

Abstract
8‐Nitrobenzothiazinones (BTZs) are a promising class of antimycobacterial agents currently under investigation in clinical trials. Starting from thiourea derivatives, a new synthetic pathway to BTZs was established. It allows the formation of the thiazinone ring system in one synthetic step and is applicable for preparation of a wide variety of BTZ analogues. The synthetic procedure furthermore facilitates the replacement of the sulphur atom in the thiazinone ring system by oxygen or nitrogen to afford the analogous benzoxazinone and quinazolinone systems. 36 BTZ analogues were prepared and tested in luminescence‐based assays for in vitro activity against Mycobacterium tuberculosis (Mtb) using the microdilution broth method and a high‐throughput macrophage infection assay.
Keywords: Mycobacterium tuberculosis, benzothiazinone, DprE1, SAR, intracellular activity
We report the synthesis of antitubercular 8‐nitrobenzothiazinones and their antimycobacterial characterization. A synthesis using thiourea intermediates was established, and 35 analogues were prepared. Their antimycobacterial potential was investigated against Mycobacterium tuberculosis: minimal inhibitory concentrations (MICs) of all derivatives were determined in broth and in a macrophage infection model. Compounds with MICs of <50 nM could be described and characterized.
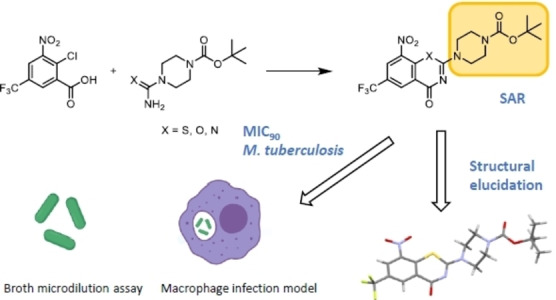
Introduction
Tuberculosis (TB) remains the most prevalent infectious disease worldwide caused by a bacterium. In 2020, 9.9 million patients developed TB and 1.5 million deaths were reported by WHO. [1] Shorter treatment duration and better therapeutic outcome against multi‐ and extensively‐drug resistant strains of Mycobacterium tuberculosis (Mtb) are important goals of TB drug development. [2] The investigation of new classes of active substances acting on targets that have not been exploited so far is an important approach to combat TB. [3]
8‐Nitrobenzothiazinones (BTZs) have proven to be a highly effective antimycobacterial class of substances [4] that inhibit the growth of Mtb even in the nanomolar concentration range by interference with arabinan biosynthesis. BTZs act as mechanism‐based inhibitors of the enzyme decaprenylphosphoryl‐β‐D‐ribofuranose‐2′‐epimerase (DprE1), which is located in the periplasmic space of Mtb. [5] DprE1 catalyses the oxidation of decaprenylphosphoryl‐β‐D‐ribofuranose (DPR) to the keto‐intermediate decaprenylphosphoryl‐2‐keto‐D‐erythropentofuranose (DPX) in the synthesis of decaprenylphosphoryl‐β‐D‐arabinose (DPA), a crucial step in the biosynthesis of arabinans. [6] Due to their high in vitro efficacy and in vivo activity, BTZs are promising drug candidates for future TB therapy. A wide variety of BTZ derivatives were synthesized and investigated in recent years,[ 4a , 4b , 7 ] but none of these surpassed the in vitro efficacy of BTZ043 and macozinone (PBTZ169) (Figure 1).
Figure 1.

Chemical diagrams of BTZ043 [4b] and macozinone[4a], currently investigated in clinical trials.
Several synthetic routes to BTZs are described in the literature (Scheme 1).[ 7a , 8 ] All start from substituted 2‐chlorobenzoic acid derivatives. In the acylisothiocyanate pathway (A), 2‐chlorobenzoyl chlorides are treated with potassium‐, sodium‐, or ammonium thiocyanate to form the intermediate acylisothiocyanates, which are after 120 minutes treated with the corresponding secondary amines to form a thiourea intermediate that undergoes ring closure by nucleophilic substitution. This pathway was initially used for the synthesis of BTZ043,[ 8b , 8c ] which was adapted from previous procedures, [9] and then further modified by Gao et al 2013 [7a] and Peng et al. 2015. [7b] In the two references mentioned, the procedures for BTZ synthesis were adapted by using phase transfer catalysis (PEG‐400) for preparation of acylisothiocyanates and changing the reaction times. As alternatives, the dithiocarbamate pathway (B) and the alkylxanthogenate (C) pathway were developed.[ 8b , 8c ] A drawback of both pathways is the use of toxic and highly flammable carbon disulfide in the synthesis of the dithiocarbamate and alkylxanthogenate reagents. The alkylsulfanyl pathway (D) [8a] can easily be adapted for combinatorial chemistry purposes since the amine moiety at position 2 is added to a stable 2‐(alkylsulfanyl)‐4H‐1,3‐benzothiazin‐4‐one intermediate. This method, however, requires carbon disulfide and carcinogenic methyl iodide. We developed a new synthetic pathway, [10] which avoids toxic reagents and intermediates with regard to Good Manufacturing Practices for pharmaceuticals [11] and green chemistry principles, [12] and prepared a panel of new BTZs and derivatives with a broad range of activity and evaluated them against Mtb.
Scheme 1.

Overview of synthetic procedures for BTZ preparation. From top to bottom: acylisothiocyanate pathway (A), dithiocarbamate pathway (B), alkylxanthogenate pathway (C) and (D) alkylsulfanyl pathway.
Despite the intensive research on DprE1 inhibitors during the last years, surprisingly few data comparing the intracellular activity of a panel of BTZs were published. Intracellular models are known to better mimic the environment of Mtb during the natural course of the disease as they include factors such as compound (in)activation, membrane permeability, removal by efflux pumps, and cytotoxicity to mammalian cells. [13] Therefore, we have studied activities of all synthesized compounds in bacterial growth medium and in a high content macrophage infection model.
Results and Discussion
A new synthetic pathway to BTZs
In this study, we describe an efficient synthesis for the preparation of BTZs using thiourea intermediates to form the thiazinone ring system in one step, which is shown in Scheme 2.
Scheme 2.

Preparation of BTZs via the thiourea pathway: (i) toluene, SOCl2, 110 °C; (ii) toluene, N,N‐dialkylthiourea, 110 °C.
The synthesis starts with 2‐chloro‐3‐nitro‐5‐(trifluoromethyl)benzoic acid, [14] activated with thionyl chloride to form the acid chloride which subsequently reacted with an N,N‐dialkyl thiourea derivative to yield the thiazinone ring system. With the thiourea reagent, it is possible for the first time to introduce the nitrogen and sulphur atoms within one synthetic step and arrive at the thiazinone ring system. Compared to the alkylsulfanyl pathway D (Scheme 1), the most recent BTZ synthesis, fewer synthetic steps are needed and toxic reagents such as carbon disulfide and methyl iodide are avoided. The alkylsulfanyl pathway also starts with the activation of the carboxylic acid, but the carboxylic acid chloride is first converted to the benzamide and is then reacted with carbon disulfide and methyl iodide to the alkylsulfanyl intermediate. The reaction of 2‐chlorobenzoic acid chloride with a thiourea reproducibly gives very high yields. For some of the BTZ derivatives described here, e. g. 3 c and 11 c, overall yields of 65–75 % were achieved, which was significantly higher than the literature data for the alkylsulfanyl pathway (30–40 %). [8a] The thiourea pathway was used to prepare a panel of 31 BTZs (Figure 2). Macozinone and BTZ043 were also synthesized via the thiourea route as a proof of concept.
Figure 2.

BTZ derivatives with secondary amine derived substituents in position 2 of the BTZ scaffold.
Synthesis of the intermediate thiourea derivatives
The crucial intermediates in the thiourea pathway to BTZs, viz. the N,N‐dialkylthioureas, were accessed by three different procedures. Thioureas were prepared from secondary amines via benzoyl isothiocyanate [15] (Scheme 3, a). In the first step of the reaction, benzoyl chloride is reacted with sodium isothiocyanate as shown in Scheme 3 (a). The reactive isothiocyanate formed immediately reacts with the corresponding secondary amine to yield the N‐benzoyl thiourea. After hydrolysis the actual thiourea is obtained. As this process needs highly reactive and toxic acyl isothiocyanates, it does not constitute a substantial improvement of the known BTZ syntheses.
Scheme 3.

Preparation of N,N‐dialkylthioureas as key intermediates of BTZ synthesis (Synthesis from benzoyl isothiocyanates (a), Synthesis under use of thiocarbonyldiimidazole (b) or trimethylsilyl isothiocyanate (c): (i) NaSCN, acetone; (ii) secondary amine, acetone (iii) 36 % HCl, 95 °C; (iv) secondary amine, THF; (v) isopropanol/water (9 : 1), reflux; (vi) secondary amine, THF; (vii) 2 N ammonia in MeOH, THF.
Trimethylsilyl isothiocyanate [16] can also serve as a reagent for the production of thioureas(Scheme 3, b). In the first reaction step, the secondary amine is reacted with trimethylsilyl isothiocyanate in anhydrous THF. This leads to the nucleophilic addition of the amino nitrogen to the CN double bond of the isothiocyanate. The N‐trimethylsilylthiourea can be converted into the corresponding thiourea by brief heating in the presence of water. This process also needs a reactive isothiocyanate.
From thiocarbonyldiimidazole, thioureas can be prepared in an efficient two‐step synthesis [17] (Scheme 3, c). Thiocarbonyldiimidazole is reacted with the secondary amine in THF, followed by a second substitution reaction with ammonia. The intermediate product formed in the first substitution is used without further purification. This robust procedure tolerates a variety of different secondary amines, even if they contain other functional groups such as tertiary nitrogen atoms, hydroxy or methoxy groups. For comparison, 4‐morpholinecarbothioamide was synthesized using the three methods mentioned above. The highest yield of 68 % was achieved using thiocarbonyldiimidazole, whereas methods a and b yielded 16 % and 41 %, respectively. Since the yields are optimised and the mild reaction conditions allow the application to a variety of secondary amines, procedure c obviously is the method of choice for the preparation of N,N‐dialkylthioureas. The use of thiocarbonyldiimidazole is limited only by substituents in close proximity to the nitrogen atom of the secondary amine. It is likely that steric hindrance prevents the substitution reaction. For example, the preparation of thiourea from 2,6‐dimethyl‐piperidine is not possible by method c.
Features of the BTZs in the panel
Two crucial sites
BTZ system substituents, most prominently the 8‐nitro group [18] known to be essential for antimycobacterial activity were assessed in this study. [18] As previous studies[ 4a , 7b , 19 ] clearly show that the side chain at position 2 of the BTZ ring system influences the efficacy of the compounds, we initially focussed on modifications of this site. Secondary amines prepared as substituents in position 2 included piperazine amides and sulfonamides, tetrahydroisoquinolines, diazabicyclononanes and piperidines. In addition, we exchanged the bivalent sulfur atom for oxygen and nitrogen (benzoxazinones BOZs, quinazolinones QZs) as discussed below.
Lipophilic to hydrophilic
Different carboxylic acids provided access to piperazine amides 1 c–9 a with straight and branched alkyl chains as well as amino acids and the drug substance ibuprofen. Thus, both lipophilic and polar BTZ derivatives were prepared to assess their influence on antimycobacterial activity. Two sulfonic acid amides (10 a and 11 c) further diversified the panel of 2‐substituents. The spatial extent of this side chain was varied in the piperidine derivatives 12 b–18 b. Additional hydroxyl groups, sulfoxides and sulfones yielded BTZ derivatives of different polarity and spatial extension. Compound 19 b has a larger piperidine‐derived side chain that is also present in the approved TB drug delamanid. [20]
Partially aromatic vs. aliphatic
The tetrahydroisoquinoline system – compounds 20 b–23 b – represents the effect of partially planar aromatic moieties in the 2‐position on antimycobacterial activity as compared to the BTZs with piperidine and piperazine and open chain substituents in this position.
Basic vs. less basic
A panel (24 b–27 b) of test compounds with a secondary or tertiary nitrogen atom in the side chain was synthesized, following the lead compound macozinone, but with varying basicity of the respective nitrogen atoms. Heterocycles introduced included the diazabicyclononane moiety (compounds 26 b–27 b) and a diazabicyclooctane (25 b).
Activity assays
The BTZs prepared were expected to cover a range of MICs from nanomolar (e. g. BTZ043, macozinone) to micromolar. The expectation is based on literature data[ 4a , 7 , 21 ] in which structure variations were made on BTZs and a large influence of the substituent at position 2 was shown: Karoli et al. [7c] used different primary and secondary amines for the preparation of BTZ analogues by the alkylsulfanyl pathway. Some of the analogues prepared in this study had similar efficacy compared to BTZ043. Gao et al. [7a] and Peng et al. [7b] used the acylisothiocyanate pathway to prepare BTZ analogues with different substituents at position 2, using piperazines and spiropiperidine derivatives. In a recent study by Zhang et al., [21] mainly spiro cyclic side chains with another tertiary nitrogen were investigated. The compounds synthesized via the alkylxanthoxygenate pathway do not reach the efficacy of the reference compound macozinone, but show improved solubility. The studies mentioned investigate the inhibitory effect of BTZ analogues only in broth and not in a macrophage infection model. The panel of test compounds prepared for this study were synthesized via the new thiourea route described above, demonstrating the suitability of the synthesis method for a variety of different derivatives. A large variety of substituents were introduced into BTZ scaffold to discover the structure‐activity relationships (in vitro/intracellular) and to find new derivatives with nanomolar activity. For compound 14 a, the required thiourea precursor was obtained in low yield by the new thiourea route using thiocarbonyldiimidazole due to steric hindrance; consequently, the traditional N‐benzoyl isocyanate synthetic pathway was employed (Scheme 1, method A).
The thiourea pathway to BTZs was modified to exchange the sulphur atom with oxygen or nitrogen. For this purpose, the carboxylic acid chloride was treated with ureas or guanidines instead of the thiourea as shown in Scheme 4. The corresponding BTZ analogues – BOZs [22] and QZs – were isolated from the reaction mixture, but in lower yields than the corresponding BTZs. For the synthesis of the BOZs, a base, in this case diisopropylethylamine (DIPEA), had to be added, and it proved advantageous to carry out the reaction under argon. The QZ 32 a was only formed when a stronger base was added; however, even with 1,8‐diazabicyclo[5.4.0]undec‐7‐ene (DBU) the yield of isolated product was 8 %.
Scheme 4.

Synthesis of BOZs and QZs by the adapted thiourea pathway: (i) toluene, SOCl2, 110 °C; (ii) Synthesis of BOZs toluene, N,N‐dialkylurea, DIPEA, 110 °C (iii); Synthesis of QZs toluene, N,N‐dialkylguanidine, DBU, 110 °C.
Structural characterisation of BTZs and analogues
Figure 3 below shows the 1H NMR spectra for analogous BTZs, BOZs and QZs, with all three compounds having the tetrahydroisoquinoline side chain.
Figure 3.

1H NMR data (400 MHz, CDCl3, S denotes the residual solvent signal) of 23 b, 31 b and 32 a.
It is noticeable that the aromatic protons of the BTZ 23 b exhibit the largest downfield shift, whereas these protons in the BOZ and QZ are shielded. While there are no differences in the 1H NMR spectrum of the aromatic ring system of tetrahydroisoquinoline, the CH2 groups of this part of the molecule show different characteristics. In the spectrum of the BTZ 23 b, these protons show signal broadening, which indicates a hindered rotation at the C−N bond. The BOZ and QZ analogues show significantly sharper signals for the CH2 groups in question, whereby a signal broadening is also recognisable for the BOZ 31 b.
Compound 1 c was structurally characterized also by X‐ray crystallography. [23] Figure 4 depicts the molecular structure in the crystal. The X‐ray analysis confirmed the structure and ruled out the formation of the 4H‐3,1‐benzothiazin‐4‐one structural isomer. [24] In the crystal, 1 c exhibits a virtually planar BTZ scaffold, as previously encountered in macozinone [25] and other BTZs[ 18b , 26 ], with the nitro group being nearly coplanar. As expected, the piperazine ring adopts a low energy chair conformation. The bond angles in the piperazine ring show some minor deviations from ideal tetrahedral values due to the planar structure at the two nitrogen atoms. It is worth noting that the opposite enantiomeric conformer of the molecular structure depicted in Figure 4 is also present in the centrosymmetric crystal structure of 1 c.
Figure 4.
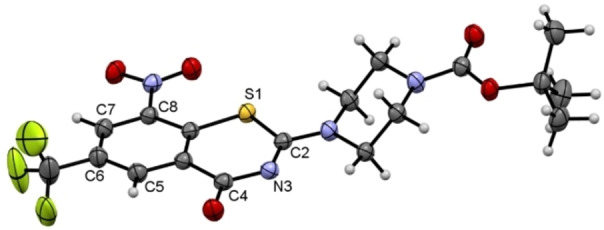
Molecular structure of 1 c in the crystal. Displacement ellipsoids are shown at the 50 % probability level. Rotational disorder of the trifluoromethyl group is omitted for clarity.
Activity profiling of BTZs, BOZs and QZs against Mtb H37Rv
BTZs and analogues prepared for this study were evaluated for their efficacy against Mtb. For the first time, a panel of numerous BTZ derivatives was investigated in a luciferase‐based macrophage infection assay. This method was developed for high‐throughput purposes [27] and used here to determine the minimum inhibitory concentrations (MIC) resulting in greater than 90 % growth inhibition in liquid culture employing supplemented 7H9 medium as well an intracellular THP‐1 macrophage infection model.
All test compounds were evaluated in vitro in a micro broth dilution assay against Mtb H37Rv and the MIC90 was determined. For this in vitro assay, a luciferase‐expressing Mtb H37Rv strain was used and mycobacterial growth was quantified using luminescence data.[ 27b , 28 ] To validate the assay, a z score was determined based on positive and negative controls. [29] The reference compounds, BTZ043 and macozinone, showed MICs of 3 and 2 nM respectively, in agreement with literature values.[ 4a , 4b ]
The BTZ 2‐substituent determines whether nanomolar or micromolar MICs are achieved
A very wide range of MICs was observed for the BTZs prepared in this study as shown in Table 1 and Figure 5A. The most active compound 1 e reached a MIC90 of 6 nM, whereas 7 c had a significantly reduced activity of 14 μM. The strong influence of the secondary amine in position 2 of the BTZ scaffold was striking. In the BTZs investigated, this substituent shifted the MIC by a factor of up to 10,000 and is thus of decisive importance for the extraordinary efficacy of particular BTZs. Comparing the BTZ macozinone and 33 b, a benzoxazinone (BOZ) with the same 2‐substituent, the strong contribution of the 2‐substituent to activity is again highlighted: 33 b has an MIC of “only” 1 μM. However, another BOZ 31 b with a tetrahydroisoquinoline side chain, analogous to BTZ 23 b, was inactive. BTZs, BOZs and QZs are discussed in comparison below.
Table 1.
Activity determination and cytotoxicity of BTZs and analogues.
|
ID |
7H9 MIC90 [μM][a] |
intracellular MIC90 [μM][b] |
cytotoxicity [μM][c] |
ID |
7H9 MIC90 [μM][a] |
intracellular MIC90 [μM][b] |
cytotoxicity [μM][c] |
|---|---|---|---|---|---|---|---|
|
1 c |
0.2 |
0.2 |
21.8 |
17 b |
1.3 |
1.3 |
28.8 |
|
1 e |
0.006 |
0.1 |
20.6 |
18 b |
5.0 |
27.7 |
2.5 |
|
2 a |
0.3 |
0.7 |
16.0 |
19 b |
0.05 |
0.4 |
4.7 |
|
3 c |
0.05 |
0.2 |
20.6 |
20 b |
0.01 |
0.2 |
2.5 |
|
4 c |
0.02 |
0.2 |
20.6 |
21 b |
0.2 |
0.9 |
>10 |
|
5 d |
3.8 |
3.8 |
21.1 |
22 b |
1.0 |
3.9 |
>21.4 |
|
6 d |
6.9 |
13.7 |
>18.3 |
23 b |
0.5 |
1.1 |
>24.5 |
|
7 c |
14.1 |
28.1 |
>18.7 |
24 b |
4.1 |
0.5 |
5.7 |
|
5 c |
1.6 |
1.6 |
>17.4 |
25 b |
0.2 |
0.9 |
2.5 |
|
8 c |
0.8 |
1.6 |
9.1 |
26 b |
1.8 |
7.5 |
>20.0 |
|
9 a |
1.6 |
0.8 |
9.1 |
26 c |
4.5 |
4.5 |
>25.0 |
|
10 c |
0.2 |
0.9 |
>19.4 |
26 d |
0.9 |
3.7 |
>20.4 |
|
11 c |
0.01 |
0.2 |
>19.7 |
27 b |
1.8 |
3.7 |
>20.4 |
|
12 b |
1.3 |
1.3 |
29.0 |
30 b |
>43.4 |
>43.4 |
>29.0 |
|
13 b |
1.3 |
2.5 |
27.8 |
31 b |
>38.3 |
19.2 |
3.2 |
|
14 a |
0.3 |
0.5 |
25.8 |
32 a |
>38.4 |
>38.4 |
25.6 |
|
15 b |
1.2 |
2.4 |
13.3 |
33 b |
1.0 |
0.2 |
22.7 |
|
16 c |
4.6 |
38.1 |
25.4 |
macozinone |
0.002 |
0.027 |
21.9 |
|
16 d |
1.1 |
2.2 |
24.4 |
BTZ043 |
0.003 |
0.014 |
23.2 |
[a] Mtb H37Rv in 7H9+OADC; [b] Mtb H37Rv during macrophage infection; [c] MTT assay cytotoxicity against THP‐1 cells.
Figure 5.

Visualization of extracellular and intracellular MICs of BTZ analogues against Mtb H37Rv (red triangle: macozinone, blue circle: BTZ043). A MICs of BTZ analogues in 7H9+OADC, B MICs of BTZ analogues in macrophage infection assay, C Quotient of extracellular and intracellular MIC (green squares: compounds with an increased intracellular activity 9 a, 24 b and 33 b).
The 2‐substituent should be rather lipophilic or weakly basic
The introduction of an N‐acyl or sulfonyl piperazine side chain also yielded derivatives with antimycobacterial activity stronger than for reported N‐acyl and N‐sulfonyl BTZs. [7b] Of note, 1 e, 4 c and 11 c all had MICs in the lower nanomolar range, with 1 e showing comparable activity to BTZ043 or macozinone with a MIC of 6 nM. Therefore, lipophilic alkyl chains with 5–7 carbon atoms on a piperazine moiety seem to be particularly suitable for profiling the efficacy of BTZ. This might have to do with the ability of a BTZ to penetrate into the outer mycobacterial membrane to where DprE1 is located, [5] but this was not investigated in this study. There is certainly an optimum because neither longer lipophilic side chains as in compound 2 a nor the introduction of heteroatoms to increase polarity are beneficial for efficacy, as reflected by significant increases in MICs to >1 μM. Secondary amines derived from piperidine only reached MICs >1 μM, with 14 a representing the optimum in this small series at 0.3 μM. It would appear that a lipophilic side chain is necessary for nanomolar MICs. In compound 24 b, the second piperidine moiety could fulfil this requirement, but the more basic second nitrogen atom counteracts the gain in lipophilicity in the side chain by being basic enough to be protonated at physiological pH. The conclusions are supported by the observation that 19 b with a lipophilic non‐basic substituent showed strong inhibition of Mtb growth (MIC: 50 nM).
Aliphatic, not aromatic far end of 2‐substituent for low nanomolarf MICs
BTZs with tetrahydroisoquinoline substituents – 20 b–23 b – show distinct activity against Mtb without achieving the outstanding inhibition values of the lead compounds. In particular, compound 20 b with a 6‐methoxy‐1,2,3,4‐tetrahydroisoquinoline substituent stands out for its in vitro MIC of 10 nM. Comparison with its isomer 21 b (7‐methoxy, MIC 0.2 μM), 22 b (6,7‐dimethoxy, MIC 1.0 μM) and with 23 b (no methoxy substituent, MIC 0.5 μM) proves that subtle differences of the positioning of just a methoxy group at the “far end” of the 2‐substituent can cause 100‐fold MIC differences. They cannot presently be accounted for drug/target interaction since firstly in vitro MICs are controlled by a number of factors, and secondly, DprE1 structures cocrystallized with BTZ display the domain next to this part of BTZs to be flexible, leading to low resolution of this part of the enzyme.[ 4a , 6c , 30 ]
Annulated aliphatic aza heterocycles in the 2‐position do not lead to nanomolar MICs
A series of test compounds with a diazabicyclooctane 25 b and diazabicyclononane 26 b, 26 c, 26 d and 27 b were evaluated for their antimycobacterial activity. Compound 25 b with the 3‐benzyl‐3,7‐diazabicyclo[3.3.0]octane side chain and a MIC of 0.2 μM proved to be the most effective here, while the other derivatives showed a significant increase in MIC compared to the lead structures.
Benzoxazinones and quinazolinones have higher MICs than benzothiazinones
Three benzoxazinones (BOZs) were prepared, with only 33 b showing efficacy against Mtb. This compound contains the methylcyclohexylpiperazine side chain of macozinone, but has a MIC of 1 μM only, corresponding to a 500× reduction of activity on exchanging sulfur for oxygen. The QZ 32 a bears the same tetrahydroisoquinoline side chain as BTZ 23 b and BOZ 31 b. While 23 b had an MICs of 0.5 μM, both 31 b and 32 a were inactive (MIC >38 μM). As shown in a previous study, [30] BOZs show reduced affinity for the target DprE1 compared to their BTZ counterparts. Conceivably, the redox potential of the nitroaromatic is influenced or BOZs have a lower affinity for initial binding to the target.
Evaluation of antimycobacterial activity in a macrophage infection model
The infection of alveolar macrophages is crucial for the pathogenesis of tuberculosis. Therefore, antitubercular agents that are effective against intracellular pathogens are highly desirable. We mimicked the process of macrophage infection in an intracellular infection assay, [27b] enabling the investigation of the efficacy of the BTZs and to determine the intracellular MICs of all compounds described in this study. Mtb H37Rv expressing the firefly luciferase gene from the hsp60 promoter was used as described before. [31] For the infection experiment, THP‐1 cells are incubated with Mtb H37Rv (MOI 10 : 1) and differentiated by phorbol‐12‐myristate‐13‐acetate (PMA). After three days of incubation, the assay medium was removed and the luciferase reagent added. Luminescence measurement can now be used to quantify mycobacterial growth, allowing the investigation of the intracellular growth of Mtb without time‐consuming colony counting. The luciferase reaction was also used to determine the MICs in broth discussed above, which ensures comparability of the results.
BTZs with slightly higher or retained MICs in the extra‐ and intracellular assay
As shown in Figure 5B, a wide range of intracellular MICs was found for the set of BTZs and analogues selected for this study. At 14 and 27 nM, the MICs of BTZ043 and macozinone were only slightly higher than in the extracellular assay. BTZ 1 c also retained the efficacy of the extracellular experiment (MIC 0.2 μM in both assays). For the BTZ derivatives 5 c, 5 d, 6 d, 7 c, 8 c, 12 b, 13 b, 14 a, 15 b, 16 d, 17 b and 26 c only a moderate increase in MIC (by a maximum factor of 2) was observed, which suggests that they can achieve a similar efficacy intracellularly as in the 7H9 assay medium.
BTZs with considerably higher MIC in the intracellular assay
Among the compounds newly prepared for this study, 1 e again proved to be most effective with a MIC of 0.1 μM in the macrophage infection model. However, compared to the extracellular assay (MIC 6 nM) the activity in the infection model was significantly reduced. A similar observation was made for the derivatives 3 c, 4 c and 11 c with an intracellular MIC of 0.2 μM each. They have a piperazine amide/sulfonamide side chain in common.
Since the BTZs discussed in the two preceding paragraphs “only” differ in the 2‐substituent, the lower intracellular MICs presumably result from reduced passage through macrophage membranes.
BTZs with lower MIC in the intracellular assay
Special attention was paid to compounds that were more effective in the macrophage infection assay. In Figure 5C the quotient of extracellular MIC and intracellular MIC is shown for better visualization. Most of the BTZs tested showed a score of <1, indicating reduced activity against intracellular mycobacteria. This behaviour was also observed for the highly effective compounds macozinone and BTZ043. A numerical value >1 indicates an enhancement of the antimycobacterial effect during macrophage infection. This was only observed for three tested compounds, viz. 9 a, 24 b and 33 b. In particular, the 8‐nitro‐BOZ 33 b, following macozinone with a cyclohexylmethylpiperazine side chain but with oxygen in place of sulfur, showed a fivefold improvement in efficacy during macrophage infection, with the MIC reduced from 1 μM to 0.2 μM. Possible explanations for this behaviour are an accumulation in the macrophage or a better metabolic stability of the compound because the presence of oxidising species in macrophages may well lead to oxidation of the sulfur atom in BTZs and follow‐up reactions as was reported by us. [32]
Evaluation of cytotoxicity
To investigate possible cytotoxicity, all derivatives were analysed in a cell viability assay against THP‐1 cells, which was photometrically evaluated after addition of the colour reagent 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT). [33] Only a few compounds showed a cytotoxic effect at concentrations <10 μM, this was observed for 8 c, 9 a, 18 b, 22 b, 24 b, 25 b, and 31 b. With the exception of compound 18 b, the intracellular MICs of these analogues were also clearly below the cytotoxic concentration, so that an evaluation of the assay data is possible. The reference compounds BTZ043 and macozinone showed low cytotoxicity, 21.9 and 23.2 μM, respectively, to macrophages. For the most potent compounds in this study – 1 e and 11 c – no cytotoxicity was detected in this assay, underlining their suitability as antimycobacterial drug candidates.
Solubility of selected BTZs analogues
Although BTZs are a very promising class of compounds, only few solubility data have been published. [21] For a selection of BTZ derivatives described in this study, the solubility in PBS (phosphate‐buffered saline, pH 7.4)was determined using a nephelometric method (Table 2). [34] The solubility of the analogues with high activity was below the detection limit and is therefore reported with <30 μM. Only the derivatives 7 c and 13 b possess an improved solubility, but both compounds show only low or modest antimycobacterial activity.
Table 2.
Solubility of selected BTZ analogues.
|
ID |
Solubility[a] [μM] |
ID |
Solubility[a] [μM] |
|---|---|---|---|
|
1 e |
<30 |
10 c |
<30 |
|
3 c |
<30 |
11 c |
<30 |
|
4 c |
<30 |
13 b |
110 |
|
5 c |
<30 |
19 b |
<30 |
|
5 d |
<30 |
20 b |
<30 |
|
6 d |
52 |
BTZ043 |
32 |
|
7 c |
92 |
macozinone |
<30 |
|
8 c |
<30 |
[a] The limit of quantification is 30 μM, below this concentration the solubility cannot be quantified by nephelometry [34] .
Conclusion
We have established a new BTZ synthesis pathway, which for the first time introduces nitrogen and sulphur of the thiazinone ring in a single step and uses thioureas as precursors. This synthetic pathway provides a robust and reproducible way to derivatize the BTZ scaffold. A selection of new BTZs, BOZs and QZs could be synthesized by the new synthetic route, which is also applicable for preparation of the lead compounds BTZ043 and macozinone. Moreover, this synthetic pathway can be easily adapted to replace the sulphur atom of the BTZs by oxygen or nitrogen yielding novel compounds which previously described BTZ syntheses do not enable. Our synthetic route also has the potential for the industrial production of the BTZ drug candidates, especially regarding the avoidance of iodomethane and carbon disulfide, compared to the alkylsulfanyl pathway.
The compounds were tested against the pathogen Mtb and the MICs were determined in microbiological growth medium and in a macrophage infection model. Through derivatization with chemically highly diverse substituents in position 2 of the BTZ scaffold, structure‐activity relationships for this substance class could be elucidated. In view of their strong antimycobacterial activity, compounds 1e and 11c proved to be the most promising. For the antimycobacterial characterisation, special attention was paid to testing all derivatives in a macrophage infection model. Most of the 8‐nitro BTZs showed a slight reduction in efficacy, but several compounds have intracellular MICs between 100–200 nM, which underlines the potential of these agents as antitubercular agents.
Experimental Section
M. tuberculosis H37Rv culture and growth medium. The M. tuberculosis strain used for the experiments was H37Rv transformed with pJAK2.A plasmid. [31] The pJAK2.A plasmid is an integrative plasmid based on the pMV361 vector, which allows high‐level expression of the firefly luciferase gene from the hsp60 promoter and can be selected using kanamycin. Bacteria were grown in 7H9 broth supplemented 10 % OADC, 0.05 % Tween 80 and 50 μg mL−1 kanamycin in a standing culture.
THP‐1 cells and culture media. The THP‐1 cells (ATCC® TIB‐202™) used are derived from human monocytes obtained from a one‐year old male infant with acute monocytic leukaemia. The cells were stored in liquid nitrogen. THP‐1 cells were grown in complete RPMI medium. The cells were grown in tissue culture flask with a minimum volume of 20 ml and maximum volume of 50 ml and were incubated in an atmosphere of 95 % air and 5 % CO2 at a temperature of 37 °C. Cell density was kept between 0.25 and 1×106 mL−1. Every two or three days the cells were counted in a microscope and diluted to 0.25×106 mL−1. The cells double every 48 h. A culture from nitrogen stock can be sub cultured for up to three months, after this time a change in morphology and growth behaviour is observed. For culturing of THP‐1 cells RPMI‐1640 medium supplemented with 5 % FBS, 2 % glutamine, 1 % non‐essential amino acids and 1 % penicillin+streptomycin was used (complete RPMI medium). For the intracellular growth inhibition assay incomplete RPMI‐1640 medium supplemented with 5 % FBS, 2 % glutamine, 1 % non‐essential amino acids is used.
In‐broth activity analysis using a luciferase‐based assay. M. tuberculosis H37Rv transformed with pJAK2.A plasmid is grown in 7H9+10 % OADC, 0.05 % Tween 80 and 50 μg mL−1 kanamycin in a standing culture to the mid‐log phase (∼0.5–0.8 OD600). The culture is diluted with the 7H9+10 % OADC and 0.05 % Tween 80 to 0.01 OD600. The compounds described in this study were diluted in 7H9+10 % OADC and 0.05 % to 2× the testing concentrations and 100 μL of each sample was aliquoted into 96‐well plates. For MIC determination a serial dilution was performed. Subsequently 100 μL of the diluted bacterial suspension into each well. The plates are incubated at 37 °C in a humidified incubator for 5 days. After incubation 50 μL of the luciferase assay reagent are added to each well. The plates are sealed with transparent, self‐adhesive plate caps after 5 min incubation at 22 °C the read‐out is performed with a luminometer (1 s per well, Applied Biosystems Tropix TR717).
High‐throughput intracellular screening using luciferase‐expressing M. tuberculosis H37Rv.[27b] The intracellular activity determination was executed according Zheng et al. [27b] A serial dilution of the test compounds was prepared in complete RPMI medium and the infected macrophages were added to the test compounds at day 0 of the incubation time.
Cytotoxicity analysis using a 3‐(4,5‐dimethylthiazol‐2‐yl)‐ 2,5‐diphenyltetrazolium bromide (MTT) assay. The MTT assay was executed according Zheng et al.[27b]
X‐ray crystallography. A crystal of 1 c suitable for single‐crystal X‐ray diffraction was obtained from a solution in dimethylformamide by slow evaporation of the solvent at room temperature. The X‐ray intensity data were collected on a STOE IPDS II diffractometer, using graphite‐monochromated Mo‐Kα radiation. A multi‐scan absorption correction was carried out with PLATON. [35] The crystal structure was solved with SHELXT [36] and refined with SHELXL‐2018/03. [37] The trifluromethyl group exhibits rotational disorder, which was refined using a split‐atom model. Standard similar distance (SADI) restraints on the 1,2‐ and 1,3‐C−F‐distances and EADP constraints and ISOR restraints on the F atoms were applied. Refinement of the ratio of occupancies by means of a free variable yielded 0.830(3):0.170(3). H atoms were placed in geometrically calculated positions with Caromatic−H=0.95 Å, Cmethylene−H=0.99 Å and Cmethyl−H=0.98 Å and refined using the appropriate riding model with U iso(H)=1.2 U eq(C) (1.5 for methyl groups). The structure picture was drawn with Mercury. [38]
Nephelometric determination of solubility.[34] Preparation of a 0.01 M stock solution of the compounds in DMSO, serial dilution of the stock solution with DMSO 1 : 2 (35 μl stock solution+35 μl DMSO), then dilution 1 : 50 in measuring buffer (245 μl PBS+5 μl of the serial dilution) in 96‐well plates, highest concentration 2×10−4 PBS (life technology, # 1720796), 6‐fold determination.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Professor Kurt Merzweiler is gratefully acknowledged for providing access to the X‐ray diffraction facility. We thank Mary Ko for administrative assistance. Open Access funding enabled and organized by Projekt DEAL.
A. Richter, G. Narula, I. Rudolph, R. W. Seidel, C. Wagner, Y. Av-Gay, P. Imming, ChemMedChem 2022, 17, e202100733.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.WHO, Global Tuberculosis Report 2021.
- 2.
- 2a. Sulis G., Centis R., Sotgiu G., D'Ambrosio L., Pontali E., Spanevello A., Matteelli A., Zumla A., Migliori G. B., Prim. Care Resp. Med. 2016, 26(1), 16078; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Dooley K. E., Hanna D., Mave V., Eisenach K., Savic R. M., PLoS Med. 2019, 16(7), e1002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shetye G. S., Franzblau S. G., Cho S., Transl. Res. 2020, 220, 68–97. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Makarov V., Lechartier B., Zhang M., Neres J., Sar A. M., Raadsen S. A., Hartkoorn R. C., Ryabova O. B., Vocat A., Decosterd L. A., Widmer N., Buclin T., Bitter W., Andries K., Pojer F., Dyson P. J., Cole S. T., EMBO Mol. Med. 2014, 6(3), 372–383; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Makarov V., Manina G., Mikusova K., Mollmann U., Ryabova O., Saint-Joanis B., Dhar N., Pasca M. R., Buroni S., Lucarelli A. P., Milano A., De Rossi E., Belanova M., Bobovska A., Dianiskova P., Kordulakova J., Sala C., Fullam E., Schneider P., Mckinney J. D., Brodin P., Christophe T., Waddell S., Butcher P., Albrethsen J., Rosenkrands I., Brosch R., Nandi V., Bharath S., Gaonkar S., Shandil R. K., Balasubramanian V., Balganesh T., Tyagi S., Grosset J., Riccardi G., Cole S. T., Science 2009, 324(5928), 801–804; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Lupien A., Vocat A., Foo C. S.-Y., Blattes E., Gillon J.-Y., Makarov V., Cole S. T., Antimicrob. Agents Chemother. 2018, 62(11); [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4d. Chauhan A., Kumar M., Kumar A., Kanchan K., Life Sci. 2021, 274, 119301. [DOI] [PubMed] [Google Scholar]
- 5. Brecik M., Centárová I., Mukherjee R., Kolly G. S., Huszár S., Bobovská A., Kilacsková E., Mokošová V., Svetlíková Z., Šarkan M., Neres J., Korduláková J., Cole S. T., Mikušová K., ACS Chem. Biol. 2015, 10(7), 1631–1636. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Trefzer C., Rengifo-Gonzalez M., Hinner M. J., Schneider P., Makarov V., Cole S. T., Johnsson K., J. Am. Chem. Soc. 2010, 132(39), 13663–13665; [DOI] [PubMed] [Google Scholar]
- 6b. Trefzer C., Škovierová H., Buroni S., Bobovská A., Nenci S., Molteni E., Pojer F., Pasca M. R., Makarov V., Cole S. T., Riccardi G., Mikušová K., Johnsson K., J. Am. Chem. Soc. 2012, 134(2), 912–915; [DOI] [PubMed] [Google Scholar]
- 6c. Neres J., Pojer F., Molteni E., Chiarelli L. R., Dhar N., Boy-Rottger S., Buroni S., Fullam E., Degiacomi G., Lucarelli A. P., Read R. J., Zanoni G., Edmondson D. E., De Rossi E., Pasca M. R., Mckinney J. D., Dyson P. J., Riccardi G., Mattevi A., Cole S. T., Binda C., Sci. Transl. Med. 2012, 4(150), 150ra121-150ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Gao C., Ye T.-H., Wang N.-Y., Zeng X.-X., Zhang L.-D., Xiong Y., You X.-Y., Xia Y., Xu Y., Peng C.-T., Zuo W.-Q., Wei Y., Yu L.-T., Bioorg. Med. Chem. Lett. 2013, 23(17), 4919–4922; [DOI] [PubMed] [Google Scholar]
- 7b. Peng C.-T., Gao C., Wang N.-Y., You X.-Y., Zhang L.-D., Zhu Y.-X., Xv Y., Zuo W.-Q., Ran K., Deng H.-X., Lei Q., Xiao K.-J., Yu L.-T., Bioorg. Med. Chem. Lett. 2015, 25(7), 1373–1376; [DOI] [PubMed] [Google Scholar]
- 7c. Karoli T., Becker B., Zuegg J., Moellmann U., Ramu S., Huang J. X., Cooper M. A., J. Med. Chem. 2012, 55(17), 7940–7944. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a.V. A. Makarov, Process for the preparation of 2-amino substituted 1,3-benzothiazine-4-ones, WO2011132070A1, Russia, 2011;
- 8b.V. Makarov, S. T. Cole, U. Moellmann, New Benzothiazinone derivatives and their use as antibacterial agents, WO2007134625A1, Germany, 2007;
- 8c.U. Moellmann, V. Makarov, S. T. Cole, New antimicrobial compounds, their synthesis and their use for treatment of mammalian infection, WO2009010163A1, Germany, 2009.
- 9.
- 9a. Imrich J., Kristian P., Collect. Czech. Chem. Commun. 1982, 47, 3268–3282; [Google Scholar]
- 9b. Koscik D., Kristian P., Gonda J., Dandarova E., Collect. Czech. Chem. Commun. 1983, 48, 3315–3328. [Google Scholar]
- 10.I. Rudolph, P. Imming, A. Richter, Preparation of benzothiazinone, thiochromenone, benzoxazinone, and dihydroquinazolinone derivatives as antimycobacterial agents, processes for their production and their use, DE 102014012546, Martin-Luther-Universitaet Halle-Wittenberg, Germany, Germany, 2014.
- 11. Nally J. D., Good Manufacturing Practices for Pharmaceuticals, CRC Press, 2007. [Google Scholar]
- 12. Lancaster M., Green Chemistry: An Introductory Text, Royal Society of Chemistry, 2016. [Google Scholar]
- 13.
- 13a. Stanley S. A., Barczak A. K., Silvis M. R., Luo S. S., Sogi K., Vokes M., Bray M.-A., Carpenter A. E., Moore C. B., Siddiqi N., Rubin E. J., Hung D. T., PLoS Pathog. 2014, 10(2), e1003946; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Mdluli K., Kaneko T., Upton A., Cold Spring Harbor Perspect. Med. 2015, 5(6), a021154–a021154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Richter A., Goddard R., Schlegel T., Imming P., Seidel R. W., Acta Crystallogr. Sect. E 2021, 77(2), 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a.G. Seybold, M. Dimmler, A. Stange, Verfahren zur Herstellung von 1,1-disubstituierten Thioharnstoffen, DE3314435A1, Germany, 1984;
- 15b.H. Hartmann, I. Reuther, Verfahren zur Herstellung von 1,1-disubstituierten Thioharnstoffen, DD100467A1, German Democratic Republic, 1973.
- 16. Narumi T., Arai H., Yoshimura K., Harada S., Nomura W., Matsushita S., Tamamura H., Bioorg. Med. Chem. Lett. 2011, 19(22), 6735–6742. [DOI] [PubMed] [Google Scholar]
- 17. Collins J. L., Blanchard S. G., Boswell G. E., Charifson P. S., Cobb J. E., Henke B. R., Hull-Ryde E. A., Kazmierski W. M., Lake D. H., Leesnitzer L. M., Lehmann J., Lenhard J. M., Orband-Miller L. A., Gray-Nunez Y., Parks D. J., Plunkett K. D., Tong W.-Q., J. Med. Chem. 1998, 41(25), 5037–5054. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Makarov V., Neres J., Hartkoorn R. C., Ryabova O. B., Kazakova E., Šarkan M., Huszár S., Piton J., Kolly G. S., Vocat A., Conroy T. M., Mikušová K., Cole S. T., Antimicrob. Agents Chemother. 2015, 59(8), 4446–4452; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18b. Nosova E. V., Batanova O. A., Lipunova G. N., Kotovskaya S. K., Slepukhin P. A., Kravchenko M. A., Charushin V. N., J. Fluorine Chem. 2019, 220, 69–77; [Google Scholar]
- 18c. Zhang G., Sheng L., Hegde P., Li Y., Aldrich C. C., Med. Chem. Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang F., Sambandan D., Halder R., Wang J., Batt S. M., Weinrick B., Ahmad I., Yang P., Zhang Y., Kim J., Proc. Natl. Acad. Sci. USA 2013. [Google Scholar]
- 20. Gler M. T., Skripconoka V., Sanchez-Garavito E., Xiao H., Cabrera-Rivero J. L., Vargas-Vasquez D. E., Gao M., Awad M., Park S.-K., Shim T. S., Suh G. Y., Danilovits M., Ogata H., Kurve A., Chang J., Suzuki K., Tupasi T., Koh W.-J., Seaworth B., Geiter L. J., Wells C. D., N. Engl. J. Med. 2012, 366(23), 2151–2160. [DOI] [PubMed] [Google Scholar]
- 21. Zhang G., Howe M., Aldrich C. C., ACS Med. Chem. Lett. 2019, 10(3), 348–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li P., Wang B., Zhang X., Batt S. M., Besra G. S., Zhang T., Ma C., Zhang D., Lin Z., Li G., Huang H., Lu Y., Eur. J. Med. Chem. 2018, 160, 157–170. [DOI] [PubMed] [Google Scholar]
- 23.Crystal data for 1 c: C18H19F3N4O5S, M r = 460.43, T = 170(2) K, λ = 0.71073 Å, triclinic, space group P-1, a = 6.4887(3), b = 9.4396(5), c = 16.7854(8) Å, α = 80.347(4), β = 82.041(4), γ = 87.310(4)°, V = 1003.50(9) Å3, Z = 2, ρ calc = 1.524 mg m−3, μ = 0.228 mm−1, F(000) = 476, crystal size 0.49×0.35×0.30 mm, θ range 2.19–29.20°, reflections collected/unique 14539/5359 (R int = 0.0455), 293 parameters, 81 restraints, S = 1.040, R1 [I > 2σ(I)] = 0.0476; wR2 = 0.1311, Δρ max, Δρ min = 0.69, −0.38 eÅ−3. CCDC 2099461 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/structures.
- 24.
- 24a. Gutschow M., Schlenk M., Gab J., Paskaleva M., Alnouri M. W., Scolari S., Iqbal J., Müller C. E., J. Med. Chem. 2012, 55(7), 3331–3341; [DOI] [PubMed] [Google Scholar]
- 24b. Stossel A., Schlenk M., Hinz S., Kuppers P., Heer J., Gütschow M., Müller C. E., J. Med. Chem. 2013, 56(11), 4580–4596. [DOI] [PubMed] [Google Scholar]
- 25. Zhang G., Aldrich C. C., Acta Crystallogr. Sect. C 2019, 75(8), 1031–1035. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Zhang G., Chen D., Wang S., Chen H., Wei N., Chen G., ChemistrySelect 2020, 5(43), 13775–13779; [Google Scholar]
- 26b. Madikizela B., Eckhardt T., Goddard R., Richter A., Lins A., Lehmann C., Imming P., Seidel R. W., Med. Chem. Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.
- 27a. Sorrentino F., Gonzalez Del Rio R., Zheng X., Presa Matilla J., Torres Gomez P., Martinez Hoyos M., Perez Herran M. E., Mendoza Losana A., Av-Gay Y., Antimicrob. Agents Chemother. 2016, 60(1), 640–645; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Zheng X., Av-Gay Y., J. Visualized Exp. 2017(122). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.
- 28a. Larsson M. C., Lerm M., Ängeby K., Nordvall M., Juréen P., Schön T., J. Microbiol. Methods 2014, 106, 146–150; [DOI] [PubMed] [Google Scholar]
- 28b. Gordon S., Chung G., Andrew P., in Mycobacteria Protocols, Humana Press, pp. 235–244. [Google Scholar]
- 29. Zhang J.-H., Chung T. D. Y., Oldenburg K. R., J. Biomol. Screening 1999, 4(2), 67–73. [DOI] [PubMed] [Google Scholar]
- 30. Richter A., Rudolph I., Möllmann U., Voigt K., Chung C.-W., Singh O. M. P., Rees M., Mendoza-Losana A., Bates R., Ballell L., Batt S., Veerapen N., Fütterer K., Besra G., Imming P., Argyrou A., Sci. Rep. 2018, 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sun J., Lau A., Wang X., Liao T.-Y. A., Zoubeidi A., Hmama Z., Plasmid 2009, 62(3), 158–165. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Eckhardt T., Goddard R., Lehmann C., Richter A., Sahile H. A., Liu R., Tiwari R., Oliver A. G., Miller M. J., Seidel R. W., Imming P., Acta Crystallogr. Sect. C 2020, 76(9), 907–913; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32b. Tiwari R., Miller P. A., Cho S., Franzblau S. G., Miller M. J., ACS Med. Chem. Lett. 2015, 6(2), 128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hansen M. B., Nielsen S. E., Berg K., J. Immunol. Methods 1989, 119(2), 203–210. [DOI] [PubMed] [Google Scholar]
- 34. Hoelke B., Gieringer S., Arlt M., Saal C., Anal. Chem. 2009, 81(8), 3165–3172. [DOI] [PubMed] [Google Scholar]
- 35. Spek A. L., Acta Crystallogr. Sect. D 2009, 65(2), 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheldrick G. M., Acta Crystallogr. Sect. A 2015, 71(Pt 1), 3–8. [Google Scholar]
- 37. Sheldrick G. M., Acta Crystallogr. Sect. C 2015, 71(Pt 1), 3–8. [Google Scholar]
- 38. Macrae C. F., Sovago I., Cottrell S. J., Galek P. T. A., McCabe P., Pidcock E., Platings M., Shields G. P., Stevens J. S., Towler M., Wood P. A., J. Appl. Crystallogr. 2020, 53(Pt 1), 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.