

Abstract
Background
Heparin‐induced thrombocytopenia (HIT) is a prothrombotic, immune‐mediated adverse drug reaction associated with high rates of thrombosis‐related morbidity and mortality caused by FcγRIIa‐activating pathogenic antibodies to PF4‐heparin. Procoagulant platelets are a platelet subset that promote thrombin generation, are clinically relevant in prothrombotic diseases, and are formed when platelet G‐protein‐coupled receptor (GPCR) and ITAM‐linked receptors are co‐stimulated.
Objectives
We examined the procoagulant platelet response of healthy donors to platelet agonists in the presence of HIT plasma and determined the contribution of FcγRIIa.
Patients/Methods
Our previously established flow cytometry‐based procoagulant platelet assay was modified to incorporate plasma samples, performed using FcγRIIa‐responsive donor platelets. Plasma samples were serotonin‐release assay–confirmed HIT (HIT+), or negative on HIT screening.
Results
In response to GPCR stimulation, only HIT+ plasma produced a heparin‐dependent sensitization that required active FcγRIIa. As a potential diagnostic tool, the procoagulant platelet assay achieved 98% accuracy in identifying clinically verified HIT when performed blinded to the diagnoses of a validation cohort. Samples inducing a higher procoagulant platelet response were more likely from patients with thrombotic complications. Thrombin stimulation markedly increased the procoagulant platelet response with HIT+ plasma that was heparin independent and only partially reversed by FcγRIIa blockade, possibly reflecting ongoing thrombotic risk after heparin cessation.
Conclusions
We demonstrate that HIT plasma together with platelet agonists increased the procoagulant platelet proportions, which may contribute to thrombotic risk in HIT. Targeting procoagulant platelet activation may represent a novel treatment strategy. This assay may be a rapid, clinically relevant functional assay for accurately detecting pathological HIT antibodies.
Keywords: FcγRIIa, flow cytometry, heparin‐induced thrombocytopenia, procoagulant platelets, thrombosis
Essentials.
Rapid and accurate diagnosis of HIT remains a challenge yet is crucial to improve outcomes.
A novel flow cytometry procoagulant platelet (PP) assay accurately identifies patients with HIT.
HIT antibodies induce heparin‐dependent changes in PP profiles with PAR‐1 stimulation.
Thrombin co‐stimulation with HIT plasma induces heparin‐independent PP, irreversible with IV.3.
1. INTRODUCTION
Heparin‐induced thrombocytopenia (HIT) is a prothrombotic, immune‐mediated adverse drug reaction to the common anticoagulant heparin, or its derivatives. HIT is associated with high rates of morbidity and mortality, with thrombotic complications affecting >50% of untreated patients. 1 HIT occurs in up to 3% of patients receiving unfractionated heparin 2 and 0.2% of patients on low molecular weight heparin. 2
Key to the pathogenesis of HIT is the formation of platelet‐activating immunoglobulin G (IgG) antibodies recognizing complexes of platelet alpha granule protein, platelet factor 4 (PF4), 3 and heparin. 4 , 5 These HIT antibodies, together with PF4‐heparin complexes, form immune complexes, which bind and cross‐link the low‐affinity IgG receptor, FcγRIIa, on platelets, monocytes, and neutrophils, causing platelet activation and aggregation through the immunoreceptor tyrosine‐based activation motif (ITAM). 6 Thrombocytopenia and thrombosis then results, augmented by increased platelet clearance, release of platelet‐derived microparticles, 7 and thrombin produced on activated monocytes. 8
HIT diagnosis and treatment remain clinical challenges. One gold standard functional laboratory test is the serotonin release assay (SRA). This test measures release of radiolabeled‐serotonin from washed platelets at low (0.1–0.3 U/mL) and high (100 U/mL) heparin doses; a positive test characterized by >20% release at therapeutic dose that is inhibited at high‐dose heparin. The high‐dose heparin confirmation step relates to the requirement for a particular stoichiometry of PF4:heparin to form stable ultra‐large complexes that activate FcγRIIa. 9 , 10 The limitations of the SRA are well documented and include requirements for washed “pedigree” donor platelets, radioactivity, and counters. These features limit its use to the rare reference laboratory; for example, a single laboratory in Australia. These restrictions delay definitive diagnosis of HIT, and treatment decisions must often be made before laboratory confirmation of either diagnosis or exclusion of HIT. Although a combination of clinical pretest probability and rapid immunoassay testing can in some cases bypass requirements for a functional assay, 10 , 11 there remains a category of patients for whom this combination does not give a definitive result and a functional assay confirmatory step remains important. 10 , 12
There remains a clinical need for improved diagnostic and therapeutic approaches for HIT driven by improved understanding of underlying mechanisms. Tomer and colleagues reported that the platelet death marker, annexin V, detection by flow cytometry can identify HIT plasmas in absence of platelet agonists. 13 Recent exploration of cell death pathways in response to KKO antibody/PF4, a model that mimics HIT, demonstrated a marked increase in cell death (apoptotic – mitochondrial membrane depolarization, up‐regulation of pro‐apoptotic protein Bax, and non‐apoptotic – calpain activation) markers in treated platelets but that procoagulant effect was related to caspase independent, calpain‐mediated platelet cell death. 14 Generated through the cyclophilin‐D–dependent necrosis pathway, procoagulant platelets are a subpopulation of platelets that promote coagulation by providing a procoagulant surface for assembly and propagation of coagulation factors leading to thrombin generation. 15 , 16 Procoagulant platelets are of clinical relevance in thrombotic conditions such as coronary artery disease and stroke. 17 , 18 , 19 , 20 , 21 Tutwiler and colleagues demonstrated that a subset of procoagulant platelets, coated platelets (expression of P‐selectin, binding of annexin V, and anti‐factor X), were induced by HIT‐like antibody KKO. 8 Thus, we hypothesized that procoagulant platelets significantly contribute to the thrombotic risk in HIT.
Given the combined stimulation of platelet G‐protein‐coupled (GPCR) (e.g., PAR‐1, PAR‐4, P2Y12) and ITAM‐linked receptors (ILR) (e.g., GPVI, CLEC2, FcγRIIa) synergistically induces procoagulant platelet formation in suspension, 22 , 23 we hypothesized that low‐level priming of platelets with GPCR agonists in donor platelets in suspension would permit identification of an FcγRIIa‐dependent pathological procoagulant response that would allow both mechanistic insight and yield a sensitive and specific assay for HIT. We have earlier shown that in healthy platelets stimulated with thrombin plus collagen, 96% of 4‐(N‐(S‐glutathionylacetyl)amino)phenylarsonous acid (GSAO)‐positive platelets compared with only 27% of platelets that express phosphatidylserine, bind active factor X, and hence support thrombin generation. 17 , 24 Here, we used a GSAO‐based procoagulant platelet assay, which is more specific for functionally relevant procoagulant platelets than annexin V, to investigate the impact of HIT antibodies on procoagulant platelet profile of healthy platelets using plasma from patients with SRA‐confirmed HIT, as compared with HIT‐negative samples. We provide insights into the mechanisms underlying HIT thrombosis and present a flow cytometry‐based procoagulant platelet assay that may be a viable functional assay with rapid turnaround time for HIT diagnosis.
2. METHODS
2.1. Study approval
Human studies were approved by Concord Repatriation General Hospital (CRGH) Human Research Ethics Committee (HREC/18/CRGH/294), WSLHD Human Research Ethics Committee #1812‐01. Blood donors gave written informed consent.
2.2. Materials
GSAO and control compound 4‐(N‐(S‐glutathionylacetyl)amino)benzoic acid were synthesized previously as described 25 and conjugated to amine‐reactive succinimidyl ester Alexa Fluor 647‐NHS (Life Technologies). Confirmation of GSAO activity and batch to batch validation testing after conjugation were performed as previously described. 17 , 26 Monomeric collagen‐related peptide (CRP, Auspep) was cross‐linked using N‐succinimidyl 3‐(2‐pyridyldithio)propionate (Sigma‐Aldrich) to produce CRP‐xL. Reagents are available on request.
2.3. Plasma samples
Citrated platelet‐poor plasma stored at −80°C were from patients with suspected HIT, referred to diagnostic laboratories in three Australian hospitals (CRGH, Royal Prince Alfred, and the Institute of Clinical Pathology and Medical Research at Westmead Hospital) for subsequent investigation by SRA at a reference laboratory. SRA was performed following initial positive immunological screening (either AcuStar HIT‐IgG(PF4‐H) chemiluminescence or STic Expert HIT lateral flow) on patients with intermediate or high pretest probability of HIT based on the 4Ts scoring system 27 , 28 , 29 or by request of treating physicians. SRA‐positive samples that fulfilled clinical criteria for HIT were considered HIT+. Clinical notes were adjudicated by independent blinded observers to verify clinical HIT. HIT negative (HIT−) samples were from inpatients with thrombocytopenia or platelet count drop >25%, intermediate/high 4Ts score with negative immunoassay testing.
2.4. Screening for high responders
Healthy donors recruited at the CRGH and ANZAC Research Institute (Sydney, Australia) were screened to identify FcγRIIa responders 30 using light transmission aggregometry response to 1.5 µg/mL of anti‐CD9, clone ALB6 (Santa Cruz Biotechnology) as previously described. 31
High responders demonstrated >80% aggregation with a time to initiation <180 s, intermediate responders: 50%–80% aggregation, or >80% but time to initiation >180 s. Of 30 healthy donors screened, eight high and two intermediate responders were included in the development study. Only high responders were used in the validation study.
2.5. Blood collection
Blood was collected from the antecubital fossa of healthy volunteers into 3.2% citrate tubes using a 21G butterfly needle. The initial 3 mL of blood was discarded.
2.6. Procoagulant platelet assay
The GSAO‐based whole blood procoagulant platelet flow cytometry assay established by our laboratory 17 , 26 was modified for testing of various platelet agonists in the presence of HIT+ or HIT− plasma. Citrated whole blood (13 μL) was diluted in Hanks' balanced salt solution (pH 7.35) and incubated with and without agonists, unfractionated heparin (Pfizer) and plasma (5 μL) for 10 min, with a final concentration of 2.5 mM Gly‐Pro‐Arg‐Pro peptide (Sigma‐Aldrich) and 2.5 mM calcium chloride. The final reaction volume was 50 μL. Agonists included CRP‐xL, ADP (Helena Laboratories), thrombin receptor‐activating peptide (SFLLRN, Auspep), and bovine thrombin (Sigma‐Aldrich). Heparin was omitted in experiments that include thrombin because of the neutralizing effect of heparin on thrombin activity. In some experiments, blood was pretreated with 10 µg/mL IV.3 (StemCell Technologies), a monoclonal antibody against FcγRIIa, for 15 min before the reaction. The reaction was stopped by further dilution with Hanks' balanced salt solution (150 μL, 1:3 v/v) followed by staining with antibodies to CD45 (HI30) (StemCell Technologies), CD41a (HIP8) (BD Biosciences), CD62P (Psel.KO2.3) (eBioscience) or isotype control (eBioscience), and GSAO or 4‐(N‐(S‐glutathionylacetyl)amino)benzoic acid. After staining, samples were fixed with PAMFix (Platelet Solutions Ltd), centrifuged, and resuspended. Samples were rested for 1 h in the dark at room temperature before analysis on a BD LSRFortessa X‐20 with acquisition of 10 000 platelet events (see Table S1 for technical settings). Occasional thrombin+HIT plasma samples were stopped before 10 000 events if there was insufficient sample. The gating strategy to identify procoagulant platelets was detailed in Tan and colleagues. 26
2.7. Two‐dimensional visualization of flow cytometry parameters
The t‐distributed stochastic neighbor embedding (t‐SNE) algorithm was used to visualize high‐dimensional, multiparameter flow cytometry data. Datasets were concatenated and the Barnes‐Hut implementation of t‐SNE was used for applying the dimensionality reduction algorithm on compensated flow cytometry parameters (forward scatter, size scatter, CD41a, CD45, GSAO, and CD62P) using FlowJo version 10.6.
2.8. Clinical adjudication
Two hematologists adjudicated HIT diagnosis and thrombotic outcomes while blinded to assay results. Electronic medical records and HIT test results were reviewed, 4T scores recalculated, and clinical outcomes in response to changes in anticoagulation assessed. A third hematologist independently adjudicated where clinical adjudication was discrepant from SRA or flow cytometry results.
2.9. Statistical analyses
Statistical analyses were performed using GraphPad Prism 9, with statistical significance set at p < 0.05.
For original data, please contact vivien.chen@sydney.edu.au.
3. RESULTS
3.1. HIT+ plasma does not increase procoagulant platelets with GPCR agonists alone or heparin alone
Procoagulant platelets were identified using a whole blood flow cytometry‐based assay as CD41a+/CD45− events marked with both the cell necrosis marker GSAO, and the platelet activation marker P‐selectin (GSAO+/CD62P+) as described. 17 We examined procoagulant platelet response to HIT+plasma in presence and absence of agonist stimulation of GPCR ligands: PAR‐1 agonist (SFLLRN) and P2Y12 receptor agonist (ADP), and ILR ligand GPVI agonist (CRP‐xL). In the absence of exogenous heparin, there was no difference in the proportion of procoagulant platelets in the presence of HIT+plasma compared with either HIT−, no plasma, or autologous plasma under resting conditions or following stimulation with SFLLRN, ADP, or CRP‐xL (Figures 1A and S1A) within our limited sample size of six to 13 data points generated from 5 HIT− and 4 HIT+plasmas tested on 10 healthy donors. There were no differences in procoagulant platelet proportion with either HIT+ or HIT− plasma, at either therapeutic concentration (0.5 U/mL) or high‐dose heparin (100 U/mL), compared with basal levels of procoagulant platelets without exogenous plasma (Figure 1B).
FIGURE 1.
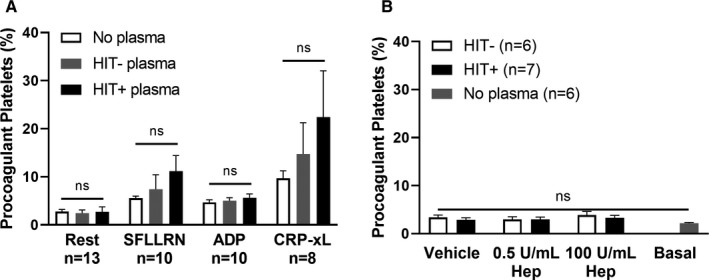
Plasma samples from HIT patients do not increase the proportion of procoagulant platelets from healthy donors when treated with either platelet agonists or heparin, alone. (A) The proportion of procoagulant platelets in whole blood samples from healthy donors (n = 8–13) did not change significantly upon exposure to HIT− (n = 5) or HIT+ (n = 4) plasma under resting conditions or treatment with platelet agonists (SFLLRN 5 µM, ADP 20 µM, CRP‐xL 2 µg/mL). Mixed‐effects analysis was performed with Tukey correction for multiple comparisons. (B) In the absence of stimulation with platelet agonist, the addition of low‐dose (0.5 U/mL) or high‐dose (100 U/mL) heparin to platelets from healthy donors (n = 6–7) exposed to HIT− (n = 3) or HIT+ (n = 4) plasma did not alter the basal proportion of procoagulant platelets observed in the absence of patient plasma. Vehicle control consisted of Hanks' balanced salt solution in place of heparin. One‐way anova was performed with Dunnett correction for multiple comparisons to basal level of procoagulant platelets. Error bars indicate mean ± SEM. ns, not significant. ADP, adenosine diphosphate; CRP‐xL, crosslinked collagen‐related peptide; Hep, heparin; HIT, heparin‐induced thrombocytopenia; SEM, standard error of the mean; SFLLRN, thrombin receptor‐activating peptide
3.2. HIT+ plasma sensitizes healthy platelets to become procoagulant with GPCR agonist stimulation in a heparin‐dependent manner and requires active FcγRIIa
HIT antibodies recognize complexes of heparin with the platelet alpha granule protein PF4, and agonist stimulation is known to release PF4. SFLLRN, ADP, or CRP‐xL stimulation, leading to >90% alpha granule release measured by P‐selectin (Figure S2). In presence of HIT− plasma, there was no difference in procoagulant platelet formation in response to SFLLRN, ADP, or CRP‐xL (Figure 2A,C,E), at either therapeutic or high heparin concentrations. However, in presence of HIT+ plasma, there was a significant difference in the proportion of procoagulant platelets at therapeutic‐concentration heparin compared with no heparin for SFLLRN (36.53 ± 7.50 vs 11.19 ± 3.23, p < 0.01) and ADP (25.22 ± 5.94 vs 5.66 ± 0.79, p < 0.05; Figure 2B,D). HIT has a well‐established stoichiometric ratio of heparin and PF4 underscoring the formation of pathogenic immune complexes, 32 and the heightened procoagulant platelet response at therapeutic‐concentration heparin was fully abrogated at high‐dose heparin for all agonists tested: SFLLRN (36.53 ± 7.50 vs 4.39 ± 0.35, p < 0.01), ADP (25.22 ± 5.94 vs 5.16 ± 0.52, p < 0.05), and CRP‐xL (26.87 ± 3.58 vs 11.70 ± 2.63, p < 0.01) (Figure 2B,D,F).
FIGURE 2.
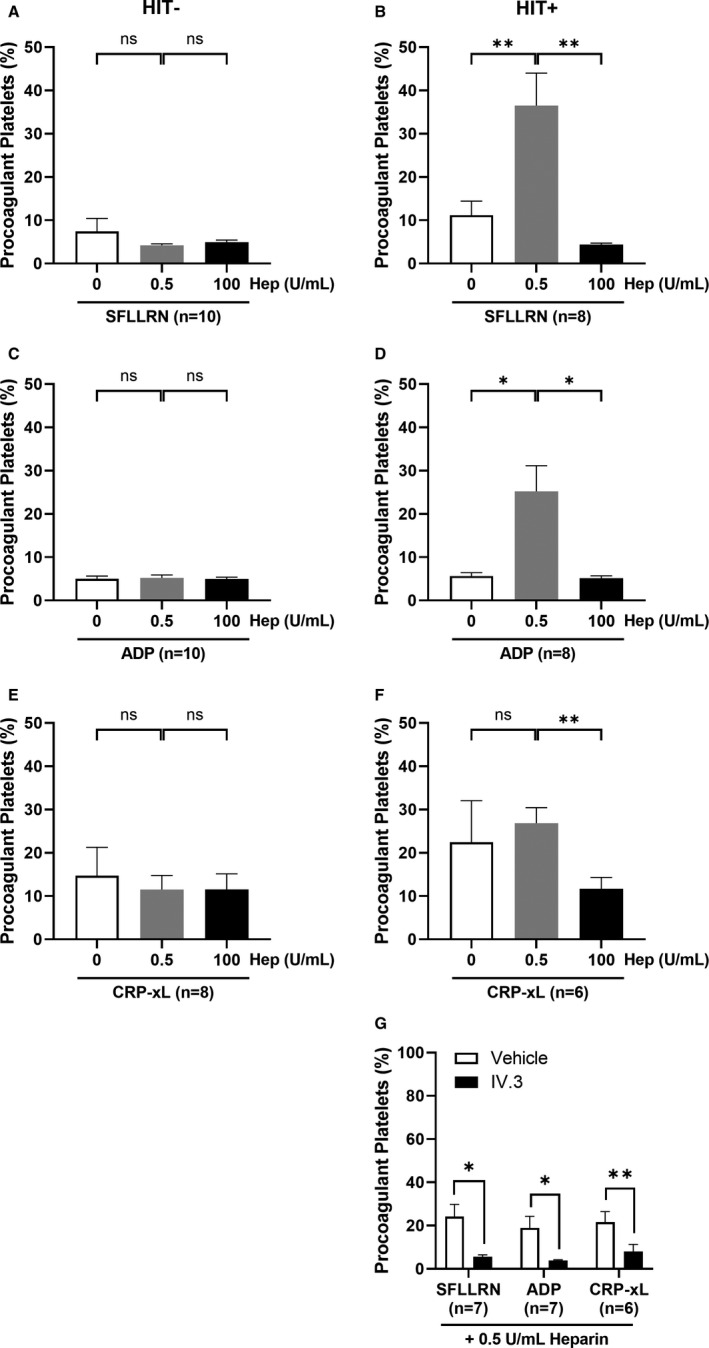
Plasma samples from SRA‐positive (HIT+) but not HIT immunoassay‐negative (HIT−) patients increased the procoagulant platelet response in donor platelets treated with platelet agonists in a heparin‐dependent manner and requires active FcγRIIa. Whole blood from healthy donors (n = 6–10) were treated with plasma samples from either HIT− (n = 5; A, C, E) or HIT+ (n = 3–4; B, D, F) patients, in the absence of heparin, or addition of low‐dose (0.5 U/mL) or high‐dose (100 U/mL) heparin and platelet agonists: (A–B) thrombin receptor‐activating peptide, SFLLRN (5 µM), or (C–D) adenosine diphosphate, ADP (20 µM), or (E–F) crosslinked collagen‐related peptide, CRP‐xL (2 µg/mL) for 10 min before staining for flow cytometry analysis of procoagulant platelets. Repeated measures anova was performed with Tukey correction for multiple comparisons. (G) Donor platelets (n = 6–7) in whole blood were pretreated with the FcγRIIa function‐blocking monoclonal antibody, IV.3 (10 µg/mL) for 15 min before exposure to a separate set of SRA‐positive HIT plasma (n = 4) and platelet agonists. In the presence of low‐dose heparin (0.5 U/mL) and platelet agonists: CRP‐xL (2 µg/mL), SFLLRN (5 µM) or ADP (20 µM), pretreatment with IV.3 abrogated the procoagulant platelet proportions compared with vehicle control. Paired t‐test was performed comparing vehicle with IV.3 treatment for each agonist. Procoagulant platelet percentages were defined by the proportion of GSAO+/CD62P+ platelet events. Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ns, not statistically significant. Hep, heparin; ADP, adenosine diphosphate; CRP‐xL, crosslinked collagen‐related peptide; HIT, heparin‐induced thrombocytopenia; SFLLRN, thrombin receptor‐activating peptide; SRA, serotonin release assay
We examined the requirement for FcγRIIa in the sensitization of procoagulant platelets by receptor‐specific agonists together with therapeutic‐concentration heparin. Pretreatment of healthy platelets with IV.3 markedly reduced the proportion of procoagulant platelets stimulated with receptor‐specific agonists in presence of therapeutic‐concentration heparin and HIT+plasma, with SFLLRN (24.24 ± 5.59 vs 5.63 ± 0.83, p < 0.05), ADP (18.98 ± 5.28 vs 3.85 ± 0.34, p < 0.05), and CRP‐xL (21.63 ± 4.82 vs 8.02 ± 3.24, p < 0.01; Figure 2G), indicating that active FcγRIIa is required for the HIT plasma‐induced and heparin‐dependent potentiation of procoagulant platelet formation.
3.3. Effect of FcγRIIa‐inhibition on multiparameter flow cytometry profile of healthy platelets
Having observed the effect of FcγRIIa inhibition on a specific platelet population, HIT‐induced procoagulant platelets, we then used the t‐SNE algorithm to visualize the HIT‐induced qualitative changes across total platelet subpopulations (Figure 3). The density in t‐SNE profiles represent relative proportions of platelet subgroupings clustered according to the flow cytometry parameters. The t‐SNE profile of healthy platelets in presence of SFLLRN was markedly altered by the addition of HIT+ plasma compared with no plasma. IV.3 pretreatment produced a visually similar t‐SNE profile to that of no plasma (Figure 3), indicating platelet changes were FcγRIIa mediated. t‐SNE profiles of healthy platelets in presence of ADP and CRP‐xL with and without HIT+plasma followed similar patterns to that of SFLLRN (Figure S3).
FIGURE 3.
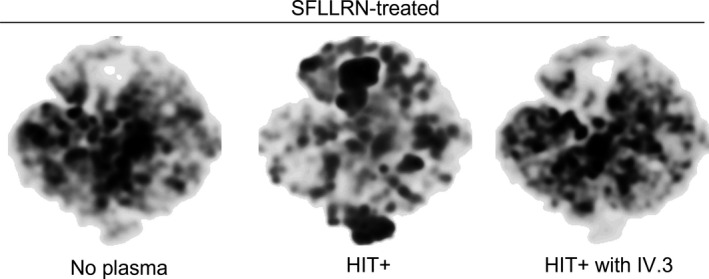
Visualization of multiparameter flow cytometry data using t‐distributed stochastic neighbor embedding (t‐SNE) algorithm. The t‐SNE algorithm was employed to compare the platelet flow cytometry profiles between healthy donor whole blood exposed to either no plasma, HIT+ or HIT+ plasma plus IV.3 pretreatment in the presence of SFLLRN (5 µM) plus heparin (0.5 U/mL) in n = 7 donors. A subset of 6000 CD41a+ events were selected from each experiment (n = 7 per condition). The downsized files were subsequently concatenated to generate files containing 168 000 SFLLRN‐treated CD41a+ events. The Barnes‐Hut implementation of t‐SNE with 1000 iterations, a perplexity value of 30, and a learning rate of 11 760 was used for applying the dimensionality reduction algorithm on compensated flow cytometry parameters. HIT, heparin‐induced thrombocytopenia; SFLLRN, thrombin receptor‐activating peptide
3.4. Procoagulant platelet formation induced by SFLLRN, therapeutic‐concentration heparin, and patient plasma has potential to differentiate thrombocytopenia secondary to HIT from other etiologies
To assess the diagnostic value of a flow cytometry‐based procoagulant platelet assay, we compared procoagulant platelet formation after SFLLRN stimulation in the presence of therapeutic‐concentration heparin using plasma from HIT+ and HIT− thrombocytopenic patients. This yielded a receiver operating characteristics (ROC) area under the curve (AUC) of 1.0 ± 0, p = 0.0004, with a procoagulant result of 7.6% demonstrating 100% sensitivity and specificity (Figure 4A, Table S2). ADP under the same conditions yielded a ROC AUC 0.93 ± 0.06, p = 0.0025, with a cutoff of 6.2%, demonstrating 100% sensitivity and 70% specificity (Figure 4B, Table S3), and CRP‐xL yielded a ROC AUC of 0.92 ± 0.08, p = 0.0098, with a cutoff of 13.5%, demonstrating 100% sensitivity and 75% specificity (Figure 4C, Table S4).
FIGURE 4.
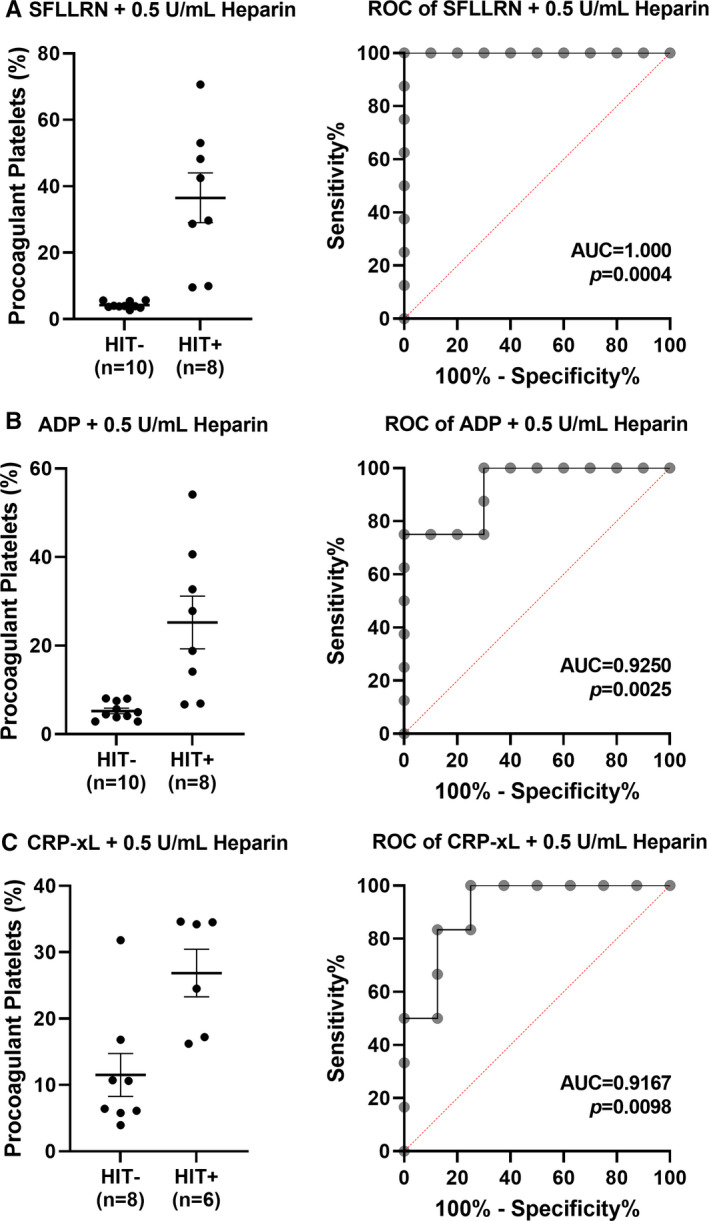
Procoagulant platelet formation in the presence of platelet agonists, low‐dose heparin, and patient plasma has potential to differentiate thrombocytopenia secondary to HIT from other etiologies. Receiver operating characteristic (ROC) curve analysis was performed to evaluate the diagnostic potential of procoagulant platelet formation in healthy donors (n = 6–10) induced by plasma from patients suspected of HIT in the presence of low‐dose heparin (0.5 U/mL) and receptor‐specific platelet agonists: (A) thrombin receptor‐activating peptide, SFLLRN (5 µM), (B) adenosine diphosphate, ADP (20 µM), or (C) crosslinked collagen‐related peptide, CRP‐xL (2 µg/mL). Plasma samples were collected from either HIT immunoassay‐negative patients (HIT−) or SRA‐confirmed HIT patients (HIT+). Error bars indicate mean ± SEM. ADP, adenosine diphosphate; AUC, area under the curve; CRP‐xL, crosslinked collagen‐related peptide; HIT, heparin‐induced thrombocytopenia; SRA, serotonin release assay. Refer to supplemental data (Tables S2–S4) for the list of sensitivity and specificity at various cutoff values
In a validation cohort of 64 patients with clinically suspected HIT, the procoagulant platelet assay with SFLLRN in presence of healthy whole blood, patient plasma, and heparin was performed while blinded to the diagnosis. SRA was again used as the gold standard for HIT+ samples. A procoagulant platelet result of >7.6% correctly identified 43 of 44 immunoassay‐ or SRA‐negative samples and 18 of 20 SRA‐positive samples (Figure 5A). This yielded a positive predictive value of 94.7%, a negative predictive value of 95.6%, and an accuracy of 95.3%. Independent blinded clinical review of the HIT diagnosis demonstrated 98% concordance with assay results and final clinical HIT diagnosis with correction of one false‐positive and one false‐negative SRA test. Notably, the assay correctly identified three of three samples that were false positive by immunoassay, but negative by SRA and negative by clinical review. Two of these were patients with a previous diagnosis of HIT. One of two samples that was SRA‐positive but negative in our assay had a final diagnosis of immune thrombocytopenia, whereas the other was clinically HIT (Table S5, Figure S4). Our assay correctly identified a false‐negative SRA sample that was clinically HIT (Figure 5A). Procoagulant platelet response induced by 13 HIT+plasma did not correlate with anti‐PF4/heparin antibody levels by chemiluminescence (Figure S5). Importantly, a higher procoagulant platelet response (p = 0.0213, Figure 5B), was observed in donor platelets treated with plasma from HIT+ patients with thrombotic outcomes compared to those without, whereas anti‐PF4/heparin antibody levels by chemiluminescence immunoassay was not associated with thrombosis (p = 0.9559, Figure 5C).
FIGURE 5.
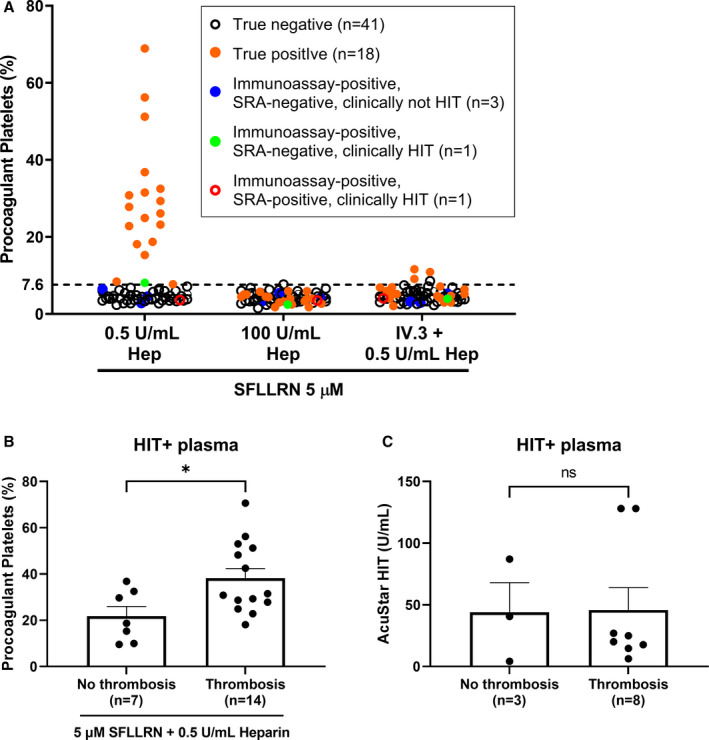
Validation of procoagulant platelet assay using plasma samples from 64 patients suspected of HIT. (A) Whole blood from healthy donors was treated with 5 µM SFLLRN and therapeutic‐concentration (0.5 U/mL) or high‐dose (100 U/mL) heparin in the presence of plasma from immunoassay‐negative or clinically not HIT patients (true negative, black open circles; n = 41) or SRA‐confirmed and clinically verified HIT patients (true positives, orange circles; n = 18). Blue circles represent plasma samples that were immunoassay‐positive but SRA‐negative and clinically adjudicated as not HIT (n = 3). The green circle represents a patient who was immunoassay‐positive and clinically verified as HIT but SRA‐negative (n = 1), whereas the red open circle represents a false negative (n = 1) on the procoagulant platelet assay. A positive result is defined by a procoagulant platelet percentage of greater than 7.6% as determined in Figure 4A. The assay was performed while blinded to the diagnosis of the patients. (B) Procoagulant platelet response of healthy donors to clinically adjudicated HIT+ plasma (n = 21) in the presence of 5 µM SFLLRN and 0.5 U/mL heparin, and (C) anti‐PF4/heparin antibody levels of 11 HIT+ plasma, measured on AcuStar HIT−IgG(PF4‐H) chemiluminescence immunoassay was compared between patients with and without thrombotic outcomes based on review of clinical notes while blinded to assay results. Unpaired t‐test was performed. Error bars indicate mean ± SEM. ns, not significant, *p < 0.05. Hep, heparin; HIT, heparin‐induced thrombocytopenia; SFLLRN, thrombin receptor‐activating peptide; SRA, serotonin release assay
3.5. HIT+plasma hypersensitizes healthy platelets to thrombin‐induced procoagulant platelet response in a heparin‐independent manner that is only partially dependent on FcγRIIa
A paradox in HIT is the enhancement of thrombin generation in presence of heparin. 33 Thrombosis can occur even after cessation of heparin and initiation of alternative anticoagulation including direct thrombin inhibitors. Hence, we explored the procoagulant platelet response induced by HIT plasma on donor platelets stimulated with thrombin ± ILR agonist in the absence of heparin. Heparin was excluded due to the neutralizing effect on the exogenous thrombin. Preincubation with HIT+ or HIT− plasma increased the proportion of procoagulant platelets in healthy donors following thrombin stimulation compared with no plasma (HIT−: 19.55 ± 4.16 vs 4.16 ± 0.24, p = 0.0021; HIT+: 18.86 ± 3.94 vs 4.16 ± 0.24, p = 0.0039; Figure 6A). There was no difference in the procoagulant platelet response between HIT+ and HIT− plasma. Blockade of FcγRIIa did not affect the thrombin‐induced procoagulant platelet response (Figure 6B,C). Combined thrombin+CRP‐xL stimulation increased the procoagulant platelet proportion generated by HIT+ plasma (69.57 ± 6.85) compared with no plasma (28.52 ± 2.17, p < 0.0001) and HIT− plasma (46.54 ± 3.53, p = 0.0020; Figure 6D). This hypersensitivity in donor platelets was not observed with autologous plasma (29.45 ± 5.08, Figure S1C). A partial inhibition of the procoagulant platelet response by IV.3 was evident with HIT+ (50.16 ± 6.82 vs 60.97 ± 8.04, p = 0.0161, Figure 6F) but not HIT− plasma (39.91 ± 3.95 vs 43.53 ± 4.20, p = 0.2259, Figure 6E), suggesting a partial role for FcγRIIa in the thrombin‐driven procoagulant platelet response generated by HIT plasma. Concordantly, inhibition of FcγRIIa in presence of HIT+ plasma did not return the t‐SNE subpopulation profile to the pattern seen in no plasma (Figure 6G), unlike the profiles generated by GPCR agonists (Figure 3), demonstrating a differential effect between GPCR agonists and thrombin stimulation.
FIGURE 6.
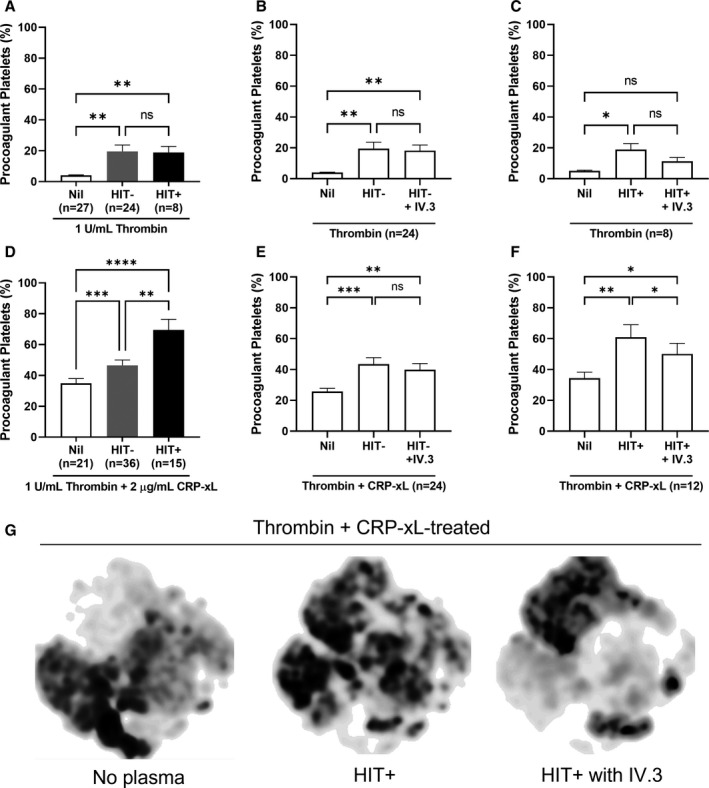
Plasma samples from SRA‐positive (HIT+) patients sensitized healthy donor platelets to form procoagulant platelets when treated with thrombin in a manner that is partially dependent on FcγRIIa. Whole blood from healthy donors were treated with plasma samples from either HIT immunoassay‐negative (HIT−) or HIT+ patients, and thrombin alone or a combination of thrombin and CRP‐xL for 10 min, then stained for flow cytometry analysis of procoagulant platelets. (A–C) The proportion of procoagulant platelets from healthy donors (n = 27) exposed to HIT− (n = 24) or HIT+ (n = 8) plasma when treated with thrombin (1 U/mL) or with the addition of FcγRIIa function‐blocking monoclonal antibody, IV.3 (10 µg/mL). (D–F) The proportion of procoagulant platelets from healthy donors (n = 21) exposed to HIT− (n = 36) or HIT+ (n = 15) plasma when treated with thrombin (1 U/mL) and CRP‐xL (2 µg/mL) or with the addition of IV.3 (10 µg/mL). (A and D) Mixed‐effects analysis was performed with Tukey correction for multiple comparisons. (B–C and E–F) Repeated measures anova was performed with Tukey correction for multiple comparisons. (G) The t‐SNE algorithm was employed to compare the platelet flow cytometry profiles between healthy donor whole blood exposed to either no plasma, HIT+, or HIT+ plasma plus IV.3 pretreatment in the presence of thrombin (1 U/mL) and CRP‐xL (2 μg/mL) in n = 6 donors demonstrating changes from baseline in presence of HIT+ plasma that was not reversed by pretreatment with monoclonal antibody IV.3. Error bars indicate mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns, not significant. CRP‐xL, crosslinked collagen‐related peptide; HIT, heparin‐induced thrombocytopenia; SEM, standard error of the mean; SRA, serotonin release assay; t‐SNE, t‐distributed stochastic neighbor embedding
4. DISCUSSION
Early and accurate detection of HIT is critical for improved patient outcomes, yet this remains a clinical challenge. An increased understanding of the mechanisms underlying HIT is helpful for improved diagnostic platforms and therapeutics. Procoagulant platelets play an important role in hemostasis by providing a surface for the assembly and propagation of coagulation factors, enabling thrombin generation and subsequent clot stabilization through fibrin formation. A well‐known paradox in HIT is that thrombin generation, demonstrable on calibrated thrombography, is enhanced instead of reduced in the presence of heparin. 33 , 34 Expanding on the work of Tutwiler and colleagues on coated platelets and HIT, 8 we confirm the marked increase in the procoagulant platelet proportion in donor platelets by HIT+ patient plasma in presence of heparin as a potential mechanism for this paradox. We demonstrate that low‐dose GPCR agonist sensitizes platelets to the mechanism of heparin‐dependent HIT plasma induction of a procoagulant platelet response in a process that is fully dependent on active FcγRIIa. Furthermore, measurement of SFLLRN‐induced procoagulant platelet formation in donor platelets under these conditions can accurately differentiate HIT from non‐HIT thrombocytopenia by detecting a heparin‐dependent, antibody‐mediated increase in procoagulant platelet proportions. These results provide important insights into the mechanisms underlying thrombosis in HIT and suggest that our flow cytometry‐based procoagulant platelet assay may be a viable strategy for a rapid yet accurate diagnosis of HIT.
Using a flow cytometry assay that identifies procoagulant platelets using combined detection of a cell death marker GSAO and platelet activation marker P‐selectin to examine the effect of SRA‐confirmed HIT+ plasma on the procoagulant profile of healthy donor platelets, we showed that HIT+ plasma‐sensitized platelets become markedly procoagulant with agonist stimulation at levels that normally cause no procoagulant platelet response. We speculate that agonist‐induced platelet PF4 release allows generation of the pathogenic complex without requirement for exogenous PF4 in this whole blood platform that avoids platelet activation from washing steps. In our hands, the increase in procoagulant platelets seen at therapeutic‐concentration heparin (0.5 U/mL) was completely abrogated when excess heparin offset the PF4:heparin (or PF4:other polyanion) molar stoichiometry required to form a stable HIT immune complex, resulting in the characteristic abrogated response. 9 , 32 , 35 , 36 Importantly, the sensitizing heparin concentration in our experiments is within the clinical therapeutic range (0.3–0.7 U/mL), 37 suggesting this heparin‐dependent sensitization plausibly contributes to the thrombotic complications seen in patients with HIT in vivo.
Using the KKO and PF4 HIT model, Tutwiler and colleagues previously speculated that the combination of PAR stimulation and ITAM signaling via FcγRIIa was required for annexin V+ “coated” platelet formation in HIT. 8 Here, the increased procoagulant platelet proportion in the presence of HIT plasma and therapeutic‐concentration heparin was seen with either SFLLRN or ADP stimulation, indicating that, although PAR signaling appears to be a potent co‐factor, stimulation of alternative GPCR pathways such as P2Y12 also induce procoagulant platelets in presence of HIT antibody‐mediated ITAM signaling. This is in agreement with previous findings that ADP and its receptor P2Y12 can induce platelet aggregation with HIT sera 38 and that P2Y12 blockade inhibits formation of pathological‐coated platelets and procoagulant platelets. 39 , 40 , 41 , 42 , 43 In contrast, co‐stimulation with CRP‐xL, an alternative ILR ligand, instead of a GPCR agonist, did not produce a synergistic procoagulant platelet response. This suggests that the marked procoagulant response in HIT is synergized by stimulation of GPCR pathways, but this is not limited to PAR‐1.
Our finding that SFLLRN priming of healthy whole blood allows measurement of heparin‐dependent FcγRIIa‐mediated procoagulant platelet response, resulted in design of a HIT diagnostic assay with high sensitivity and specificity and a ROC curve of 1.0. Considering the limitations of current diagnostic tests, a new functional assay that maintains the specificity and sensitivity of SRA without use of radioactivity is potentially useful. 28 A flow cytometry donor platelet, annexin V‐based assay for diagnosis of HIT was first proposed by Tomer and colleagues, 13 , 44 and PF4‐dependent P‐selectin expression was proposed by Padmanabhan and colleagues. 45 Functional platforms like the Multiplate aggregation and immunoassays like the chemiluminescence AcuStar HIT‐IgG(PF4‐H) offer alternatives. 28 , 29 , 31 Our assay may have some advantages over other functional assays: the use of whole blood rather than washed platelets, minimal volume of plasma required (5 μL per patient), low‐dose agonist stimulation rather than PF4, and use of a standard diagnostic flow cytometer rather than dedicated platform, increase practicality.
Importantly, this platform distinguished clinically adjudicated, SRA‐confirmed true HIT patients from hospitalized non‐HIT patients referred for HIT testing after 4Ts screening, including patients with sepsis, non‐heparin drug‐induced thrombocytopenia, consumptive thrombocytopenia, and anti‐phospholipid syndrome (Table S5). Plasma from non‐HIT thrombocytopenic patients on occasion induced a procoagulant response in donor platelets but were differentiated from HIT by the low‐ and high‐dose heparin conditions and response to IV.3. The high sensitivity and specificity in the development cohort were confirmed in the validation cohort with an overall performance of this assay achieving 98% accuracy when compared with clinical adjudication of final HIT diagnosis. Furthermore, it identified the chemiluminescence immunoassay false positives. This platform could potentially be established in any diagnostic flow cytometry laboratory, thereby shortening time to definitive diagnosis and allowing timely cessation of alternative anticoagulants in patients who do not have activating HIT antibodies. Up to 44% of patients eventually demonstrated to be HIT– experience major bleeding when commenced on alternative anticoagulation while awaiting confirmatory studies. 46 This platform has recently been adapted for diagnosis of another FcγRIIa‐mediated thrombotic condition, vaccine‐induced immune‐thrombotic thrombocytopenia and adopted for vaccine‐induced immune‐thrombotic thrombocytopenia diagnosis in Australia. 47
Ramstrom and colleagues showed that procoagulant platelet formation is limited to a subpopulation of platelets, even with strong dual agonist stimulation, thus our assay has a readout with a wide measurement range. 48 In addition, because low‐dose SFLLRN causes platelets to become activated but not procoagulant, 49 procoagulant platelet numbers in this assay are completely driven by the potency of the HIT antibody. Since higher procoagulant platelet levels in our assay were seen with plasma from HIT patients with thrombosis compared with HIT patients with thrombocytopenia alone, we speculate that the ability of HIT antibody to generate procoagulant platelets may play a pathogenic role that relates to clinical thrombotic risk. 17
This study offers mechanistic insight. Our results suggest that in HIT patients treated with therapeutic heparin, small physiological increases in agonist concentrations, plausibly caused by presence of atherosclerotic plaque, stenotic vasculature, or surgical trauma, could have a large contribution to procoagulant platelet formation and consequent risk of thrombosis. Indeed, surgical patients are three to four times more likely to develop HIT than medical patients. 50 , 51 , 52 , 53 We speculate that inflammation may likewise prime platelets for FcγRIIa signaling. For example, chemoattractant proteins released during inflammation including tumor necrosis factor‐α, interleukin‐1β, and interleukin‐6 via endothelial cells and tissue factor positive macrophages may promote endothelial‐platelet and leukocyte‐platelet interactions, and low‐level thrombin generation, therefore reducing the threshold for procoagulant platelet formation in the presence of HIT antibody. 54 In addition, PAR‐1 can be cleaved by metalloproteinases, cathepsin G, or neutrophil elastase, which are released during inflammation. It is possible that non‐thrombin GPCR priming could result in a similar increase in procoagulant platelet formation with co‐stimulation of FcγRIIa. Our experiments, performed in whole blood, do not directly investigate the contribution of platelet FcγRIIa signaling compared with signaling via leukocytes. It is therefore possible that the procoagulant platelet response observed here is augmented by leukocyte FcγRIIa signaling because platelet transactivation by monocytes is implicated in thrombotic complications in HIT. 8 Targeting FcγRIIa signaling at an early stage of HIT may be viable. 8 FcγRIIa does not play a major role in platelet adhesion; thus, a therapeutic inhibitor is less likely to cause the increased bleeding seen with non‐heparin anticoagulants. A humanized version of IV.3, VIB9600, has been developed and assessed in preclinical studies for treatment of immune‐mediated proinflammatory conditions like sepsis, 55 and appears to have an acceptable safety profile in primates.
t‐SNE is an unsupervised, nonlinear technique for visualizing cellular subpopulations clustered according to similar flow cytometry characteristics, previously used to visualize platelet activation profile differences in patients with inherited platelet disorders. 56 We used this technique to visualize the FcγRIIa blockade mediated reversal of platelet subpopulation changes induced by HIT plasma and demonstrate a clear difference between the reversibility of platelet subpopulation changes induced by PAR‐1 compared with thrombin stimulation in the presence of HIT antibody. This difference may be due to non‐PAR‐1 effects of thrombin. Upon thrombin stimulation, PAR‐1 forms heterodimers with PAR‐4, and PAR‐4 forms heterodimers with P2Y12, likely leading to enhanced cleavage of PAR‐4 and downstream AKT activation. PAR‐4 cleavage is associated with sustained intracellular calcium flux triggering procoagulant activity that cannot be dissociated from platelet activation. Thus, although single GPCR agonists are not able to generate procoagulant platelets in the absence of the HIT antibody, thrombin (including exogenous thrombin), will directly generate a proportion of procoagulant platelets that can support further thrombin formation in a feed‐forward loop. Procoagulant components in both HIT+ and HIT− plasma appear to potentiate this (Figure 6), with the HIT antibody providing an additional procoagulant driver (identified through the partial reversal of procoagulant platelets in the presence of IV.3 antibody with thrombin+CRP‐xL). We suggest that this thrombin‐driven, heparin‐independent procoagulant response (Figure 6) may be a mechanism for the known ongoing thrombotic risk after cessation of heparin when direct thrombin inhibition is not commenced. Furthermore, these data suggest that application of FcγRIIa blockade is likely to be most effective before excess thrombin generation. Investigation of the effects of inhibiting thrombin pathways, such as the PAR‐1 and PAR‐4 receptors, on procoagulant platelet response in HIT antibody‐sensitized platelets, would help to further define the mechanisms by which thrombin acts in HIT, and could give rise to further therapeutic options.
This study has limitations. A larger validation cohort of HIT+ plasmas would be useful; however, the samples used were multicenter and well‐characterized, and HIT diagnosis was independently verified by expert clinicians blinded to assay results. This study looked only at HIT plasma effect on healthy donor platelets. Future studies could directly examine procoagulant platelet responses in platelets from patients with HIT, both at time of diagnosis and upon resolution. Although we determined the contribution of FcγRIIa to procoagulant platelet response following stimulation with agonists together with HIT antibody, we did not explore the relative contribution of platelet compared with leukocyte FcγRIIa signaling, nor the impact of inhibiting downstream signaling targets, like spleen tyrosine kinase, Src family kinase, and Bruton's tyrosine kinase. A strength of the study is that plasmas used for the validation cohort were well‐characterized because they had previously been used in published comparisons of chemiluminescence, ELISA, and SRA platforms. 28 , 29
In conclusion, we expanded on current literature to show plasma from HIT patients, in presence of PAR‐1 agonist and therapeutic‐concentration heparin, markedly increased the procoagulant platelet proportions in donor platelets mediated through FcγRIIa. This mechanism may contribute to the hypercoagulability that causes the high thrombotic morbidity and mortality associated with HIT. Moreover, we showed that our flow cytometry‐based procoagulant platelet assay can accurately differentiate HIT from thrombocytopenia because of other causes, laying the foundations for a novel, rapid, yet accurate HIT diagnostic assay that can be adapted for diagnosis of other immune‐thrombotic conditions.
CONFLICT OF INTEREST
V.M.C. and L.P. hold a US patent “Selective targeting of procoagulant platelets” US15/521435. V.M.C. and C.S.M.L. hold Australian Provisional Patent Applications 2020903828, 2020903834 and 2021901983. All other authors declare no conflicts of interests.
AUTHOR CONTRIBUTIONS
Christine S. M. Lee and Maria V. Selvadurai performed experiments, analyzed data, and wrote the manuscript; Leonardo Pasalic reviewed clinical notes; James Yeung performed experiments, analyzed data, and reviewed clinical notes; Maria Konda, Geoffrey W. Kershaw, and Emmanuel J. Favaloro provided characterization and stored plasma samples; Vivien M. Chen designed and supervised the study, analyzed data and wrote the manuscript; and all authors helped revise the manuscript and approved its submission.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Heather Campbell for technical assistance, Freda Passam and Grace Wolyncewicz for reviewing clinical notes, and Diane Criminale and the staff at CRGH Blood Collection Unit for assistance in blood collection. Screening for high responders was performed at NSW Health Pathology, Royal Prince Alfred Hospital, Sydney, Australia. Open access publishing facilitated by Monash University, as part of the Wiley ‐ Monash University agreement via the Council of Australian University Librarians. Open Access publishing facilitated by The University of Sydney as a part of Wiley‐ The University of Sydney Agreement via the Council of Australian University Librarians. [Correction added on 26 May 2022, after first online publication: CAUL funding statement has been added.]
Lee CSM, Selvadurai MV, Pasalic L, et al. Measurement of procoagulant platelets provides mechanistic insight and diagnostic potential in heparin‐induced thrombocytopenia. J Thromb Haemost. 2022;20:975–988. 10.1111/jth.15650
Manuscript handled by: Matthew T. Rondina
Final decision: Matthew T. Rondina, 13 January, 2022
REFERENCES
- 1. Greinacher A, Farner B, Kroll H, Kohlmann T, Warkentin TE, Eichler P. Clinical features of heparin‐induced thrombocytopenia including risk factors for thrombosis. A retrospective analysis of 408 patients. Thromb Haemost. 2005;94:132‐135. doi: 10.1160/th04-12-0825 [DOI] [PubMed] [Google Scholar]
- 2. Martel N, Lee J, Wells PS. Risk for heparin‐induced thrombocytopenia with unfractionated and low‐molecular‐weight heparin thromboprophylaxis: a meta‐analysis. Blood. 2005;106:2710‐2715. doi: 10.1182/blood-2005-04-1546 [DOI] [PubMed] [Google Scholar]
- 3. Giddings JC, Brookes LR, Piovella F, Bloom AL. Immunohistological comparison of platelet factor 4 (PF4), fibronectin (Fn) and factor VIII related antigen (VIIIR:Ag) in human platelet granules. Br J Haematol. 1982;52:79‐88. doi: 10.1111/j.1365-2141.1982.tb03863.x [DOI] [PubMed] [Google Scholar]
- 4. Amiral J, Bridey F, Dreyfus M, et al. Platelet factor 4 complexed to heparin is the target for antibodies generated in heparin‐induced thrombocytopenia. Thromb Haemost. 1992;68:95‐96. [PubMed] [Google Scholar]
- 5. Kelton JG, Smith JW, Warkentin TE, Hayward CP, Denomme GA, Horsewood P. Immunoglobulin G from patients with heparin‐induced thrombocytopenia binds to a complex of heparin and platelet factor 4. Blood. 1994;83:3232‐3239. [PubMed] [Google Scholar]
- 6. Kelton JG, Sheridan D, Santos A, et al. Heparin‐induced thrombocytopenia: laboratory studies. Blood. 1988;72:925‐930. [PubMed] [Google Scholar]
- 7. Warkentin TE, Hayward CP, Boshkov LK, et al. Sera from patients with heparin‐induced thrombocytopenia generate platelet‐derived microparticles with procoagulant activity: an explanation for the thrombotic complications of heparin‐induced thrombocytopenia. Blood. 1994;84:3691‐3699. [PubMed] [Google Scholar]
- 8. Tutwiler V, Madeeva D, Ahn HS, et al. Platelet transactivation by monocytes promotes thrombosis in heparin‐induced thrombocytopenia. Blood. 2016;127:464‐472. doi: 10.1182/blood-2013-11-539262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suvarna S, Espinasse B, Qi R, et al. Determinants of PF4/heparin immunogenicity. Blood. 2007;110:4253‐4260. doi: 10.1182/blood-2007-08-105098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arepally GM. Heparin‐induced thrombocytopenia. Blood. 2017;129:2864‐2872. doi: 10.1182/blood-2016-11-709873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Marchetti M, Barelli S, Zermatten MG, et al. Rapid and accurate Bayesian diagnosis of heparin‐induced thrombocytopenia. Blood. 2020;135:1171‐1184. doi: 10.1182/blood.2019002845 [DOI] [PubMed] [Google Scholar]
- 12. Nellen V, Sulzer I, Barizzi G, Lämmle B, Alberio L. Rapid exclusion or confirmation of heparin‐induced thrombocytopenia: a single‐center experience with 1,291 patients. Haematologica. 2012;97:89‐97. doi: 10.3324/haematol.2011.048074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tomer A, Masalunga C, Abshire TC. Determination of heparin‐induced thrombocytopenia: a rapid flow cytometric assay for direct demonstration of antibody‐mediated platelet activation. Am J Hematol. 1999;61:53‐61. doi: 10.1002/(sici)1096-8652(199905)61:1<53:aid-ajh10>3.0.co;2-f [DOI] [PubMed] [Google Scholar]
- 14. Mordakhanova E, Nevzorova T, Synbulatova G, Rauova L, Weisel J, Litvinov R. Platelet activation in heparin‐induced thrombocytopenia is followed by platelet death via complex apoptotic and non‐apoptotic pathways. Int J Mol Sci. 2020;21:2556. doi: 10.3390/ijms21072556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Agbani EO, Poole AW. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. 2017;130:2171‐2179. doi: 10.1182/blood-2017-05-787259 [DOI] [PubMed] [Google Scholar]
- 16. Tohidi‐Esfahani I, Lee CSM, Liang HPH, Chen VMY. Procoagulant platelets: laboratory detection and clinical significance. Int J Lab Hematol. 2020;42:59‐67. [DOI] [PubMed] [Google Scholar]
- 17. Pasalic L, Wing‐Lun E, Lau JK, et al. Novel assay demonstrates that coronary artery disease patients have heightened procoagulant platelet response. J Thromb Haemost. 2018;16:1198‐1210. doi: 10.1111/jth.14008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Denorme F, Manne BK, Portier I, et al. Platelet necrosis mediates ischemic stroke outcome in mice. Blood. 2020;135:429‐440. doi: 10.1182/blood.2019002124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Prodan CI, Dale GL. Coated‐platelets in ischemic stroke ‐ potential insight into the etiology of stroke subtypes. Int J Stroke. 2008;3:249‐250. doi: 10.1111/j.1747-4949.2008.00223.x [DOI] [PubMed] [Google Scholar]
- 20. Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Increased platelet procoagulant potential predicts recurrent stroke and TIA after lacunar infarction. J Thromb Haemost. 2020;18:660‐668. doi: 10.1111/jth.14714 [DOI] [PubMed] [Google Scholar]
- 21. Kirkpatrick AC, Stoner JA, Dale GL, Rabadi M, Prodan CI. Higher coated‐platelet levels in acute stroke are associated with lower cognitive scores at three months post infarction. J Stroke Cerebrovasc Dis. 2019;28:2398‐2406. doi: 10.1016/j.jstrokecerebrovasdis.2019.06.033 [DOI] [PubMed] [Google Scholar]
- 22. Batar P, Dale GL. Simultaneous engagement of thrombin and Fc gamma RIIA receptors results in platelets expressing high levels of procoagulant proteins. J Lab Clin Med. 2001;138:393‐402. doi: 10.1067/mlc.2001.120049 [DOI] [PubMed] [Google Scholar]
- 23. Fernández DI, Kuijpers MJE, Heemskerk JWM. Platelet calcium signaling by G‐protein coupled and ITAM‐linked receptors regulating anoctamin‐6 and procoagulant activity. Platelets. 2021;32(7):863‐871. doi: 10.1080/09537104.2020.1859103 [DOI] [PubMed] [Google Scholar]
- 24. Hua VM, Abeynaike L, Glaros E, et al. Necrotic platelets provide a procoagulant surface during thrombosis. Blood. 2015;126:2852‐2862. doi: 10.1182/blood-2015-08-663005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dick LJ, Gray A, Ram A, et al. Elimination of the antimicrobial action of the organoarsenical cancer therapeutic, 4‐(N‐(S‐glutathionylacetyl)amino) phenylarsonous acid, before finished product sterility testing. J Pharm Pharmacol. 2013;65:1664‐1669. doi: 10.1111/jphp.12143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tan CW, Bourcy M, Pasalic L, Chen VM. Flow cytometry assessment of procoagulant platelets using a dithiol‐reactive probe. Methods Mol Biol. 2019;1967:305‐321. doi: 10.1007/978-1-4939-9187-7_20 [DOI] [PubMed] [Google Scholar]
- 27. Lo GK, Juhl D, Warkentin TE, Sigouin CS, Eichler P, Greinacher A. Evaluation of pretest clinical score (4 T's) for the diagnosis of heparin‐induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4:759‐765. doi: 10.1111/j.1538-7836.2006.01787.x [DOI] [PubMed] [Google Scholar]
- 28. Favaloro EJ, McCaughan G, Mohammed S, et al. HIT or miss? A comprehensive contemporary investigation of laboratory tests for heparin induced thrombocytopenia. Pathology. 2018;50:426‐436. doi: 10.1016/j.pathol.2017.11.089 [DOI] [PubMed] [Google Scholar]
- 29. Favaloro EJ, Mohammed S, Donikian D, et al. A multicentre assessment of contemporary laboratory assays for heparin induced thrombocytopenia. Pathology. 2021;53:247‐256. doi: 10.1016/j.pathol.2020.07.012 [DOI] [PubMed] [Google Scholar]
- 30. Worthington RE, Carroll RC, Boucheix C. Platelet activation by CD9 monoclonal antibodies is mediated by the Fc gamma II receptor. Br J Haematol. 1990;74:216‐222. doi: 10.1111/j.1365-2141.1990.tb02568.x [DOI] [PubMed] [Google Scholar]
- 31. Morel‐Kopp M‐C, Mullier F, Gkalea V, Bakchoul T, Minet V, Elalamy I. Ward CM, immunology tsop. Heparin‐induced multi‐electrode aggregometry method for heparin‐induced thrombocytopenia testing: communication from the SSC of the ISTH. J Thromb Haemost. 2016;14:2548‐2552. doi: 10.1111/jth.13516 [DOI] [PubMed] [Google Scholar]
- 32. Rauova L, Poncz M, McKenzie SE, et al. Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin‐induced thrombocytopenia. Blood. 2005;105:131‐138. doi: 10.1182/blood-2004-04-1544 [DOI] [PubMed] [Google Scholar]
- 33. Tardy‐Poncet B, Piot M, Chapelle C, et al. Thrombin generation and heparin‐induced thrombocytopenia. J Thromb Haemost. 2009;7:1474‐1481. doi: 10.1111/j.1538-7836.2009.03514.x [DOI] [PubMed] [Google Scholar]
- 34. Chilver‐Stainer L, Lämmle B, Alberio L. Titre of anti‐heparin/PF4‐antibodies and extent of in vivo activation of the coagulation and fibrinolytic systems. Thromb Haemost. 2004;91:276‐282. doi: 10.1160/th03-07-0454 [DOI] [PubMed] [Google Scholar]
- 35. Cines DB, Yarovoi SV, Zaitsev SV, et al. Polyphosphate/platelet factor 4 complexes can mediate heparin‐independent platelet activation in heparin‐induced thrombocytopenia. Blood Adv. 2016;1:62‐74. doi: 10.1182/bloodadvances.2016000877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cai Z, Zhu Z, Greene MI, Cines DB. Atomic features of an autoantigen in heparin‐induced thrombocytopenia (HIT). Autoimmun Rev. 2016;15:752‐755. doi: 10.1016/j.autrev.2016.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Baluwala I, Favaloro EJ, Pasalic L. Therapeutic monitoring of unfractionated heparin ‐ trials and tribulations. Expert Rev Hematol. 2017;10:595‐605. doi: 10.1080/17474086.2017.1345306 [DOI] [PubMed] [Google Scholar]
- 38. Polgár J, Eichler P, Greinacher A, Clemetson KJ. Adenosine diphosphate (ADP) and ADP receptor play a major role in platelet activation/aggregation induced by sera from heparin‐induced thrombocytopenia patients. Blood. 1998;91:549‐554. [PubMed] [Google Scholar]
- 39. Norgard NB, Saya S, Hann CL, Hennebry TA, Schechter E, Dale GL. Clopidogrel attenuates coated‐platelet production in patients undergoing elective coronary catheterization. J Cardiovasc Pharmacol. 2008;52:536‐539. doi: 10.1097/FJC.0b013e3181907390 [DOI] [PubMed] [Google Scholar]
- 40. Norgard NB, Hann CL, Dale GL. Cangrelor attenuates coated‐platelet formation. Clin Appl Thromb Hemost. 2009;15:177‐182. doi: 10.1177/1076029608321437 [DOI] [PubMed] [Google Scholar]
- 41. Kirkpatrick AC, Vincent AS, Dale GL, Prodan CI. Clopidogrel use and smoking cessation result in lower coated‐platelet levels after stroke. Platelets. 2020;31:236‐241. doi: 10.1080/09537104.2019.1609661 [DOI] [PubMed] [Google Scholar]
- 42. Judge HM, Buckland RJ, Sugidachi A, Jakubowski JA, Storey RF. The active metabolite of prasugrel effectively blocks the platelet P2Y12 receptor and inhibits procoagulant and pro‐inflammatory platelet responses. Platelets. 2008;19:125‐133. doi: 10.1080/09537100701694144 [DOI] [PubMed] [Google Scholar]
- 43. Kotova YN, Ataullakhanov FI, Panteleev MA. Formation of coated platelets is regulated by the dense granule secretion of adenosine 5'diphosphate acting via the P2Y12 receptor. J Thromb Haemost. 2008;6:1603‐1605. doi: 10.1111/j.1538-7836.2008.03052.x [DOI] [PubMed] [Google Scholar]
- 44. Tomer A. A sensitive and specific functional flow cytometric assay for the diagnosis of heparin‐induced thrombocytopenia. Br J Haematol. 1997;98:648‐656. doi: 10.1046/j.1365-2141.1997.2613077.x [DOI] [PubMed] [Google Scholar]
- 45. Padmanabhan A, Jones CG, Bougie DW, et al. A modified PF4‐dependent, CD62p expression assay selectively detects serotonin‐releasing antibodies in patients suspected of HIT. Thromb Haemost. 2015;114:1322‐1323. doi: 10.1160/th15-02-0175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pishko AM, Lefler DS, Gimotty P, et al. The risk of major bleeding in patients with suspected heparin‐induced thrombocytopenia. J Thromb Haemost. 2019;17:1956‐1965. doi: 10.1111/jth.14587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lee CSM, Dey A, Campbell H, et al. Flow cytometric detection of procoagulant properties of plasma from patients with clinically confirmed vaccine‐induced immune thrombotic thrombocytopenia. Blood. 2021;138:3211. doi: 10.1182/blood-2021-148261 [DOI] [Google Scholar]
- 48. Södergren AL, Ramström S. Platelet subpopulations remain despite strong dual agonist stimulation and can be characterised using a novel six‐colour flow cytometry protocol. Sci Rep. 2018;8:1441. doi: 10.1038/s41598-017-19126-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Abbasian N, Millington‐Burgess SL, Chabra S, Malcor JD, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv. 2020;4:154‐164. doi: 10.1182/bloodadvances.2019000182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Linkins LA, Warkentin TE. Heparin‐induced thrombocytopenia: real‐world issues. Semin Thromb Hemost. 2011;37:653‐663. doi: 10.1055/s-0031-1291375 [DOI] [PubMed] [Google Scholar]
- 51. Smythe MA, Koerber JM, Mattson JC. The incidence of recognized heparin‐induced thrombocytopenia in a large, tertiary care teaching hospital. Chest. 2007;131:1644‐1649. doi: 10.1378/chest.06-2109 [DOI] [PubMed] [Google Scholar]
- 52. Warkentin TE, Eikelboom JW. Who is (still) getting HIT? Chest. 2007;131:1620‐1622. doi: 10.1378/chest.07-0425 [DOI] [PubMed] [Google Scholar]
- 53. Cuker A, Arepally GM, Chong BH, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin‐induced thrombocytopenia. Blood Adv. 2018;2:3360‐3392. doi: 10.1182/bloodadvances.2018024489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wagner DD, Burger PC. Platelets in inflammation and thrombosis. Arterioscler Thromb Vasc Biol. 2003;23:2131‐2137. doi: 10.1161/01.ATV.0000095974.95122.EC [DOI] [PubMed] [Google Scholar]
- 55. Chen B, Vousden KA, Naiman B, et al. Humanised effector‐null FcgammaRIIA antibody inhibits immune complex‐mediated proinflammatory responses. Ann Rheum Dis. 2019;78:228‐237. doi: 10.1136/annrheumdis-2018-213523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Blair TA, Michelson AD, Frelinger AL 3rd. Mass cytometry reveals distinct platelet subtypes in healthy subjects and novel alterations in surface glycoproteins in Glanzmann thrombasthenia. Sci Rep. 2018;8:10300. doi: 10.1038/s41598-018-28211-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material