3′-endo, 5 | O2′ donates in a water-mediated, intra-nucleotide H-bond to N3 of guanine, 1.8 and 2.2 Å, resp. Water also donates in another H-bond to O3′ from neighboring molecule (1.6 Å). |
| A62 | −18 | b (p) | C3′-endo, 14 | Water-mediated H-bonds to O2′ from neighboring molecule (2 Å/1.8 Å) and N3 of G69# (2.0 Å/1.9 Å), and direct H-bond to O2 of C52# from the same neighbor (2.3 Å). |
| G63 | −38 | b (p,b) | C3′-endo, 10 | Donates in water-mediated H-bond to N3 of its base (1.7 Å/2.2 Å) and a second water links the first one to N2 of G63 and N6 of A62. |
| G64 | +81 | p (p) | C3′-endo, 10 | H-bonds to N6 of A57 and O4′ of A65 (2.0 and 1.8 Å, resp.). |
| A65 | −48 | b (p!) | C3′-endo, 2 | H-bonds to water that links to a phosphate from a neighboring molecule (2.6 Å/1.7 Å) and N3 of its adenine (2 Å). |
| C66 | +79 | p (p!) | C3′-endo, 8 | Intra-nucleotide H-bond to O3′ (2.3 Å), accepts in an H-bond to water (2 Å) and donates in a further H-bond to phosphate from a neighboring molecule (2.3 Å). |
| C67 | −12 | b (p!) | C3′-endo, 14 | O2′ accepts H-bond from water that sits between O3′ and O2′ (2.2 Å and 2.5 Å, resp.) and donates in an H-bond to a water that is part of a tandem of waters that connects to O2 of its own base (1.7 Å/2.0 Å/2.3 Å). |
| G68 | +54 | p (p!) | C3′-endo, 12 | No clear interaction; O3′ is quite far away (D…O3′ 2.7 Å). |
| G69 | −57 | b (p!) | C3′-endo, 11 | Donates in H-bond to water tandem, the first linking O2′ to N3 (1.8 Å/1.9 Å) and the second water linking the first water to N2 (2 Å/2 Å). |
| A70 | +110 | p (p!) | C3′-endo, 14 | Intra-nucleotide H-bond to O3′ (2.3 Å). |
| G71 | +26 | p (p!) | C2′-exo, 358 | No close contact. |
| U72 | +108 | p (p!) | C3′-endo, 14 | Intra-nucleotide H-bond to O3′ (2.2 Å) and O2′ accepts in an H-bond to water that forms a bridge to N2 of G48 (2.4 Å/2.3 Å). G48 and U72 form a wobble pair such that N2 of the former sticks into the minor groove. This is likely a conserved motif. |
| G73 | −33 | b (p,b) | C3′-endo, 26 | No close contact. |
*κ is defined as the H2′-C2′-O2′-DO2′ torsion angle (Figure 1A). Sector is similar to the parameter cx as defined by Auffinger and Westhof (19) and used by both Denning and MacKerell (17) and Darré et al. and Orozco (18), i.e. for a C3′-endo pucker c1 ( = p) includes the sc+ range with DO2′ directed toward backbone O3′, c2 ( = s) is ap with DO2′ roughly directed toward sugar O4′, and c3 ( = b) includes the sc– range with DO2′ directed toward the base. This is similar to the directions of water clusters around O2′ atoms in the high-resolution crystal structure of the (CCCCGGGG)2 duplex (4). Residues and atoms from symmetry mates are marked by #. κ values from the MD simulation are listed in parentheses; in cases where more than one sector is listed, the most common value is given first, e.g. (b,p) indicates base orientation with significant population of backbone sector values as well. Listings followed by an ‘!’ indicate significant sampling of all three sectors during the simulation, with the most common value listed. Entries for residues adopting the less common ribose pucker are highlighted in boldface.
The κ torsion angles observed in the simulation are also presented in Table 2. The κ values fluctuate dramatically during the simulation (Supplementary Figure S2), so it is difficult to establish any simple correlations between the solution phase results and the neutron diffraction structure. The sugar pucker values observed in the simulation are generally in excellent agreement with the experimental observations, with some minor exceptions. The sugar pucker for U47 and G73 fluctuates frequently between C2′-endo and C3′-endo conformations, consistent with the highly dynamic behavior of these terminal residues. The A60 sugar pucker behavior is unique in that it populates primarily C3′-endo, as observed in the crystal structure, but also significantly samples C2′-endo and C2′-exo conformations (Supplementary Figure S3). There is no obvious explanation for the more ‘dynamic’ behavior of the A60 sugar pucker, except that it is the most ‘exposed’ base at the tip of the SRL loop structure, so the extensive hydration at this site may partially explain the more facile sugar pucker transitions.
2′-OH orientation in a cross-strand water-mediated interaction between nucleobase and ribose
The G48•U72 pair at the base of the SRL stem exhibits the usual sheared geometry with guanine shifted into the minor groove. This exposes the exocyclic amino group in the center of that groove and results in formation of H-bonds between N1D (G) and O2 (U) and O6 (G) and N3D (U) (Figure 7). The 2′-hydroxyl group of the uridine ribose assumes a backbone orientation and therefore points away from the N2 amino group of G. However, a water molecule bridges the two and the neutron analysis shows that D2O is the acceptor and donor in the H-bonds to N2(D)2 (G) and 2′-OD (U), respectively (Figure 7). A similar water-mediated interaction involving a hydroxyl group with its O–H bond directed toward the base would require the water to act as acceptor in both H-bonds. In the major groove, a water molecule bridges O6 (G) and O4 (U), although the distance to the former is somewhat long. Undoubtedly, these water-mediated base-sugar and base-base interactions contribute to the stability of G•U pairs and likely constitute a conserved hydration pattern around the wobble pair. As pointed out above, the adjacent G73•U47 pair is disrupted; G73 is stacked underneath the G48•U72 pair and U47 loops away (Figure 7). A water molecule also bridges the guanine base to the ribose of uridine in this case. However, the water molecule accepts in H-bonds to N1D and N2(D)2 (G) and then donates in a somewhat long contact to the ribose O4′ (U).
Figure 7.
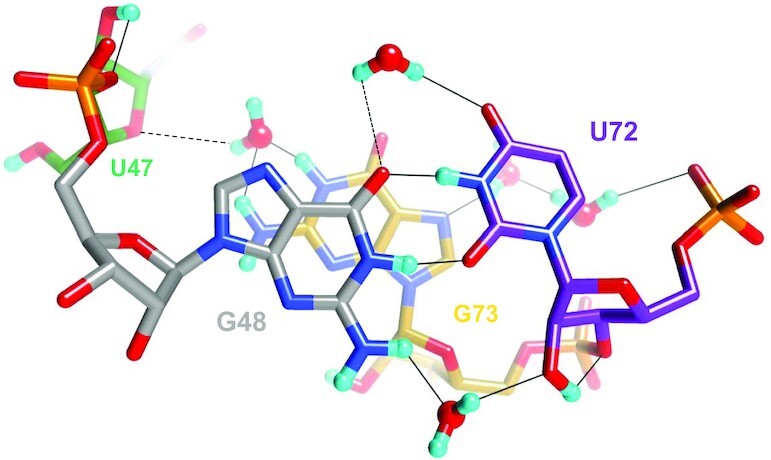
A water-mediated cross-strand ribose-base interaction stabilizes the G•U wobble pair in the minor groove. The D2O in the lower foreground links G48 and U72 by accepting and donating H-bonds to N2(D)2 and 2′-OD, respectively. The color code is identical to that in Figure 3A, H-bonds are drawn with thin solid lines and residues are labeled. Somewhat long H-bonds are indicated with dashed lines. One of them occurs between a water that sits adjacent to O4 of U72 and O6 of G48. The other occurs between a water bound to N1D and N2(D)2 of G73 and O4′ of U47.
The G48•U72 base pair geometry is generally well preserved in the MD simulation, although the U72 2′-hydroxyl group orientation fluctuates freely, as noted in the discussion of κ angles above. Water molecules in the first hydration shell are dynamic and exchange readily with bulk solvent during the simulation. Nonetheless, there is usually a water molecule present that forms a bridging H-bond between the G48 N2 amino group and the U72 2′-OH. The interaction observed in the crystal structure, where the bridging water molecule accepts an H-bond from the G48 amino group and donates an H-bond to the U72 2′-OH group is the most common motif, but other patterns are also seen. In a few cases (<5% of conformations), the bridging water molecule serves as the acceptor for H-bonds with both the amino and 2′-OH groups. In rare cases, no water molecule is in a favorable orientation to form bridging H-bonds and we observe an intriguing shift of G48 toward the major groove, altering the base pair interactions with U72 and retracting the guanine amino group from the minor groove (Figure 8).
Figure 8.
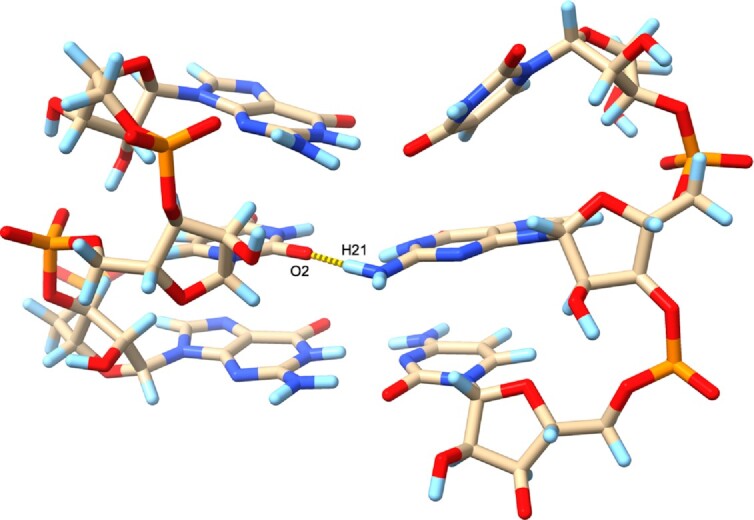
A simulation snapshot displaying a rare situation where there is no minor groove water molecule that forms a bridging H-bond interaction with the G48•U72 wobble base pair. In this situation, G48 slides back toward the major groove and the G48 exocyclic amino group is retracted from the minor groove, instead H-bonding with atom O2 of U72, as displayed with the yellow dashed line. Carbon atoms are colored beige, nitrogen atoms blue, oxygen atoms red, phosphorous atoms orange and hydrogen atoms light blue.
A prominent example of a G•U pair is found in the tRNAAla acceptor stem and serves as the key signal for recognition of this tRNA by alanyl-tRNA synthetase (AlaRS). The N2 group of that G is shifted into the minor groove in the wobble pair, i.e. as in the case seen in the SRL, with water bridging 2′-OH of U and N2 of G with various H-bond donor-acceptor patterns. However, in the crystal of the complex between tRNAAla and AlaRS, a protein backbone keto group takes the place of water to mediate the interaction between O2′ (U) and N2 (G) (Supplementary Figure S4). Both, RNA atoms (e.g. O2′ (19)) or protein atoms (e.g. in the case discussed here) can replace water that mediates a contact between 2′-OH and a base edge in the minor groove.
Even with stringent hydrogen bond geometry criteria (a donor-acceptor distance of 2.8–3.2 Å and an H-bond angle of 20°), we determine that a bridging water molecule forms good H-bonds at least 75% of the time during the simulation, independent of the exact κ angle for U72. We also observe that a water molecule is almost always present to accept an H-bond from the G48 amino group, even if a bridging H-bond to U72 2′-OH is not possible. The major groove region near the G48•U72 base pair is extensively hydrated during the simulation, so we observe good water H-bonds with both the G48 O6 and U72 O4 atoms essentially 100% of the time. The water-mediated H-bonds with G48 and U72 do appear to stabilize this wobble base pair, as we only observe the G48 base shift and ‘retraction’ of the G48 amino group from the minor groove on those rare occasions when no water molecule can form a bridging interaction (Figure 8).
The SRL structure displays multiple direct interactions between nucleobase atoms and either phosphate or ribose 2′-OH of the ‘pairing’ strand. Examples include A54 (N6) and phosphate (C66), G55 (N1/N2) and phosphate (A65), A57 (N6) and 2′-OH (G64) and G59 (N2, O2′) and phosphate as well as 2′-OH (A62). Most of these direct interactions are also observed in the simulation, although they are often transient and water molecules compete with these interactions, especially those that involve 2′-OH groups. In canonical RNA and DNA duplexes, direct contacts between nucleobase atoms from one strand and the sugar-phosphate backbone of the opposite strand are not possible. However, we observed a water-mediated contact between the base of incoming dATP or dGTP and the phosphate of the template strand with an abasic site in ternary complexes of the Y-family DNA polymerase η (60).
2′-OH groups with a base orientation
Many 2′-OD moieties are directed toward the base and engage in unique interactions with water and/or adjacent RNA atoms. Selected examples are depicted in Figure 9 and the orientations of all SRL hydroxyl groups and brief descriptions of the contacts they engage in are listed in Table 2. In the G59–A60–G61–A62 tetraloop, the 2′-OD of the first residue forms an H-bond with N7 of G61 (Figure 9A). A60, G61 and A62 form a continuous stack whereby the base of A60 constitutes the tip of the SRL and A62 stacks onto the ribose of G63 at the other end (Figure 3A). C8 of G61 is directed toward its phosphate group, a feature shared with other purine nucleotides in the SRL structure, and the hydrogen has been partially exchanged (D occupancy 0.4). The distance between D8 and O5′ is 2.38 Å (Figure 9A). G55, G63, G64 and G68 are other purines that have undergone partial H-D exchange (42%, 7%, 20% and 30%, respectively). With the exception of G55 for which the C8-D/H8 bond juts into the solvent, guanine C8 and the 5′-phosphate group are closely spaced, resulting in an intra-nucleotide H-bond between C8-D/H and either O5′ or a non-bridging phosphate oxygen. These interactions are generally well preserved in the simulation, with occasional fluctuations to longer C8-H…O5′ distances than those observed in the crystal structure.
Figure 9.

Interactions by selected SRL 2′-OD groups with a base orientation and associated water structure: (A) G59, (B) G61, (C) G69 and (D) C67. The color code matches that in Figure 3A, except for phosphate groups which are colored orange (P) and red (O). Residues are labeled, H atoms are colored in white and H-bonds are drawn with thin solid lines. Panel D includes a sulfate ion at the upper left-hand corner.
The 2′-OD of the third residue in the GAGA tetraloop, G61, is also directed toward the base and O2′ donates in a water-mediated, intra-nucleotide H-bond to N3 of guanine (Figure 9B). The Watson–Crick edge of the G61 guanine is exposed on the surface and both N1D and N2(D)2 form H-bonds to the same water. A somewhat similar water structure around the minor groove edge under participation of the ribose hydroxyl group as in the case of G61 is also seen with G69. Here, a first water molecule links 2′-OD to N3 and is then H-bonded to a second water molecule that itself accepts an H-bond from the exocyclic amino group of G (Figure 9C). The water-mediated, intra-nucleotide H-bond between G61 2′-OH and N3 is not preserved during the simulation, as the G61 κ angle fluctuates extensively and the 2′-OH often exhibits a backbone or sugar orientation. We always observe individual water molecules forming H-bonds with G61 N3 and 2′-OH, but a bridging water H-bond between G61 N3 and 2′-OH is less common, present in only ∼15% of conformations when the κ angle is favorable.
A chain of water molecules across the minor groove links the 2′-OD moieties of C67 and U53 that form a mismatch pair (Figure 9D). The 2′-OD of C67 adopts a base orientation and sustains a three-water bridge to O2 and N3 (the Watson-Crick edge of this cytosine is turned toward the minor groove). A sulfate ion then mediates contacts to waters that are H-bonded to N4(D)2 of C and 2′-OD of U53. The latter is in a backbone orientation and a water-mediated contact between 2′-OD and phosphate group completes the crossing of the minor groove (Figure 10C). Unlike purine residues that feature single water bridges between N3 and 2′-OD in the base orientation (e.g. G61, G69; Figure 9B, C), more than one water is typically needed to link 2′-OD and O2 of C or U. Besides the aforementioned example of C67, a tandem of waters bridges 2′-OD and O2 of residue U50. Each snapshot from the MD simulation exhibits many more water molecules in the first hydration shell than are resolved in the crystal structures, so it is unsurprising that bridging water interactions observed for C67 with U53 and around U50 are generally well preserved throughout the simulation.
Figure 10.
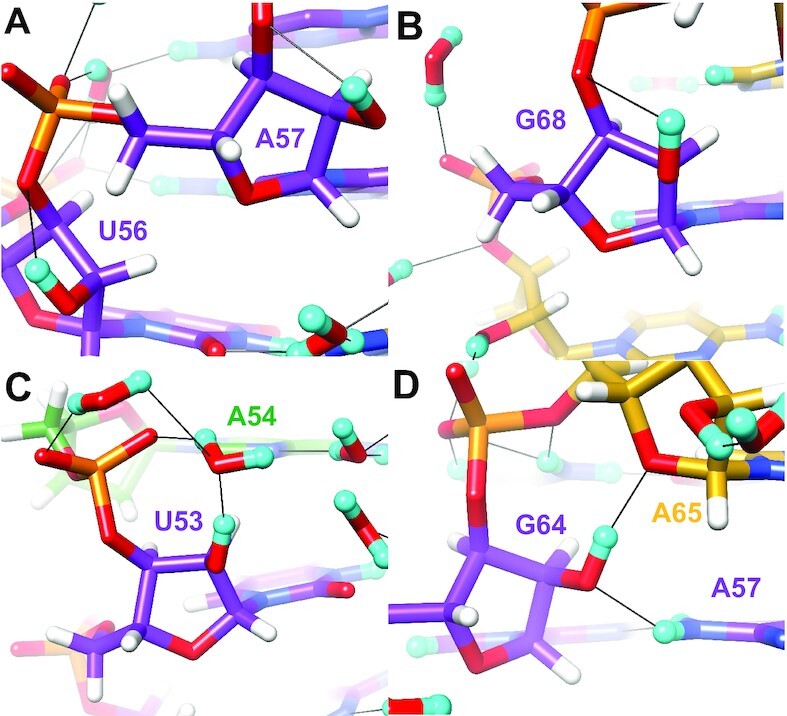
Interactions by selected SRL 2′-OD groups with a backbone orientation. (A) U56 and A57, (B) G68, (C) U53 and (D) G64. The color code matches that in Figure 3A, except for phosphate groups which are colored orange (P) and red (O). Residues are labeled, H atoms are colored in white and H-bonds are drawn with thin solid lines.
2′-OH groups with a phosphate orientation
Ribose hydroxyl groups that adopt a backbone orientation typically direct the hydrogen toward O3′ and the observed (O2′)D…O3′ distances vary between ca. 2.1 Å and 2.7 Å. Shorter distances occur for κ angles that lie just beyond the higher end of the sc+ range. Examples of nucleotides with a backbone orientation of the hydroxyl group are depicted in Figure 10. Thus, U56 (κ = +102°) and A57 (κ = +110°) exhibit relatively tight (O2′)D…O3′ contacts, 2.2 and 2.1 Å, respectively (Figure 10A). Neither hydroxyl group appears to engage in water-mediated bridges to base or phosphate oxygens. For G68, the (O2′)D…O3′ distance is longer, 2.7 Å, consistent with the contracted κ angle of +54° (Figure 10B). For even smaller positive κ angles, the distance between O2′ and O3′ is increased further, thereby weakening the electrostatically favorable interaction between 2′-hydroxyl group and the bridging 3′-oxygen. Rather than simply engaging in this backbone contact, the 2′-OD of U53 stabilizes a tandem water bridge to the 3′-phosphate group (Figure 10C). U53 donates an H-bond to the first water; this water then donates a direct H-bond to OP1 but is also H-bonded to a second water that links it to OP2. This interaction between U53 2′-OH and phosphate oxygens mediated by bridging water molecules is generally retained when the κ angle has a backbone orientation, but is not feasible when κ assumes base or sugar orientations during the simulation. Instead, when κ assumes a base orientation during the simulation, a water-mediated H-bond between U53 2′-OH and C67 N3 is more likely. On rare occasions, the U53•C67 and A54•C66 base pairs ‘break’, facilitating formation of a direct H-bond between U53 2′-OH and A54 N7. At the G64pA65 dimer step, the latter nucleotide exhibits a non-standard backbone conformation (torsion angles α to γ, Table 3). This results in a direct H-bond between the 2′-OD of G64 that adopts a backbone orientation and the 4′-oxygen of A65 (Figure 10D). In addition, O2′ of G64 also accepts an H-bond with N6(D)2 of A57. The unique backbone geometry at G64pA65 is generally preserved in the simulation and these specific interactions are likewise preserved. The κ angle for G64 exhibits by far the least fluctuation during the simulation of any κ angle in the SRL structure (Supplementary Figure S2), perhaps because the unique backbone geometry may help ‘shield’ the G64 2′-OH from close solvent approach. In turn, the stable G64 κ angle facilitates formation of the observed H-bonds with A65 O4′ and A57 N6 amino group.
Table 3.
Backbone and glycosidic torsion angles* and κ angle sectors in the SRL RNA. The standard A-form backbone torsion angle ranges are sc−, ap, sc+, sc+, ap, sc− (α to ζ) and the χ angle average is −160°. Numbers in italicized font represent non-standard angle ranges, ζ-α pairs highlighted in bold font adopt the non-standard ap/sc- conformation around phosphates, and residues in italicized font are part of the A-form SRL stem region (Figure 1C). Residues G48 and C49 exhibit dual occupancy and torsion angles are listed separately for the two conformers A and B, including angles α to δ for U50 (Supplementary Figure S1)
| Residue | α [°] | β [°] | γ [°] | δ [°] | ϵ [°] | ζ [°] | χ [°] | κ sector | Remarks |
|---|---|---|---|---|---|---|---|---|---|
| U47 | – | – | 59.0 | 137.3 | −99.9 | 88.2 | −140.9 | s | |
| G48 | −97.8 | −166.7 | 41.5 | 83.7 | −155.0 | −66.8 | −154.6 | – | A conf. |
| 170.3 | 96.7 | 85.7 | −138.2 | −63.1 | −175.0 | – | B conf. | ||
| C49 | −50.0 | 160.9 | 56.8 | 78.9 | −148.4 | −75.0 | −160.0 | p | A conf. |
| −70.0 | 177.7 | 47.9 | 79.1 | −160.7 | −64.0 | −154.5 | b | B conf. | |
| U50 | −73.5 | 168.3 | 65.2 | 79.3 | −142.4 | −71.7 | −165.4 | b | A conf. |
| 138.6 | −175.7 | −161.1 | 87.5 | B conf. | |||||
| C51 | −65.8 | 160.0 | 62.7 | 77.5 | −155.2 | −69.8 | −164.2 | p | |
| C52 | −68.3 | 179.8 | 54.0 | 80.4 | −146.9 | −59.5 | −157.9 | p | |
| U53 | −67.5 | 171.9 | 52.5 | 82.2 | −159.2 | 59.7 | −147.5 | p | |
| A54 | 166.9 | 143.0 | 50.2 | 148.4 | −86.9 | 152.9 | −133.3 | s | |
| G55 | −92.4 | 84.4 | −177.1 | 150.6 | −167.4 | 140.2 | −88.8 | s | |
| U56 | −71.4 | 150.9 | 42.7 | 86.2 | −138.3 | −66.4 | −177.1 | p | |
| A57 | −72.7 | −179.4 | 52.1 | 79.2 | −143.1 | −52.0 | −166.3 | p | |
| C58 | −72.5 | 173.8 | 56.9 | 77.8 | −147.1 | −63.6 | −163.2 | p | |
| G59 | −74.5 | 175.3 | 54.3 | 75.2 | −125.2 | −67.1 | −171.6 | b | |
| A60 | 172.0 | 137.4 | 56.7 | 83.2 | −126.3 | −66.9 | −161.0 | b | |
| G61 | −78.1 | 140.3 | 84.2 | 79.0 | −149.7 | −62.0 | −163.8 | b | |
| A62 | −68.8 | 179.5 | 48.9 | 80.8 | −141.4 | −65.9 | −150.2 | b | |
| G63 | 144.6 | −133.0 | 178.0 | 81.2 | −138.9 | −56.9 | −174.3 | b | |
| G64 | −67.8 | 174.9 | 58.2 | 76.6 | −158.8 | −77.1 | −164.5 | p | |
| A65 | −106.7 | 78.2 | 165.8 | 78.9 | −148.1 | −74.3 | 179.7 | b | |
| C66 | −56.2 | 169.2 | 65.7 | 81.0 | −129.2 | −68.8 | −168.3 | p | |
| C67 | −62.4 | 161.6 | 54.7 | 85.1 | −140.8 | −57.6 | −165.6 | b | |
| G68 | −76.4 | 174.9 | 58.5 | 72.6 | −145.3 | −73.4 | −169.8 | p | |
| G69 | −53.8 | 161.5 | 60.4 | 78.7 | −142.0 | −79.5 | −171.2 | b | |
| A70 | −67.9 | 171.5 | 56.1 | 74.6 | −162.1 | −75.4 | −162.5 | p | |
| G71 | 148.9 | −168.2 | −171.0 | 90.7 | −130.3 | −68.5 | −171.1 | p | |
| U72 | −64.1 | 165.3 | 52.2 | 79.1 | −158.2 | −67.5 | −159.5 | p | |
| G73 | −74.1 | 175.4 | 62.6 | 73.3 | – | – | −159.2 | b |
*Definition of backbone and χ torsion angles: α = O3′(i− 1)–P–O5′–C5′, β = P–O5′–C5′–C4′, γ = O5′–C5′–C4′–C3′, δ = C5′–C4′–C3′–O3′, ϵ = C4′–C3′–O3′–P(i + 1), ζ = C3′–O3′–P(i + 1)–O5′(i + 1), χ for pyrimidine = O4′–C1′–N1–C2, χ for purine = O4′–C1′–N9–C4.
2′-OH groups with a sugar orientation
Three riboses in the SRL crystal structure feature 2′-OD groups with a sugar orientation, i.e. a κ angle that is in the ap range (U47, A54, G55). All of them also adopt a C2′-endo sugar pucker. The U47 residue is at the 5′-terminus of the construct used for crystallization and its 2′-OD is not engaged in any interactions with water or nucleotides from a neighboring molecule in the crystal structure. The U47 κ angle samples base, phosphate and sugar orientations and its sugar pucker fluctuates readily between C2′-endo and C3′-endo conformations throughout the simulation. This is typical behavior for a terminal oligonucleotide residue in MD simulations, so it is unsurprising that there is poor correlation with the diffraction structure. In the case of A54, a tandem water bridge links the 2′-OD to N3 of the base, with additional H-bonds between 2′-OD and O3′ (Figure 11A). The A54 κ angle adopts a backbone orientation during most of the simulation, but A54 maintains a C2′-endo sugar pucker, consistent with diffraction results. The 2′-oxygen of G55 is about equidistant from OP1 and OP2 and the 2′-OD forms an H-bond to OP2 and the bridging oxygen of the 3′-phosphate. Thus, O2′ adopts an almost ideal in-line O2′…P-O5′ orientation (Figure 11B). However, the absence of a cofactor like a properly spaced metal ion or a base moiety that could activate the 2′-OD for nucleophilic attack precludes a cleavage reaction. The G55 κ angle fluctuates between backbone and sugar orientations frequently but this residue also maintains a C2′-endo sugar pucker throughout the simulation. Based on the observation that the three residues in the SRL with an unusual C2′-endo pucker all have the HO2′ hydrogen directed toward the sugar in the neutron diffraction structure, we conclude that 2′-hydroxyl orientation directly affects sugar conformation such that an antiperiplanar κ angle is associated with a DNA-like ribose conformation. Further, a 2′-OH sugar orientation allows H-bond interactions between the hydroxyl group and both backbone (bridging and/or non-bridging 3′-phosphate oxygens) and - water-mediated - nucleobase atoms.
Figure 11.
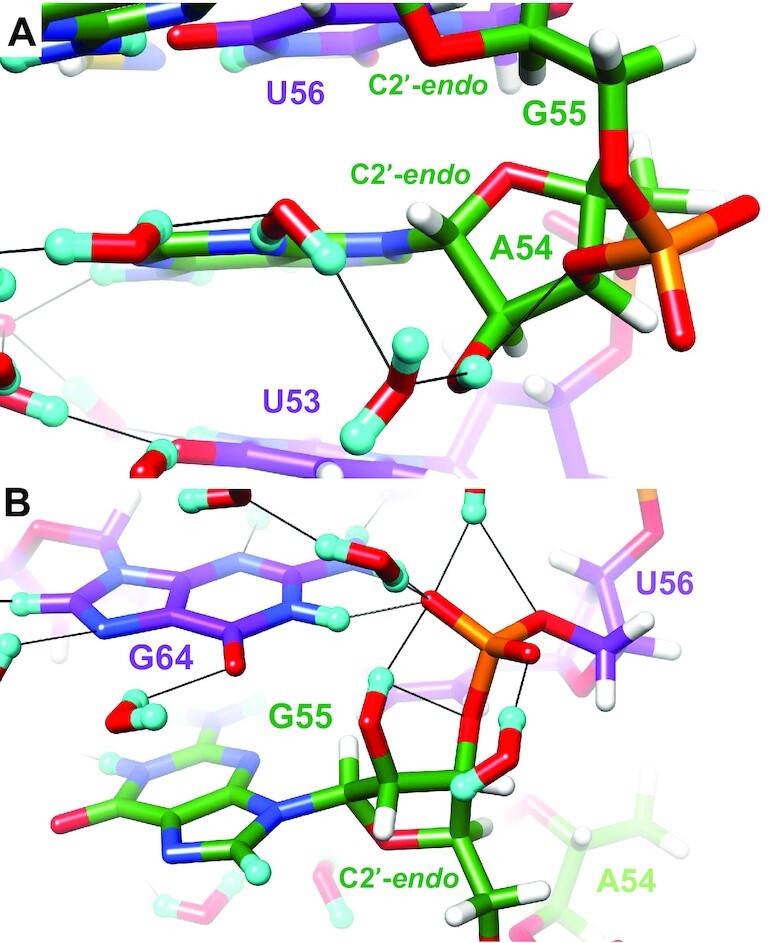
Interactions of two SRL 2′-OD groups in a sugar orientation. (A) A54 and (B) G55. The color code matches that in Figure 3A, except for phosphate groups which are colored orange (P) and red (O). Residues are labeled, H atoms are colored in white and H-bonds are drawn with thin solid lines. The riboses of both A54 and G55 adopt the unusual C2′-endo pucker.
Phosphate hydration
Intra-strand phosphate groups in RNA are more closely spaced than in DNA, owing to the predominant C3′-endo pucker of the ribose in canonical stem and also non-standard hairpin loop and bulged regions, among others. Thus, OP2 non-bridging oxygens of adjacent phosphodiester moieties along the major groove are typically separated by ca. 4.7 Å, but the spacing can be as tight as 4.3 Å (e.g. in the case of G59 and A60 that initiate the SRL GAGA tetraloop). The corresponding P…P distances are ca. 5.5 Å, but amount to just 5.2 Å in the case of residues G59 and A60. Thus, a single water molecule that serves as the donor in both H-bonds can bridge adjacent phosphate oxygens. Examples of this hydration pattern are seen in portions of the backbone that comprises nucleotides G55 to A62 (Figure 12). OP2 oxygens of G59 and G61 in the loop are separated by just 6 Å and two water molecules bridge that gap such that the first also forms an H-bond with OP2 of A60. The OP2 oxygen of the G61 phosphate stacks onto the guanine plane of G59 (30) and the close proximity of phosphate and base prevents a tandem water bridge between OP2 atoms of G61 and A62, although they are only separated by 6.3 Å. Instead, the first of these two waters is near OP1, but the distance between water oxygen and phosphate oxygen is too long for an effective H-bond (4 Å, dashed line in Figure 12). The second water forms an H-bond to OP2 of A62 and also participates in a three-water bridge that crosses the open major groove and ends at OP2 of U56, thereby completing the continuous chain of nine water molecules that links phosphates near the apex of the SRL.
Figure 12.
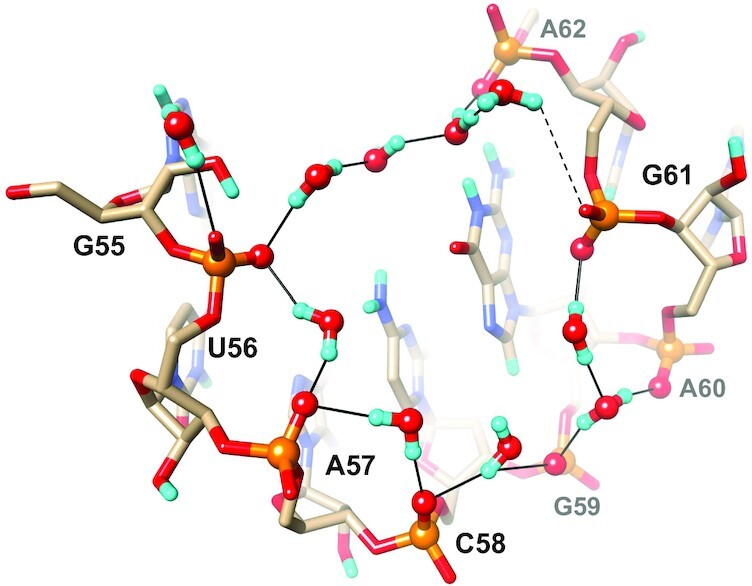
Phosphate hydration involving eight nucleotides including the tetraloop G59−A60−G61−A62. Water molecules link adjacent OP2 oxygens and then cross the major groove, whereby phosphate groups of U56 and A62 serve as bridgeheads. Water molecules (O red, D cyan) and OP2 oxygens are highlighted in ball-and-stick mode, residues are labeled and H-bonds are drawn as thin solid lines. A somewhat long distance between OP1 of G61 and the nearest water is indicated by a dashed line.
DISCUSSION
The 2′-hydroxyl group plays fundamental roles in RNA folding, conformation, stability, activity and hydration. Its involvement in all aspects of RNA structure and function can be tied to a simple observation: the 2′-OH is the only H-bond donor within a sphere of ca. 5.5 Å centered at the oxygen. The closest donor is the exocyclic amino group of guanine which is positioned at a distance of almost 6 Å, in the center of the minor groove. Thus, the 2′-OH is surrounded by a sea of negatively charged oxygen (OP1, OP2) or polarized oxygen (O3′, O4′, O5′, O2(U/C)) and nitrogen atoms N3(A/G) which can only act as H-bond acceptors. This unique ability makes the hydroxyl group indispensable for folding and packing. Its deprotonation affords a nucleophile that is at the heart of phosphodiester cleavage reactions catalyzed by ribozymes. The 2′-OH also undergoes facile H-D exchange in D2O that allows the visualization of deuterium and therefore the 2′-OH orientation in RNA crystals by neutron diffraction. To our knowledge, this is the first report of a single crystal RNA neutron structure determination. The joint cryo X-ray/neutron refinement of the SRL RNA 27mer is based on X-ray and neutron diffraction data from the same crystal with resolutions of 1 Å and 2.2 Å, respectively. It is notable that nearly all aspects of SRL structural features are well preserved in the solution phase, 300K MD simulation, even though the simulation does sample, at least transiently, a number of dramatically different structures with large local deviations from the crystal structures. These simulation results suggest that most of the structural features observed in the 100K diffraction structures are not merely a consequence of crystal packing effects and/or low temperature.
The importance of the H-bond donor ability of the ribose hydroxyl group is demonstrated by a simple comparison between the number of observations in the SRL structure of 2′-OH acting as an H-bond donor or acceptor in intra-nucleotide, inter-nucleotide, inter-SRL and/or water interactions. Thus, the donor: acceptor ratio for 39 observations is almost 6:1! This ratio trends even larger in the simulation because of the vastly greater number of water molecules present; while the 2′-OH certainly does accept H-bonds from surrounding water molecules, it appears to function more typically as a donor, particularly when the water molecules serve as bridging H-bond partners with either phosphate backbone or RNA bases. The illustrations in Figures 9 to 12 clearly attest to this preference in that individual panels show 2′-OH groups exclusively donating H-bonds with just a single exception (Figure 10D). Even in that case the hydroxyl is both donor and acceptor. Trying to assign potential donor-acceptor patterns to 2′-OH groups vis-à-vis adjacent H-bond donor and acceptor moieties including water based on the 2′-oxygen positions in X-ray structures is tempting but quite futile. The SRL neutron structure reveals RNA’s rich repertoire of interactions that involve the 2′-OH moiety. Observed contacts include bifurcated H-bonds and direct interactions with phosphate groups and nucleobases, as well as water-mediated interactions. The latter feature water bridges that can entail a single or two or three solvent molecules. Although a single 2′-OH has the potential to donate and accept in multiple H-bonds, such cases are rare. And even in the few examples of this type seen in the SRL structure, an interaction mode that relies on bifurcated H-bonds (e.g. Figure 11B) is more common than one where a 2′-OH establishes three discrete interactions.
The κ angle behavior is the one area of somewhat limited agreement between the neutron diffraction structure and MD simulation results, suggesting that κ angle behavior may be the structural feature most impacted by low temperature in the diffraction experiments. While general κ angle trends in the simulation correspond reasonably well with the crystal structure, the simulation exhibits numerous deviations from the neutron diffraction structure values. However, it is easy to rationalize this discordance. The simulation represents a solution-phase system at 300K rather than a rigorous crystal lattice simulation at 100K. Additionally, the 2′-OH groups have hydrogen rather than deuterium atoms in the simulation. We have shown previously that many features in protein simulations can be quite different in solution phase versus crystal lattice simulations (61,62), and there is no reason to expect that this should not be true for oligonucleotides as well. The κ angle behavior in our simulations is consistent with the small energy barrier (∼1 kcal mol–1) reported for the C2′-O2′ torsion angle (18), and would be more appropriately compared to solution phase NMR experiments. While the dynamic behavior of the κ angles makes it difficult to draw any conclusions from the simulation results regarding correlations between κ preferences and other structural properties, it is clear from the simulation that the 2′-OH group plays a significant role in modulating intra-molecular interactions in the RNA molecule, and thus in influencing tertiary structure. This is true regardless of the specific details observed in any individual simulation snapshot, due to the key point noted above: the 2′-OH group is the only H-bond donor available to potential acceptors in the nearby phosphate backbone, ribose groups and bases.
The SRL RNA structure allows a better understanding of the presumably different functions of 2′-OH groups that assume either base or backbone orientations. Thus, a backbone orientation is not limited to a close favorable contact with the adjacent 3′-oxygen (Figure 10A, B), but can stabilize a water-mediated interaction between 2′-OH and a nucleobase O/N atom (Figure 9D, U53). Alternatively, a 2′-OH in a backbone orientation participates in a water-mediated interaction with non-bridging phosphate oxygens (Figure 10C). A 2′-OH directed toward the backbone can also form an H-bond to O4′ and a direct H-bond to a nucleobase (Figure 10D). In this case, the interaction pattern is associated with a non-standard backbone geometry as illustrated by the torsion angle analysis (Table 3). Similar to the complex range of interactions that are enabled by 2′-OH moieties with a phosphate orientation, a 2′-OH with a base orientation may be energetically favored based on the intrinsic energetics of the phosphodiester backbone without consideration of water or additional interactions with the surrounding RNA (17,18,20), but can certainly be associated with non-canonical backbone geometries as well. For example, backbone orientations of 2′-OH groups in the SRL stem are more common than base orientations (Figures 1C, 3A). The SRL GAGA loop offers even more convincing evidence that base-oriented 2′-OH groups can be associated with non-canonical regions: Thus, all four nucleotides in the tetraloop exhibit a base orientation (Figure 9A, B). Although the 2′-OH is the only H-bond donor in the RNA backbone, all four bases feature H-bond donor moieties that can be used to establish tertiary structural contacts. The electrostatically favorable interaction between 2′-OH and O3′ when the former adopts a backbone orientation must be a staple of RNA folding and stability and a key enabler of the expansive RNA fold space. Separating the roles of 2′-OH and nucleobases in inducing complex RNA tertiary folding patterns remains difficult, however, although the lack of the 2′-OH in DNA and its repetitive structural behavior relative to RNA seem to lend support for a more dominant role of the hydroxyl group relative to the bases in this regard.
The neutron structure of the SRL provides experimental evidence for an important role of the 2′-OH orientation in flipping the sugar pucker between the standard C3′-endo and atypical C2′-endo modes. In three nucleotides, U47, A54 and G55, the κ angle is in the antiperiplanar range and the ribose adopts the less common C2′-endo pucker. Other torsion angles in these regions of the backbone also assume non-standard conformations (Table 3). The role of the 2′-OH as an arbiter of not just backbone conformation and RNA tertiary structure but also the ribose pucker itself can be rationalized using steric and electrostatic arguments (Figure 13). Accordingly, an ap orientation of the 2′-OH for a ribose in the C3′-endo pucker creates a short contact between the HO2′ hydrogen and H4′ (around the sum of the vdW radii, 2.0 Å minimum) while simultaneously preventing an H-bond with O3′ (Figure 13B). Flipping the pucker to the C2′-endo type relieves the short contact and leads to a favorable interaction with O3′ (Figure 13A). Interestingly, the sugar orientation of the 2′-hydroxyl group allows both backbone contacts (O3′ and non-bridging phosphate oxygens) and water-mediated interactions with base atoms (Figure 11).
Figure 13.
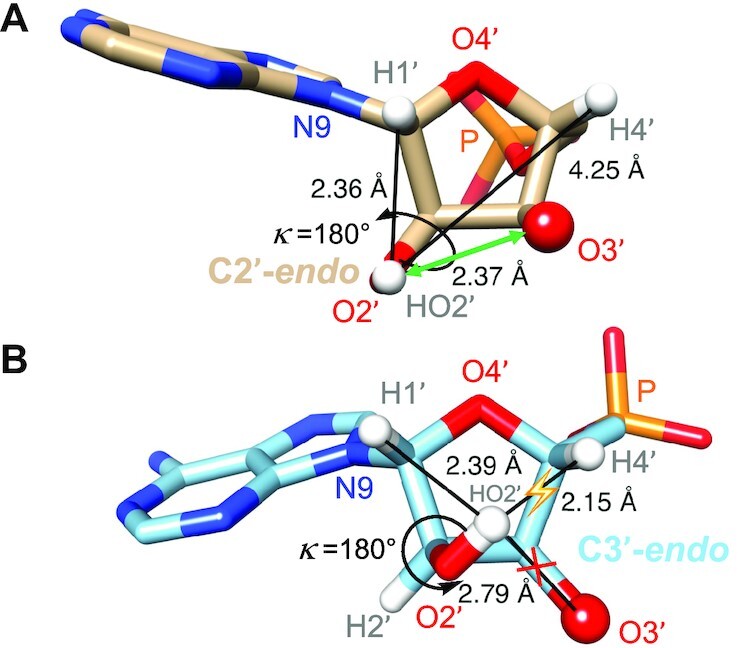
Relationship between 2′-OH orientation and ribose pucker. (A) A 2′-OH with a ‘sugar orientation’ (shown here is a κ angle in an ideal 180° ap conformation) combined with a C2′-endo pucker precludes short contacts to ribose H atoms and allows H-bonding between 2′-OH and O3′ (green double arrow). (B) A 2′-OH with a ‘sugar orientation’ (as in panel A, the κ angle is in an ideal 180° ap conformation) combined with a C3′-endo pucker results in a short contact (yellow flash) and precludes H-bonding between 2′-OH and O3′ (red cross). Selected atoms are labeled and relevant distances are shown in Å. Please see also the correlation between sugar pucker and 2′-OH orientation in an early MD study of RNA (Figure 1, C3′-endo, and Figure 5, C2′-endo (19)).
The correlation between the κ angle in the ap range and adoption of a C2′-endo ribose pucker in the SRL structure also seen in a previous MD simulation of RNA (19) allows us to make predictions as to the whereabouts of the HO2′ hydrogen. In the SRL structure, the maximum observed value of κ is +172° (G55, Table 2). Increasing the size of the angle further will weaken the interaction with O3′ and create a clash with H1′ (Figure 13A). Therefore, κ angles in a 40° range from ca. +170° to −150° can be expected to be disfavored. We analyzed the yeast tRNAPhe crystal structure (PDB ID 1EHZ) (63) in regard to the ribose conformations with the program PROSIT (64) (https://cactus.nci.nih.gov/prosit/) and found 10 nucleotides out of 76 that adopt a C2′-endo pucker: U7, A9, G18, G19, 7meG45, C47, U48, 1meA58, C60 and A76 (3′-terminus). We calculated the positions of H2′ and HO2′ hydrogen atoms in all cases except for the 3′-terminal residue and adjusted the κ angle to + 160° (the average of the κ angle in residues A54 and G55 of the SRL structure). As expected the HO2′ hydrogen engaged in at least two geometrically reasonable H-bonds to O3′, non-bridging phosphate oxygens, base atoms and/or water molecules in all nine cases. An illustration for five tRNA ribonucleotides is depicted in Supplementary Figure S5.
RNA-binding proteins can sculpt RNA backbone conformation and potentially set the sugar pucker or at least lower the energy barrier that separates the C3′-endo and C2′-endo conformations by influencing the orientation of the 2′-hydroxyl group. In the crystal structure of human Argonaute2 (Ago2) bound to miR-20a (PDB ID 4F3T) (65) two nucleotides of the micro-RNA exhibit a C2′-endo pucker. These residues are 5′P-U1 that is lodged in the MID domain binding pocket and G18 that resides right outside the PAZ domain that captures the 3′-terminal G19 and G20. In both cases, the 2′-OH is contacted by an Ago2 side chain. The Oϵ amide oxygen of Gln-548 is located at 3.03 Å from O2′ (U1) and acts as an H-bond acceptor. Further, the O2′ is located at 2.55 Å from the adjacent phosphate (OP1) that links U1 and A2. Thus, the HO2′ hydrogen is most likely shared between Gln-548 and the phosphate (Supplementary Figure S6a). At the opposite end of the RNA, Arg-315 lies at 2.8 Å from the G19 phosphate (NϵH…OP1) and 3.4 Å from O3′ of that phosphodiester as well as 3.4 Å from O2′ of G18 (NH2…O2′/O3′). Here, the arginine acts as an H-bond donor in all three cases (Supplementary Figure S6b). Setting the κ angle to 160° for both riboses produces a satisfactory model of the H-bond interactions and is again in accordance with the H2′−C2′−O2′−HO2′ torsion angle range for riboses that adopt a C2′-endo conformation.
Interactions between two 2′-hydroxyl groups are fairly common in larger RNA folding motifs and are among intermolecular interactions that stabilize RNA crystals. Consecutive occurrences of such H-bonded 2′-OH pairs are called ribose zippers (5–9). In X-ray crystal structures two 2′-oxygen atoms separated by less than 3 Å can reasonably be expected to be H-bonded. However, without knowledge of the HO2′ positions we cannot be sure that such hydroxyl pairs actually form an H-bond. In the SRL crystal structure there are no ribose zippers, but there are two examples of O2′ pairs from adjacent RNA molecules where oxygen atoms are separated by a distance consistent with H-bond formation (ca. 2.7 Å; Figure 14). The neutron data reveal that this is indeed the case for one pair, but that the deuterium atoms of the other O2′ pair are used to engage in alternative H-bonds. Thus, 2′-OH of C51 and 2′-OH of C58# are H-bonded. Conversely, the 2′-OH of C52 is directed toward O3′ and the 2′-OH of A62# forms a bifurcated H-bond to O2 of C52 and a water molecule that is also H-bonded to N3 and 2′-OH of G69 as well as another water (Figure 14).
Figure 14.
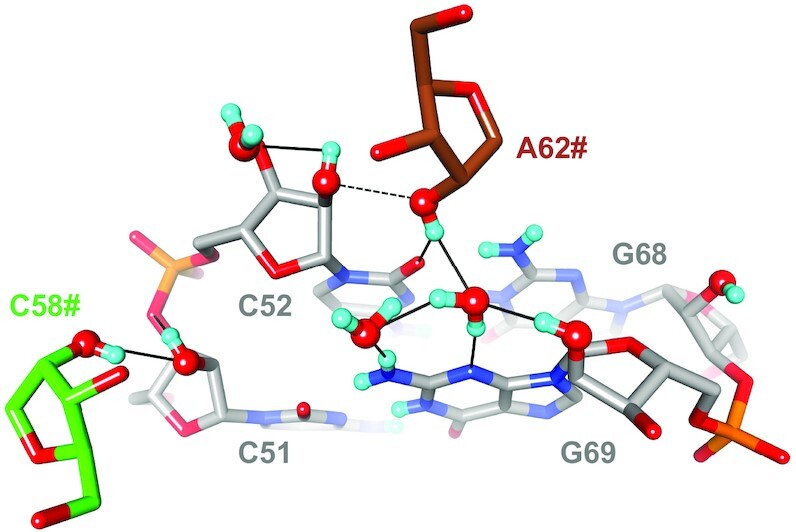
Neutron diffraction data reveal H-bonding between pairs of 2′-hydroxyl groups as well as the absence thereof. The SRL C51pC52:G68pG69 base pair step (gray carbon atoms) is viewed from the minor groove side along with the ribose moieties of nucleotides C58# (green carbons) and A62# (brown carbons) from symmetry mates. The O2′ atoms of C51 and C58# and C52 and A62# are in both cases separated by ca. 2.7 Å, but only the former pair is H-bonded (solid line, left) whereas the latter is not (dashed line).
CONCLUSION AND ETIOLOGICAL CONSIDERATIONS
Much has been written and conjectured about nature's choice of phosphate in the nucleic acids backbone (66,67). Similarly, we can try and rationalize nature's choice of a ribose-based pairing system. Investigations of artificial nucleic acids that combine the phosphodiester moiety and four standard bases with many alternative sugars have demonstrated that stable pairing is by no means a unique attribute of RNA (and DNA) (68–73). Many of the explored systems were able to cross-pair with DNA and RNA. Others form autonomous pairing systems. Examples of such systems are pyranosyl-RNA (p-RNA (74)) and 2′,3′-dideoxyglucopyranosyl nucleic acid (homo-DNA (75)). Hexose-based nucleic acids lack conformational plasticity and the sugar moieties are restricted to the chair conformation. Threofuranosyl nucleic acid (TNA) constitutes a stable pairing system that also forms hybrids with DNA and RNA. However, it too lacks conformational plasticity as its pucker is limited to the C4′-exo type. Unlike RNA, TNA lacks an H-bond donor in its backbone and it is unlikely that TNA can exceed RNA in terms of the complexity of its fold space. Arabinonucleic acid (ANA) with the opposite C2′ stereochemistry compared to RNA lacks conformational flexibility and its sugar pucker is limited to the Southeastern region of the pseudorotation phase cycle (72). No reports regarding ANA self-cleavage reactions exist, but the analog has been used for in vitro selection experiments (Selex) and an ANAzyme was reported to catalyze RNA cleavage (76). Similarly, Selex afforded a TNA enzyme ‘threozyme’ with RNA ligase activity (77), and base-modified TNA aptamers ‘threomers’ that bind protein targets with KD values in the low nM range (78). Still, the search to date for alternative nucleic acids has not uncovered a system that can mimic RNA structurally, chemically and/or functionally.
The conversion of the sugar conformation between the North and South types appears to be unique for DNA and RNA (3). Unlike DNA that can undergo reversible transitions between the A- and B-form duplexes depending on ionic strength and level of humidity, the RNA 2′-OH group can apparently set the pucker solely as a result of its orientation. Thanks to its versatile H-bond donor and acceptor qualities and ability to act as a nucleophile, RNA genotype and phenotype are intricately linked to the 2′-OH group. As the SRL RNA neutron structure shows, the orientation of the 2′-OH is important for RNA conformation, stability and water structure. Just focusing on the 2′-oxygen positions from X-ray crystallography and ignoring the whereabouts of the HO2′ hydrogen may lead us to overlook important information that can be gained from a neutron experiment. The possibility to do so with relatively small crystals and using higher flux spallation sources in combination with cryo data collection now renders this approach attractive for much larger RNAs as well. Furthermore, the generally good agreement between the 100K diffraction structures and the 300K solution phase MD simulation results suggests that many structural features we observe in the crystal structures are NOT merely artifacts due to low temperature or crystal packing effects, and may thus be more broadly relevant.
DATA AVAILABILITY
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data Bank under accession number 7UCR.
Supplementary Material
ACKNOWLEDGEMENTS
The research at O.R.N.L.’s Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, US Department of Energy.
Contributor Information
Joel M Harp, Department of Biochemistry, Vanderbilt University, Nashville, TN 37232, USA; Center for Structural Biology, Vanderbilt University, Nashville, TN 37232, USA.
Terry P Lybrand, Center for Structural Biology, Vanderbilt University, Nashville, TN 37232, USA; Department of Chemistry, Vanderbilt University, Nashville, TN 37235, USA.
Pradeep S Pallan, Department of Biochemistry, Vanderbilt University, Nashville, TN 37232, USA; Center for Structural Biology, Vanderbilt University, Nashville, TN 37232, USA.
Leighton Coates, Neutron Scattering Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN 37831, USA.
Brendan Sullivan, Neutron Scattering Division, Oak Ridge National Laboratory, 1 Bethel Valley Road, Oak Ridge, TN 37831, USA.
Martin Egli, Department of Biochemistry, Vanderbilt University, Nashville, TN 37232, USA; Center for Structural Biology, Vanderbilt University, Nashville, TN 37232, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Vanderbilt University Advanced Computing Center for Research and Education (supported in part by NIH) [S10 OD020154 and S10 RR031634]; Alnylam Pharmaceuticals (to M.E.); Vanderbilt University (to M.E.); Volkswagen Stiftung (to M.E.); US National Science Foundation [NSF 2224897 to M.E.]; US National Institutes of Health [NIH R01 GM71939 to L.C.]. The open access publication charge for this paper has been waived by Oxford University Press - NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Altona C., Sundaralingam M.. Conformational analysis of the sugar ring in nucleosides and nucleotides. New description using the concept of pseudorotation. J. Am. Chem. Soc. 1972; 94:8205–8212. [DOI] [PubMed] [Google Scholar]
- 2. Thibaudeau C., Chattopadhyaya J.. Stereoelectronic Effects in Nucleosides and Nucleotides and Their Structural Implications. 1999; Uppsala, Sweden: Uppsala University Press. [Google Scholar]
- 3. Rich A. The double helix: a tale of two puckers. Nat. Struct. Mol. Biol. 2003; 10:247–249. [DOI] [PubMed] [Google Scholar]
- 4. Egli M., Portmann S., Usman N.. RNA hydration: a detailed look. Biochemistry. 1996; 35:8489–8494. [DOI] [PubMed] [Google Scholar]
- 5. Quigley G.J., Rich A.. Structural domains of transfer RNA molecules. Science. 1976; 194:796–806. [DOI] [PubMed] [Google Scholar]
- 6. Cate J.H., Gooding A.R., Podell E., Zhou K., Golden B.L., Kundrot C.E., Cech T.R., Doudna J.A.. Crystal structure of a group I ribozyme domain: principles of RNA packing. Science. 1996; 273:1678–1685. [DOI] [PubMed] [Google Scholar]
- 7. Berger I., Egli M.. The role of backbone oxygen atoms in the organization of nucleic acid tertiary structure: zippers, networks, clamps, and C-H···O hydrogen bonds. Chem. Eur. J. 1997; 3:1400–1404. [Google Scholar]
- 8. Tamura M., Holbrook S.R.. Sequence and structural conservation in RNA ribose zippers. J. Mol. Biol. 2002; 320:455–474. [DOI] [PubMed] [Google Scholar]
- 9. Butcher S.E., Pyle A.M.. The molecular interactions that stabilize RNA tertiary structure: RNA motifs, patterns, and networks. Acc. Chem. Res. 2011; 44:1302–1311. [DOI] [PubMed] [Google Scholar]
- 10. Shi Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017; 18:655–670. [DOI] [PubMed] [Google Scholar]
- 11. Fica S.M., Nagai K.. Cryo-electron microscopy snapshots of the spliceosome: structural insights into a dynamic ribonucleoprotein machine. Nat. Struct. Mol. Biol. 2017; 24:791–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Peebles C.L., Perlman P.S., Mecklenburg K.L., Petrillo M.L., Tabor J.H., Jarrell K.A., Cheng H.L.. A self-splicing RNA excises an intron lariat. Cell. 1986; 44:213–223. [DOI] [PubMed] [Google Scholar]
- 13. Qin P.Z., Pyle. A.M. The architectural organization and mechanistic function of group II intron structural elements. Curr. Opin. Struct. Biol. 1998; 8:301–308. [DOI] [PubMed] [Google Scholar]
- 14. Cech T.R. The chemistry of self-splicing RNA and RNA enzymes. Science. 1987; 236:1532–1539. [DOI] [PubMed] [Google Scholar]
- 15. Mitchell Emilsson G., Nakamura S., Roth A., Breaker R.R.. Ribozyme speed limits. RNA. 2003; 9:907–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. O’Rourke S.M., Scott W.G.. Structural simplicity and mechanistic complexity in the hammerhead ribozyme. Prog. Mol. Biol. Transl. Sci. 2018; 159:177–202. [DOI] [PubMed] [Google Scholar]
- 17. Denning E.J., MacKerell A.D. Jr. Intrinsic contribution of the 2′-hydroxyl group to RNA conformational heterogeneity. J. Am. Chem. Soc. 2012; 134:2800–2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Darré L., Ivani I., Dans P.D., Gómez H., Hospital A., Orozco M.. Small details matter: the 2′-hydroxyl as a conformational switch in RNA. J. Am. Chem. Soc. 2016; 138:16355–16363. [DOI] [PubMed] [Google Scholar]
- 19. Auffinger P., Westhof E.. Rules governing the orientation of the 2′-hydroxyl group in RNA. J. Mol. Biol. 1997; 274:54–63. [DOI] [PubMed] [Google Scholar]
- 20. Fohrer J., Hennig M., Carlomagno T.. Influence of the 2′-hydroxyl group conformation on the stability of A-form helices in RNA. J. Mol. Biol. 2006; 356:280–287. [DOI] [PubMed] [Google Scholar]
- 21. Ying J., Bax A.. 2′-Hydroxyl proton positions in helical RNA from simultaneously measured heteonuclear scalar couplings and NOEs. J. Am. Chem. Soc. 2006; 128:8372–8373. [DOI] [PubMed] [Google Scholar]
- 22. Barnwal R.P., Yang F., Varani G.. Applications of NMR to structure determination of RNAs large and small. Arch. Biochem. Biophys. 2017; 628:42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Egli M., Tereshko V., Teplova M., Minasov G., Joachimiak A., Sanishvili R., Weeks C.M., Miller R., Maier M.A., An H.et al.. X-ray crystallographic analysis of the hydration of A- and B-Form DNA at atomic resolution. Biopolymers (Nucleic Acid Sci.). 2000; 48:234–252. [DOI] [PubMed] [Google Scholar]
- 24. Bancroft D., Williams L.D., Rich A., Egli M.. The low temperature crystal structure of the pure-spermine form of Z-DNA reveals binding of a spermine molecule in the minor groove. Biochemistry. 1994; 33:1073–1086. [DOI] [PubMed] [Google Scholar]
- 25. Brzezinski K., Brzuszkiewicz A., Dauter M., Kubicki M., Jaskolski M., Dauter Z.. High regularity of Z-DNA revealed by ultra high-resolution crystal structure at 0.55 A. Nucleic Acids Res. 2011; 39:6238–6248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Szewczak A.A., Moore P.B., Chang Y.L., Wool I.G.. The conformation of the sarcin/ricin loop from 28S ribosomal RNA. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:9581–9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Glück A., Endo Y., Wool I.G.. The ribosomal RNA identity elements for ricin and for α-sarcin: mutations in the putative CG pair that closes a GAGA tetraloop. Nucleic Acids Res. 1994; 22:321–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Correll C.C., Wool I.G., Munishkin A.. The two faces of the Escherichia coli 23 s rRNA sarcin/ricin domain: the structure at 1.11 Å resolution. J. Mol. Biol. 1999; 292:275–287. [DOI] [PubMed] [Google Scholar]
- 29. Olieric V., Rieder U., Lang K., Serganov A., Schulze-Briese C., Micura R., Dumas P., Ennifar E.. A fast selenium derivatization strategy for crystallization and phasing of RNA structures. RNA. 2009; 15:707–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pallan P.S., Lybrand T.P., Schlegel M.K., Harp J.M., Jahns H., Manoharan M., Egli M.. Incorporating a thiophosphate modification into a common RNA tetraloop motif causes an unanticipated stability boost. Biochemistry. 2020; 59:4627–4637. [DOI] [PubMed] [Google Scholar]
- 31. Coates L., Tomanicek S., Schrader T.E., Weiss K.L., Ng J.D., Jüttner P., Ostermann A.. Cryogenic neutron protein crystallography: routine methods and potential benefits. J. Appl. Cryst. 2014; 47:1431–1434. [Google Scholar]
- 32. Kwon H., Langan P.S., Coates L., Raven E.L., Moody P.C.E.. The rise of neutron cryo-crystallography. Acta Cryst. D. 2018; 74:792–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Azadmanesh J., Lutz W.E., Coates L., Weiss K., Borgstahl G.E.O.. Cryotrapping peroxide in the active site of human mitochondrial manganese superoxide dismutase crystals for neutron diffraction. Acta Cryst. F. 2022; 78:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harp J., Coates L., Sullivan B., Egli M.. Cryo-neutron crystallographic data collection and preliminary refinement of left-handed Z-DNA d(CGCGCG). Acta Cryst. F. 2018; 74:603–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harp J.M., Coates L., Sullivan B., Egli M.. Water structure around a left-handed Z-DNA fragment analyzed by cryo neutron crystallography. Nucleic Acids Res. 2021; 49:4782–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coates L., Cuneo M.J., Frost M.J., He J.H., Weiss K.L., Tomanicek S.J., McFeeters H., Vandavasi V.G., Langan P., Iverson E.B.. The macromolecular neutron diffractometer MaNDi at the spallation neutron source. J. Appl. Cryst. 2015; 48:1302–1306. [Google Scholar]
- 37. Coates L., Cao H.B., Chakoumakos B.C., Frontzek M.D., Hoffmann C., Kovalevsky A.Y., Liu Y., Meilleur F., dos Santos A.M., Myles D.A.A.et al.. A suite-level review of the neutron single-crystal diffraction instruments at Oak Ridge National Laboratory. Rev. Sci. Instrum. 2018; 89:092802. [DOI] [PubMed] [Google Scholar]
- 38. Arnold O., Bilheux J.C., Borreguero J.M., Buts A., Campbell S.I., Chapon L., Doucet M., Draper N., Leal R.F., Gigg M.A.et al.. Mantid - Data analysis and visualization package for neutron scattering and µ SR experiments. Nuclear Instrum. Methods A. 2014; 764:156–166. [Google Scholar]
- 39. Sullivan B., Archibald R., Vandavasi V., Langan P., Langan P.S., Coates L., Azadmanesh J., Lynch V.E.. BraggNet: integrating Bragg peaks using neural networks. J. Appl. Cryst. 2019; 52:854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Campbell J.W., Hao Q., Harding M.M., Nguti N.D., Wilkinson C.. LAUEGEN version 6.0 and INTLDM. J. Appl. Cryst. 1998; 31:496–502. [Google Scholar]
- 41. Helliwell J.R., Habash J., Cruickshank D.W.J., Harding M.M., Greenhough T.J., Campbell J.W., Clifton I.J., Elder M., Machin P.A., Papiz M.Z.et al.. The recording and analysis of synchrotron X-radiation Laue diffraction photographs. J. Appl. Cryst. 1989; 22:483–497. [Google Scholar]
- 42. Adams P.D., Afonine P.V., Bunkoczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. D. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Afonine P.V., Mustyakimov M., Grosse-Kunstleve R.W., Moriarty N.W., Langan P., Adams P.D.. Joint X-ray and neutron refinement with phenix.refine. Acta Cryst. D. 2010; 66:1153–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Cryst. D. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eriksson A., Caldararu O., Ryde U., Oksanen E.. Automated orientation of water molecules in neutron crystallographic structures of proteins. Acta Cryst. D. 2020; 76:1025–1032. [DOI] [PubMed] [Google Scholar]
- 46. Murakawa T., Kurihara K., Adachi M., Kusaka K., Tanizawa K., Okajima T.. Re-evaluation of protein neutron crystallography with and without X-ray/neutron joint refinement. IUCrJ. 2022; 9:342–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Case D.A., Cheatham T.E. 3rd, Darden T., Gohlke H., Luo R., Merz K.M. Jr, Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aytenfisu A.H., Spasic A., Grossfield A., Stern H.A., Mathews D.H.. Dihedral parameters for the AMBER force field improve RNA molecular dynamics. J. Chem. Theory Comput. 2017; 13:900–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jorgensen W.L., Chandrasekhar J., Madura J., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926–935. [Google Scholar]
- 50. Joung I.S., Cheatham T.E. III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008; 112:9020–9041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Cerutti D.S., Duke R.E., Darden T.A., Lybrand T.P.. Staggered mesh ewald: an extension of the smooth particle-mesh ewald method adding great versatility. J. Chem. Theory Comp. 2009; 5:2322–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Izaguirre J.A., Catarello D.P., Wozniak J.M., Skeel R.D.. Langevin stabilization of molecular dynamics. J. Phys. Chem. 2001; 114:2090–2098. [Google Scholar]
- 53. Ryckaert J.P., Ciccotti G., Berendsen H.J.C., Hirasawa K.. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977; 23:327–341. [Google Scholar]
- 54. Miyamoto S., Kollman P.A.. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J. Comput. Chem. 1992; 13:952–962. [Google Scholar]
- 55. Roe D.R., Cheatham T.E. III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013; 9:3084–3095. [DOI] [PubMed] [Google Scholar]
- 56. Petterson E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 57. Shi X., Khade P.K., Sanbonmatsu K.Y., Joseph S.. Functional role of the sarcin-ricin loop of the 23S rRNA in the elongation cycle of protein synthesis. J. Mol. Biol. 2012; 419:125–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Garcia-Moreno E.B. Electrostatics and hydrogen bonding. Adv. Mol. Cell Biol. 1997; 22:109–132. [Google Scholar]
- 59. Chen S.-J. RNA folding: conformational statistics, folding kinetics, and ion electrostatics. Annu. Rev. Biophys. 2008; 37:197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Patra A., Zhang Q., Lei L., Su Y., Egli M., Guengerich F.P.. Structural and kinetic analysis of nucleoside triphosphate incorporation opposite an abasic site by human translesion DNA polymerase η. J. Biol. Chem. 2015; 290:8028–8038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cerutti D.S., Le Trong I., Stenkamp R.E., Lybrand T.P.. Simulations of a protein crystal: explicit treatment of crystallization conditions links theory and experiment in the streptavidin-biotin complex. Biochemistry. 2008; 47:12065–12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Cerutti D.S., Le Trong I., Stenkamp R.E., Lybrand T.P.. Dynamics of the streptavidin-biotin complex in solution and in its crystal lattice: distinct behavior revealed by molecular simulations. J. Phys. Chem. B. 2009; 113:6971–6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Shi H., Moore P.B.. The crystal structure of yeast phenylalanine tRNA at 1.93 Å resolution: a classic structure revisited. RNA. 2000; 6:1091–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun G., Voigt J.H., Filippov I.V., Marquez V.E., Nicklaus M.C.. PROSIT: Pseudo-rotational online service and interactive tool, applied to a conformational survey of nucleosides and nucleotides. J. Chem. Inf. Comput. Sci. 2004; 44:1752–1762. [DOI] [PubMed] [Google Scholar]
- 65. Elkayam E., Kuhn C.D., Tocilj A., Haase A.D., Greene E.M., Hannon G.J., Joshua-Tor L.. The structure of human argonaute-2 in complex with miR-20a. Cell. 2012; 150:100–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Westheimer F. Why nature chose phosphate. Science. 1987; 235:1173–1178. [DOI] [PubMed] [Google Scholar]
- 67. Kamerlin S.C., Sharma P.K., Prasad R.B., Warshel A.. Why nature really chose phosphate. Q. Rev. Biophys. 2013; 46:1–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eschenmoser A. Chemical etiology of nucleic acid structure. Science. 1999; 284:2118–2124. [DOI] [PubMed] [Google Scholar]
- 69. Eschenmoser A. The quest for the chemical roots of life. Chem. Commun. 2004; 40:1247–1252. [DOI] [PubMed] [Google Scholar]
- 70. Eschenmoser A. The TNA-family of nucleic acid systems: properties and prospects. Origins Life Evol. Biospheres. 2004; 34:277–306. [DOI] [PubMed] [Google Scholar]
- 71. Egli M., Herdewijn P.. Chemistry and Biology of Artificial Nucleic Acids. 2012; Weinheim: Wiley-VCH Publishers. [Google Scholar]
- 72. Anosova I., Kowal E.A., Dunn M.R., Chaput J.C., van Horn W.D., Egli M.. The structural diversity of artificial genetic polymers. Nucleic Acids Res. 2016; 44:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Devine K.G., Jheeta S.. De novo nucleic acids: a review of synthetic alternatives to DNA and RNA that could act as bio-information storage molecules. Life. 2020; 10:346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Schlönvogt I., Pitsch S., Lesueur C., Eschenmoser A., Jaun B., Wolf R.M.. Pyranosyl-RNA (p-RNA): NMR and molecular-dynamics study of the duplex formed by self-pairing of ribopyranosyl-(C-G-A-A-T-T-C-G). Helv. Chim. Acta. 1996; 79:2316–2349. [Google Scholar]
- 75. Egli M., Pallan P.S., Pattanayek R., Wilds C.J., Lubini P., Minasov G., Dobler M., Leumann C.J., Eschenmoser A.. Crystal structure of homo-DNA and nature’s choice of pentose over hexose in the genetic system. J. Am. Chem. Soc. 2006; 128:10847–10856. [DOI] [PubMed] [Google Scholar]
- 76. Taylor A.I., Pinheiro V.B., Smola M.J., Morgunov A.S., Peak-Chew S., Cozens C., Weeks K.M., Herdewijn P., Holliger P. Catalysts from synthetic genetic polymers. Nature. 2015; 518:427–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang Y., Wang Y., Song D., Sun X., Zhang Z., Li X., Li Z., Yu H.. A threose nucleic acid enzyme with RNA ligase activity. J. Am. Chem. Soc. 2021; 143:8154–8163. [DOI] [PubMed] [Google Scholar]
- 78. McCloskey C.M., Li Q., Yik E.J., Chim N., Ngor A.K., Medina E., Grubisic I., Co Ting Keh L., Poplin R., Chaput J.C.. Evolution of functionally enhanced α-L-threofuranosyl nucleic acid aptamers. ACS Synth. Biol. 2021; 10:3190–3199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structure have been deposited with the Protein Data Bank under accession number 7UCR.