


Abstract
Advances in genome sequencing have produced hundreds of thousands of bacterial genome sequences, many of which have integrated prophages derived from temperate bacteriophages. These prophages play key roles by influencing bacterial metabolism, pathogenicity, antibiotic resistance, and defense against viral attack. However, they vary considerably even among related bacterial strains, and they are challenging to identify computationally and to extract precisely for comparative genomic analyses. Here, we describe DEPhT, a multimodal tool for prophage discovery and extraction. It has three run modes that facilitate rapid screening of large numbers of bacterial genomes, precise extraction of prophage sequences, and prophage annotation. DEPhT uses genomic architectural features that discriminate between phage and bacterial sequences for efficient prophage discovery, and targeted homology searches for precise prophage extraction. DEPhT is designed for prophage discovery in Mycobacterium genomes but can be adapted broadly to other bacteria. We deploy DEPhT to demonstrate that prophages are prevalent in Mycobacterium strains but are absent not only from the few well-characterized Mycobacterium tuberculosis strains, but also are absent from all ∼30 000 sequenced M. tuberculosis strains.
INTRODUCTION
The explosion in bacterial genome sequencing and the accumulated vast data sets present bioinformatic challenges to the identification and analysis of their prophages. Prophages are the genomes of temperate bacteriophages either integrated into bacterial chromosomes or maintained as extrachromosomal replicons. Temperate phages are a substantial proportion of the phage population—plausibly the majority—and prophages are common residents of sequenced bacterial genomes (1). Bacteriophage genomics has revealed them to be highly diverse genetically with extensive mosaicism being a hallmark of phage genome architecture (2). A high proportion (∼70%) of bacteriophage genes are of unknown function, and phages likely represent the biggest reservoir of unexplored sequences in the biosphere (3). When present, a prophage genome typically accounts for only about 1% of the length of bacterial genomes, ∼20–80 kb, coding for 20–100 genes. Temperate phages and their prophages commonly include genes that are lysogenically expressed and influence host physiology, including virulence genes, metabolic genes, and phage defense genes (4,5). Identifying and characterizing prophages is therefore critical to understanding bacterial pathogenesis and microbial dynamics.
Although highly diverse, there is substantial heterogeneity among the types of phages infecting any particular bacterial host. Over 2000 phages infecting Mycobacterium smegmatis have been completely sequenced, and these can be grouped into ‘clusters’ according to their overall sequence relationships (6–8). Using a threshold of 35% shared gene content (9), these represent 31 clusters (Clusters A–Z, AA–AE) and seven singletons each of which has no close relative (10). However, these are heterogeneously represented such that there are nearly 700 Cluster A phages, but 10 clusters have fewer than five phages each. Phages of other hosts can be grouped similarly (11–13), and this heterogeneity likely emerges from unequal sampling of the underlying phage population which has a continuum of diversity created by exchange of mosaic components, often single genes (14,15).
The proportion of phages that are lytic versus temperate varies depending on the bacterial host, although the biological basis for this is not known. This is illustrated by bacteria within the phylum Actinobacteria; in Gordonia, Streptomyces and Mycobacteria temperate phages are prevalent and constitute a majority of the phage clusters (10). In contrast, temperate phages are a minority in Arthrobacter (16) and are rare in Microbacterium (17). These differences are likely reflected in the prevalence of prophages in bacterial genomes, such that prophages are expected to be rare in Microbacterium genomes but common in Gordonia, Streptomyces and Mycobacterium. In general, Actinobacterial genomes mostly carry intact prophages capable of spontaneous induction and growth as lytic phages (18) although many bacteria also carry defective or cryptic phages that have lost genes and are no longer viable. Some may be quite small and less easy to identify, although likely play important roles in microbial evolution (19,20).
Although some prophages replicate extrachromosomally (21), by far the majority integrate into the host chromosome. Integration is site-specific and is mediated by integrases that catalyze site-specific recombination between bacterial and phage attachment sites, attB and attP, respectively (22). Two types of integrases are used, tyrosine- and serine-integrases (Int-Y and Int-S, respectively) with different recombination mechanisms, although both catalyze strand exchange within a short conserved common core sequence shared by the attachment sites (23). Integrated prophages are flanked by attachment junctions attL and attR, which are used for excisive recombination in prophage induction. The attB site for Int-Y enzymes commonly overlaps a host tRNA gene (24), and the common core is 20–45 bp such that the tRNA gene is reconstituted following integration (25). In contrast, Int-S phages often integrate within open reading frames, resulting in insertional inactivation and phenotypic consequences (26–28); the common core sequence is usually smaller, 8–12 bp (29). There are, however, instances where the common core can be only 3–5 bp (18,30). Some bacterial genera also have transposable phages that integrate into multiple different chromosomal locations (31).
A number of phage and prophage identification tools have been developed, including Phage_Finder (32), PhageBoost (33), PhiSpy (34), PHASTER (35), ProphET (36), ProPhinder (37), Seeker (38), VirFinder (39) and VirSorter2 (40). Many of these rely on sequence similarity searches, gene predictions, or nucleotide k-mer distributions, and most are computationally intensive and relatively slow, presenting challenges for searching large numbers of sequenced genomes. VirSorter2 and PhageBoost use machine learning algorithms to speed up processing, and Seeker uses Long Short-Term Memory (LSTM) models to distinguish phage from bacterial sequences. Seeker can effectively discern phage from bacterial DNA in environmental metagenomic data using fast alignment-free strategies but is not designed to identify prophages in bacterial genomes (38). All of these tools were developed with the goal of being broadly applicable to bacterial genomes, but for many genomes they perform poorly at precise prediction of the prophage boundaries. Many are also computationally intensive, limiting their utility for screening vast number of sequenced bacterial genomes [currently ∼500 000 (41)]. Thus, there is a need for new approaches for fast searching of large numbers of bacterial genomes and for precise extraction of prophage sequences. We have used an alternative approach by developing prophage discovery tools that are genera-specific, focusing initially on Mycobacterium. A large number of Mycobacterium prophages have been previously identified and manually curated (28), providing a rare but valuable dataset for validation of the tools and performance comparison with other prophage discovery approaches.
The Discovery and Extraction of Phages Tool (DEPhT) is a multimodal program trained to identify genera-specific prophages. DEPhT is initially trained to investigate Mycobacterium genomes, as prophages are prevalent among non-tuberculous mycobacteria (NTM) (28,42,43), In contrast, full-length intact prophages may be rare in Mycobacterium tuberculosis, but large numbers of sequenced genomes [∼30 000 sequenced strains in the PATRIC database (44)] present a computational challenge for prophage identification. We show that DEPhT is sufficiently efficient in fast mode to analyze tens of thousands of sequenced genomes, and sufficiently precise in its other modes for extraction of prophage sequences with little to no manual revision. We also show that DEPhT can be trained to work with other bacterial genera, including Pseudomonas and Gordonia.
MATERIALS AND METHODS
DEPhT programming
DEPhT is a Python package compatible with Python 3.7 and above, and uses several non-Python packages including Prodigal (45) and Aragorn (46) for genome annotation, MMseqs2 (47) for identification of shell/accessory genes, HHsuite3 (48) for phage gene homology detection, and BLAST to make a best approximation of attachment site location. DEPhT can be obtained from GitHub (https://github.com/chg60/DEPhT.git) or the Python Package Index (https://pypi.org/project/depht/), and non-Python dependencies can be installed with Anaconda, or compiled manually. Python dependencies are installed automatically if DEPhT is obtained from PyPI.
Selection of Mycobacterium genomes for training
DEPhT uses sets of bacterial and phage genomes for training on specific bacterial genera. Many Mycobacterium genomes have been sequenced, and prophages have been defined and the precise prophage sequences extracted from M. abscessus genomes (28). A subset of these known to be prophage-free as well as a subset of prophage-containing genomes were used as training sets (Supplementary Tables S1 and S2). To create a diverse dataset of phage and prophage genomes, single representatives of each temperate mycobacteriophage subcluster (6) were selected by identifying genomes with the highest pairwise gene content similarity (GCS) (49) to other members of that subcluster, yielding 60 temperate mycobacteriophages (Supplementary Table S2). Similarly, a single representative was selected from each subcluster of M. abscessus prophages (28) choosing prophages from as few bacterial genomes as possible to reserve as many genomes as possible for DEPhT validation; additional prophages present in these bacteria were also used in the prophage training dataset (Supplementary Table S2); these prophages were added to the 60 temperate phage genomes.
Training of the gene size and transcription directional change (tdc) classifier
The temperate phage and prophage genomes in Supplementary Table S2, and the indicated Mycobacterium genomes in Supplementary Table S1 were retrieved in FASTA format. Prodigal (45) was used to annotate protein-coding genes in all genomes with full motif scanning (-n) and closed ends (-c) specified. Genes were examined in centered windows to derive each gene's local average gene size and number of tdc's. Separate histograms were constructed for both phage and bacterial genes, with respect to both gene size—using 10 bp-sized bins—and tdc's using integer counts; for both features, histogram values were normalized to frequencies. The phage and bacterial histograms were combined to create probability distributions describing the relative probability that a bin value is derived from a prophage. These probability distributions are retained by the classifier and used with equal weight to predict prophage regions.
Construction of bacterial shell gene content and nucleotide sequence databases
Completely sequenced Mycobacterium genomes from existing databases at NCBI and PATRIC were sorted into subclades based on shared gene content using a custom clustering pipeline anchored on the DBSCAN algorithm (50) (see Figure 3A). Using the data from genomes in each subclade, a nucleotide sequence database was constructed using makeblastdb from the BLAST command line toolkit (51). A protein sequence cluster database was created using the predicted products from all the retrieved genomes using the cluster pipeline from MMseqs2 (47), with parameters of 50% minimum identity and 80% minimum coverage. Protein sequence clusters that were represented at least once in 60% or more of the genomes in a subclade were assigned as a shell of protein orthogroups for that subclade (52). Subclade shell set membership for each protein orthogroup is stored as a bit array, where the length of the bit array corresponds to the number of given subclades. The indices in the array map to individual subclades and the value at an index i in the array is a Boolean where a value of 1 indicates an orthogroup's membership in the subclade shell represented by the array's ith index. The bit arrays were then compacted and converted into a hexadecimal value for efficient data storage. For each predicted product, their protein sequence and the hexadecimal value representing their subclade membership were stored in an indexed FASTA database.
Figure 3.

DEPhT employs MMseqs2 to identify and mask shell mycobacterial genes from predicted prophage regions. (A) The gene content of prophage-free Mycobacterium genomes was compared using MMseqs2 and sorted into four clades corresponding to M. abscessus (MAB), M. tuberculosis (MTB), M. avium-intracellulare (MAI) complexes, and rapid growing Mycobacterium (RGM) as depicted by an unrooted phylogenetic tree. Separate MMseqs2 databases were constructed for each clade for use in the shell/accessory classifier. (B) Schematic representation of the shell/accessory classifier which catalogues genes as being either bacterial orthologs or accessory genes. Bacterial genes, red; prophage genes, blue. (C) Validation of the shell/accessory classifier using 10 M. abscessus genomes (Supplementary Table S4) shows the proportion of either known bacterial or known prophage genes as being classified as shell or accessory genes. (D) Distribution of shell and accessory gene island sizes across regions known to be either prophage or bacterial. For every identified shell gene from content contributed by a prophage in 10 M. abscessus genomes (Supplementary Table S4), the size of the gene island it belongs to was plotted as a histogram (left; red), where the height of each bin in the histogram corresponds to the percentage of the total prophage genes. A similar histogram (left; blue) was plotted in the adjacent spaces for accessory gene content contributed by a prophage. The right histograms are the same representations for gene content not contributed by a prophage, where again a histogram (right; red) is plotted for shell gene content and an adjacent histogram (right; blue) is plotted for accessory gene content.
Construction of phage gene homolog HMM databases
The MySQL Actinobacteriophage genome database, Actino_Draft, available at http://databases.hatfull.org was retrieved, and GenBank-formatted flat files for a set of 1,889 mycobacteriophage genomes stored in Actino_Draft were created, using the get_db and export pipeline from the pdm_utils toolkit (8), respectively. The annotated protein product sequences for each genome were assembled into non-redundant sequence clusters, or ‘phams’, using the phamerate pipeline from the pdm_utils toolkit (53). Each pham containing a consensus annotation of some variation of ‘terminase’, ‘major capsid protein’, ‘portal protein’, ‘lysin’, ‘integrase’ or ‘immunity repressor’ with five or more protein sequence members were assembled into an HMM database using a custom python package and the HHsuite3 toolkit (48). Similarly, an HMM database was constructed for each pham with 10 or more members that contained a consensus annotation other than ‘hypothetical protein’ or those included in the primary database. Entries for all created HMM databases were labelled with their consensus annotation for easy identification.
Automated annotation of microbial sequences
Protein-coding genes were annotated in microbial sequences using Prodigal (45) with closed ends (-c) and full motif scanning (-n) specified, in metagenomic mode (-p meta) for contigs shorter than 100 000 bp; tRNA and tmRNA genes were predicted using ARAGORN (46). Contigs shorter than 20 000 bp are not processed further, based on our observation that Prodigal's gene calling accuracy performs poorly below that length. For regions in the genome with prophage signal, the translations for annotated open reading frames (ORFs) were written to FASTA files and searched in parallel against the phage gene homolog database described above using the HHsearch pipeline from the HHsuite3 toolkit. The sequence-HMM alignments produced were parsed with a custom python script, and alignments with better than 90% probability, 50% bidirectional coverage and e-values of 10–4 were retained. For each input protein sequence, a predicted functional annotation was assigned from the consensus annotation of the highest probability aligned HMM. Protein sequences with no significant HMM-HMM alignments were labelled as ‘hypothetical protein’.
Identification of accessory genomic islands
Annotated protein sequences of the input microbial genome were clustered with shell mycobacterial orthologous protein sequences using the linclust pipeline from the MMseqs2 toolkit, with parameters of 50% minimum identity and 80% minimum coverage (47). For each sequence cluster, the previously described subclade hexadecimal values associated with each database entry were converted back into bit arrays, and an OR operation was performed with the collective bit arrays. Input protein sequences were assigned their respective cluster's cumulative bit array. The values for every ith position were summed from the assigned bit arrays of all protein sequences associated with the entered microbial genome. The position(s) i with the greatest sum, if that sum is greater than half the total entered protein sequences, is used to create a bit array mask, with a value of 1 at the position(s) i and a value of 0 at every other position. The created bit mask is used in an AND operation with the bit arrays for each entered protein sequence. Those sequences where this operation yields a bit array with a 1 at any position are labelled as part of the shell set of orthologous gene content, and the remaining are labelled as accessory gene content.
Attachment site prediction
The gene size/tdc and shell-accessory classifiers yield predicted prophage regions, but with imprecise prophage boundaries. For att site prediction, these boundaries were extended by 5000 bp on either side to create upstream and downstream, ‘left’ and ‘right’, nucleotide regions to search for sequence repeats. The left and right sequences are extended inward toward the prophage signal by a user-definable multiplier parameter S (default = 7) and subsequently aligned using the BLASTN tool from the BLAST command line toolkit, with word-size = 5 and E value = 5000 × S. The left and the right regions are searched with BLASTN against the bacterial nucleotide sequence database described above, and results that indicate an overlap in coverage of the left and right regions in the reference strain are cataloged. Sequence repeat pairs generated from the BLASTN results of the left and the right regions (i.e. left-right pairs) are then scored with a feature-based algorithm, where the pair with the highest score is considered the prophage region's attachment site and the boundary coordinates adjusted to the ends of the pair. With this algorithm, sequence repeat pairs are scored on a 0 to 1 scale for the z-score of their alignment bitscore, their proximity to an integrase-like homolog encapsulated within the area they demarcate, and the coverage of this area against the initial putative prophage region. The scores produced from each of these feature analyses are weighted and summed to a total score of 3, where the weights were optimized against the training data set according to the sum of z-scores for metrics of the average score of manually determined attachment sites, the average score difference between manually determined attachment sites and the greatest scoring sequence repeat noise, the worst score of manually determined attachment sites, and the worst score difference between manually determined attachment sites and the greatest scoring sequence repeat noise. Finally, any left-right pairs that align to overlapping regions of a reference bacterium (suggesting identification of attB) are given a score bonus of up to 1.5 points, based on the bitscore of the aligned regions.
Prediction of gene size and tdc discriminators for other bacterial genera
All complete phage genomes were downloaded from RefSeq (accessed 29 August 2021) and binned according to their infected host's genus. These genomes were filtered with two criteria: they need to encode at least 55 protein-coding genes and be shorter than 100 kb. The first criterion allowed us to use DEPhT’s processing workflow to examine gene size and tdc's, while the second criterion was employed to reduce the likelihood of contaminating the analysis by inclusion of larger phages, many of which are lytic and do not form prophages. For host genera with at least five phage genomes after filtering, we downloaded all the complete bacterial genomes from RefSeq; genera without complete sequences were not analyzed further. DEPhT was used to analyze the gene size and tdc properties of 2064 phage and 14 233 bacterial genomes from 41 genera (including Mycobacterium), in 55-gene windows. For each phage genome, the 95th percentile gene size and tdc windows were retained. For each bacterial genome, the 5th percentile gene size and tdc windows were retained. Within each host genus the phage 95th percentile values were averaged, and the bacterial 5th percentile values were averaged. Then, these averages were expressed as a ratio of the average bacterial 5th percentile to the average phage 95th percentile. This ratio serves as a surrogate for the degree of overlap of the phage and bacterial distributions; values greater than one suggest good separation of the distributions, while values less than one suggest that the distributions are less well separated.
Training models for Gordonia and Pseudomonas genomes
DEPhT models for Gordonia and Pseudomonas were trained similarly to the Mycobacterium model.
Illustrations and graphics
The DEPhT graphical output showing prophage predictions is generated with custom python scripts utilizing python packages DNAFeaturesViewer (54), pretty-html-table (https://pypi.org/project/pretty-html-table), and bokeh (https://bokeh.org). Figures and illustrations were generated with custom scripts that utilize the Plotly (https://plotly.com) python package, and R packages BioCircos (55) and ggplot2 (https://ggplot2.tidyverse.org/index.html). For genome representation, prophages identified and extracted by DEPhT were added to a Phamerator (56) database ‘Mycobacterium_Prophages’ (Version 8, available at http://databases.hatfull.org) containing 180 individual prophages, of which 88 were identified by DEPhT; the other prophages have been described previously (28). An additional 36 prophages identified by DEPhT were not included in the Phamerator database as they are exact duplicates of other prophages in the dataset.
RESULTS
Design of DEPhT for multimodal prophage discovery
DEPhT is designed to combine two key functionalities: prophage discovery, and precise extraction of intact prophage sequences (Figure 1). To provide both speed and accuracy in prophage detection, DEPhT was designed with multiple run modes, ‘normal’, ‘fast’ and ‘sensitive’ (Figure 1). The discovery component is shared by all modes, and the modes differ in the precision with which the prophages are extracted. The discovery component avoids time-consuming extensive homology searching. It takes advantage of three architectural features that distinguish phage/prophage and bacterial genomes: (i) phage genes are on average shorter than bacterial genes (and more densely packed), (ii) phage genes are commonly arranged into multigene operons that on average are longer than bacterial operons with fewer transcription directional changes (tdc's) and (iii) they are part of the ‘accessory’ genome that varies among related bacterial strains; HHsearch-based phage gene homology detection is limited to the normal and sensitive modes, and then only to the subset of genes found by the discovery component. Because this approach is computationally efficient, DEPhT has the capacity to discover prophages in tens of thousands of genomes relatively quickly, particularly when run in fast mode where precision in prophage extraction is sacrificed for speed in prophage discovery (Figure 1). The normal and sensitive modes of DEPhT use targeted protein-based homology searches to improve the accuracy of prophage discovery and to identify the precise location of the attachment sites at prophage boundaries (Figure 1). The sensitive mode provides a more detailed prophage annotation.
Figure 1.
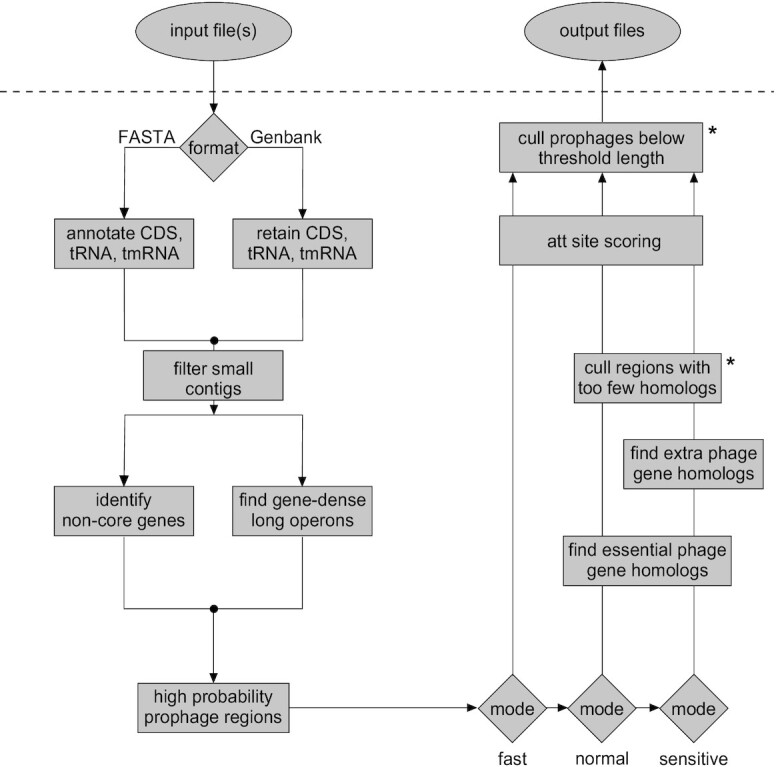
Workflow for DEPhT. Input files may be in either FASTA or Genbank flat file format. For FASTA files, contigs are parsed and CDS, tRNA and tmRNA genes are annotated. For Genbank files, contigs are parsed and annotations are reduced to CDS, tRNA and tmRNA genes. Annotated CDS genes are examined for (i) global homology to conserved mycobacterial genes and (ii) size, spacing, and transcription directionality change frequency. Genomic islands with gene architecture similar to that seen in temperate phages are identified as high likelihood prophage regions. In normal or sensitive run modes, the CDS genes in these regions are searched against a database of HMMs of genes performing phage-specific functions such as integrase, lysin, capsid, and terminase. In sensitive run mode genes are searched against a larger HMM database that includes any additional functionally annotated gene families. The sensitive mode thus yields a reasonably complete prophage sequence annotation. These limited homology searches help to distinguish true intact prophages from false positive prophage-like regions. Remaining prophage regions are searched for the phage attachment sites (attL/attR) before output files are generated. Asterisks indicate components that use thresholds that may be adjusted by users.
DEPhT fast mode predicts prophages using phage genome architectural features
To examine the discriminatory and predictive value of prophage gene size and tdc's we first constructed two datasets, one with known bacterial genes and one with known phage genes. The bacterial training dataset was obtained by identifying 25 Mycobacterium genomes in the PATRIC (41) dataset (designated as being either ‘representative’ or from RefSeq) that are prophage free, as determined using PHASTER and careful manual inspection (Supplementary Table S1). The phage training dataset contains 95 phage genomes (Supplementary Table S2) representing temperate mycobacteriophages from phagesdb.org (6) together with representatives from clusters of recently reported M. abscessus prophages (18,28). Protein-coding genes in both datasets were annotated using Prodigal (45), then a custom Naïve-Bayes-type classifier was trained on both the average gene density (gene size calculated with a sliding window) and tdc frequency using windows ranging from 5 to 55 genes (with the upper limit imposed by the size of the smallest mycobacteriophage genomes in the training dataset). At all window sizes tested there are evident differences in the size distributions of bacterial and phage genes (Figure 2A), and we note the bimodal distribution of phage genes which likely reflects the differences between virion structure and assembly genes (which are relatively large), and non-structural genes (which are relatively small). Likewise, there are distinct bacterial and prophage distributions of the frequencies of tdc's (Figure 2B) reflecting the preponderance for longer operons in phage genomes. For both measures, increasing window size improves the discrimination between bacterial and prophage genes and reduces the proportion that fall into both categories (Figure 2, Supplementary Table S3). The 55-gene window yields the most accurate predictions, and we use this window size for all subsequent analyses.
Figure 2.

DEPhT uses gene size and transcription direction changes (tdc's) in multi-gene windows to identify prophage regions sensitively. (A) The percentage of bacterial (red) and prophage (blue) genes corresponding to average gene size is plotted using window sizes of 25, 40 and 55 genes, as indicated. Increasing window size improves separation of bacterial and prophage gene size distributions. (B) The percentage of bacterial (red) and prophage (blue) genes corresponding to the average number of transcription directionality changes is plotted using window sizes of 25, 40 and 55 genes, as indicated. Increasing window size improves separation of bacterial and prophage distributions for transcription direction changes. (C) The cumulative percentage of bacterial (red) and prophage (blue) genes is plotted as a function of the classifier probability, which combines the gene size and tdc parameters. All prophage genes have a classifier probability above 25%, and only 1.5% of bacterial genes have classifier probabilities above 25%. (D) Schematic representation of the challenges in identifying prophage boundaries. Using a 25% threshold value the gene size/tdc classifier readily identifies prophage regions (dashed line represents raw classifier probabilities), but the prophage boundaries are ill-defined due to the large window size. Inclusion of shell gene analysis can align the signal over regions determined to be accessory parts of the genome, thus improving specificity.
DEPhT uses the 55-gene window distributions (Figure 2) to calculate probabilities for these two parameters (gene size and tdc's) for each gene, and then combines these with equal weight into a single probability score indicating if that gene is prophage-derived; these probabilities are then designated as being either above or below a threshold value (25%; Figure 2C). The discriminatory behavior is greatly enhanced by the relatively large window size, and whereas >99% phage genes score above a threshold value of 25% (and >90% phage genes have probability scores >60%, Figure 2C), bacterial genes rarely score above the 25% cutoff value (<1.5%), rendering this a powerful and fast approach to prophage identification. We note that a subset of prophages – particularly those within Cluster MabE – have average gene scores as low as 45–50%, although these are still well above the 25% threshold value. Values approaching 25% are only rarely observed in complete prophages, and manual inspection suggests these typically correspond to genes adjacent to the phage attL or attR sites.
Discriminating between shell and accessory genes enhances prophage boundary predictions
A disadvantage of using a relatively large window size for prophage prediction is that the prophage boundaries are ill-defined (Figure 2D). To correct for this, DEPhT scores each gene of a bacterial query sequence to classify it as either being part of the bacterial shell gene content (gene present in 60% genomes within a clade), or an accessory gene (Figure 1). DEPhT does this in a four-step process. First, a dataset of predicted proteins encoded by 34 Mycobacterium genomes (Supplementary Table S1) are grouped into ‘phamilies’ using a k-mer based approach with MMseqs2 (47). Secondly, the genomes are assorted into clusters (clades) of related genomes, and for Mycobacterium we assigned four major clades (Figure 3A). Thirdly, protein sequences of a bacterial query sequence are assembled with the bacterial dataset using MMseqs2, and the query genome is assigned as belonging to one of the pre-determined clades. Lastly, each gene is assigned as part of the shell bacterial genome if it is present in 60% of the genomes constituting a clade. If not, it is assigned as an accessory gene (Figure 3B). These data are used to mask the probability values generated from the gene size/tdc classifier to quickly eliminate prophage signal from regions in the genome that are common among similar bacteria and therefore unlikely to contain active prophages (see Figure 2D). This helps to demarcate gene content contributed by a prophage, as only a small fraction of prophage genes is masked in this way; as a corollary most of the prophage-like signal in non-phage gene content is substantially dampened (Figure 3C). Because phage genomes are architecturally asymmetric—with the larger virion structural genes in one half and the smaller genes of unknown function in other half—the phage signature derived from the gene size/tdc classifier is skewed to one side of the prophage. The shell/accessory classifier reconciles this by centering the prophage signal on the identified genomic island, and trims signal toward the attL/attR of prophages (Figure 2D). Within the gene content contributed by prophages from M. abscessus genomes in our validation set (Supplementary Table S4), shell gene content within prophage boundaries that might deform the signal from valid prophages is not only a small fraction of the total gene content in these regions but also occurs solely as isolated or paired interruptions, not as large gene-blocks (Figure 3C).
Enhanced accuracy of prophage discovery using limited homology searches in normal mode
In the prior analysis of M. abscessus prophages, all the prophages identified are likely intact and functional (18,28), but there is the expectation that there may be defective or cryptic prophages that have lost essential genes and are undergoing decay. To discriminate between intact and defective prophages, the normal and sensitive DEPhT modes were designed to quantify phage genes within the prophage-like signal. In these modes, protein-coding genes in the prophage-like regions are searched using HHsearch against a collection of HMMs built from genes that are characteristically phage-derived, and absent from the bacterial shell; these include integrases, lysins, major capsid proteins and terminases (see Materials and Methods). In the normal mode, a limited set of ∼400 HMMs are used, whereas in sensitive mode a total of ∼800 HMMs are used. To determine suitable threshold values for the number of positive matches, we examined the profiles of matches in 10 M. abscessus genomes (Supplementary Table S4, Figure S1) and chose values of 5 and 10 for normal and sensitive modes, respectively (Supplementary Figure S1).
The integration of the gene size/tdc and shell/accessory classifiers together with the limited homology searches provides powerful prophage prediction capabilities (Figure 4A, B). In Figure 4A and B, it is evident that the gene size/tdc classifier and the shell/accessory classifier identify the true prophage regions, but also have signals in other parts of the genomes that are not prophage. In M. abscessus GD91, the five prophages are correctly identified, as is the single prophage in M. abscessus GD68A (Figure 4A, B). However, in each genome there is a region identified by both classifiers, and has some homology hits, although fewer than the threshold value (five). As such, these two regions are reported as false positive predictions in DEPhT fast mode, but not in normal mode (Table 1). However, such false positive predictions in fast mode are relatively uncommon, and these two are the only events in the 10 genomes (containing 27 prophages) we tested (Table 1). Further examination shows that this region in GD91 contains a defective piece of a MabI-like prophage in which about 55 kb of the ∼80 kb genome has been lost, and the only phage-like homology-based hits are to the terminase, portal and integrase proteins (Figure 4C). The additional region in M. abscessus GD68A (Figure 4D) is unlikely to be phage-derived, as none of the protein-coding genes in this region have significant similarity to any known phage genes, but it has some phage-like features including an abundance of small genes. It is unusual in having a large array of 35 closely linked tRNA genes (Supplementary Table S5), greatly expanding the extant set of ∼49 M. abscessus tRNA genes. DEPhT does not identify this region in normal mode due to the lack of phage homologues. The origins and roles of this unusual part of the accessory gene content are not known, although BLASTN searching shows that there are several other M. abscessus genomes with similar regions. We note that some phages encode large tRNA sets, including MabI prophages and Cluster M mycobacteriophages which can have up to 24 tRNA genes (28,57), and we cannot exclude the possibility that at least part of this island is phage derived.
Figure 4.

Phage gene homology detection in normal mode eliminates false positives from fast mode. (A) A circular genome diagram for M. abscessus GD91, with five known prophages, illustrated with green (forward oriented) or red (reverse oriented) boxes outside the circle. The outermost track (cyan) shows the location of genes determined not to be part of the M. abscessus shell genome. The next track (purple) shows the location of genes with size/tdc similar to temperate phages. The innermost track (red dots) shows the location of genes with strong hits to one or more HMMs in the limited database used in normal mode. Regions with overlapping cyan and purple signal are identified as prophages in fast mode. Any such regions with fewer than 5 phage gene homologs are removed from the output in normal mode. (B) The same as (A), but for M. abscessus GD68A, which has one known prophage. (C) Close inspection of the false positive from GD91 reveals that it is a defective phage with many of the genes related to Cluster MabI prophages, but 55 kb of the 80 kb MabI length missing, including all tail assembly genes. (D) Inspection of a false positive from GD68A reveals an abundance of short genes arranged in long operons with few tdc's, and an array of 35 predicted tRNA genes that nearly doubles the species normal ∼49 tRNA genes (Supplementary Table S5); nonetheless the region contains no compelling hits to any phage proteins.
Table 1.
Comparison of prophage discovery programs
| Program | Prophages Identifieda | False positives | Prophages missed | Prophages split |
|---|---|---|---|---|
| PHASTER | 45 | 8 | 0 | 10 |
| VirSorter2+ CheckV | 43 | 15 | 0 | 1 |
| PhageBoost | 63 | 17 | 3 | 14 |
| DEPhT (fast mode) | 29 | 2 | 0 | 0 |
| DEPhT (normal mode) | 27 | 0 | 0 | 0 |
aThe total number of prophage or prophage segments reported for each program using a test-set of 10 completely sequenced M. abscessus genomes. A total of 25 prophages was previously reported for these genomes, and the two additional prophages discovered with DEPhT in the standard mode were checked by manual inspection, and were clearly missed in the prior analysis.
Attachment site detection
Precise extraction of the prophage sequences typically requires identification of the sequences shared by the common core sites of attL and attR (and also attP and attB). If these common core sequences are relatively large (>20 bp) they are comparatively easy to identify bioinformatically. However, detection is more challenging for smaller att core sequences, or where there are base pair mismatches within the core. In most phages the attP site is located close (typically within 500 bp) to the integrase gene, such that one of the att junctions (attL or attR) is near int, and this is helpful for att site prediction. However, there are notable exceptions—including Cluster M mycobacteriophages and Cluster MabI prophage—where attP is located more than 8 kb away from int.
DEPhT uses BLASTN to search for sequence similarities targeted to up to 35 kb regions spanning the prophage junctions identified in the discovery process; a high e-value threshold (scaled to the length of the searched region) is used such that most BLASTN hits of at least 5 bp length will be retained for subsequent analysis (Figure 5). In the normal and sensitive modes, each BLASTN pair of hits is then assigned a score based on (i) z-score of bitscore, (ii) distance between the hits (as a fraction of the predicted prophage region) for that BLASTN pair and (iii) proximity of one member of the BLASTN pair to a putative integrase gene, identified in the homology searches described above (see Figure 1). In fast mode, DEPhT uses only the first two parameters. Additionally, each pair receives a score bonus if a BLASTN search reveals that the pair coincides with a single sequence in a prophage-free region in a related strain, suggesting a putative attB site. BLASTN pairs are sorted on their final scores, and the top scoring pair above a threshold value is selected as the predicted attachment site. If no BLASTN pairs score above threshold, the entire prophage region predicted by the discovery process is reported. Typically, a single pair of hits scores discernably higher than all other pairs and is readily identified as corresponding to the attL and attR sites (Figure 5).
Figure 5.
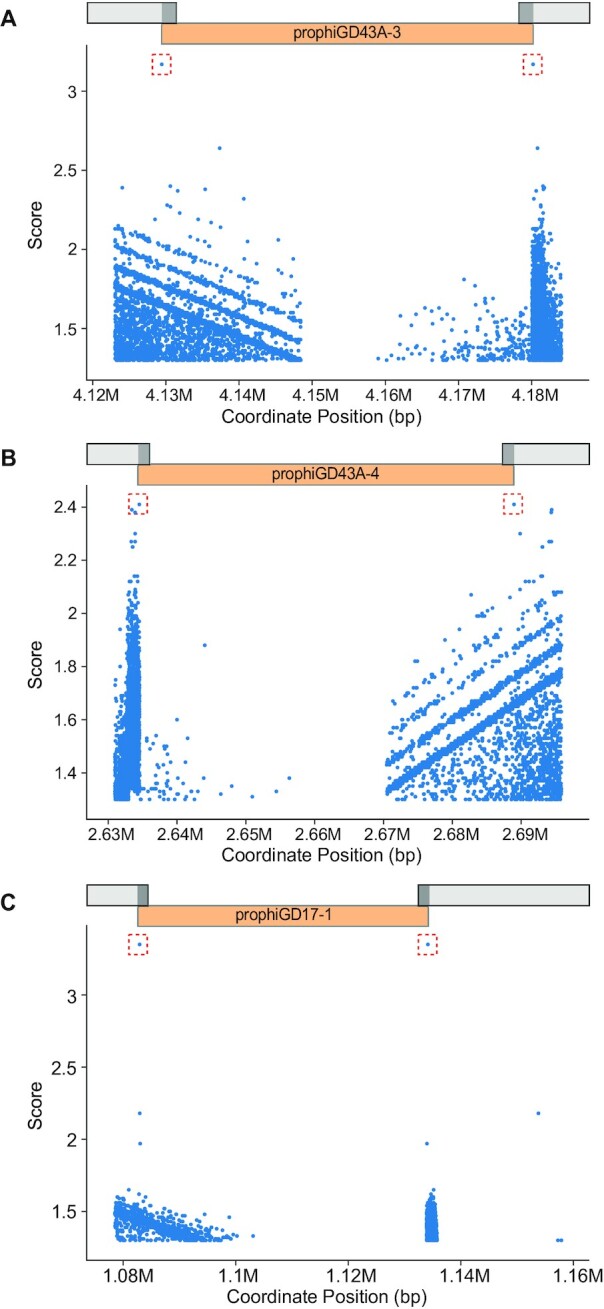
DEPhT identifies attachment sites at prophage boundaries. DEPhT uses BLASTN to identify all sequence repeats present at both the left and right sides of predicted prophages. All left/right pairs identified by BLASTN are scored as described (see Methods) and the highest scoring sites are shown in a red box. The position of the prophage is shown. Three examples are presented: (A) prophage prophiGD43A-3 that uses a tyrosine integrase and has a 17 bp att site common core with a single mismatch between attL and attR, (B) prophage prophiGD43A-4 which uses a serine integrase and has an 8 bp common core at attL and attR, and (C) prophiGD17-1 which uses a tyrosine integrase and has a 39 bp common core without mismatches at attL and attR.
Validation of DEPhT and comparison with other prophage prediction programs
We previously described a set of 82 M. abscessus genomes with a rich and varied set of integrated prophages (18,28), which were identified using PHASTER, BLAST, and careful manual curation. We used a set of 10 with completely sequenced genomes (Supplementary Table S4) and at least one prophage to validate DEPhT and to compare it to other prophage prediction programs; the total number of prophages previously reported was 25 (18,28). In normal mode, DEPhT identifies all these plus two additional prophages (designated prophiGD91-5 and prophiGD05-4) which on closer inspection are true prophages and not false positives (Table 1, Figure 4A, B). The likely reason these were missed in the prior analysis is that in both cases there is a closely related prophage (including extensive nucleotide sequence identity) in the same genome (prophiGD91-2 and GD05-2, respectively; Supplementary Figures S2 and S3). In fast mode, DEPhT does not miss any prophages, but gives two false positive hits, as described above. In normal mode, DEPhT predicts all the known prophages with no false positive hits.
We used the same set of genomes to perform prophage predictions using PHASTER, ProphET, VirSorter2+CheckV (58) and PhageBoost (Table 1). PHASTER and VirSorter2+CheckV did not miss any prophages, although PhageBoost failed to identify three prophages. However, all three programs identified many more prophage regions than DEPhT (Table 1), either because they falsely identified some candidate prophage regions, or because prophages were split into more than one piece due to poor boundary definition. Manual inspection of a subset of the false positive regions confirmed they are not bona fide prophages missed both in the prior analysis and by DEPhT. We determined the positive predicted values (PPV) for PhageBoost, PHASTER and VirSorter2, all of which were poorer for this genome set than DEPhT, even in fast mode (Figure 6A).
Figure 6.

DEPhT discovers, discriminates, and extracts prophage signal deftly. (A) The positive predictive value (PPV) of the outputs for PHASTER, VirSorter2 supplemented with CheckV, PhageBoost, DEPhT (fast mode) and DEPhT (normal mode) for prophage discovery is displayed as a bar graph. PPV for prophage discovery was calculated as the number of manually identified prophages discovered by a program divided by the total number of prophage-like regions reported. (B) The sensitivity, PPV, accuracy, and Matthew's correlation coefficient were determined on a nucleotide basis for the same outputs and displayed as a multi-bar graph, using the same color scheme. For prophage extraction, true positives (TP) are calculated as the total nucleotide base pairs (bp) reported within a prophage region that belong to a manually identified prophage, true negatives (TN) is calculated as the total bp not reported in a prophage region that do not belong to a manually identified prophage, false positives (FP) are calculated as the total bp that is reported within a prophage region that belong to a manually identified prophage, and false negatives (FN) are calculated as the total bp not reported in a prophage region that do not belong to a manually identified prophage. Sensitivity is calculated as 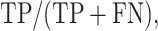 positive predictive value (PPV) is calculated as
positive predictive value (PPV) is calculated as 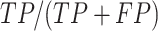 , Accuracy is calculated as
, Accuracy is calculated as 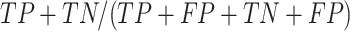 , and Matthew's Correlation Coefficient is calculated as (
, and Matthew's Correlation Coefficient is calculated as (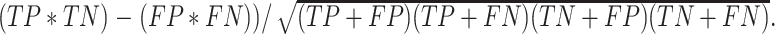 (C) A schematic representation of how predicted prophage regions align to true prophages is shown. At the bottom three known prophage are depicted of different lengths, and these are length normalized, to a scale of 0–100%, together with flanking sequences corresponding to 25% of genome length at each side. Examples of how predicted prophage regions may correspond to the actual prophage are shown above, including good alignment and multiple ways in which the alignment is imperfect. To quantify these alignments, each sequence is divided into bins, with the normalized true prophages forming 150 bins. If the predicted sequence aligns with a bin then it receives a positive score, and these score are summed and represented as a heat map, as shown in panel D. (D) Prophage discovery length-normalized coverage for binned regions of manually identified prophages from 10 M. abscessus strains (Supplementary Table S4) were plotted as a heatmap for the outputs of PHASTER, VirSorter2 supplemented with CheckV, PhageBoost and DEPhT, using the method depicted in panel C. Coverage for a particular nucleotide region bin was assigned if the cumulative output for the whole region was recognized as part of a prophage at least once, where coverage is represented on a red to blue color gradient with blue representing the most coverage.
(C) A schematic representation of how predicted prophage regions align to true prophages is shown. At the bottom three known prophage are depicted of different lengths, and these are length normalized, to a scale of 0–100%, together with flanking sequences corresponding to 25% of genome length at each side. Examples of how predicted prophage regions may correspond to the actual prophage are shown above, including good alignment and multiple ways in which the alignment is imperfect. To quantify these alignments, each sequence is divided into bins, with the normalized true prophages forming 150 bins. If the predicted sequence aligns with a bin then it receives a positive score, and these score are summed and represented as a heat map, as shown in panel D. (D) Prophage discovery length-normalized coverage for binned regions of manually identified prophages from 10 M. abscessus strains (Supplementary Table S4) were plotted as a heatmap for the outputs of PHASTER, VirSorter2 supplemented with CheckV, PhageBoost and DEPhT, using the method depicted in panel C. Coverage for a particular nucleotide region bin was assigned if the cumulative output for the whole region was recognized as part of a prophage at least once, where coverage is represented on a red to blue color gradient with blue representing the most coverage.
To determine the accuracy of the prophage prediction (and boundary definitions) rather than number of prophages, we also collated the total size of the prophage regions determined by DEPhT and the other programs (Table 2). The total size of the true prophages identified by DEPhT differs from the prior analysis by only 157 bp, including loss of one nucleotide for one prophage, and inclusion of 156 bp from prophages where DEPhT identifies an overextended attachment site sequence from the manually determined attB (Supplementary Table S6). However, it predicts 13 of the 27 prophages with 100% precision (both attL and attR extremities identified correctly), and in four prophages it identified one end correctly, and overextended the other by no more than 10 bp (Supplementary Table S6). No prophage was predicted to be more than 34 bp longer than expected, and the longest att overcall was 21 bp (Supplementary Table S6). There is no evident correlation between the accuracy of the prediction and the specific attB site used (Supplementary Table S6). In contrast to DEPhT, all of the other programs included not only false positive signals, but also missed substantial parts of the prophages, even though PHASTER and VirSorter2+CheckV did not miss any prophages entirely (Tables 1 and 2). And when the prophage predicted sizes including false positive and false negatives values are accounted for, DEPhT performs substantially better than the other prediction programs in terms of sensitivity, PPV, and by Matthews correlation coefficient (MCC, Figure 6B). Thus, at least for this validation genome set, DEPhT performs well in both the accuracy of prophage prediction as well as prophage extraction.
Table 2.
Single-nucleotide error rate comparison of prophage detection programs
| Program | True positive (bp)a | False positive (bp)b | True negative (bp)c | False negative (bp)d |
|---|---|---|---|---|
| PhageBoost | 628256 | 240902 | 44072113 | 896160 |
| VirSorter2 + CheckV | 1391420 | 908285 | 43404730 | 132996 |
| PHASTER | 1382161 | 253163 | 44059852 | 142255 |
| DEPhT (fast) | 1524416 | 864719 | 43320915 | 0 |
| DEPhT (normal) | 1524415 | 156 | 44312859 | 1 |
aTrue positive values are determined as the number of nucleotides in each prophage prediction that are within a manually validated prophage.
bFalse positive values are determined as the number of nucleotides in each prophage prediction that are not within a manually validated prophage.
cTrue negative values are determined as the number of nucleotides that are not within a manually validated prophage and are not part of a prophage prediction.
dFalse negative values are determined as the number of nucleotides that are within a manually validated prophage, and are not part of a prophage prediction.
We then compared PhageBoost, PHASTER and VirSorter with DEPhT on the consistency of prophage prediction. As DEPhT discovers all prophages and determines their boundaries precisely or within a few base pairs, the output of DEPhT is largely centered and totally encompassing of the prophage region. A heatmap illustrates the collective alignment of prophage regions determined by DEPhT and other programs with manually identified prophages in the validation genome set (Figure 6C). This prophage coverage, together with the other metrics, shows that DEPhT consistently reports the entirety of a prophage region with no large relative variation, while other programs tend to skew or truncate prophage signal. And although the output from PHASTER more consistently encompasses most of a prophage region, the bimodal distribution of its output indicates that this seemingly global coverage is sometimes the result of reporting two separate regions.
Finally, we evaluated the output of DEPhT in fast mode and normal mode against a second validation set of 45 completely sequenced M. abscessus genomes, which we also used to manually identify 60 prophages for this study (Supplementary Table S7). DEPhT in normal mode and fast mode perform similarly and do not miss any of these prophages, except one dilysogen where both outputs report the presence of one large prophage instead of two. In addition, DEPhT in fast mode included 11 false positive hits and DEPhT in normal mode included one likely false-positive hit, which is similar to previously described prophages but with large foreign and disrupting insertions (Supplementary Table S8). With respect to extraction accuracy, DEPhT performs similarly well on this dataset, completely encompassing all manually identified prophages and mostly with close proximity to the manually determined boundaries (Supplementary Figure S4).
DEPhT performance with whole genome sequence (WGS) data
The vast majority of sequenced bacterial genomes in the public databases are not assembled beyond the WGS level, and the overall quality of these assemblies varies greatly, as shown by the number of contigs and the N50 values. Seventy-two of the M. abscessus genomes described previously (18,28) are WGS assemblies containing more than contig, whereas all of the genomes in our validation set (Table 1) are completely sequenced (i.e. in one contig per replicon). We therefore tested the ability of DEPhT to detect prophages in WGS projects by re-assembly of eight genomes (Supplementary Table S9) that had previously been completely assembled and shown to contain 1–6 prophages. To do so, varying numbers of the Illumina sequence reads were used to re-assemble the genomes yielding a broad span of contig numbers, coverage, and N50 values (Supplementary Table S9). When all available reads were used, we were able to identify all the prophages in each genome—even though they are in multiple contigs—with two notable exceptions, M. abscessus GD05 and M. abscessus GD91 (Supplementary Table S9), in which two prophages were not identified. The reason for this is seemingly not a DEPhT deficiency, but an assembly issue that arises when a genome contains two closely related prophages whose nucleotide sequence identity interferes with contig assembly (Supplementary Figures S2 and S3). These are also the same two genomes in which prophages were missed in the prior characterization (18,28). However, this circumstance is not common—at least among M. abscessus genomes—and of the 82 genomes we have characterized, only these two strains each contain two related prophages.
The re-assembled genomes with fewer reads and overall lower quality vary in the ability of DEPhT to detect prophages, especially when average contig size is sufficiently small that the prophages are no longer represented in a single contig. For M. abscessus GD22 and GD26 DEPhT identified the single prophage in each using as few as 300 000 and 400 000 reads (Supplementary Table S9), respectively, giving 23–30-fold coverage. For M. abscessus GD43A and GD21, which contain 6 and 4 prophages, respectively, DEPhT failed to identify one of the prophages when only 500 000 reads were used, or when coverage was 34–36-fold. Overall, we predict that DEPhT will accurately identify most of the prophages in WGS genome projects that are assembled into fewer than 100 contigs, and with average coverage greater than 35-fold. DEPhT can identify prophages in lower quality assemblies, but in poorer assemblies it may not identify all of them.
We used DEPhT in both fast and normal mode to screen 28 WGS M. abscessus genome assembles not used in any previous training or validation steps (Supplementary Table S10). In normal mode it identified all 15 prophages present as complete regions (i.e. the entire prophage is wholly within a single contig), and identified 23 of 25 incomplete prophages (which are typically located in two or more contigs and at contig ends; Supplementary Table S10). DEPhT performed similarly in fast mode, but also reported 22 false positive regions. We note that for relatively small numbers of genomes such as in this data set, fast mode only offers a modest time-saving, and at the expense of more false positive regions identified.
M. tuberculosis does not contain integrated prophages
We used DEPhT in normal mode to predict and extract prophages in sequenced Mycobacteriaceae genomes; M. tuberculosis genomes were excluded as there are a large number and they are analyzed separately below. Searching the 2498 genomes with fewer than 100 contigs (1799 of which are M. abscessus) identified 2630 prophages present in 1479 genomes. The proportion of prophage-containing genomes (60%) is somewhat higher than reported for a set of 235 NTM genomes (34%) (42), but the larger data set has a high proportion of M. abscessus genomes (72%), a larger proportion of which—up to 85% (28,42)—contain prophages. More targeted searches of either complete genome sequences (249) or those assembled into 9 contigs (496) or fewer identified 125 and 614 prophages, respectively. In general, the proportion of prophage-containing Mycobacterium genomes and the numbers of prophages identified are in good agreement with prior reports (28,42). Visualization of prophage genomes shows good alignment with previously identified prophages, as illustrated by Cluster MabA prophages (Figure 7). DEPhT is thus a powerful tool for discovery and extraction of large numbers of Mycobacterium prophages. The detailed comparative genomic analyses of these will be reported elsewhere (CHG, LA and GFH, manuscript in preparation), but these analyses suggest a potential wealth of biological insights. The prophages can be grouped into more than 200 sequence-related groups (Clusters/Subclusters/Singletons), and among the observations we note polymorphic toxin-immunity cassettes are prevalent among prophages of M. abscessus (28) but generally absent from prophages of other Mycobacterium species, suggesting they contribute to the behaviors of this important pathogen.
Figure 7.

Genome maps of Cluster MabA M. abscessus prophages. DEPhT running in normal mode was used to search all single-contig M. abscessus genome assemblies in PATRIC, prophages were identified and extracted, and—together with previously identified prophages (28)—were used to build a Phamerator database (56). The genome maps for a subset of related genomes grouped in Cluster MabA are displayed, with each genome represented as a ruler with predicted genes shown as colored boxes above or below each genome, reflecting rightwards and leftwards transcription, respectively. Pairwise nucleotide sequence similarly is displayed by spectrum colored shading between genomes, with violet being the most similar. Prophages previously described are labeled as prophiGDxx at the right, and the DEPhT-derived sequences are labeled with the strain name and subspecies shown in parentheses, where known (a, abscessus; b. bolettii, m, massiliense). The genome alignments show good identification of prophage boundaries and illustrate their diversity and variation. We note that strain FLAC013 has a similar transposon insertion as prophiGD15-1 and prophiGD20-1 reported previously (28) giving longer genomes. The prophage in M. bolettii 1518 appears to have undergone deletions at both the left and right ends and is likely non-functional.
Unlike in M. abscessus, no prophages have been reported in M. tuberculosis genomes although two small (∼10 kb) prophage-like elements (ϕRv1 and ϕRv2) have been described in M. tuberculosis H37Rv (59). This is somewhat surprising as a number of temperate phages (or mutants of them) have been described for M. tuberculosis (60,61) and M. tuberculosis genomes have variable CRISPR arrays suggesting they have been defending themselves against phage infection in their recent evolutionary past (62). However, there is a large number of M. tuberculosis genome sequences (29 279 in the PATRIC database as of 26 July 2021) raising the question as to whether integrated prophages are present in any of these strains, not just the few that have been closely examined to-date. DEPhT provides the opportunity to address this question, as it can screen large numbers of bacterial genomes relatively quickly, with modest false positive but very low false negative outcomes, at least in those genomes with good coverage and assembly using WGS data.
The 29 279 M. tuberculosis genomes were downloaded (Supplementary Table S11), binned according to the number of contigs (Table 3), and DEPhT was used in normal mode to screen all 29 279 genomes (requiring a run time of ∼60 h using all 8 cores of a workstation equipped with an AMD Ryzen 7 3700X processor). Only 523 of the input genomes are assembled into single contigs, although over 6600 are in fewer than 100 contigs for which DEPhT is anticipated to identify all of the prophages if there are any; another 11 750 are in 100–249 contigs, and DEPhT is expected to identify many prophages in this assembly-range (Supplementary Table S9). We note that some genomes tagged as ‘M. tuberculosis’ may be either misidentified or contain contigs from contaminating bacterial species.
Table 3.
Genomic assembly profiles of M. tuberculosis
| Groupa | # Contigsb | # Genomesc | % ‘Good’d | Total contigse | Usable contigsf | Median N50g |
|---|---|---|---|---|---|---|
| 1 | 1 | 523 | 100 | 523 | 523 | 4411217 |
| 2 | 2-9 | 788 | 99.5 | 4434 | 3186 | 2575182 |
| 3 | 10-99 | 5301 | 99.9 | 322785 | 171895 | 132057 |
| 4 | 100-249 | 11750 | 99.9 | 1995446 | 677407 | 81101 |
| 5 | 250-499 | 7026 | 99.5 | 2335349 | 450625 | 64634 |
| 6 | 500-749 | 1208 | 97.4 | 724170 | 77638 | 61910 |
| 7 | 750-999 | 584 | 94.0 | 501681 | 34738 | 64010 |
| 8 | 1000-9999 | 1915 | 0.05 | 5393139 | 117683 | 44747 |
| 9 | 10000+ | 184 | 0.00 | 3571408 | 13010 | 1516 |
aEach group is a bin containing the number of M. tuberculosis genome projects with different contig numbers and the overall quality. All data are from PATRIC (https://www.patricbrc.org).
bThe range of contig numbers for M. tuberculosis genome projects in each group
cThe number of M. tuberculosis genome projects containing the range of contig numbers
dThe percentage of sequencing projects designated as ‘good’ quality by PATRIC
eThe total number of contigs from all projects in that group
fThe total number of contigs above threshold length (20 kb) for DEPhT analysis
gMedian of N50 for each group, which is the sequence length of the shortest contig at 50% of the total genome length.
In this M. tuberculosis data set, DEPhT identified a total of 27 prophages in 25 genomes (Table 4). However, closer inspection and BLASTN analysis of individual contigs shows that most of these are in non-M. tuberculosis contigs derived from other bacteria; closer inspection of some of these indicates they are accurate prophage predictions. Of the two bona fide M. tuberculosis strains, one (M. tuberculosis CG24) was isolated as a mutant that is resistant to phage Fred313_cpm in vitro and was shown previously to contain large segments of Fred313 integrated into the genome (60). Although DEPhT identified this as expected, it does not reflect a naturally occurring integrated prophage. The second hit was to a contig in M. tuberculosis (PATRIC ID: 1773.19498) that corresponds to a phage virion genome with defined termini, and genes that are most closely related to Streptomyces phages. It seems likely that this phage was a contaminant in the DNA preparation, although we cannot exclude the possibility that it is a true M. tuberculosis phage replicating in the M. tuberculosis strain. In summary, we discovered no integrated prophages in any M. tuberculosis strain, and we conclude that naturally occurring M. tuberculosis isolates are devoid of intact integrated prophages. We note that the ϕRv1 and ϕRv2 prophage-like elements are too small (∼10 kb) to be reported by DEPhT.
Table 4.
Predicted prophages identified in genome projects
| GenomeIDa | SRA | PATRIC speciesb | #contigs | %GCc | Size (Mbp)d | # Prophagese | Prophage contig matchf |
|---|---|---|---|---|---|---|---|
| 1733.4750 | ERR2514035 | M. tuberculosis | 1047 | 55.0 | 8.03 | 1 | Enterococcus casseliflavus |
| 1733.8175 | ERR2517475 | M. tuberculosis | 11,273 | 59.1 | 13.1 | 1 | M. abscessus |
| 1733.8957 | SRR5709926 | M. tuberculosis | 2074 | 67.2 | 11.5 | 2 | Actinomycetales |
| 1733.10987 | SRR6153015 | M. tuberculosis | 1025 | 53.2 | 8.99 | 1 | Clostridium; Bacillus ssp. |
| 1765.193 | n/a | M. bovis | 576 | 59.4 | 11.95 | 1 | Bacillus ssp. |
| 1773.5318 | SRR2101661 | M. tuberculosis | 635 | 21.1 | 5.29 | 1 | Bacillus licheniformis |
| 1773.13291 | n/a | M. tuberculosis | 650 | 67.3 | 10.38 | 1 | M. avium |
| 1773.13467 | n/a | M. tuberculosis | 1683 | 53.3 | 6.99 | 1 | Staphylococcus epidermitis |
| 1773.13508 | n/a | M. tuberculosis | 2572 | 53.6 | 7.31 | 1 | Staphylococcus epidermitis |
| 1773.13517 | n/a | M. tuberculosis | 5330 | 52.5 | 7.39 | 1 | Staphylococcus epidermitis |
| 1773.14094 | n/a | M. tuberculosis | 1025 | 53.2 | 8.99 | 1 | Clostridium; Bacillus ssp. |
| 1773.14969 | SRR6397654 | M. tuberculosis | 390 | 67.3 | 10.35 | 1 | M. avium |
| 1773.15146 | SRR6397396 | M. tuberculosis | 330 | 53.4 | 6.86 | 1 | Staphylococcus epidermitis |
| 1773.15187 | SRR6397568 | M. tuberculosis | 334 | 53.4 | 6.86 | 1 | Staphylococcus epidermitis |
| 1773.15196 | SRR6397721 | M. tuberculosis | 363 | 53.2 | 6.92 | 1 | Staphylococcus epidermitis |
| 1773.15773 | SRR6153015 | M. tuberculosis | 308 | 53.2 | 8.89 | 1 | Clostridium; Bacillus ssp. |
| 1773.17698 | ERR751414 | M. tuberculosis | 167 | 58.6 | 8.40 | 1 | Morganella morganii |
| 1773.18195 | ERR1873557 | M. tuberculosis | 199 | 62.3 | 10.36 | 1 | Pseudomonas ssp. |
| 1773.18223 | ERR1873445 | M. tuberculosis | 214 | 62.2 | 10.34 | 1 | Pseudomonas ssp. |
| 1773.18344 | ERR1035327 | M. tuberculosis | 177 | 56.2 | 6.26 | 1 | Lactobacillus |
| 1773.18754 | ERR036242 | M. tuberculosis | 385 | 59.2 | 9.62 | 2 | Paenibacillus |
| 1773.18812 | ERR037510 | M. tuberculosis | 1051 | 50.5 | 8.78 | 1 | Clostridium |
| 1773.19498 | ERR181684 | M. tuberculosis | 87 | 65.7 | 4.42 | 1* | M. tuberculosis |
| 1773.20520 | ERR1035082 | M. tuberculosis | 746 | 50.3 | 9.44 | 1 | Bacillus ssp. |
| 1773.25614 | n/a | M. tuberculosis | 1 | 65.6 | 4.46 | 1** | M. tuberculosis |
aPATRIC GenomeID.
bSpecies designated in PATRIC.
cAverage GC% M. tuberculosis GC% is 65.52 ± 0.39%, and values outside this suggest non-M. tuberculosis or mixed assemblies.
dAverage M. tuberculosis genome size is 4.41± 0.4 Mb. Projects outside this likely reflect mixed assemblies.
eNumber of prophages predicted using DEPhT in fast mode. *, phage virion DNA, probably not M. tuberculosis; **, scrambled prophage in phage resistant mutant.
fClosest genome(s) to prophage-containing contig by BLASTN.
Adaptation of DEPhT to non-Mycobacterium genomes
While DEPhT was developed with the goal of quickly and accurately extracting intact prophages from Mycobacterium genomes, we wanted to investigate whether the prophage identification and extraction strategy is applicable to other bacterial genera. First, we tested the discriminatory power of the phage and host gene size and tdc compositions for 40 additional host genera (Figure 8). Each genus has at least five phage genomes and at least one bacterial genome in RefSeq. The gene size and tdc parameters were determined for each of these using a 55-gene window size (as for Mycobacterium), and the ratios of the fifth percentile of bacterial values to 95% percentiles of phage values within each genus were determined; this approximates the regions of overlap in Figure 2A and B (Figure 8). Although these values vary considerably for different genera, Mycobacterium is close to the median of both parameters. Thus, although DEPhT may have reduced predictive efficacy for bacterial genera at the lower extremities of this plot, we predict DEPhT will be broadly applicable to a large number of bacterial systems; training models will, however, need to be established for other bacteria.
Figure 8.

Gene size and transcription directional change parameters for 41 bacterial genera and their phages. Gene size and tdc distributions were examined for the complete phage and bacterial genomes available in RefSeq (see Materials and Methods). The intra-genus ratio of the average bacterial genome 5th percentile to the average phage genome 95th percentile for both gene size and tdc, was calculated and plotted. Each point on the plot indicates the calculated gene size and tdc ratios for a single genus. Genera are colored according to the phylum (or family for the Proteobacteria) that the genus belongs to. A value of one or greater reflects minimal overlap of the phage and bacterial distributions.
Second, we explored the application of DEPhT to Pseudomonas and Gordonia genomes by developing training models for each; both of these have environmental or clinical importance and for which many temperate phages have been identified. For Gordonia, we retrieved all 28 of the complete, non-redundant genome sequences available in PATRIC (Supplementary Table S12), and 110 diverse phages from phagesdb.org (Supplementary Table S13). For Pseudomonas, we retrieved 53 representative bacterial genomes from Genbank (Supplementary Table S14), and all 214 Pseudomonas phage genomes from RefSeq (Supplementary Table S15). As with the Mycobacteria, the goal was to retrieve as many diverse and representative sequences as possible while keeping the total number of genomes relatively low. The gene size and tdc classifiers and shell gene content for Gordonia and Pseudomonas were trained as described above for Mycobacteria.
The gene size and tdc classifier for Gordonia (Supplementary Figure S5) performs similarly as for Mycobacterium (Figure 2) with good discrimination between bacterial and phage genomes at larger window sizes, and when combined with equal weight, the classifier achieves an MCC of 0.941 on the training data (similar to 0.955 for Mycobacterium). Also similar to Mycobacterium, Gordonia prophages are primarily composed of genes lacking homology to the bacterial shell genome (Supplementary Figure S6A). However, the distribution of shell gene block sizes identified by DEPhT is somewhat different (Supplementary Figure S6B) which may reflect different overall diversity of the Gordonia genomes.
DEPhT running in normal and sensitive modes showed high precision in extracting 9 of the 12 Gordonia prophages when compared to PHASTER (Supplementary Table S16), extracting them either perfectly or to within a few dozen nucleotides of the manually determined boundaries (Supplementary Figures S7 and S8). The other three prophages are identified in their entirety but are less precisely extracted, likely reflecting their use of serine integrases, and the Gordonia genome diversity constrains attB identification using BLAST against the reference genomes. In fast mode, DEPhT identifies two additional ‘prophages’ in plasmids, which do not appear to correspond with true prophages, intact or otherwise.
For Pseudomonas, the bacterial and phage gene size and tdc distributions do not discern between host and phage sequences as clearly as for Mycobacterium and Gordonia, although increasing the window size to 55 genes improves the separation (Supplementary Figure S9). For both features, the Pseudomonas phage distributions have longer tails that increase the overlaps in the distributions, and the optimized classifier achieves an MCC of only 0.759. However, the shell and phage gene profiles align with the bacterial and prophage genomes (Supplementary Figure S10) because Pseudomonas has been deeply sampled and individual species (or clades) tend to have well-defined shell genomes, as identified by DEPhT using MMseqs2. Prophages in Pseudomonas are primarily composed of genes lacking homologs in the bacterial shell genome (∼98%), and the vast majority (∼85%) of non-prophage genes are recognizable as part of the shell genome (Supplementary Figure S10A). Although Pseudomonas genomes have more accessory genes than Mycobacteria, the distribution of the size of genomic islands is similar in the two genera (Supplementary Figure S10B).
Because many Pseudomonas phages are poorly annotated, DEPhT normal and sensitive modes provide relatively little additional resolution than fast mode, compared to the Mycobacterium and Gordonia examples. Furthermore, many of the Pseudomonas phages are either very closely related, or so distantly related as to share few if any genes, such that the HMMs that DEPhT uses to identify homologs of functionally characterized phage genes are either too small or too homogeneous to provide much sensitivity. As a consequence, HHsearch contributes only modestly to the identification of phage regions and the discrimination against false positive predictions. Nonetheless, DEPhT is largely successful in identifying Pseudomonas prophages (Supplementary Figures S11 and S12), with precision and accuracy comparable to PHASTER (Supplementary Tables S17 and S18), but substantially faster. We note that DEPhT is somewhat less precise at extracting prophages from Pseudomonas than from Mycobacterium or Gordonia, perhaps reflecting in part the prevalence of transposable phages, which use multiple integration sites in the chromosome.
DISCUSSION
Advances in sequencing technology and the reduction in sequencing costs have resulted in the availability of hundreds of thousands of bacterial sequencing projects. This represents a substantial challenge for computational tools for prophage discovery and extraction of the prophage sequences for comparative genomic analyses. DEPhT is a multimodal tool that in fast mode enables the search for prophages in large numbers of genomes relatively quickly, albeit with some imprecision in identification of prophage boundaries, although no more so than other prophage identification tools. In normal mode, DEPhT may be up to 3 times slower than fast mode but can extract prophages with considerable precision. Sensitive mode is not as fast but gives a more detailed genome annotation.
The development of DEPhT benefitted from a genus-specific approach, focusing on prophages in Mycobacterium genomes. The conclusion that there are no prophages in M. tuberculosis is an important one, as it suggests that M. tuberculosis is not accessible by phages in its environment. There are temperate phages that infect M. tuberculosis in vitro but this does not appear to occur in vivo, or else we would expect to find at least some integrated prophages in the thousands of M. tuberculosis genomes. This could be explained by the propensity for M. tuberculosis to live and grow intracellularly within macrophages, and within granulomatous structures. However, in a severely ill TB patient there are large numbers of extracellular bacteria, which are transmissible to other patients, and the bacteria are likely to be exposed to environmental phages in this process. These are important considerations when contemplating the therapeutic use of bacteriophages as TB control measure (60). DEPhT will be an effective and powerful tool for identification and extraction of the potentially hundreds or thousands of prophages in sequenced NTM genomes, and which may be key drivers of physiology and virulence (18,28).
Although DEPhT was developed with Mycobacterium genomes in mind, it can be adapted to other bacteria. In support of this, we determined the predicted discriminatory effectiveness of the DEPhT gene size and tdc parameters for 40 additional bacterial genera, and although there is considerable variation, Mycobacterium is not an outlier. DEPhT is thus expected to be broadly although perhaps not universally applicable. We evaluated DEPhT on Pseudomonas and Gordonia spp. by developing the required training models, notwithstanding the more limited knowledge of the prophage content in these strains. DEPhT performs similarly for Gordonia as it does for Mycobacterium but is not as precise in prophage extraction for Pseudomonas, largely due to the preponderance of transposable phages. Nonetheless, DEPhT performs at least as well as PHASTER (Supplementary Tables S18 and S19). It is also likely that DEPhT benefits from larger, more diverse, or better annotated genomic datasets. Training of DEPhT for additional bacterial genera will further inform us of the applicability and limitations of DEPhT.
In normal mode, DEPhT can identify and precisely extract the sequences for about half of the Mycobacterium prophages; the other half have a few additional base pairs at one or both att sites (Supplementary Table S6). This imprecision occurs primarily because of the need to accommodate mismatches in the common core sequences at the attachment sites, but rarely are base pairs omitted. Perfect precision for all prophages is likely not an achievable goal and may not be required for many subsequent analyses. If precision is needed, then the prophages may require manual inspection and correction, but with little or no additional sequence searching, and revision of only a few base pairs. This may be important if attempting to design and synthetically reconstruct a lytic phage from the prophage sequence.
We note that whereas DEPhT performed well when benchmarked for the given set of M. abscessus genomes and out-performed other prophage-prediction programs, this may not be replicated for all other bacterial genomes, even after re-training for phylum-specific searches. There are thus likely to be utilities for the spectrum of prophage prediction programs depending on project goals, and usage of multiple programs in combination may be desirable for some projects. However, the considerable speed of DEPhT in fast mode is especially effective in screening large numbers of genomes for putative prophages, followed by DEPhT in normal or sensitive mode or one of the other programs for extraction of the prophage sequences. We also note that Mycobacterium prophages appear to be most intact and capable of growing lytically (18), and there are few defective or cryptic prophages. These may be more prevalent in other bacteria, and may not be recognized by DEPhT, especially if they have lost attachment junctions or integrase genes.
DEPhT has some evident limitations. First, in fast mode it identifies some non-phage accessory parts of genomes as false-positive hits, thus fast mode is most useful for screening genomes for those that lack prophages, and as a rapid pre-screen for genomes to input into DEPhT normal or sensitive mode. Also, some of these false-positive non-phage regions may be of general interest, especially those contributing large numbers of tRNA genes. Second, att site identification in normal and sensitive mode is strengthened by the assumption that one att junction is closely linked to the integrase gene. Although this is true for many phages, there are exceptions including the Cluster M mycobacteriophages and Cluster MabI prophages (28,57), and DEPhT may struggle to predict the correct att sites. Transposons inserted either within prophages or close to prophages can confound att site prediction, as transposon genes are typically grouped within the accessory genome component, but this occurs with low frequency. Third, some prophages in non-Mycobacterium strains may be as small as 15 kb, requiring a more stringent set of DEPhT parameters to identify these correctly. Fourth, prophage boundaries may be especially challenging to predict accurately for transposable phages, and generally less accurate for prophages encoding serine-integrase than those with tyrosine integrases. Lastly, DEPhT may not correctly identify regions in genomes where two prophages are integrated at the same site, or dilysogens with two copies of the same prophage. These can usually be readily resolved by manual curation. Overall, DEPhT provides an effective multimodal tool for prophage discovery and extraction.
DATA AVAILABILITY
The code for the DEPhT Python package is publicly available with installation and usage instructions at https://github.com/chg60/DEPhT.git and https://pypi.org/project/depht/. The models we trained for this study are available at https://osf.io/zt4n3. The training and testing data accessions are available in Supplementary Tables.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Haley Aull, Deborah Jacobs-Sera, Carlos Guerrero and Rebekah Dedrick for helpful insights and discussions.
Contributor Information
Christian H Gauthier, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Lawrence Abad, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Ananya K Venbakkam, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Julia Malnak, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Daniel A Russell, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
Graham F Hatfull, Department of Biological Sciences, University of Pittsburgh, Pittsburgh, PA 15260, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NIH [GM131729]; Howard Hughes Medical Institute [GT12053]; Fowler Fund for Phage Research. Funding for open access charge: NIH [GM131729].
Conflict of interest statement. G.F.H. is a compensated consultant for Tessera and for Janssen Inc. All other authors have no conflicts to declare.
REFERENCES
- 1. Bernheim A., Sorek R.. Viruses cooperate to defeat bacteria. Nature. 2018; 559:482–484. [DOI] [PubMed] [Google Scholar]
- 2. Hatfull G.F., Hendrix R.W.. Bacteriophages and their Genomes. Curr. Opin. Virol. 2011; 1:298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hendrix R.W. Knipe D.M., Howley P.M.. Fields Virology. 2013; 6th ednPhiladelphia: Lippincott Williams & Wilkins. [Google Scholar]
- 4. Bondy-Denomy J., Qian J., Westra E.R., Buckling A., Guttman D.S., Davidson A.R., Maxwell K.L.. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016; 10:2854–2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dedrick R.M., Jacobs-Sera D., Bustamante C.A., Garlena R.A., Mavrich T.N., Pope W.H., Reyes J.C., Russell D.A., Adair T., Alvey R.et al.. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017; 2:16251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Russell D.A., Hatfull G.F.. PhagesDB: the actinobacteriophage database. Bioinformatics. 2017; 33:784–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hatfull G.F., Jacobs-Sera D., Lawrence J.G., Pope W.H., Russell D.A., Ko C.C., Weber R.J., Patel M.C., Germane K.L., Edgar R.H.et al.. Comparative genomic analysis of 60 mycobacteriophage genomes: genome clustering, gene acquisition, and gene size. J. Mol. Biol. 2010; 397:119–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hatfull G.F., Pedulla M.L., Jacobs-Sera D., Cichon P.M., Foley A., Ford M.E., Gonda R.M., Houtz J.M., Hryckowian A.J., Kelchner V.A.et al.. Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2006; 2:e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pope W.H., Mavrich T.N., Garlena R.A., Guerrero-Bustamante C.A., Jacobs-Sera D., Montgomery M.T., Russell D.A., Warner M.H., Hatfull G.F.Science Education Alliance-Phage Hunters Advancing Genomics and Evolutionary Science . Bacteriophages of Gordonia spp. display a spectrum of diversity and genetic relationships. MBio. 2017; 8:e01069-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hatfull G.F. Actinobacteriophages: genomics, dynamics, and applications. Annu. Rev. Virol. 2020; 7:37–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grose J.H., Casjens S.R.. Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology. 2014; 468-470:421–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xu Z.W., Wei Y.L., Ji X.L.. Progress on phage genomics of Pseudomonas spp. Yi Chuan. 2020; 42:752–759. [DOI] [PubMed] [Google Scholar]
- 13. McShan W.M., McCullor K.A., Nguyen S.V.. The bacteriophages of Streptococcus pyogenes. Microbiol. Spectr. 2019; 7: 10.1128/microbiolspec.GPP3-0059-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pope W.H., Bowman C.A., Russell D.A., Jacobs-Sera D., Asai D.J., Cresawn S.G., Jacobs W.R., Hendrix R.W., Lawrence J.G., Hatfull G.F.et al.. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife. 2015; 4:e06416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pedulla M.L., Ford M.E., Houtz J.M., Karthikeyan T., Wadsworth C., Lewis J.A., Jacobs-Sera D., Falbo J., Gross J., Pannunzio N.R.et al.. Origins of highly mosaic mycobacteriophage genomes. Cell. 2003; 113:171–182. [DOI] [PubMed] [Google Scholar]
- 16. Klyczek K.K., Bonilla J.A., Jacobs-Sera D., Adair T.L., Afram P., Allen K.G., Archambault M.L., Aziz R.M., Bagnasco F.G., Ball S.L.et al.. Tales of diversity: Genomic and morphological characteristics of forty-six Arthrobacter phages. PLoS One. 2017; 12:e0180517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jacobs-Sera D., Abad L.A., Alvey R.M., Anders K.R., Aull H.G., Bhalla S.S., Blumer L.S., Bollivar D.W., Bonilla J.A., Butela K.A.et al.. Genomic diversity of bacteriophages infecting Microbacterium spp. PLoS One. 2020; 15:e0234636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dedrick R.M., Smith B.E., Garlena R.A., Russell D.A., Aull H.G., Mahalingam V., Divens A.M., Guerrero-Bustamante C.A., Zack K.M., Abad L.et al.. Mycobacterium abscessus strain morphotype determines phage susceptibility, the repertoire of therapeutically useful phages, and phage resistance. mBio. 2021; 12:e03431-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Redfield R.J., Campbell A.M.. Structure of cryptic lambda prophages. J. Mol. Biol. 1987; 198:393–404. [DOI] [PubMed] [Google Scholar]
- 20. Wang X., Wood T.K.. Cryptic prophages as targets for drug development. Drug Resist. Updat. 2016; 27:30–38. [DOI] [PubMed] [Google Scholar]
- 21. Wetzel K.S., Aull H.G., Zack K.M., Garlena R.A., Hatfull G.F.. Protein-mediated and RNA-based origins of replication of extrachromosomal mycobacterial prophages. mBio. 2020; 11:e00385-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grindley N.D., Whiteson K.L., Rice P.A.. Mechanisms of site-specific recombination. Annu. Rev. Biochem. 2006; 75:567–605. [DOI] [PubMed] [Google Scholar]
- 23. Landy A. The lambda integrase site-specific recombination pathway. Microbiol. Spectr. 2015; 3:MDNA3-0051-2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Williams K.P. Integration sites for genetic elements in prokaryotic tRNA and tmRNA genes: sublocation preference of integrase subfamilies. Nucleic Acids Res. 2002; 30:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mantri Y., Williams K.P.. Islander: a database of integrative islands in prokaryotic genomes, the associated integrases and their DNA site specificities. Nucleic. Acids. Res. 2004; 32:D55–D58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kim A.I., Ghosh P., Aaron M.A., Bibb L.A., Jain S., Hatfull G.F.. Mycobacteriophage Bxb1 integrates into the Mycobacterium smegmatis groEL1 gene. Mol. Microbiol. 2003; 50:463–473. [DOI] [PubMed] [Google Scholar]
- 27. Ojha A., Anand M., Bhatt A., Kremer L., Jacobs W.R., Hatfull G.F. GroEL1: a dedicated chaperone involved in mycolic acid biosynthesis during biofilm formation in mycobacteria. Cell. 2005; 123:861–873. [DOI] [PubMed] [Google Scholar]
- 28. Dedrick R.M., Aull H.G., Jacobs-Sera D., Garlena R.A., Russell D.A., Smith B.E., Mahalingam V., Abad L., Gauthier C.H., Hatfull G.F.. The prophage and plasmid mobilome as a likely driver of Mycobacterium abscessus diversity. mBio. 2021; 12:e03441-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Smith M.C., Brown W.R., McEwan A.R., Rowley P.A.. Site-specific recombination by phiC31 integrase and other large serine recombinases. Biochem. Soc. Trans. 2010; 38:388–394. [DOI] [PubMed] [Google Scholar]
- 30. Smith M.C.M. Phage-encoded serine integrases and other large serine recombinases. Microbiol. Spectr. 2015; 3: 10.1128/microbiolspec.mdna3-0059-2014. [DOI] [PubMed] [Google Scholar]
- 31. Toussaint A., Rice P.A.. Transposable phages, DNA reorganization and transfer. Curr. Opin. Microbiol. 2017; 38:88–94. [DOI] [PubMed] [Google Scholar]
- 32. Fouts D.E. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006; 34:5839–5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Siren K., Millard A., Petersen B., Gilbert M.T.P., Clokie M.R.J., Sicheritz-Ponten T.. Rapid discovery of novel prophages using biological feature engineering and machine learning. NAR Genom Bioinform. 2021; 3:lqaa109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akhter S., Aziz R.K., Edwards R.A.. PhiSpy: a novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012; 40:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arndt D., Grant J.R., Marcu A., Sajed T., Pon A., Liang Y., Wishart D.S.. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016; 44:W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reis-Cunha J.L., Bartholomeu D.C., Manson A.L., Earl A.M., Cerqueira G.C.. ProphET, prophage estimation tool: a stand-alone prophage sequence prediction tool with self-updating reference database. PLoS One. 2019; 14:e0223364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lima-Mendez G., Van Helden J., Toussaint A., Leplae R.. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics. 2008; 24:863–865. [DOI] [PubMed] [Google Scholar]
- 38. Auslander N., Gussow A.B., Benler S., Wolf Y.I., Koonin E.V.. Seeker: alignment-free identification of bacteriophage genomes by deep learning. Nucleic Acids Res. 2020; 48:e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ren J., Ahlgren N.A., Lu Y.Y., Fuhrman J.A., Sun F.. VirFinder: a novel k-mer based tool for identifying viral sequences from assembled metagenomic data. Microbiome. 2017; 5:69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guo J., Bolduc B., Zayed A.A., Varsani A., Dominguez-Huerta G., Delmont T.O., Pratama A.A., Gazitua M.C., Vik D., Sullivan M.B.et al.. VirSorter2: a multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome. 2021; 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Davis J.J., Wattam A.R., Aziz R.K., Brettin T., Butler R., Butler R.M., Chlenski P., Conrad N., Dickerman A., Dietrich E.M.et al.. The PATRIC Bioinformatics Resource Center: expanding data and analysis capabilities. Nucleic Acids Res. 2020; 48:D606–D612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Glickman C., Kammlade S.M., Hasan N.A., Epperson L.E., Davidson R.M., Strong M.. Characterization of integrated prophages within diverse species of clinical nontuberculous mycobacteria. Virol J. 2020; 17:124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Fan X., Xie L., Li W., Xie J.. Prophage-like elements present in Mycobacterium genomes. BMC Genomics. 2014; 15:243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gillespie J.J., Wattam A.R., Cammer S.A., Gabbard J.L., Shukla M.P., Dalay O., Driscoll T., Hix D., Mane S.P., Mao C.et al.. PATRIC: the comprehensive bacterial bioinformatics resource with a focus on human pathogenic species. Infect. Immun. 2011; 79:4286–4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hyatt D., Chen G.L., Locascio P.F., Land M.L., Larimer F.W., Hauser L.J.. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinf. 2010; 11:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laslett D., Canback B.. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004; 32:11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steinegger M., Soding J.. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017; 35:1026–1028. [DOI] [PubMed] [Google Scholar]
- 48. Steinegger M., Meier M., Mirdita M., Vohringer H., Haunsberger S.J., Soding J.. HH-suite3 for fast remote homology detection and deep protein annotation. BMC Bioinf. 2019; 20:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mavrich T.N., Hatfull G.F.. Bacteriophage evolution differs by host, lifestyle and genome. Nat. Microbiol. 2017; 2:17112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ester M., Kriegel H.-P., Sander J., Xu X.. Simoudis E., Han J., Fayyad U.M.. Proceedings of the Second International Conference on Knowledge Discovery and Data Mining (KDD-96). 1996; AAAI Press; 226–231. [Google Scholar]
- 51. Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T.L.. BLAST+: architecture and applications. BMC Bioinf. 2009; 10:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Koonin E.V., Wolf Y.I.. Genomics of bacteria and archaea: the emerging dynamic view of the prokaryotic world. Nucleic Acids Res. 2008; 36:6688–6719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mavrich T.N., Gauthier C., Abad L., Bowman C.A., Cresawn S.G., Hatfull G.F.. pdm_utils: a SEA-PHAGES MySQL phage database management toolkit. Bioinformatics. 2021; 37:2464–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zulkower V., Rosser S.. DNA Features Viewer: a sequence annotation formatting and plotting library for Python. Bioinformatics. 2020; 36:4350–4352. [DOI] [PubMed] [Google Scholar]
- 55. Cui Y., Chen X., Luo H., Fan Z., Luo J., He S., Yue H., Zhang P., Chen R.. BioCircos.js: an interactive Circos JavaScript library for biological data visualization on web applications. Bioinformatics. 2016; 32:1740–1742. [DOI] [PubMed] [Google Scholar]
- 56. Cresawn S.G., Bogel M., Day N., Jacobs-Sera D., Hendrix R.W., Hatfull G.F.. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinf. 2011; 12:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pope W.H., Anders K.R., Baird M., Bowman C.A., Boyle M.M., Broussard G.W., Chow T., Clase K.L., Cooper S., Cornely K.A.et al.. Cluster M Mycobacteriophages Bongo, PegLeg, and Rey with unusually large repertoires of tRNA isotypes. J. Virol. 2014; 88:2461–2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nayfach S., Camargo A.P., Schulz F., Eloe-Fadrosh E., Roux S., Kyrpides N.C.. CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 2021; 39:578–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hendrix R.W., Smith M.C., Burns R.N., Ford M.E., Hatfull G.F.. Evolutionary relationships among diverse bacteriophages and prophages: all the world's a phage. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:2192–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Guerrero-Bustamante C.A., Dedrick R.M., Garlena R.A., Russell D.A., Hatfull G.F.. Toward a phage cocktail for tuberculosis: susceptibility and tuberculocidal action of Mycobacteriophages against diverse Mycobacterium tuberculosis strains. mBio. 2021; 12:e00973-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jacobs-Sera D., Marinelli L.J., Bowman C., Broussard G.W., Guerrero Bustamante C., Boyle M.M., Petrova Z.O., Dedrick R.M., Pope W.H.Science Education Alliance Phage Hunters Advancing, G. et al.. On the nature of mycobacteriophage diversity and host preference. Virology. 2012; 434:187–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gruschow S., Athukoralage J.S., Graham S., Hoogeboom T., White M.F.. Cyclic oligoadenylate signalling mediates Mycobacterium tuberculosis CRISPR defence. Nucleic Acids Res. 2019; 47:9259–9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The code for the DEPhT Python package is publicly available with installation and usage instructions at https://github.com/chg60/DEPhT.git and https://pypi.org/project/depht/. The models we trained for this study are available at https://osf.io/zt4n3. The training and testing data accessions are available in Supplementary Tables.