

Abstract
Cutaneous malignant melanoma is the fastest growing and the most aggressive form of skin cancer that is diagnosed. However, its incidence is relatively scarce compared to the highest mortality rate of all skin cancers. The much more common skin cancers include nonmelanoma malignant skin cancers. Moreover, over the past several decades, the frequency of all skin cancers has increased much more dynamically than that of almost any other type of cancer. Among the available therapeutic options for skin cancers, chemotherapy used immediately after the surgical intervention has been an essential element. Unfortunately, the main problem with conventional chemopreventive regimens involves the lack of response to treatment and the associated side effects. Hence, there is a need for much more effective anticancer drugs. Correspondingly, the targeted alternatives have involved phytochemicals, which are safer chemotherapeutic agents and exhibit competitive anticancer activity with high therapeutic efficacy. Among polyphenolic compounds, some flavonoids and their derivatives, which are mostly found in medicinal plants, have been demonstrated to influence the modulation of signaling pathways at each stage of the carcinogenesis process, which is also important in the context of skin cancers. Hence, this review focuses on an exhaustive overview of the therapeutic effects of luteolin and its derivatives in the treatment and prevention of skin cancers. The bioavailability and structure–activity relationships of luteolin derivatives are also discussed. This review is the first such complete account of all of the scientific reports concerning this particular group of natural compounds that target a specific area of neoplastic diseases.
Keywords: luteolin, melanoma, phytotherapy, skin cancer
Abbreviations
- ADAMTS
a disintegrin and metalloproteinase with thrombospondin motifs
- AhR
aryl hydrocarbon receptors
- AIDS
acquired immunodeficiency syndrome
- AK
actinic keratoses
- AKT
protein kinase B
- AP‐1
activator protein‐1
- ATP
adenosine triphosphate
- Bax
Bcl‐2‐associated X
- BCC
basal cell carcinoma
- Bcl‐2
B‐cell lymphoma 2
- BRAF
v‐raf murine sarcoma viral oncogene homolog B
- cAMP
cyclic adenosine monophosphate
- CDK4
cyclin‐dependent kinase 4
- CDKN2A
cyclin‑dependent kinase inhibitor 2A
- CDKN2B
cyclin‑dependent kinase inhibitor 2B
- CHOP
CCAAT/enhancer‐binding protein‐homologous protein;
- CMM
cutaneous malignant melanoma
- COX‐2
cyclooxygenase‐2
- CREB
cAMP response element‐binding protein
- CTLA‐4
cytotoxic T‐lymphocyte‐associated protein 4
- CTS
cathepsins
- c‐KIT
tyrosine‐protein kinase Kit
- DCT
dopachrome tautomerase
- DDB2
damage specific DNA binding protein 2
- DNMTs
DNA methyltransferases
- DTIC
dacarbazine
- ECM
extracellular matrix
- EDF
European Dermatology Forum
- EGF
epidermal growth factor
- EGFR
EGF receptor
- EMT
epithelial–mesenchymal transition
- ER
endoplasmic reticulum
- ERK
extracellularly‐regulated kinase
- EZH2
enhancer of zeste homolog 2
- FAK
focal adhesion kinase
- FDA
U.S. Food and Drug Administration
- FGF10
fibroblast growth factor 10
- FN1
fibronectin 1
- GANAB
glucosidase II alpha subunit
- GDP
guanosine diphosphate
- GM‐CSF
granulocyte macrophage colony‐stimulating factor
- GTP
guanosine triphosphate
- H3K27me3
trimethylation in histone H3 at lysine 27
- HPV
β human papillomavirus;
- IARC
International Agency for Research on Cancer
- ICAM1
intercellular adhesion molecule‐1
- IFN‐γ
interferon‐γ
- IFN‐ α
interferon‐α
- IKK
inhibitory‐κB kinase
- IL‐1α
interleukin 1α
- IL‐1β
interleukin 1β
- IL‐2
interleukin 2
- IL‐36
interleukin 36
- IL‐6
interleukin 6
- ITGα2B
integrin α2B
- ITGβ3
integrin β3
- JNK
c‐Jun N‐terminal kinase
- KIT
type III transmembrane receptor tyrosine kinase
- KSR2
kinase suppressor of RAS 2
- LAMA1
laminin subunit alpha 1
- MAPK
the mitogen‐activated protein kinase
- MC1R
melanocortin‐1‐receptor
- MEK
mitogen‐activated protein kinase
- MHC
major histocompatibility complex
- MITF
microphthalmia‐associated transcription factor
- MMP
matrix metalloproteinase
- MTT
3‐(4,5‐dimethylthiazol‐2‐yl)−2,5‐diphenyltetrazolium bromide
- NDPK
nucleoside diphosphate kinase
- NF1
neurofibromin type 1
- NF‐κB
nuclear factor kappa‐light‐chain‐enhancer of activated B;
- NMSC
non‐melanoma malignant skin cancers
- NRAS
neuroblastoma RAS viral oncogene homolog
- Nrf2
nuclear factor 2 associated erythroid 2
- PDIA3
protein disulfide‐isomerase A3
- PD‐1
programmed death‐1
- PD‐L1
programmed death ligand‐1
- PI3K
phosphoinositol‑3‑kinase
- PKCε
protein kinase Cε
- PTEN
phosphatase and tensin homolog
- RAF
rapidly accelerated fibrosarcoma
- RAS
rat sarcoma viral oncogene
- ROS
reactive oxygen species
- RTK
tyrosine kinase receptors
- SAR
structure–activity relationship
- SCC
squamous cell carcinoma
- SHH
sonic hedgehog
- SRB
sulforhodamine B
- Src
steroid receptor coactivator
- STAT1
signal transducer and activator of transcription 1
- STAT3
signal transducer and activator of transcription 3
- TET1
ten‐eleven translocation‐1
- TGF
tumor growth factor
- TIMP
tissue inhibitor of MMP
- TLR
toll‐like receptor
- TNF
tumor necrosis factor
- TNF‐α
tumor necrosis factor α
- TRAIL
TNF‐α ‐related apoptosis‐inducing ligand
- TRP
transient receptor potential
- TYR
tyrosinase
- UV
ultraviolet
- VEGF
vascular endothelial growth factor
- XTT
2,3‐bis‐(2‐methoxy‐4‐nitro‐5‐sulfophenyl)−2H‐tetrazolium‐5‐carboxanilide.
- α‐MSH
α‐melanocyte stimulating hormone
1. INTRODUCTION
Cutaneous malignant melanoma (CMM) is the fastest growing cancer in the fair‐skinned Caucasian population and the most aggressive form of diagnosed skin cancer. Its incidence is less than 5% per year, which is relatively low compared to its high mortality rate, which is the highest of all skin cancers. However, over the past several decades, the incidence of CMM has increased much more rapidly than that of almost any other cancer. 1 According to the GLOBOCAN 2020 database (https://gco.iarc.fr/today/home) published by the International Agency for Research on Cancer (IARC), non‐melanoma malignant skin cancers (NMSCs) are the most common skin cancers, accounting for 30% of the cancer burden, with an estimated incidence of over 350,000 cases per year only in Europe, making them the most common malignant neoplasms in white populations each year. The term NMSC encompasses basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), which account for 90% of the tumors of this type. 2 , 3 The term is also used in reference to adnexal tumors, cutaneous lymphomas, Merkel cell carcinoma, and other rare primary skin cancers. The incidence of NMSC (BCC and SCC) is 18‐ to 20‐fold higher than that of malignant melanoma. 4 , 5 , 6 , 7 , 8
The reported dramatic increase in skin cancer incidence is mainly attributed to chronic ultraviolet (UV) exposure and to skin type, which are the dominant risk factors. Additionally, personal factors such as age, sex, and genetic background contribute to CMM susceptibility and are mainly attributed to the melanin content in skin layers. Another important aspect is the inheritance of skin cancer susceptibility associated with low and high penetrance genes described in the following sections. 3 The incidence of melanoma increases with age, as evidenced by the data showing that the average age of diagnosis is approximately 60 years. The risk of occurrence is also closely related to sex. The incidence of melanoma in men is 1.5‐fold greater than it is in women. The relationship between incidence and age becomes very clear in people older than 75 years, when the incident coefficient increases twofold. Additionally, geographic zone, common or atypical nevi, and chronic sun exposure, especially in childhood, are suggested to be major environmental risk factors for melanoma. 6 , 9 , 10 The relationship is evident in the correlation between the risk of BCC and the “history” of UV ray overexposure, particularly sunburn, especially in childhood. However, these factors do not translate into SCC, the risk of which is closely related to long‐term UV exposure. 11 In contrast to that of melanoma, the incidence of NMSC has been proven to be closely related to age. At an early age, people of either sex show a similar prevalence for acquiring either NMSC. The situation changes for men older than 45 years, because NMSC affects this group of men 2‐ to 3‐fold more frequently than women. 3 , 5 , 6 , 12 , 13 , 14 , 15
Among the available therapeutic options for skin cancer, chemotherapy administered immediately after surgical intervention has been an essential element of the available anticancer therapies for decades. Unfortunately, the main problem with conventional chemopreventive regimens is the lack of response to treatment. Hence, there is a need for much more effective anticancer drugs. Moreover, inherent side effects are other problems with currently available chemotherapeutics. Therefore, alternatives based on phytochemicals have been used because they are safer than traditional chemotherapeutic agents and exhibit competitive anticancer activity with high therapeutic efficacy. 16 Currently, of all cancer therapeutics approved by the U.S. Food and Drug Administration (FDA), as many as 40% are directly or indirectly related to natural sources. 17 Notably, cytostatic compounds including vincristine, vinblastine, vinorelbine, paclitaxel, docetaxel, topotecan, irinotecan, and others are only some examples of the anticancer drugs and plant‐derived agents approved for clinical use. 18 , 19 , 20 The current state of knowledge supports evidence of the beneficial effects of combination therapies consisting of conventional anticancer drugs and natural compounds. 21 Medicinal plants and their bioactive compounds have been successfully used for years as complementary therapies. In addition, the research performed with multiple cancer cell lines and animal models including skin cancer proves that these combinations can suppress many stages of development and progression of cancer cells by influencing a number of mechanisms, such as the cell cycle, and by inhibiting angiogenesis and proliferation or activating proapoptotic and pro‐survival proteins. 22 , 23 , 24 , 25 Currently, various formulations in the market contain compounds of natural origin for use in skin cancer or precancerous conditions such as Birch Bark ointment, Curaderm®, or Cansema®. 26 Among the groups of phytochemicals studied thus far, flavonoids constitute a class of secondary plant metabolites showing potent anticancer activity, particularly in the context of skin cancer, by modulating signaling pathways at each stage of carcinogenesis. 18 , 27 However, in a search of literature published worldwide, only general reviews of the broadly understood anticancer activity of flavonoids can be found. Contrary to similar manuscripts, our review describes the therapeutic effects of luteolin 22 and its derivatives in treating and preventing skin cancers, both NMSCs and melanoma, which significantly distinguishes our review from others. The bioavailability and structure–activity relationships (SARs) of luteolin and its derivatives are described, making this report the first comprehensive and complete account of all scientific reports concerning this particular group of natural compounds targeting specific neoplastic diseases.
2. METHODOLOGY/SEARCH STRATEGY
A comprehensive search analysis was performed to identify relevant scientific literature on the basis of an appropriate search string entered in relevant subject databases. The electronic databases of SCOPUS, Google Scholar, PubMed/MEDLINE, Web of Science (SCI‐EXPANDED), Taylor & Francis Online, Wiley Online Library, EBSCO Discovery Service (EDS), REAXYS Database, and Science Direct/ELSEVIER were extensively searched in the preparation of this review. Clinical trial information was retrieved from the ClinicalTrials.gov database. Chemical structures were confirmed with entries in the PubChem and REAXYS databases. Titles, abstracts, and keywords (TITLE‐ABS‐KEY) contained in these databases were searched using the following terms separately or in various combinations taking into account the requirements or limitations of the databases searched in the first screening step: “luteolin,” OR “luteolin derivatives,” OR “natural compounds,” OR “flavonoids,” OR “melanoma,” OR “skin cancer,” OR “cutaneous melanoma,” OR “skin melanoma,” OR “nonmelanoma malignant skin cancers,” OR “basal cell carcinoma,” OR “squamous cell carcinoma,” OR “skin basal cell carcinoma,” OR “skin squamous cell carcinoma,” OR “SCC,” OR “BCC,” OR “anticancer activity,” OR “chemopreventive activity.” The search was restricted to studies written in the English language. Only articles published between 1994 and 2021, which includes the first scientific report on the activity of the tested group of compounds in the context of skin cancer therapy or prevention, were retained. The second screening was based on full texts. Additional papers were identified from the review articles and reference lists identified in the initial literature searches.
2.1. Inclusion and exclusion criteria
Studies conducted on models of skin melanoma and other skin neoplasms treated with luteolin and/or its derivatives and that included an evaluation of the preventive and/or antitumor effects of these natural compounds were retained. The inclusion criteria of the published studies were (1) research model criteria were adopted with in vitro and/or in vivo models and/or clinical trials of skin cancer treatments, (2) preventive and/or antitumor effects of pure compounds (luteolin and/or its derivatives) and/or plant extracts rich in luteolin and/or its derivatives were administered to the adopted models, (3) the criteria for the antitumor response were defined for each documented experiment (with an IC50 value above the concentration of the tested compounds) affecting the proliferation of cancer cells, (4) the research was reported in full, (5) the paper was published in the English language, and (6) the article was published after the first scientific report on the activity of the tested group of compounds and/or plant extracts containing the tested compounds on an adopted research model and before July 2021.
The exclusion criteria for the published articles were (1) studies on melanoma but not skin melanoma, (2) studies on cancers such as squamous cell carcinoma, SCC or basal cell carcinoma, or BCC but not in skin, (3) studies that reported preventive and/or antitumor activity of natural compounds on the adopted models but not of luteolin or a luteolin derivative, (4) studies that reported preventive and/or antitumor activity of flavonoids on the adopted models but not luteolin or a luteolin derivative, (5) studies that reported preventive and/or antitumor activity of synthetic drugs on the adopted models, (6) papers not published in the English language, and (7) study results presented in the form of a letter to the editor, a commentary, a preface, an abstract without full accompanying paper, a conference paper or a book review.
3. SKIN CANCERS
3.1. Non‐melanoma malignant skin cancers
3.1.1. Basal cell carcinomas
BCC, the most common skin cancer, is characterized by low malignancy and limited local invasiveness. 3 , 28 However, the term semi‐malignant appears in the context of BCC, particular with respect to rare metastases, occasional aggressive growth with tissue destruction, and involvement of lymph nodes. 29 , 30 BCC develops de novo from cutaneous keratinocytes and is caused by UV‐induced mutations in basal layer cells of the epidermis and its appendages.
Most often, BCC is located in areas exposed to direct sunlight, such as facial skin, especially above the line connecting the angle of the mouth with the opening of the external auditory canals, as well as the backs of the hands; however, it can occur anywhere on the body. Early cases of BCC usually appear as a translucent or pearly small papule, sometimes with visible telangiectasia. The BBC growth rate is slow, and its metastasis is sporadic. BCC consists of the following subtypes, which are distinguished on the basis of physical characteristics and histological findings: nodular, superficial, and morphea forms, accounting for 60%, 30%, and 5%–10% of cases, respectively. 5 , 7 , 8 , 31
3.1.2. Squamous cell carcinomas
SCC is caused by malignant neoplasia of epidermal keratinocytes with variable squamous differentiation, local infiltration, and invasion into surrounding tissues. 3 , 28 In contrast to BCC, SCC can arise from precursor lesions, including actinic keratoses (AKs) and SCC in situ, a condition called Bowen's disease, which is much more invasive and metastatic than BCC, mainly spreading to lymph nodes. SCC metastasis is the result of a complex process leading to the migration of cancer cells through the extracellular matrix (ECM), which is also degraded by proteases. AKs progress to SCC in 1%–10% of cases, a percentage that increases with the number of lesions on the patient, and the incidence of Bowen's disease derived from AKs is 3%–5%.
SCC lesions are most often located on sun‐exposed skin areas, such as the back of the hands, ears, scalp, and central part of the face, which results in photodamage of the skin. Only in rare cases, they develop elsewhere on the body. 2 Characteristic SCC lesions are erythematous papules with well‐limited edges. The first symptom of malignancy is induration, which is common to all SCCs. 5 , 8 , 32
3.2. Melanoma
The European Dermatology Forum (EDF) Society defines melanoma as the most malignant skin cancer arising from pigmented nevi, mainly in atypical or unchanged skin, and tends toward early metastasis. 1 , 33 , 34 The melanocytes located in the basal layer of the epidermis break away, causing uncontrolled malignant proliferation.
The clinicopathological classification of invasive melanoma is based on Clark and McGovern's proposal: superficial, lentigo maligna, nodular, and acral lentiginous melanoma. 28 , 35 The most common type, accounting for 75% of all malignant melanomas, is superficial melanoma. A characteristic histological feature is the radial growth of the plaque into deeper layers of tissue past the papillary dermis and the dermal layer via single‐cell dispersion. The most common localization of superficial melanoma is the surface of the back in men and the lower extremities in women and manifest, where the lesions feature irregular borders and asymmetrical shapes. In addition, the lesions have no characteristic color; melanoma can present as white, gray, blue, red, brown, and black. 4 , 36 , 37 Lentigo maligna melanoma is the second most common type. Most often, this form develops in areas exposed to direct sunlight, showing itself as a small macule with an asymmetric shape with an irregular border. Its size and color change as the tumor grows. Nodular melanoma constitutes 15%–30% of aggressive melanomas. It manifests as dark polypoid or pedunculated nodules with a rather small surface area because it grows rapidly deep into the skin layers and relatively slowly in width on the exposed layer. This growth pattern makes late diagnosis likely. The least common melanoma is acral lentiginous melanoma, accounting for only 5% of cases. Acral lentiginous lesions most often appear on the back of the hands, on the palmar side, and in subungual areas, where it appears as a change in pigmentation in the nail plate to dark brown or black. 5 , 38 , 39 Despite the information obtained through the continuous development of clinical, histological, and biochemical methods, the course of melanoma remains very unpredictable. Melanoma is an aggressive malignant neoplasm that, despite favorable survival rates upon detection at an early stage, metastasizes beyond the primary site. In cases of metastases, the therapy options are quite limited because of the resistance to treatment, and the prognosis for patients is bleak. 1 , 30 , 40 , 41 , 42
4. CAUSATIVE FACTORS AND PATHOGENESIS OF SKIN CANCERS
The complex process of skin carcinogenesis resulting from the clonal spread of mutated cells begins with neoplasm initiation and continues with the promotion and progression of neoplastic cells. In the irreversible stage of initiation, genotoxic effects influence normal cells, and in the next stage, the proliferation of initiated cells is reversible, but these cells ultimately progress through a subsequent stage of irreversible malignant transformation characterized by specific karyotypic instability; id est, the distinct stages involve promotion and progression of the malignancy, respectively. 43 Cells that undergo malignant transformation during cancer progression are predisposed to angiogenic responses and unlimited proliferation with the involvement of surrounding tissues and metastases, simultaneously triggering protective mechanisms against therapeutic proliferation‐limiting pathways. 44 , 45 This process is induced via complicated interactions between genetic and environmental factors such as UV radiation, genetic mutations, oncogene activation, malignancy suppressor gene deactivation, and DNA repair process disorder (Figure 1). 3 , 5 In addition to UV radiation, many triggers are associated with skin cancers. Melanoma is defined as an immunogenic neoplasm, as evidenced by the high morbidity in people with treatment‐induced immunosuppression or with acquired immunodeficiency syndrome (AIDS). Additionally, patients taking immunosuppressants are also at high risk for developing SCC. In the case of BCC, chronic exposure to arsenic or a diagnosis of basal cell nevus syndrome (Gorlin syndrome) is a factor, and in the case of SCC, epidermolysis bullosa syndromes, chronic inflammation caused by damage to the skin, and mechanical irritation such as burn scars are factors. SCCs located within genital organs are often associated with the presence of the potentially oncogenic β human papillomavirus (HPV). HPV impairs the DNA repair process, driving carcinogenesis. 4 , 5 , 46 , 47 , 48 , 49 , 50 , 51
Figure 1.
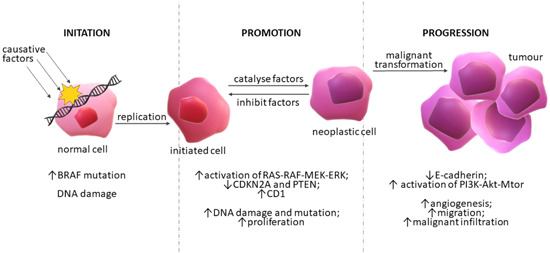
Skin carcinogenesis. Schematic representation of three steps in the process of carcinogenesis initiated by causative factors and a proposed mechanism of melanoma progression. Each stage of the depicted process includes associated mutations in key genes, changes in the cells, and the reversibility of the process [Color figure can be viewed at wileyonlinelibrary.com]
The multifactorial pathogenesis of skin cancers in the greatest number of cases begins with UV radiation exposure, resulting in a cascade of direct and indirect effects, such as DNA damage, gene mutations, immunosuppression, formation of cyclobutane pyrimidine dimers, oxidative stress, and inflammation. Chronic inflammation is characteristic of the pathogenesis of neoplasms, including skin cancers. Inflammation caused by exposure to UV radiation occurs through several mechanisms. In one mechanism, the levels of reactive oxygen species (ROS); cytokines, such as tumor necrosis factor α (TNF‐α), interleukin 6 (IL‐6), interleukin 1β (IL‐1β), cyclooxygenase‐2 (COX‐2); and prostaglandin metabolites are increased. 52 , 53 , 54 In another mechanism aryl hydrocarbon receptors (AhR) in keratinocytes and melanocytes are activated after binding of polycyclic aromatic hydrocarbons and organic environmental pollutants, leading to AhR‐mediated induction of monooxygenases cytochrome P450 (CYP), CYP1A1 and CYP1A2, important enzymes in the metabolism of xenobiotics and often results in excessive generation of ROS, causing oxidative stress, inflammation, and carcinogenesis. 55 , 56 , 57 Moreover, it has been shown that UV radiation exposure influences the course of all the above‐mentioned stages of skin carcinogenesis, although the exact mechanism of action in the promotion and progression stages is unclear. 45 , 58 UV rays degrade keratinocytes and melanocytes, causing malignant mutations, which are especially prevalent in fair skin, which has a low level of dark pigment (eumelanin) to block UV radiation. A correlation has been found between the occurrence of melanoma and increased melanogenesis and overexpression of melanogenic enzymes such as tyrosinase (TYR). 59 , 60 Hence, blood TYR is measured as a marker in the diagnosis of melanoma. 61 , 62 In addition to skin complexion, the harmful mutational UV effect is exacerbated by ozone depletion, latitude, and other factors. 54 , 63 , 64 UVB radiation is much more mutagenic than UVA radiation; it induces changes in adjacent pyrimidines, contributing to the formation of mutagenic cyclopyrimidine dimers and pyrimidine‐pyrimidine adducts. UVA changes DNA through oxidative stress. 8 , 65 , 66 As mentioned, low‐ and high‐risk genes are essential in the process of skin cancer formation. The former includes the melanocortin‐1‐receptor (MC1R), which is closely related to the repair of UV‐damaged DNA and the adaptive pigmentation response by encoding the α‐melanocyte‐stimulating hormone (α‐MSH) receptor critical for melanin synthesis. 63 Key elements in skin cancer and its clinical evaluation are apoptotic pathways such as the tumor suppressors p53, tumor necrosis factor (TNF)‐related apoptosis‐inducing ligand (TRAIL), COX‐2, nuclear factor kappa‐light‐chain‐enhancer of activated B (NF‐κB), epidermal growth factor (EGF) receptor (EGFR), mitogen‐activated protein kinase (MAPK) pathways and the sonic hedgehog (SHH) signaling. 8 , 63 , 64 , 67
A significant portion of NMSC cases, as many as 90% SCC and 50% BCC cases, present with mutation in the p53 suppressor gene inherently involved in cell cycle regulation, apoptosis, and DNA repair through its effect on genes such as p21, Fas, and damage specific DNA binding protein 2 (DDB2). 68 , 69 p53 mutation is rarely found in melanoma. UV ray‐induced mutation results in the induction of NMSC through resistance to apoptosis and clonal keratinocyte expression. Additionally, in SCC and melanoma, p53 has been observed to upregulate Fas/FasL pathway component expression, inducing apoptosis after binding of the Fas receptor to the FasL under physiological conditions. UV radiation exposure inhibits the expression of the death receptor characteristic in TRAIL, as observed in AK and SCC. 8 , 63 , 70 Equally important in the pathogenesis of skin cancers, specific binding of EGFR, belonging to the tyrosine kinase receptor (RTK) family, and involved in most cell signaling processes, such as growth, proliferation, migration, differentiation and cell apoptosis, is changed in a manner measurable by immunohistochemistry. EGFR changes are detected in most cases of NMSC. Overexpression of EGFR in SCC cells contributes to the acquisition of the aggressive phenotype. Activation of EGFR is mediated not only by EGF but also by heparin binding and is a consequence of tumor growth factor (TGF) and amphiregulin activation. Active EGFR forms complexes with signaling proteins including Shc, steroid receptor coactivator (Src), leading to activation of MAPK and the phosphoinositol‑3‑kinase (PI3K) pathways, ultimately leading to cell proliferation, apoptosis, tumor development, cancer cell migration, and metastasis. 63 , 71 , 72 , 73 , 74
Other RTKs in NMSC are also associated with disruptions to the signaling of the complex NF‐κB pathway induced directly by free radicals, carcinogens, X‐rays, and UV radiation or indirectly by binding of cytokines to plasma membrane receptors under the influence of UV radiation. 68 NF‐κB controls many physiological processes and cell proliferation by regulating the cell cycle, apoptosis, and inflammation. As a result of a cascade of mechanisms, activated NF‐κB is translocated to the nucleus, where it promotes the transcription of apoptotic pro‐inflammatory genes and genes targeting cytokine, including interferon, production pathways. 63 , 75 , 76
Activation of the NF‐κB pathway, as well as the MAPK and PI3K pathways, may also be a consequence of the overexpression of COX‐2 induced by UVB radiation. COX‐2 induces in this way inflammation, and cancer cell grows by induction of IL‐6 and catalyzation of the formation of Prostaglandin E2 (PGE2) that is known to bind and activate its G protein‐coupled receptors, prostaglandin E2 receptors (EPs) 1–4 (known as EP1, EP2, EP3, and EP4). The relationship between premalignant lesions and NMSC development and COX‐2 activity has been proven repeatedly, and inhibition of the EP receptors pathways has the potential to prevent cutaneous SCCs 77 ; in addition, COX‐2 inhibitors have been used in the therapy of SCC and BCC. 63 , 78 , 79 , 80 Additionally, the SHH signaling pathway, consisting of transmembrane proteins Ptch1, Smo, and Shh, is important for sporadic and hereditary BCC. However, it is not associated with SCC. Activation of SHH is an underlying mechanism of BCC triggered by point mutation‐induced inactivation of the Ptch1 component. 30 , 63
4.1. The role of microbiome in skin cancers
The microbiome is another aspect correlated with skin cancer, changes to which may relate to mechanisms that increase or decrease the risk of skin cancer. As already mentioned, tissue damage and chronic skin inflammation are closely related to the occurrence of skin cancer. Modulation of inflammatory and immunological processes in the skin occurs through diverse external microbiome environments. Disruption of the normal skin microflora occurs due to environmental exposure, UV radiation, the influence of antibiotics as well as immunosuppressive drugs. Commensal skin bacteria have been shown to reduce inflammation during wound healing by regulating the inflammation‐dependent toll‐like receptors (TLRs) expressed in skin cells directly involved in neoplastic transformation (keratinocytes and melanocytes). Uncontrolled activation of TLRs is closely associated with chronic inflammation and increased likelihood of skin cancer. Hence, a strong correlation between a normal skin microbiome, adequate TLR receptor signaling, and the process of carcinogenesis is noted. 81 Additionally, some commensal HPVs protect against UV‐induced carcinogenesis. On the other hand, Staphylococcus aureus is strongly associated with both AK and SCC, implicating the carcinogenic process by inducing the release of pro‐inflammatory cytokines (interleukin 1α (IL‐1α), and interleukin 36 (IL‐36)) in keratinocytes and thus promoting chronic inflammation. Furthermore, other pro‐inflammatory cytokine‐dependent cytokines regulate the cutaneous colonization of these microorganisms, maintaining the inflammatory loop and ultimately triggering tumor progression. In addition to S. aureus, other bacteria such as S. epidermidis, Escherichia coli, and Pseudomonas aeruginosa modulate inflammatory processes in keratinocytes that underlie oncogenesis. However, another strain, Malassezia reduces excessive colonization of S. aureus while preventing SCC. 82 Moreover, the gut microbiome, in addition to its well‐documented effects on gastrointestinal cancers, also influences dermatoses such as acne vulgaris, atopic dermatitis, psoriasis as well as skin cancers by modulating immune function. The cutaneous immunomodulatory effects of Lactobacillus paracasei have been documented. 81 , 83 , 84 Although there are direct indications linking the skin microbiota and the immune system, the role of the skin microflora both in direct skin carcinogenesis and in modulating the immune system still needs to be clarified.
5. MOLECULAR PATHOGENETIC PATHWAYS IN THE GENESIS OF SKIN CANCER ESPECIALLY MELANOMA
5.1. The mitogen‐activated protein kinase pathway
Currently, many molecular pathways that accompany the transformation of normal melanocytes into benign or melanoma cells, as well as the progression and malignancy of melanomas, are known. Compared to many other human cancers, melanoma is more closely associated with somatic alterations. 85 The most common somatic mutations involve oncogenes neuroblastoma rat sarcoma viral (RAS) viral oncogene homolog (NRAS), v‐raf murine sarcoma viral oncogene homolog B (BRAF), and neurofibromin type 1 (NF1) and suppressor genes phosphatase and tensin homolog (PTEN), p53 and others. These mutations affect cellular processes such as cell proliferation, growth and metabolism, apoptosis, and the cell cycle. The genomic changes induce impairments to the activation of fundamental signaling pathways, namely, the PI3K pathway and the RAS/rapidly accelerated fibrosarcoma (RAF)/mitogen‐activated protein kinase (MEK)/extracellular‐regulated kinase (ERK) signaling cascade, also known as the MAPK pathway (Figure 2). Mutations in the MAPK protein kinase pathway are the most frequently observed, and they have been found in 75%–90% of melanoma cases. The primary mutations affect BRAF (in 60%–80% of cases) and NRAS (in 15%–30% of cases), which are in the same pathway, although they are infrequently mutated at the same time. 86 , 87 , 88 , 89
Figure 2.
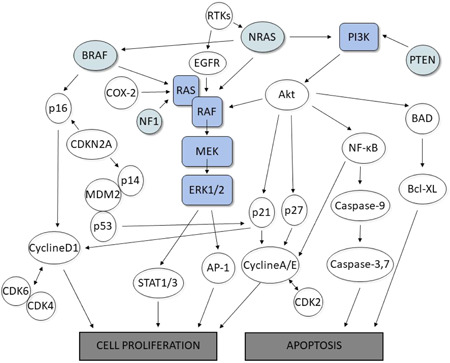
The molecular pathways in skin cancers. Objects highlighted in blue color symbolize RAS/RAF/MEK/ERK signaling cascade, also known as the mitogen‐activated protein kinase (MAPK) pathway. The oncogenes outlined in the green have been identified as the most common oncogenes with somatic mutations in skin cancer, especially melanoma. Presented pathways and steps, connected by arrows showing interdependencies, represent complex signaling pathways leading to cell proliferation or apoptosis [Color figure can be viewed at wileyonlinelibrary.com]
The intracellular MAPK pathway transmits extracellular signals to the nucleus, thereby regulating proliferation, differentiation, and apoptosis. In addition, it is the central platform for the development of melanoma and may enable its initiation or propagation. The mechanism underlying dysregulated signaling involves somatic BRAF mutations, a proto‐oncogene encoding a serine‐threonine protein kinase in the MAPK pro‐growth signaling pathway. These mutations induce genomic instability, enhance cell replicative potential and angiogenesis, inhibit apoptosis, and drive uncontrolled cell proliferation, which plays an important role in the development of melanoma. 31 , 88 Typically, BRAF mutants are results of missense mutations, particularly an amino acid substitution at valine 600, such as V600E, V600K, and V600D, leading to abrogated encoded valine and increased glutamic acid, lysine, aspartic acid, and arginine residues, respectively. 86 , 90 , 91 The BRAF protein has three domains, with two regulatory and one catalytic, involved in the phosphorylation of MEK and adenosine triphosphate (ATP) binding through a hydrophobic interaction with the “glycine‐rich” loop and the activation segment of the catalytic domain in MEK. BRAF mutations generate the replacement of hydrophobic valine with polar and hydrophilic glutamic acid, BRAFV600E. This abnormal domain inversion results in a constitutively active conformation with very high kinase activity, driving melanoma progression. 86 , 92 BRAF mutations are observed not only in cases of metastatic melanomas but also in more than one‐half of benign nevi. They are crucial elements not only in the formation of melanocytic neoplasm but also in cancer progression. 88 , 93 , 94 , 95
The second source of molecular changes related to the activation of the RAS‐RAF‐mitogen in the MAPK pathway is the NRAS oncogene, which is associated with guanosine triphosphate (GTP) binding and regulation of the cellular response to soluble growth factors. NRAS mutations are reported in 15%–30% of melanoma cases, most of which are missense changes in codons 12/13 or 60/61, leading to prolonged NRAS signaling along with activated MAPK and PI3K pathways. 31 , 86 , 88 , 89 As in the case of BRAF mutations, NRAS mutations are observed in patients with metastatic melanomas but also with benign nevi. Both of the described mutations in the MAPK pathway are associated with uncontrolled proliferation and metastatic development of primary melanoma, but these mutants may be used as targets for the development of anti‐melanoma drugs. 88 , 93 , 96 , 97
The least common MAPK mutations include those correlated with suppressor gene NF1. The NF1 protein inhibits RAS signaling by inactivating RAS‐GTP to RAS‐guanosine diphosphate (RAS‐GDP). Ultimately, the NF1 mutation leads to increased activity of NRAS and hence activated MAPK and PI3K pathways. Moreover, the integral cellular component tyrosine‐protein kinase Kit (c‐KIT) receptor, belonging to the previously mentioned RTK family, is also involved in these pathways due to multiple docking sites for proteins such as PI3K, leading to the activation of MAPK signaling pathway. 74 , 98 c‐KIT receptor activation leads to the proliferation and migration of melanoma cells or melanocytes, contributing to melanogenesis and the formation of tumors. 72 , 74 , 86 , 88 , 99 , 100
5.2. The PI3K pathway
A distinct phosphoinositol‑3‑kinase (PI3K) pathway is involved in melanoma cell proliferation and metastasis. Overactivation of the PI3K pathway may be an indirect result of NRAS mutation, as described above, or loss of PTEN function. The PI3K pathway plays a role in inhibiting apoptosis, and in melanoma cells, its increased activity is associated with acquired resistance in melanoma treated with BRAF inhibitors. 101 , 102 Under physiological conditions, PTEN, a suppressor gene, is closely related to the progression of the cell cycle. Additionally, as a protein subject to dephosphorylation and able to regulate cell‐to‐cell adhesion, PTEN deactivates the PI3K pathway and suppresses MAPK signaling. Detectable changes in the PI3K pathway and in PTEN expression are results of chromosomal deletions, missense point mutations, epigenetic mechanisms, or microRNA action. 31 , 86 , 88 , 103
5.3. Oncogenes CDK4 and CDKN2A
Oncogene cyclin‐dependent kinase 4 (CDK4) and the cyclin‑dependent kinase inhibitor 2A (CDKN2A), which encodes p16INK4A, which is expressed in a cyclin‐dependent kinase‑dependent manner, are not only involved in the development of in situ melanoma (familial melanoma) but are also correlated with other malignancies, such as breast and pancreatic cancer. 31 , 97 , 104 , 105 Both CDK4 and CDKN2A participate in the regulation of the cell cycle and regulate the transition of tumor cells from the G1 to S phase and thus can cause uncontrolled proliferation (Figure 2). Cyclin D1 activates the proto‐oncogene CDK4, while p16INK4a has the opposite effect by inhibiting abnormal melanoma cell division. 45 , 86 , 88 , 106 In addition, mutations in a tumor suppressor gene cyclin‑dependent kinase inhibitor 2B (CDKN2B) in benign melanocytic nevi can lead to melanoma development. 31 , 107
6. AVAILABLE THERAPY FOR SKIN CANCERS
As stated by the EDF Society, the mainstay of NMSC treatment is surgery followed by the histological examination of tumor margins, which is required to ensure treatment success and complete removal of the NMSC lesion. 8 , 108 For radical excision intervention, the size, and depth of the infiltrating neoplastic lesion should be taken into consideration. 3 In the case of high‐risk tumors, in addition to surgical excision, Mohs micrographic surgery and radiotherapy are performed. 63 Mohs micrographic surgery is a highly efficient procedure for complete resection of both primary and recurrent BCC and SCC lesions, enabling the identification and complete removal of the tumor. Radiation therapy is used as complementary and palliative therapy in NMSC, but its effectiveness is limited by the inability to introduce it into the therapeutic management of recurrent BCC. 8 Ablative techniques such as electrodesiccation, curettage, and cryotherapy are also recommended for low‐risk NMSC. 63 Electrodesiccation with curettage is a frequent therapeutic method characterized by high effectiveness but limited to use in poorly defined BCC and SCC tumors posing increased risk and presenting with a recurrent nature. 8 Therefore, in the case of SCC with an increased risk of metastases, surgical excision or Mohs surgery, not electrodesiccation or curettage, is recommended. 5 Liquid nitrogen cryotherapy involving cold‐induced NMSC destruction and precise CO2 laser ablation are effective methods for the treatment of low‐risk SCC and BCC. However, tumor removal using a CO2 laser is a rarely used method. 8 For all of the abovementioned methods, it is suggested that chemotherapy and/or immunotherapy be used as supplementary treatment or monotherapy. 63 , 109 Topical therapy with 5% imiquimod is acceptable for application in BCC and Bowen's disease when used with 5‐fluorouracil for regulating key cell receptors. 3 , 5
Primary melanomas detected in the early stages may respond effectively to local therapy involving surgical excision of the neoplastic skin lesion, with a 92% overall survival rate, with marginal cases depending on the pathological staging of the melanoma on the basis of Breslow classification. 3 , 5 , 110 , 111 Additionally, sentinel lymph node dissection and radical removal of surrounding lymph nodes are recommended. 95 , 112 , 113 The use of these methods of treatment at an early stage ensures a high survival rate. However, the prognosis becomes less encouraging with nodal involvement or metastasis, declining to only a 10% chance of 5‐year survival. In the treatment of inoperable melanoma in the advanced stage of this disease, radiotherapy has also been ineffective. 5 , 88 , 114 In recent years, the therapeutic options for metastatic melanoma have significantly expanded and include chemotherapy, immunotherapy, and targeted therapy. 95 , 108 , 115
6.1. Chemotherapy
In the treatment of metastatic melanoma, chemotherapy has been the "standard" for more than 40 years, targeting the pathological pathways of apoptosis or their absence in cancer cells. Monotherapy of melanoma with dacarbazine (DTIC) is mainly a palliative therapy. DTIC is converted to the active alkylating metabolite 3‐methyl‐[triazen‐1‐yl]‐imidazole‐4‐carboxamide. Although DTIC, the first chemotherapeutic treatment approved by the FDA for metastatic melanoma, 116 is the most effective of all available methods of chemotherapy, it is largely ineffective, often inducing no therapeutic response. As a result, DTIC is recommended for use in combination therapy with other cytostatics: vindesine, vinblastine, cisplatin, carboplatin, taxane, carboplatin, or nitrosoureas, such as carmustine, lomustine, or fotemustine. These chemotherapeutic agents exhibit little effect when used as a single chemotherapeutic agent, with the exception of nitrosourea, whose activity is comparable to that of DTIC. However, combination therapy produces a slightly better response but with significant side effects. Currently, chemotherapy is considered a treatment of last resort for patients with resistance to more effective therapies (immunotherapies and targeted therapies) or in countries where access to new more‐effective drugs is limited 3 , 63 , 108 , 114 (Figure 3).
Figure 3.
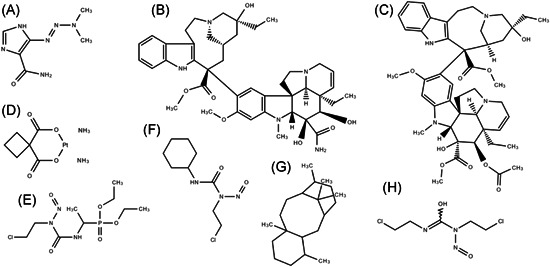
Chemical structures of clinical chemotherapeutics used in skin cancer treatment: dacarbazine (A), vindesine (B), vinblastine (C), carboplatin (D), fotemustine (E), lomustine (F), taxane (G), and carmustine (H)
6.2. Immunotherapy
Immunological treatment, which is one of the basic systemic therapies, is based on the manipulation of targeted immune system responses to melanoma cells. Upon immunostimulation of interleukin 2 (IL‐2) through receptors composed of IL‐2Rα, IL‐2Rβ, and IL‐2Rγ subunits, proliferation and the function of T lymphocytes and natural killer cells are activated; these cells search for melanoma cells expressing a major histocompatibility complex (MHC) molecule absent in all but melanoma cells and then lysing them, inhibiting tumor growth and immune checkpoints. 88 , 114 Despite its effectiveness, IL‐2 therapy is associated with numerous undesirable side effects. Inflammatory reactions, nausea, diarrhea, and capillary leak syndrome are observed in 16%–17% of patients receiving an intravenous infusion of IL‐2. 3 , 117 Interferon‐α (IFN‐α) administered after surgical excision as an adjunct therapy in patients with metastatic melanoma to inhibit the proliferation of residual melanoma cells continuously stimulates the activity of lymphocytes such as CD4+ and the secretion of interferon‐ γ (IFN‐γ) and IL‐2, leading to a long‐term immune response. 118 Unfortunately, patients in greatly advanced stages of melanoma show a low response to this therapy and, similar to IL‐2 therapy, IFN‐α induces cytotoxicity, especially during long‐term treatment. IFN‐α and IL‐2 constitute the main immunotherapies used with melanoma patients. The response rate to these treatments increases after biochemotherapy administration because of the combination of both immunotherapeutic agents and cytotoxic chemotherapeutic agents, such as DTIC, which is the third element of this therapy. 88 , 114 A breakthrough in the development of novel immunotherapies has led to a therapy based on suppressing the immune response to the tumor microenvironment. Monoclonal antibodies, such as ipilimumab, nivolumab, and pembrolizumab, against the programmed death‐1 (PD‐1)/programmed death ligand‐1 (PD‐L1) inhibitory pathway that exists in the immune system to prevent the immune cells destroy normal host cells (autoimmunity), resulting in modulation of T lymphocyte activity and consequently to the immune‐mediated tumor destruction and thus improving the overall survival time, even for people with advanced melanoma. 3 , 63 , 86 In summary, IL‐2 immunostimulation, tumor‐blockades of T cell proliferation, and immune checkpoints are major targets of the immune response in melanoma immunotherapy. 95 , 108 , 119 , 120
6.3. Targeted therapy
According to The European Interdisciplinary Guideline developed by the EDF society, targeted therapy takes advantage of the frequent mutations in MAPK pathway genes in melanoma patients by introducing highly selective BRAFV600 inhibitors, such as vemurafenib and dabrafenib. 3 , 95 , 108 Despite previous results of clinical trials confirming high efficacy of vemurafenib and dabrafenib treatment, improved survival, and high tolerance, a large proportion of the patients developed resistance to treatment with BRAF inhibitors, which in turn caused reactivation of the MAPK pathway. Hence, regimens of combination therapies that include MEK inhibitors important in the MAPK cascade, such as cobimetinib and trametinib, are being explored. 3 , 86 , 95 , 119 , 121 The effectiveness of targeting this therapeutic route has been confirmed in preclinical studies proving increased apoptosis and delayed onset of treatment resistance. 3 , 121 The combination of BRAF inhibitors and MEK inhibitors administered to melanoma patients with an activating BRAFV600E mutation resulted in a significant improvement in survival and responses. 86 , 122 Despite the success of novel therapies based on MAPK pathway inhibitors, a number of undesirable effects have also been revealed. Notably, therapies that include vemurafenib and dabrafenib can lead to SCC. 3 , 86 , 88 In cases of some melanomas with an activating type III transmembrane receptor tyrosine kinase (KIT) mutation, a different targeted therapy may be an alternative approach. However, KIT therapy with imatinib is considered controversial and is still undergoing improvements. 3 , 63
7. BIOACTIVITY OF LUTEOLIN AND ITS DERIVATIVES ON SKIN CANCER
The antitumor activity of flavonoids, in general, has been extensively described and documented thus far. 74 , 80 , 123 , 124 , 125 , 126 , 127 , 128 Therefore, we present in detail our findings on the modulation of oncogenic skin cancer pathways by luteolin and its derivatives. Luteolin, a natural flavonoid commonly found in many plant raw materials, exhibits multiple biological effects, including anti‐inflammatory, antioxidant, antiallergic, and anticancer properties. Therefore, it seems to be a promising source with preventive and therapeutic potential for the treatment of various cancers, including skin cancers. 22 , 58 , 129 , 130 , 131 , 132 , 133 , 134 Moreover, because luteolin increases the therapeutic response of cancer cells, luteolin can be used as a complementary therapy. 45 , 135 , 136
The antitumor activity of luteolin has been found as a result of inhibited induction of apoptosis, disruption of the cell cycle, inhibition of cell proliferation and/or migration, and/or angiogenesis associated with increased invasiveness and tumor development. Notably, the proliferation and development of neoplastic cells in vitro are inhibited through a number of different pathways and the expression of many diverse genes. 45 , 123 , 134 , 137 , 138 , 139 , 140 , 141 , 142 , 143 , 144 , 145 Furthermore, another important aspect is the ability of luteolin to multidimensionally regulate epigenetics layers in the case of cancers, affecting the apoptotic effect. This occurs via inhibition of DNA methylation and trimethylation in histone H3 at lysine 27 (H3K27me3), activation of nuclear factor 2 associated erythroid 2 (Nrf2) demethylation, as well as inhibition of DNA methyltransferases (DNMTs), enhancer of zeste homolog 2 (EZH2), p53 and expression of key genes in cell cycle. 24 , 146 , 147 In addition to reducing methylation of the Nrf2 promoter region, binding of (ten‐eleven translocation‐1) TET1 to the Nrf2 promoter and formation of a complex between p53 and Nrf2, are determined as the subsequent molecular mechanism underlying such proapoptotic activity of luteolin. 148 Luteolin induced both intracellular and extracellular apoptosis of C32 human amelanotic melanoma cells, which was confirmed by its effect on mitochondrial potential and the activity of caspase‐3, caspase‐8, caspase‐9, and caspase‐10 while stimulating autophagy. 149 It was also reported to downregulate the PI3K/protein kinase B (AKT) axis through downregulation of oncogenes fibroblast growth factor 10 (FGF10) and fibronectin 1 (FN1), and matrix metalloproteinases (MMPs) MMP‐2 and MMP‐9, and upregulation of tissue inhibitors of MMPs (TIMPs) TIMP‐1 and TIMP‐2, thereby inhibiting migration, inducing apoptosis, disrupting cell integrity and reducing the invasive potential of A375 human malignant melanoma cells. Furthermore, its inhibitory effect was also reported in an in vivo model. 150 , 151 , 152 , 153 However, in vivo, the tumor showed adaptation and acquired resistance to luteolin treatment. Changes in A375 cells under the influence of luteolin, particularly FGF10 and FN1 genes, strongly influenced the expression of kinase suppressor of RAS 2 (KSR2), suggesting inhibition of the RAS pathway and downregulation of a number of components involved in ECM modifications (intercellular adhesion molecule‐1 (ICAM1), laminin subunit alpha 1 (LAMA1), integrin alpha 2b (ITGA2B), and FN1) and proteinases such as a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTSs) ADAMTS1 and ADAMTS18, MMP‐1, MMP‐10, cathepsins (CTS) CTSG, CTSK, and CTSV). Luteolin exerts a significant influence on the proliferation and invasion of melanoma cells by disrupting the ECM pathway by suppressing, for instance, MMP‐1, MMP‐2, or MMP‐10 expression in other human malignant melanoma lines (SK‐MEL‐2, SK‐MEL‐28, and WM3211 cells), regardless of whether or not BRAF was mutated. In the case of the WM3211 cell line (wild‐type for NRAS and BRAF), luteolin induced the rather rare downregulation of KIT expression, which was crucial for inhibiting the growth of these cells. 151 Moreover, strong cytotoxicity and proapoptotic activity induced in these cells through arrest to the cell cycle and an accumulated number of these cells in the G0/G1 phase were demonstrated. 16 It has been documented that luteolin can inhibit cell proliferation through cell cycle arrest in the G1 phase as a result of the inhibition of CDK2 activity, enhanced expression of CDKN1A, and regulation of CDK inhibitors in A375 and C32 cells. 149 , 151
Luteolin can have both anti‐ or pro‐oxidant activity, which is at least partly determined by the cellular milieu. In malignant cells, which often contain already an increased oxidative stress level because of their upregulated metabolism, luteolin induces apoptosis by endoplasmic reticulum (ER) stress via increasing ROS levels. However, in healthy cells, luteolin shows antioxidative effects are described. 154 The relationship between cell proliferation, apoptosis, and the induction of luteolin‐induced ROS levels, expression of ER stress, and CCAAT/enhancer‐binding protein‐homologous protein (CHOP) protein has been demonstrated in A2058 human metastatic melanoma cells. 155 On the other hand, Schomberg and co‐authors examining four different melanoma lines, SK‐MEL‐2, SK‐MEL‐28, A375, and WM3211 cells, led to diametrically opposed conclusions. They hypothesized that it was not the increase in luteolin‐induced ROS production that directly caused the inhibition of cell growth but it was probably the synergism of simultaneous modifying effects on multiple pathways, including aforementioned pathways associated with the ECM, the oncogenic signaling pathway, and immune response pathways. 151 Interestingly, according to the results of studies comparing the effects of luteolin on two different melanoma lines, the SK‐MEL‐1 human metastatic melanoma cell line shows greater resistance to treatment than the B16F10 mouse primary melanoma cell line; however, the differences are insignificant, and it would be appropriate to further investigate the resistance of cells according to their origin. 156 , 157
Luteolin inhibited the invasive epithelial–mesenchymal transition (EMT) process, which induces morphological changes in melanoma cells and is involved in melanoma progression. 158 , 159 This effect is a result of the reduced expression of MMP‐9, reversed cadherin switching (downregulated N‐cadherin and upregulated E‐cadherin) in human epidermoid carcinoma and melanoma cells (A431‐III, A431, A375, and B16F10 cells), 160 , 161 , 162 , 163 and reduced expression of the E‐cadherin gene in WM3211 cells, 151 contributing to the reduction in the invasive abilities of these cells, as well as inhibited tumor growth and progression. Moreover, integrin β3 (ITGβ3) inhibition, changes in EMT signaling, and suppressed metastasis caused by luteolin treatment were also observed in an in vivo C57BL/6 mouse model established with B16F10 cells. 153 , 161 The S100A7 protein may mediate EMT activation, leading to the emergence of additional neoplastic lesions. Luteolin decreased the signaling of Src/focal adhesion kinase (FAK), Src/signal transducer and activator of transcription 3 (STAT3), and S100A7 protein, thereby reducing the migratory abilities of A431‐III cells. 164 , 165 , 166 High invasiveness is a particular feature of A431‐III cells that overexpress MMP‐9 that is not evident in primary A431 cells. The effect of luteolin on MMP‐9 may result from inhibition of Akt phosphorylation, while inhibition of N‐cadherin expression may result in inhibition of the expression of the MAPK‐ERK pathway. 160 It has been observed that luteolin induces apoptosis by regulating B‐cell lymphoma 2 (Bcl‐2) and Bcl‐2‐associated X (Bax) proteins in B16F10 cells, 167 inhibiting the secretion of MMP‐2 and MMP‐9 and changing the phosphorylation level of components of the EGFR signaling pathway in A431 cells, and the metastatic potential of these cells may be realized upon EGFR inhibition. 162 Luteolin also exerts a strong chemopreventive effect against melanoma by targeting protein kinase Cε (PKCε) and Src. It has been proven that treatment of the JB6 P+ mouse melanoma cell line that this flavone leads to suppressed expression of PKCε and Src kinase and inhibition of the UVB‐induced activity of COX‐2, activator protein‐1 (AP‐1) and NF‐κB. 152
The process of melanogenesis can be described in two ways. Melanogenesis is a physiological mechanism conferring protection against the harmful effects of UV radiation. Hence, it prevents the malignant transformation causing skin cancer. 168 On the other hand, excessive melanogenesis, melanin deposition, and the related potential cytotoxic risk are associated with melanogenesis and, hence, melanoma. 169 , 170 Hence, melanogenesis can be considered a target for therapy aimed at the elimination of malignant melanocytes and, at the same time, a target for chemopreventive action. 60 , 61
It has been documented that luteolin inhibits melanogenesis in B16F10 cells but not by reducing the level of TYR protein, as might be expected because luteolin exerts the opposite effect in the absence and presence of α‐MSH. The antimelanogenic activity is attributed to the ability of luteolin to inhibit the catalytic activity of TYR and the expression of exogenous human TYR regulated through a pathway‐dependent cyclic adenosine monophosphate (cAMP). 171 , 172 However, in HMV‐II human vaginal melanoma cells, the antimelanogenic effect was based on the opposing mechanism: Luteolin promoted melanin production by stimulating the activity of intracellular TYR. 173 Yamauchi et al. compared the proliferation of B16F10 cells and the extent of melanogenesis inhibition under the influence of luteolin and concluded that luteolin exhibits inhibitory activity only on extracellular melanogenesis and not on intracellular melanogenesis, as previously expected. 174 , 175 Melanogenesis is related to the microphthalmia‐associated transcription factor (MITF) a transcription factor of melanogenic enzymes, that is influenced by c‐Jun N‐terminal kinase (JNK), which together with p53 activates the apoptotic pathway; hence, the inhibition of B16F10 cell melanogenesis confirms the previously described proapoptotic effect of luteolin. 176 , 177
The ability to inhibit the melanogenesis of melanoma precursor cells has also been documented in the case of luteolin derivatives. The inhibitory potential of eight luteolin derivatives on extracellular melanogenesis and B16F10 cell proliferation was compared, with the results demonstrating the dependence of the antitumor effect on the length of the hydrocarbon 7‐O‐ substitution. Hence, luteolin showed the lowest activity of the compounds tested, followed by 7‐O‐methylluteolin, 7‐O‐ethylluteolin, 7‐O‐propylluteolin, 7‐O‐butylluteolin, 7‐O‐pentylluteolin, and 7‐O‐hexylluteolin, which showed the highest activity. However, the bulkiness of the substituent at position 7 did not have a significant effect on the inhibition of the compared process; that is, 7‐O‐(1‐methyl)propylluteolin and 7‐O‐methylcyclohexylluteolin showed activity similar to that of 7‐O‐ethylluteolin, 7‐O‐propylluteolin, 7‐O‐butylluteolin, and 7‐O‐pentylluteolin. 174 Another notable derivative in the context of melanogenesis inhibition is luteolin 7‐sulfate, which simultaneously inhibited the synthesis of new TYR proteins and the catalytic activity of existing TYR proteins in B16F10 cells. The inhibition of TYR gene expression was related to the signaling pathway mediated by cAMP response element‐binding protein (CREB) and MITF, which in this case, may explain the antimelanogenic activity of luteolin 7‐sulfate; its antimelanogenic action is several dozen folds greater than that of the known melanogenesis inhibitor arbutin and higher than the activity of luteolin itself. 178 , 179 The results of the antitumor effects of luteolin and its derivatives are summarized in Table 1.
Table 1.
Antitumor activities of luteolin derivatives in relation to skin cancer
| Luteolin derivative | Cell line | Inhibition of proliferation | Effect of action | Molecular target | Refs. | |
|---|---|---|---|---|---|---|
| IC50 (μg/ml) | Incubation time | |||||
| Luteolin | B16F10 | 0.7 | 72 h | ↓Proliferation | Not evaluated | 140 |
| >14.3 | 24 h | ↓Proliferation | Not evaluated | 180 | ||
| >14.3 | 72 h | |||||
| Not detected | ↑Melanogenesis | ↑TYR, ↑ CREB | 175 | |||
| >28.6 | 72 h | ↓Proliferation, ↓melanogenesis | ↓TYR, ↓ cAMP | 172 | ||
| Not detected | ↓Metastasis, ↓invasion, ↓progression, ↓EMT | ↓FAK, ↓ N‐cadherin, ↑E‐cadherin | 161 | |||
| >14.3 | 24 h | ↓Proliferation | Not evaluated | 157 | ||
| >14.3 | 72 h | |||||
| >14.3 | 24 h | ↓Proliferation | Not evaluated | 156 | ||
| >14.3 | 72 h | |||||
| >57.3 | 48 h | ↓Melanogenesis | ↓TYR | 171 | ||
| >28.6 | 24 h | ↓Melanogenesis | ↓TYR | 181 | ||
| 3.5 | 24 h | ↓Proliferation | Not evaluated | 182 | ||
| 9.8 | 72 h | ↓Extracellular melanogenesis, ↓proliferation | Not evaluated | 174 | ||
| 1.6 | 48 h | ↓Melanogenesis, ↓proliferation | Not evaluated | 141 | ||
| 5 | 48 h | ↓Melanogenesis, ↓proliferation | Not evaluated | 178 | ||
| 6 | – | ↓Proliferation | Not evaluated | 142 | ||
| 8.1 | 24 h | ↓Melanogenesis, ↓proliferation | Not evaluated | 143 | ||
| 4.3 | 48 h | ↓Proliferation | Not evaluated | 183 | ||
| 7 | 48 h | ↓Melanogenesis, ↓proliferation | ↓TYR | 179 | ||
| 41.2 | 24 h | ↓Proliferation, ↓migration, ↓invasion, ↓adhesion, ↓metastasis, ↓EMT | ↓N‐cadherin, ↑E‐cadherin, ↓MMP‐2, ↓MMP‐9, ↓p‐Akt, ↓HIF‐1α, ↓VEGF‐A, ↓ p‐VEGFR‐2 | 163 | ||
| 18.4 | 48 h | |||||
| 15.8 | 72 h | |||||
| A375 | 40.3 | 24 h | ↓Proliferation, ↓migration, ↓invasion, ↓adhesion, ↓metastasis, ↓EMT | ↓N‐cadherin, ↑E‐cadherin, ↓MMP‐2, ↓MMP‐9, ↓p‐Akt, ↓HIF‐1α, ↓VEGF‐A, ↓ p‐VEGFR‐2 | 163 | |
| 18.6 | 48 h | |||||
| 12.7 | 72 h | |||||
| 10.4 | 24 h | ↓Proliferation, ↑apoptosis, ↓migration, ↓invasion | ↓MMP‐2, ↓MMP‐9, ↑TIMP‐1, ↑TIMP‐2, ↓pAkt1, ↓PI3K, ↓ PI3K/Akt | 150 | ||
| 5.3 | 48 h | |||||
| 32.9 | 24 h | ↓Proliferation, ↑apoptosis, ↑G0/G1 phase | Not evaluated | 16 | ||
| 3.6 | 72 h |
↓Proliferation, ↑apoptosis, ↓invasion, ↑G1 phase |
↓CSF2RA, ↓ ANGPT1, ↓ FGF10, ↓ FN1, ↓ MAPK, ↓ PI3K, ↑ KSR2, ↓ RAS, ↑ CDKN1A, ↓ KRAS, ↓ BRAF, ↓ MAP2K2 (MEK2), ↓ CD274, ↓ IL24, ↓ CXCL8, ↓ NGFR, ↓ MMP‐1, ↓MMP‐10, ↓ECM | 151 | ||
| 5.2 | 24 h | ↓Proliferation | Not evaluated | 139 | ||
| 9.7 | 24 h | ↓Proliferation | Not evaluated | 144 | ||
| 6.5 | 48 h | |||||
| 5.8 | 72 h | |||||
| C32 | 95.1 | 24 h |
↓Proliferation, ↑autophagy, ↑apoptosis, ↓mitochondrial membrane potential, ↑G2/M phase, ↑S phase, ↓ G1 phase |
↑Caspase‐3, ↑caspase‐8, ↑caspase‐9, ↑caspase‐10 | 149 | |
| 2.4 | 48 h | ↓Proliferation | Not evaluated | 139 | ||
| A2058 | 35 | 48 h |
↓Proliferation, ↑apoptosis, ↑ER stress, ↑chemopreventive effect, ↑intracellular ROS |
↑Phospho PERK, ↑ phospho eIF2α, ↑ATF6, ↑ CHOP, ↑ caspase‐12 | 155 | |
| Colo829 | 2.1 | 72 h | ↓Proliferation | Not evaluated | 151 | |
| SK‐MEL‐1 | >14.3 | 24 h | ↓Proliferation | Not evaluated | 157 | |
| >14.3 | 72 h | |||||
| >14.3 | 24 h | ↓Proliferation | Not evaluated | 156 | ||
| >14.3 | 72 h | |||||
| SK‐MEL‐2 | 4.8 | 72 h | ↓Proliferation, ↑apoptosis, ↓invasion | ↓BRAF, ↓ HBEGF, ↓ Src, ↓NF1, ↓ NRTN, ↓ SPRED, ↓ MAPK, ↓ JAK3, ↓ MMP‐1, ↓MMP‐2, ↓MMP‐10, ↓ECM, ↓ CDH1 | 151 | |
| SK‐MEL‐28 | 3.4 | 72 h | ↓Proliferation, ↑apoptosis, ↓invasion, ↑chemopreventive effect, ↑intracellular ROS | ↓GDNF, ↓ MAPK, ↓ SHC2, ↓ DLC1, ↓ RASAL1, ↓ JAK3, ↓ MMP‐1, ↓ MMP‐2, ↓ ECM | ||
| SK‐MEL‐5 | 9.2 | 48 h | ↓Proliferation | Not evaluated | 184 | |
| A431 | >14.3 | 72 h | ↓Proliferation | Not evaluated | 185 | |
| 5.4 | 24 h | ↓Proliferation, ↑apoptosis, ↓metastasis | ↓EGFR, ↓ EGF, ↓ MMP‐2, ↓MMP‐9 | 162 | ||
| 25.6 | 24 h | ↓Proliferation | Not evaluated | 182 | ||
| Not detected | ↓Migration, ↓invasion, ↓progression, ↓EMT | ↓MMP‐9, ↓EGFR | 160 | |||
| A431‐III | Not detected | ↓Migration, ↓invasion, ↓progression, ↓EMT | ↓N‐cadherin, ↑E‐cadherin, ↓MMP‐9 | |||
| 7.5 | 24 h | ↓Proliferation, ↓metastasis, ↓migration, ↓invasion, ↓EMT | ↓p‐Src, ↓pSTAT3, ↓S100A7, ↓ Src/FAK ↓ Src/STAT3/S100A7, ↓ ECM, ↓ MMP, ↓ RPS12, ↓ RPS19, ↓ Akt/mTOR/c‐Myc | 164 | ||
| Not detected | 186 | |||||
| 15.9 | 24 h | 187 | ||||
| Not detected | 165 | |||||
| WM3211 | 1.9 | 72 h | ↓Proliferation, ↑apoptosis, ↓invasion, ↑chemopreventive effect, ↑intracellular ROS | ↓KIT, ↑ NRAS, ↓ MAP2K2, ↓ IL24, ↓ NGFR, ↓ MMP‐1, ↓MMP‐2, ↓MMP‐10, ↓ECM | 151 | |
| MDA‐MB‐435 | 8.7 | 48 h | ↓Proliferation | Not evaluated | 145 | |
| HMV‐II | Not detected | ↑Intracellular melanogenesis | ↑Intracellular TYR | 173 | ||
| HMB‐2 | 7 | 24 h | ↑Chemopreventive effect, ↓proliferation, ↓ROS | Not evaluated | 137 | |
| JB6 P+ | Not detected | ↑Chemopreventive effect | ↓PKCε, ↓Src, ↓COX‐2, ↓AP‐1, ↓NF‐κB | 152 | ||
| UACC‐62 | 2.9 | 48 h | ↓Proliferation | Not evaluated | 188 | |
| Luteolin 6‐glucoside | B16F10 | >44.8 | 48 h | ↓Invasion, ↓melanogenesis | Not evaluated | 141 |
| >89.7 | 48 h | ↓Melanogenesis | ↓TYR, ↓ TRP1, ↓ DCT, ↓ MITF, ↓ CREB, ↓ cAMP | 170 | ||
| Luteolin 7‐sulfate | 43.5 | 48 h | ↓Melanogenesis, ↓proliferation | ↓TYR, ↓ MITF, ↓ CREB | 178 | |
| 69.1 | 48 h | ↓Melanogenesis, ↓proliferation | ↓TYR | 179 | ||
| Luteolin 7‐methyl ether | 8.4 | 72 h | ↓Extracellular melanogenesis, ↓proliferation, ↑apoptosis | Not evaluated | 174 | |
| Luteolin 7‐ethyl ether | >15.7 | 72 h | ||||
| Luteolin 7‐propyl ether | 5.3 | 72 h | ||||
| Luteolin 7‐butyl ether | 4.6 | 72 h | ||||
| Luteolin 7‐pentyl ether | 3.6 | 72 h | ||||
| Luteolin 7‐hexyl ether | 2.4 | 72 h | ||||
| Luteolin 7‐(1‐methylpropyl) ether | 4.4 | 72 h | ||||
| Luteolin 7‐methylcyclohexyl ether | 5.2 | 72 h | ||||
| Ugonin J | >21.1 | 72 h | ↓Extracellular melanogenesis, ↓proliferation | ↓TYR | 189 | |
| Ugonin K | >21.8 | 72 h | ||||
| Ugonin L | >21.8 | 72 h | Not demonstrated | Not demonstrated | ||
| Luteolin 3′‐methyl ether | B16F10 | 17 | 24 h | ↓Proliferation | Not demonstrated | 182 |
| A431 | 15.4 | 24 h | ↓Proliferation | Not demonstrated | ||
| Luteolin 4′‐methyl ether | B16F10 | >20 | 24 h | ↓Proliferation, ↑apoptosis, ↓invasion | ↑Caspase‐3 | 190 |
| Luteolin 4′,5,7‐trimethyl ether | UACC‐62 | >250 | 48 h | Not demonstrated | Not evaluated | 138 |
| Luteolin 7‐sambubioside | C32 | >300 | 24 h | ↓Proliferation | Not evaluated | 149 |
| Luteolin 7‐glucoside | >300 | 24 h | ||||
| 12.5 | 48 h | ↓Proliferation | Not evaluated | 139 | ||
| A375 | 13.1 | 48 h | Not evaluated | |||
| UACC‐62 | 9.4 | 48 h | ↓Proliferation | Not evaluated | 188 | |
| B16F10 | >31.4 | 48 h | ↓Proliferation | Not evaluated | 191 | |
| >100 | 24 h | ↓Melanogenesis | Not evaluated | 192 | ||
7.1. Chemoprevention action
Skin cancer prevention is based on several schemes based on primary and secondary prevention. According to the National Cancer Institute, both schemes are based on public education: The primary prevention effort is based on the principles of photoprotection and the effects of increased exposure to sunlight, and secondary effort for skin cancer prevention is based on screening precancerous lesions for early diagnosis and detection. 23 , 193 , 194 , 195 Chemopreventive agents for melanoma are used not only to prevent the occurrence of neoplastic lesions but also to inhibit their development and promote remission. However, because of the complexity of the transformation of melanocytes under the influence of UV radiation and the poor patient response to chemopreventive agents in clinical trials, chemoprevention of melanoma is a purely perfunctory scheme. 58 , 196 , 197
A large body of evidence suggests that luteolin, due to its antioxidant and anti‐inflammatory properties, may also play an important role in the reduction of skin cancer progression and photocarcinogenesis and thus has a significant impact on the prevention of skin cancer. As described in the previous sections, oxidative stress is inextricably linked to the processes of tumor formation. Luteolin, due to its structure of 3‐OH, 4′‐OH, and the double bond between carbons C2‐C3 and a carbonyl group on C4, removes ROS through its own oxidation, which has been confirmed by studies on cell‐free systems. 198 , 199 Moreover, it blocks ROS‐producing oxidases, which are components in the lipoxygenase reaction involving the chelation of transition metal ions, and protects endogenous antioxidants, thus enhancing their action. 200 These antioxidant abilities distinguish the ability of luteolin and its derivatives to protect against ROS‐induced activation of MAPK, NF‐κB, and COX‐2, as well as damage to lipids, DNA, and proteins, as confirmed not only through studies of cell‐free systems but also in vitro and in vivo experiments, 137 , 172 , 201 , 202 , 203 thus preventing the development of cancer. 131 , 204 , 205 Moreover, the ability to induce the apoptosis of neoplastic cells, including melanoma cells, has been attributed to the pro‐oxidative property of this flavone. 155 However, as previously mentioned, Schomberg et al., 141 examining five lines of cutaneous melanoma, came to the opposite conclusion, suggesting that the induction of ROS is a negligible side effect of luteolin treatment of melanoma while confirming its proapoptotic effect. 151 ROS‐related apoptosis is most likely due to cytotoxicity‐related suppression of the NF‐κB pathway and activation of JNK, and this activation relationship has been confirmed in lung cancer; however, in melanoma, this relationship is based only on speculation. 45 , 174 , 206 Indeed, the antioxidant activity may be correlated with NF‐κB, as confirmed with studies of the JB6 P+ melanoma cell line showing the potential of luteolin to suppress UVB radiation‐induced NF‐κB, COX‐2, and AP‐1 expression, mainly by targeting PKCε and Src. 152
Additionally, luteolin has been shown to directly inhibit PKCε/Src activity and prevent UVB‐induced DNA damage in keratinocyte cells. There is evidence for the preventive potential of luteolin in skin cancers associated with, inter alia skin photodamage as well as Nrf2 activity, closely related to oxidative stress. Luteolin suppresses the expression of COX‐2, AP‐1, and NF‐κB, regulates antioxidant enzymes, and prevents ROS accumulation and activation of MAKP and NF‐κB signaling pathways. 207 The antioxidant activity of luteolin is a partial contribution to its anti‐inflammatory effect, which is also related to the prevention of the carcinogenesis process due to the convergence of chronic inflammation and cancer. 45 , 56 This luteolin effect is mediated by cells such as neutrophils and lymphocytes; TNF‐α and IL‐6 release‐related inhibition; and signaling pathways involving these factors as previously described. Luteolin also blocks the production of the aforementioned cytokines due to the inhibition of NF‐κB and kinases involved in the MAPK signaling pathways and activation of inhibitory‐κB kinase (IKK). 208 , 209 , 210 Furthermore, luteolin was found to inhibit UVB radiation‐induced MMP‐1 expression in the human keratinocyte cell line HaCaT, as well as UVB radiation‐induced activation of AP‐1, a well‐known transcription factor mediating inflammation and proliferation, as well as the MMP‐1 promoter, c‐Fos and c‐Jun, which make up the AP‐1 complex. 45 , 76 , 211 The photoprotective effect of luteolin on keratinocytes was also demonstrated by Verschooten and co‐authors, proving an increase in the resistance of normal cells to UVB radiation‐induced apoptosis, with an adverse effect on malignant keratinocytes. 212 In an in vivo model, the anti‐inflammatory effect of luteolin 7‐O‐glucoside was demonstrated by inhibiting the synthesis of COX‐2, IL‐1β, and TNF‐α, which are closely related to inflammation upon exposure to UVB radiation. 213 , 214
7.2. The structure–activity relationship of luteolin derivatives
SAR analysis offers the possibility to isolate the chemical groups and structures critical for induced biological response and to correlate structural features to their activity. Unfortunately, due to the use of different cell lines, different analysis conditions, and measurement methods, it is not possible to carry out SAR analysis on the basis of documented IC50 values obtained through heterogeneous techniques and the many accidents. Reports on the growth inhibitory effects against different skin cancer cell lines have not always been the same, indicating differences in the sensitivity of melanoma cells to the tested compounds. Despite these difficulties, a correlation between the position, number, and nature of substituents in the structure of luteolin and its derivatives and their antiproliferative activity has been identified.
The high antiproliferative activity of luteolin was first identified with the presence of a C2‐C3 double bond in the C ring, the presence of hydroxyl groups at C5 and C7 in the A ring, and a catechol group containing two adjacent phenolic OH groups (3′,4′‐di‐OH). Moreover, it has been shown that the C ring with an oxo group function at position C4 contributes to the high activity of compounds in this class. All the described structural elements determine and are required for high anticancer activity. 140 , 142 , 156 , 157 , 162 , 215 However, certain kinds of structural modifications improve or eliminate this activity. Special attention has been directed to the number and O‐methylation, and O‐glycosylation status of the free hydroxyl groups at the C7 position in the A ring.
During the comparison of the cytotoxic and/or antiproliferative activity of flavonoids, it was documented that, in addition to the saturation of the C2‐C3 bond, the presence of a methyl group in the structure of these compounds significantly enhances their effect. In particular, the 7‐O‐methoxyl group on the A ring of luteolin is associated with this effect. Moreover, the length of the linear hydrocarbon substituent at the C7 position of the A ring has a directly proportional effect on the antiproliferative activity (Figure 4). Additionally, it has been hypothesized that a group of these substituents attached to luteolin may stimulate the activation of JNK, which is strongly involved in melanogenesis, in addition to apoptosis, as confirmed by Yamauchi et al. 174 However, the mass of the substituent at position C7 has no significant effect on the inhibitory effect of the compound on proliferation or melanogenesis. 174 , 175 The location of the O‐methyl substituent is substantial. The attachment of a methyl group at C3′ reduces the antiproliferative activity compared to the occupation of the C7 position, which confirms that 3′‐OH moiety in the structure of luteolin plays a vital role in determining high anticancer activity. The analogous situation with the participation of the O‐methylation of 4′‐OH results in a deepening of the abolition of the discussed antitumor activity. Moreover, simultaneous attachment of methyl groups at positions 5 and 7 of the A ring and at position 4′ of the B ring significantly attenuates the antiproliferative effect, suggesting that the introduction of a higher number of methoxyl groups in the B ring (C4′) and A ring (C5) leads to decreased antitumor activity. Hence, the O‐methylation of 7‐OH seems to be crucial, and increasing the length of the linear hydrocarbon substituent in the A ring increases the activity. Although the 7‐O‐methoxy group in the A ring of luteolin shows satisfactory activity, the attachment of other functional groups at C7, such as a sulfo group, abolishes the antiproliferative effect.
Figure 4.
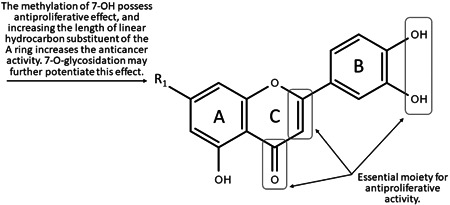
Chemical groups responsible for luteolin derivatives activity (SAR)
As previously mentioned, the presence of hydroxyl groups at C7 in the A ring significantly affects the antiproliferative activity of luteolin. O‐glycosylation at this position may further potentiate this effect. 139 , 188 However, C‐glycosylation at the C6 position significantly reduces cytotoxicity induced in melanoma cells with concomitant inhibited melanogenesis, as observed for luteolin 6‐C‐glucoside (isoorientin) in the B16F10 cell line. 141 , 170 Moreover, not only C‐glycosylation of 6‐OH result in a lack of antiproliferative activity, as observed for ugonins J, K, and L. 189
The lack of ‐OH substitution on C3 in the structure of luteolin creates the possibility of competitive binding of luteolin to the ATP‐binding site important to the activity of kinases (including PKCε, EGFR, and FAK), which may play an essential role in skin tumors, and more specifically, in the case of luteolin treatment, inhibiting kinase action. 126
The influence of substituents in the B ring is not sufficiently understood. It has been suggested that the 3′‐OH moiety in the structure of luteolin, which, among other actions, determines the high anticancer activity of the compound, probably influences cell cycle arrest to a critical level. 215 However, in the case of modification to the structure of luteolin, we have data only in the case of O‐methyl group introduction, instead of 4′‐OH, with simultaneous attachment of 5,7‐di‐OH. The antiproliferative activity of 4’,5,7‐trimethoxyluteolin is not significant, but it is not possible to state clearly what aspect of the structure determines this activity.
8. ROLE OF LUTEOLIN AND ITS DERIVATIVE‐RICH EXTRACTS IN SKIN CANCER TREATMENTS
It has been reported that the hydroalcoholic extract of Rosmarinus officinalis, of which luteolin is one of the main compounds present at 0.2%, has an antiproliferative effect through cytotoxic and cytostatic mechanisms resulting in the induction of apoptosis and cell cycle arrest of A375 cells. The accumulation of apoptotic cells in the sub‐G0 phase and arrest in the G0/G1 and G2/M phases was observed, which is consistent with the action of known anticancer substances. 144 A similar effect of Jasione montana diethyl ether extract in the C32 cell line has been described, and the main component critical for the activity was presumed to be luteolin, which was confirmed by studies on the activity of this compound alone. 149 Studies on both J. montana or Phyllodium elegans extracts in the C32 and A375 lines, respectively, proved the apoptotic potential of the extracts on the reduction mitochondria membrane potential as well as the increase of caspase‐3 and caspase‐9 activation. 216 It has been established that the antioxidant activity of natural compounds is not correlated with their antiproliferative activity in cancer cell lines; their pro‐oxidative properties are critical for their effects. 217 However, Cattaneo and co‐authors found that the pro‐oxidative effect of rosemary extract does not directly mediate its cytotoxic activity. The inhibition of the expression of proteins crucial for maintaining cellular homeostases, such as protein disulfide‐isomerase A3 (PDIA3), glucosidase II alpha subunit (GANAB), PCB1, and PCB2, causing ER stress, was identified as the molecular mechanism underlying the induced cytotoxicity. Although luteolin is one of the main components of this extract, it did not directly influence the antiproliferative activity but probably had a synergistic effect in the multicomponent activity. 144
The effects of Petroselinum crispum and Matricaria chamomilla extracts, which are abundant in polyphenols and flavonoids, including luteolin and its 7‐O‐glucoside, were tested in A375 cells. Despite the negligible antiproliferative activity and minor effect on the cell cycle distribution, P. crispum extract showed proapoptotic potential by increasing the expression of caspase‐3, which is the executioner caspase in the process of apoptosis and DNA damage, 219 , 220 and caspase‐2, as confirmed by staining with Annexin V‐PI. The M. chamomilla extract, containing 8% of the content of luteolin and its derivative, was found to have slightly weaker activity. However, these dominant compounds in both extracts were apigenin and its glycosidic forms, with a predominance of apigenin glucoside in the M. chamomilla extract. The content of luteolin and its derivative was only 5% of the polyphenol content in the P. crispum extract. 220 However, in the case of the J. montana extract, the effect on caspase‐3 activation as well as on caspase‐8, caspase‐9, and caspase‐10 is attributed to luteolin, the predominant component in the extract. 149 The aqueous extract of Olea europaea leaves also was found to be proapoptotic in B16F10 cells, with its effect mediated via ERK1/2 and p53 activating pathways. However, Majumder and co‐authors suggested that the underlying mechanism of ERK1/2 receptor interference is most likely triggered by oleuropein, which is one of the main components of this extract, not luteolin. However, due to the wide range of plant matrices, it can be assumed that this effect may be caused by multicomponent synergism in this fraction. 25
In the B16F10 line, an apoptosis‐stimulating effect was also shown by the hydromethanolic extract of Biophytum sensitivum, participating in the regulation of Bcl‐2 and p53 genes and catalyzing the expression of the aforementioned caspase‐3. In addition, Guruvayoorappan and co‐authors reported that an extract rich in flavonoids is characterized by an antimetastatic nature and the inhibition of pro‐inflammatory cytokines that play a significant role in chemoprevention, such as TNF‐α, IL‐1β, IL‐6, and granulocyte‐macrophage colony‐stimulating factor (GM‐CSF). 221 In addition, it has been proven that the extract of B. sensitivum as well as Daphne gnidium reduces the invasion and mobility of B16F10 cells in models of C57BL/6 and Balb/C mice in vivo. 191 Furthermore, it activates the expression of the tissue metalloproteinase inhibitors TIMP‐1 and TIMP‐2 and subsequently limiting the expression of MMP‐2 and MMP‐9. Changes in pro‐inflammatory cytokines had been tested in an in vitro model, 221 as well as ERK1/2 and signal transducer and activator of transcription‐1 (STAT1) pathway expression, and prolyl hydroxylase, lysyl oxidase, nucleoside diphosphate kinase (NDPK), and vascular endothelial growth factor (VEGF) levels. It can be assumed that a number of flavonoids, including luteolin 7‐methyl ester and isoorientin, are critical for the antimetastatic and anti‐malignant activity in neoplastic cells. 222 , 223
It has been suggested that the biological activity of the water extract of Gentiana veitchiorum flowers is caused by isoorientin, a main flavonoid constituting 0.4% of this extract. Due to the presence of this derivative, the extract significantly suppresses the melanin content of B16F10 cells by inhibiting the mRNA expression of TYR, transient receptor potential (TRP), and dopachrome tautomerase (DCT), as well as inhibiting MITF transcription and CREB phosphorylation. Additionally, it is suspected that the activity of isoorientin causes the arrest of the intracellular cAMP pathway. 170 A similar outcome has been observed with Phyllospadix iwatensis extract, except that luteolin 7‐sulfate was critical for its antimelanogenic activity. 179 Moreover, high activity of TYR inhibition and melanogenesis, correlating with the high content of luteolin and its derivatives, is observed in the case of Asphodelus microcarpus extracts. 224 The results of the antitumor effects of species‐rich in luteolin and its derivatives are summarized in Table 2.
Table 2.
Antitumor activities of luteolin and its derivative‐rich species in relation to skin cancer
| Species | Cell line | Inhibition of proliferation | Effect of action | Molecular target | Refs. | |
|---|---|---|---|---|---|---|
| IC50 (µg/ml) | Incubation time | |||||
| Ailanthus excelsa | C32 | 36.5 | 48 h | ↓Proliferation | Not evaluated | 139 |
| A375 | 78.4 5 | 48 h | ||||
| Ajuga chamaepitys | B16F10 | 406.7 | 24 h | ↓Proliferation | ↓NF‐κB | 225 |
| Ajuga genevensis | B16F10 | 741.4 | 24 h | |||
| Ajuga laxmannii | B16F10 | 236.8 | 24 h | |||
| Anastatica hierochuntica | B16F10 | Not detected | ↓Melanogenesis | Not evaluated | 181 | |
| Artemisia princeps | B16F10 (MM) | 80.6 | 48 h | ↓Melanogenesis, ↓proliferation | Not evaluated | 141 |
| Arthrophytum scoparium | B16F10 | >100 | 48 h | ↓Melanogenesis | Not evaluated | 226 |
| Asphodelus microcarpus | B16F10 | 400 | 48 h | ↓Melanogenesis, ↓proliferation | ↓TYR | 224 |
| Biophytum sensitivum | B16F10 | >10 | 24 h |
↓Metastasis, ↓invasion, ↑apoptosis, ↓proliferation |
↓MMP‐2, ↓MMP‐9, ↓ERK‐1, ↓ERK‐2, ↓VEGF, ↓ IL‐1β, ↓TNF‐α, ↓IL‐6, ↓GM‐CSF, ↓ Bcl‐2, ↑p53, ↑caspase‐3 | 221 , 222 |
| Citrus volkameriana | UACC62 | >100 | 48 h | ↓Proliferation | Not evaluated | 227 |
| Daphne gnidium | B16F10 | Not detected | ↑Apoptosis | ↓CTL | 191 | |
| Gentiana veitchiorum | B16F10 | >2 000 | 48 h | ↓Melanogenesis | ↓TYR, ↓ TRP1, ↓ DCT, ↓ MITF, ↓ CREB, ↓ cAMP | 170 |
| Hyssopus seravshanicus | B16F10 | Not detected | ↑Melanogenesis | Not evaluated | 228 | |
| Jasione montana | C32 | 119.7 | 24 h |
↓Proliferation, ↑autophagy, ↑apoptosis, ↓mitochondrial membrane potential, ↑G2/M phase, ↑S phase, ↓ G1 phase |
↑Caspase‐3, ↑caspase‐8, ↑caspase‐9, ↑caspase‐10 | 149 |
| Jatropha tanjorensis | A431 | 58.5 | 48 h | ↓Proliferation | Not evaluated | 229 |
| Matricaria chamomilla | A375 | >60 | 72 h | ↑Apoptosis, ↑G1 phase | ↑Caspase‐2, ↑caspase‐3 | 220 |
| Olea europaea | B16F10 | 91.8 | 24 h | ↓Proliferation, ↑apoptosis | ↑ERK1/2, ↑p53 | 25 |
| HTB‐140 | >50 | 24 h | Not evaluated | Not evaluated | 230 | |
| WM793 | >50 | 24 h | ||||
| A375 | >100 | 24 h | ↓Proliferation | Not evaluated | 231 | |
| Penthorum chinense | B16F10 | >100 | 24 h | ↓Proliferation, ↓melanogenesis, ↑autophagy | ↓TYR, ↓ MITF, ↑ LC3B | 232 |
| Petroselinum crispum | A375 | >60 | 72 h | ↑Apoptosis, ↑sub‐G1 phase | ↑Caspase‐2, ↑caspase‐3 | 220 |
| Phyllodium elegans | A375 | 117.2 | 24 h | ↓Proliferation, ↓metastasis, ↑apoptosis, ↓mitochondrial membrane potential | ↑Caspase‐3, ↑caspase‐9, ↑MuD | 216 |
| Phyllospadix iwatensis | B16F10 | >300 | 48 h | ↓Melanogenesis | ↓TYR | 179 |
| Pinus koraiensis | A375 | >1000 | 48 h | Not demonstrated | Not evaluated | 233 |
| Plantago lagopus | UACC‐62 | 66.1 | 48 h | ↓Proliferation | Not evaluated | 188 |
| Rosmarinus officinalis | A375 | 63.0 | 72 h | ↓Proliferation, ↑apoptosis, ↑sub‐G0 and ↓G0/G1 phases, ↓intracellular ROS | ↓PDIA3, ↓ GANAB, ↓ PCB1, ↓ PCB2 | 144 |
| Sonneratia caseolaris | B16F10 | >100 | 24 h | ↓Melanogenesis | Not evaluated | 192 , 234 |
9. LIMITATIONS AND CHALLENGES
When comparing the IC50 values of luteolin and its derivatives on the viability of cells in the same line, the method used, and incubation time, significant discrepancies between the extreme values can be observed. Therefore, the authors of this review analyzed the conditions of the experiments performed and the criteria of the methods used for determining the IC50 values for the compounds shown in Table 1. For example, the difference between IC50 values in the range of 5.2–32.9 μg/ml in studies describing the activity of luteolin in the A375 cell line after 24‐h incubation seems to be related to the use of different methods for assessing cell viability. The sulforhodamine B (SRB) assay used by Said et al. (IC50 = 5.2 μg/ml) is widely used to test cytotoxicity in cell‐based studies but is not based on measurement of metabolic activity, in contrast to the 2,3‐bis‐(2‐methoxy‐4‐nitro‐5‐sulfophenyl)−2H‐tetrazolium‐5‐carboxanilide (XTT) assay used by George et al. (IC50 = 32.9 μg/ml), in which XTT was metabolically reduced by the enzyme mitochondrial dehydrogenase in living cells to the water‐soluble product formazan. In both of these cases, the same type of medium (DMEM) was used, which differed from that used in other studies with the same cell line, such as the studies performed by Yao and co‐authors as well as Cattaneo and co‐authors, who used RPMI medium. The results of the studies by Yao and co‐authors and by Cattaneo and co‐authors were evaluated by another tetrazolium salt, substitute for XTT, 3‐(4,5‐dimethylthiazol‐2‐yl)−2,5‐diphenyltetrazolium bromide (MTT) assay performed under the same analysis conditions and showed very similar IC50 values of 10.4 and 9.7 μg/ml, respectively. Hence, the choice of not only the method to determine activity level but also of the culture medium seems to be important. Moreover, in many cases, it has been observed that the incubation time plays a critical role in determining the IC50 value, as shown in the results from luteolin treatment of, for example, C32 cells (Table 1). Notably, cell proliferation involves many biochemical processes, many of which depend on each other, and the studied natural compounds can influence the processes at different stages or in different ways. Moreover, the proven antiproliferative potential of luteolin definitively varies according to the diversity and complexity of the gene expressed in different cell lines. Therefore, certain mutations in different in vitro cell models can render a cell line resistant to the action of this flavone, as perfectly illustrated by the differences in the B16F10 and A375 cell lines shown in Table 1. It can be assumed that the differences between these extreme results are due to the use of different solvents with the test compound or the use of different cell media enriched with various substances. In these cases, it is difficult to standardize and compare IC50 values reflecting a proliferation‐depressing effect, as additional factors appear to influence the outcomes. In addition, cell lines should not be passaged too many times or cultured for too long, and the test substances, despite their high purity, are subject to degradation, especially when dissolved in solvent and stored under unsuitable conditions.
In vitro and in vivo, luteolin and its derivatives exhibit a diverse antitumor response depending on the model adopted. Although encouraging results render luteolin and its derivatives as potential anticancer agents for the treatment of skin cancer, the effects of these natural compounds in therapeutic intervention are quite complex due to the genetically related mechanisms underlying their activity. Therefore, the molecular mechanisms in each cell need to be understood, and the selectivity, efficacy, pharmacological and toxicological properties of luteolin and its derivatives need to be analyzed, which will be crucial in translating them from laboratory studies to clinical trials. Extensive clinical studies are needed to determine whether luteolin, as well as its derivatives, can act in a manner similar to that described in various in vitro and in vivo models. Currently, only 16 studies are listed in the ClinicalTrials.gov database (http://www.clinicaltrials.gov), of which none is addressing the use of luteolin in skin cancer. However, clinical trials of luteolin intervention for tongue SCC can be found, offering potential hope for topical application in skin cancer as well. Considering the low bioavailability of natural compounds in general and of luteolin and its derivatives in particular and because oral administration is challenging, topical administration seems to be an alternative. 235 Due to its lipophilicity, luteolin is able to penetrate deep into human skin. This effect has been confirmed with in vivo studies demonstrating its ability not only to absorb into the skin surface but also to penetrate deeper skin layers, thus providing potential for use in topical therapy of skin cancer. 131 , 132 However, there is still a need to develop a suitable formulation. The hope is for new pharmaceutical systems that provide, in addition to higher drug solubility, increased biochemical stability, and bioavailability, as well as controlled release of the drug in the target tissue. Particularly noteworthy are nanosystems such as liposomes, polymerosomes, dendrimers, nanotubes, quantum dots, nano micelles, nanogels, polymer nanoparticles, nanospheres, magnetic nanoparticles, solid lipid nanoparticles, nanostructured lipid carriers, which provide impressive advances in skin cancer therapy by efficiently transporting the therapeutic agent through the stratum corneum and delivering it to deeper skin layers. They have been proven to have high skin penetration, controlled reaching to the specific tumor site, bioavailability, and their efficacy and specificity. Moreover, recent research is focused on developing new proper nanotechnology‐based forms of skin cancer therapy. 236 , 237 , 238 , 239 Additionally, the therapeutic efficacy of nanoparticles like liposomes has also been confirmed for other cancers as potential drug delivery systems also approved for clinical use. 240 , 241 , 242 However, a prerequisite for a therapeutic effect are realistic conditions with effective concentrations at the target site, for which quantitative studies are necessary. Although the dermal penetration of luteolin offers hope for its use in topical therapy, further scientific study on luteolin and its derivatives is still needed to obtain a clear description of its dermal penetration, dosing strategies, development of a suitable pharmaceutical formulation, prolongation of topical drug release, stability, and optimal dose. In addition, it is crucial to determine the full safety and bioavailability of these compounds in patient studies because, despite their relative safety and use in children with autism, 151 , 243 cases of exacerbated chemical colitis in mice have been reported after oral administration. 244
The anticancer efficacy of luteolin and its derivatives supports further research aimed at the development of new treatment options for skin cancer. Further work is needed to evaluate their use in preclinical and clinical studies to obtain a clear picture of the effects of these natural compounds from a biochemical point of view.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
ACKNOWLEDGMENT
The study was funded by Project no. POWR.03.02.00‐00‐I051/16 from European Union funds, POWER 2014–2020, Grant no. 05/IMSD/G/2019.
Juszczak AM, Wöelfle U, Končić MZ, Tomczyk M. Skin cancer, including related pathways and therapy and the role of luteolin derivatives as potential therapeutics. Med Res Rev. 2022;42:1423‐1462. 10.1002/med.21880
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Matthews NH, Li W‐Q, Qureshi AA, Weinstock MA, Cho E. Epidemiology of melanoma. In: Ward WH, Farma JM, eds. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017:3‐23. [PubMed] [Google Scholar]
- 2. Girschik J, Fritschi L, Threlfall T, Slevin T. Deaths from non‐melanoma skin cancer in Western Australia. Cancer Causes Control. 2008;19(8):879‐885. [DOI] [PubMed] [Google Scholar]
- 3. Orthaber K, Pristovnik M, Skok K, Perić B, Maver U. Skin cancer and its treatment: novel treatment approaches with emphasis on nanotechnology. J Nanomater. 2017;2017(2):1‐20. [Google Scholar]
- 4. Diepgen TL, Mahler V. The epidemiology of skin cancer. Br J Dermatol. 2002;146(s61):S1‐S6. [DOI] [PubMed] [Google Scholar]
- 5. Linares MA, Zakaria A, Nizran P. Skin cancer. Prim Care. 2015;42(4):645‐659. [DOI] [PubMed] [Google Scholar]
- 6. Apalla Z, Lallas A, Sotiriou E, Lazaridou E, Ioannides D. Epidemiological trends in skin cancer. Dermatol Pract Concept. 2017;7(2):1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katalinic A, Kunze U, Schafer T. Epidemiology of cutaneous melanoma and non‐melanoma skin cancer in Schleswig‐Holstein, Germany: incidence, clinical subtypes. Br J Dermatol. 2003;149(6):1200‐1206. [DOI] [PubMed] [Google Scholar]
- 8. Madan V, Lear JT, Szeimies RM. Non‐melanoma skin cancer. Lancet. 2010;375(9715):673‐685. [DOI] [PubMed] [Google Scholar]
- 9. Rastrelli M, Tropea S, Rossi CR, Alaibac M. Melanoma: epidemiology, risk factors, pathogenesis, diagnosis and classification. In Vivo. 2014;28(6):1005‐1011. [PubMed] [Google Scholar]
- 10. Chen ST, Geller AC, Tsao H. Update on the epidemiology of melanoma. Curr Dermatol Rep. 2013;2(1):24‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reichrath J, Rass K. Ultraviolet damage, DNA repair and vitamin D in nonmelanoma skin cancer and in malignant melanoma: an update. In: Reichrath J, ed. Sunlight, Vitamin D and Skin Cancer. Springer; 2014:208‐233. [DOI] [PubMed] [Google Scholar]
- 12. Leiter U, Garbe C. Epidemiology of melanoma and nonmelanoma skin cancer‐the role of sunlight. Adv Exp Med Biol. 2008;624:89‐103. [DOI] [PubMed] [Google Scholar]
- 13. Fransen M, Karahalios A, Sharma N, English DR, Giles GG, Sinclair RD. Non‐melanoma skin cancer in Australia. Med J Aust. 2012;197(10):565‐568. [DOI] [PubMed] [Google Scholar]
- 14. Rubin AI, Chen EH, Ratner D. Basal‐cell carcinoma. N Engl J Med. 2005;353(21):2262‐2269. [DOI] [PubMed] [Google Scholar]
- 15. Apalla Z, Calzavara‐Pinton P, Lallas A, et al. Histopathological study of perilesional skin in patients diagnosed with nonmelanoma skin cancer. Clin Exp Dermatol. 2016;41(1):21‐25. [DOI] [PubMed] [Google Scholar]
- 16. George VC, Kumar DRN, Suresh PK, Kumar S, Kumar RA. Comparative studies to evaluate relative in vitro potency of luteolin in inducing cell cycle arrest and apoptosis in HaCat and A375 cells. Asian Pac J Cancer Prev. 2013;14(2):631‐637. [DOI] [PubMed] [Google Scholar]
- 17. Newman DJ, Cragg GM. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J Nat Prod. 2012;75(3):311‐335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chinembiri TN, Du Plessis LH, Gerber M, Hamman JH, Du Plessis J. Review of natural compounds for potential skin cancer treatment. Molecules. 2014;19(8):11679‐11721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. da Rocha AB, Lopes RM, Schwartsmann G. Natural products in anticancer therapy. Curr Opin Pharmacol. 2001;1(4):364‐369. [DOI] [PubMed] [Google Scholar]
- 20. Cragg GM, Newman DJ. Plants as a source of anti‐cancer agents. J Ethnopharmacol. 2005;100(1–2):72‐79. [DOI] [PubMed] [Google Scholar]
- 21. Aumeeruddy MZ, Mahomoodally MF. Combating breast cancer using combination therapy with 3 phytochemicals: piperine, sulforaphane, and thymoquinone. Cancer. 2019;125(10):1600‐1611. [DOI] [PubMed] [Google Scholar]
- 22. Imran M, Rauf A, Abu‐Izneid T, et al. Luteolin, a flavonoid, as an anticancer agent: a review. Biomed Pharmacother. 2019;112:108612. [DOI] [PubMed] [Google Scholar]
- 23. Iqbal J, Abbasi BA, Ahmad R, et al. Potential phytochemicals in the fight against skin cancer: current landscape and future perspectives. Biomed Pharmacother. 2019;109:1381‐1393. [DOI] [PubMed] [Google Scholar]
- 24. Penta D, Somashekar BS, Meeran SM. Epigenetics of skin cancer: interventions by selected bioactive phytochemicals. Photodermatol Photoimmunol Photomed. 2018;34(1):42‐49. [DOI] [PubMed] [Google Scholar]
- 25. Majumder D, Debnath M, Libin Kumar KV, et al. Metabolic profiling and investigations on crude extract of Olea europaea L. leaves as a potential therapeutic agent against skin cancer. J Funct Foods. 2019;58:266‐274. [Google Scholar]
- 26. Ijaz S, Akhtar N, Khan MS, et al. Plant derived anticancer agents: a green approach towards skin cancers. Biomed Pharmacother. 2018;103:1643‐1651. [DOI] [PubMed] [Google Scholar]
- 27. Chahar MK, Sharma N, Dobhal MP, Joshi YC. Flavonoids: a versatile source of anticancer drugs. Pharmacogn Rev. 2011;5(9):1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Müller CSL, Reichrath J. Histology of melanoma and nonmelanoma skin cancer. In: Reichrath J, ed. Sunlight, Vitamin D and Skin Cancer. Springer; 2014:215‐226. [Google Scholar]
- 29. Emmert S, Schön MP, Haenssle A. Molecular biology of basal and squamous cell carcinomas. In: Reichrath J, ed. Sunlight, Vitamin D and Skin Cancer. Springer; 2014:171‐191. [Google Scholar]
- 30. Khavari PA. Modelling cancer in human skin tissue. Nat Rev Cancer. 2006;6(4):270‐280. [DOI] [PubMed] [Google Scholar]
- 31. Pal HC, Hunt KM, Diamond A, Elmets A, Afaq C, F. Phytochemicals for the management of melanoma. Mini Rev Med Chem. 2016;16(12):953‐979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Röwert‐Huber J, Patel MJ, Forschner T, et al. Actinic keratosis is an early in situ squamous cell carcinoma: a proposal for reclassification. Br J Dermatol. 2007;156(s3):8‐12. [DOI] [PubMed] [Google Scholar]
- 33. Tsatmali M, Ancans J, Thody AJ. Melanocyte function and its control by melanocortin peptides. J Histochem Cytochem. 2002;50(2):125‐133. [DOI] [PubMed] [Google Scholar]
- 34. Garbe C, Amaral T, Peris K, et al. European consensus‐based interdisciplinary guideline for melanoma. Part 1: diagnostics – update 2019. Eur J Cancer. 2020;126:141‐158. [DOI] [PubMed] [Google Scholar]
- 35. Goyanna R, Torres ET, Broders AC. Histological grading of malignant tumors: Broder's method. Hospital. 1951;39(6):791‐818. [PubMed] [Google Scholar]
- 36. Abbasi NR, Shaw HM, Rigel DS, et al. Early diagnosis of cutaneous melanoma: revisiting the ABCD criteria. JAMA. 2004;292(22):2771‐2776. [DOI] [PubMed] [Google Scholar]
- 37. Vestergaard ME, Macaskill P, Holt PE, Menzies SW. Dermoscopy compared with naked eye examination for the diagnosis of primary melanoma: a meta‐analysis of studies performed in a clinical setting. Br J Dermatol. 2008;159(3):669‐676. [DOI] [PubMed] [Google Scholar]
- 38. Park HS, Cho KH. Acral lentiginous melanoma in situ: a diagnostic and management challenge. Cancers. 2010;2(2):642‐652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ward WH, Lambreton F, Goel N, Yu JQ, Farma JM. Clinical presentation and staging of melanoma. In: Ward WH, Farma JM, eds. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017:79‐91. [PubMed] [Google Scholar]
- 40. Belter B, Haase‐Kohn C, Pietzsch J. Biomarkers in malignant melanoma: recent trends and critical perspective. In: Ward WH, Farma JM, eds. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017:39‐57. [PubMed] [Google Scholar]
- 41. Erdei E, Torres SM. A new understanding in the epidemiology of melanoma. Expert Rev Anticancer Ther. 2010;10(11):1811‐1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Califano J, Nance M. Malignant melanoma. Facial Plast Surg Clin North Am. 2009;17(3):337‐348. [DOI] [PubMed] [Google Scholar]
- 43. Pitot HC. Multistage carcinogenesis – genetic and epigenetic mechanisms in relation to cancer prevention. Cancer Detect Prev. 1993;17(6):567‐573. [PubMed] [Google Scholar]
- 44. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57‐70. [DOI] [PubMed] [Google Scholar]
- 45. Lin Y, Shi R, Wang X, Shen H‐M. Luteolin, a flavonoid with potentials for cancer prevention and therapy. Curr Cancer Drug Targets. 2008;8(7):634‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Walter SD, King WD, Marrett LD. Association of cutaneous malignant melanoma with intermittent exposure to ultraviolet radiation: results of a case‐control study in Ontario, Canada. Int J Epidemiol. 1999;28(3):418‐427. [DOI] [PubMed] [Google Scholar]
- 47. Gilchrest BA, Eller MS, Geller AC, Yaar M. The pathogenesis of melanoma induced by ultraviolet radiation. N Engl J Med. 1999;340(17):1341‐1348. [DOI] [PubMed] [Google Scholar]
- 48. National Cancer Institute (NIH) Skin Cancer Treatment (PDQ®)–Health Professional Version. Accessed July 13, 2021. https://www.cancer.gov/types/skin/hp/skin-treatment-pdq
- 49. Koh HK. Cutaneous melanoma. N Engl J Med. 1991;325(3):171‐182. [DOI] [PubMed] [Google Scholar]
- 50. Preston DS, Stern RS. Nonmelanoma cancers of the skin. N Engl J Med. 1992;327(23):1649‐1662. [DOI] [PubMed] [Google Scholar]
- 51. English DR, Armstrong BK, Kricker A, Winter MG, Heenan PJ, Randell PL. Case‐control study of sun exposure and squamous cell carcinoma of the skin. Int J Cancer. 1998;77(3):347‐353. [DOI] [PubMed] [Google Scholar]
- 52. Hensler S, Mueller MM. Inflammation and skin cancer: old pals telling new stories. Cancer J. 2013;19(6):517‐524. [DOI] [PubMed] [Google Scholar]
- 53. Jiao J, Mikulec C, Ishikawa T, et al. Cell‐type‐specific roles for COX‐2 in UVB‐induced skin cancer. Carcinogenesis. 2014;35(6):1310‐1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. D'Orazio J, Jarrett S, Amaro‐Ortiz A, Scott T. UV Radiation and the skin. Int J Mol Sci. 2013;14(6):12222‐12248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dupont E, Gomez J, Bilodeau D. Beyond UV radiation: a skin under challenge. Int J Cosmet Sci. 2013;35(3):224‐232. [DOI] [PubMed] [Google Scholar]
- 56. George VC, Vijesh VV, Amararathna DIM, et al. Mechanism of action of flavonoids in prevention of inflammation‐associated skin cancer. Curr Med Chem. 2016;23(32):3697‐3716. [DOI] [PubMed] [Google Scholar]
- 57. Vogel CFA, Van Winkle LS, Esser C, Haarmann‐Stemmann T. The aryl hydrocarbon receptor as a target of environmental stressors – implications for pollution mediated stress and inflammatory responses. Redox Biol. 2020;34:101530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chhabra G, Ndiaye MA, Garcia‐Peterson LM, Ahmad N. Melanoma chemoprevention: current status and future prospects. Photochem Photobiol. 2017;93(4):975‐989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hara H, Lee MH, Chen H, Luo D, Jimbow K. Role of gene expression and protein synthesis of tyrosinase, TRP‐1, lamp‐1, and CD63 in UVB‐induced melanogenesis in human melanomas. J Invest Dermatol. 1994;102(4):495‐500. [DOI] [PubMed] [Google Scholar]
- 60. Riley PA. Melanogenesis: a realistic target for antimelanoma therapy? Eur J Cancer. 1991;27(9):1172‐1177. [DOI] [PubMed] [Google Scholar]
- 61. Riley PA. Melanogenesis and melanoma. Pigment Cell Res. 2003;16(5):548‐552. [DOI] [PubMed] [Google Scholar]
- 62. Prichard RS, Dijkstra B, McDermott EW, Hill ADK, O'Higgins NJ. The role of molecular staging in malignant melanoma. Eur J Surg Oncol. 2003;29(4):306‐314. [DOI] [PubMed] [Google Scholar]
- 63. Simões MCF, Sousa JJS, Pais AACC. Skin cancer and new treatment perspectives: a review. Cancer Lett. 2015;357(1):8‐42. [DOI] [PubMed] [Google Scholar]
- 64. Nikolaou V, Stratigos AJ. Emerging trends in the epidemiology of melanoma. Br J Dermatol. 2014;170(1):11‐19. [DOI] [PubMed] [Google Scholar]
- 65. Rünger TM. How different wavelengths of the ultraviolet spectrum contribute to skin carcinogenesis: the role of cellular damage responses. J Invest Dermatol. 2007;127(9):2103‐2105. [DOI] [PubMed] [Google Scholar]
- 66. Ridley AJ, Whiteside JR, McMillan TJ, Allinson SL. Cellular and sub‐cellular responses to UVA in relation to carcinogenesis. Int J Radiat Biol. 2009;85(3):177‐195. [DOI] [PubMed] [Google Scholar]
- 67. Bouwes JNB, Plasmeijer EI, Feltkamp MCW. Beta‐papillomavirus infection and skin cancer. J Invest Dermatol. 2008;128(6):1355‐1358. [DOI] [PubMed] [Google Scholar]
- 68. Amin ARMR, Kucuk O, Khuri FR, Shin DM. Perspectives for cancer prevention with natural compounds. J Clin Oncol. 2009;27(16):2712‐2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Bode AM, Dong Z. Post‐translational modification of p53 in tumorigenesis. Nat Rev Cancer. 2004;4(10):793‐805. [DOI] [PubMed] [Google Scholar]
- 70. Nickoloff BJ, Qin J‐Z, Chaturvedi V, Bacon P, Panella J, Denning MF. Life and death signaling pathways contributing to skin cancer. J Investig Dermatol Symp Proc. 2002;7(1):27‐35. [DOI] [PubMed] [Google Scholar]
- 71. Uribe P, Gonzalez S. Epidermal growth factor receptor (EGFR) and squamous cell carcinoma of the skin: molecular bases for EGFR‐targeted therapy. Pathol Res Pract. 2011;207(6):337‐342. [DOI] [PubMed] [Google Scholar]
- 72. Hubbard SR, Miller WT. Receptor tyrosine kinases: mechanisms of activation and signaling. Curr Opin Cell Biol. 2007;19(2):117‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Oda K, Matsuoka Y, Funahashi A, Kitano H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol Syst Biol. 2005;1:2005.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Teillet F, Boumendjel A, Boutonnat J, Ronot X. Flavonoids as RTK inhibitors and potential anticancer agents. Med Res Rev. 2008;28(6):715‐745. [DOI] [PubMed] [Google Scholar]
- 75. Feehan RP, Shantz LM. Molecular signaling cascades involved in nonmelanoma skin carcinogenesis. Biochem J. 2016;473(19):2973‐2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Oeckinghaus A, Ghosh S. The NF‐kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect Biol. 2009;1(4):a000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Moon H, White AC, Borowsky AD. New insights into the functions of COX‐2 in skin and esophageal malignancies. Exp Mol Med. 2020;52(4):538‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Maglio DHG, Paz M, Cela E, Leoni J. Cyclooxygenase‐2 overexpression in non‐melanoma skin cancer: molecular pathways involved as targets for prevention and treatment. In: La Porta C, ed. Skin Cancers – Risk Factors, Prevention and Therapy. IntechOpen Limited; 2011:272. [Google Scholar]
- 79. Buckman SY, Gresham A, Hale P, et al. COX‐2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19(5):723‐729. [DOI] [PubMed] [Google Scholar]
- 80. Nichols JA, Katiyar SK. Skin photoprotection by natural polyphenols: anti‐inflammatory, antioxidant and DNA repair mechanisms. Arch Dermatol Res. 2010;302(2):71‐83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sherwani MA, Tufail S, Muzaffar AF, Yusuf N. The skin microbiome and immune system: potential target for chemoprevention? Photodermatol Photoimmunol Photomed. 2018;34(1):25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Squarzanti DF, Zavattaro E, Pizzimenti S, Amoruso A, Savoia P, Azzimonti B. Non‐melanoma skin cancer: news from microbiota research. Crit Rev Microbiol. 2020;46(4):433‐449. [DOI] [PubMed] [Google Scholar]
- 83. Yu Y, Champer J, Beynet D, Kim J, Friedman AJ. The role of the cutaneous microbiome in skin cancer: lessons learned from the gut. J Drugs Dermatol. 2015;14(5):461‐465. [PubMed] [Google Scholar]
- 84. Rodríguez‐Daza MC, Pulido‐Mateos EC, Lupien‐Meilleur J, Guyonnet D, Desjardins Y, Roy D. Polyphenol‐mediated gut microbiota modulation: toward prebiotics and further. Front Nutr. 2021;8:689456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Akbani R, Akdemir KC, Aksoy BA, et al. Genomic classification of cutaneous melanoma. Cell. 2015;161(7):1681‐1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Leonardi GC, Falzone L, Salemi R, et al. Cutaneous melanoma: from pathogenesis to therapy (Review). Int J Oncol. 2018;52(4):1071‐1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chappell WH, Steelman LS, Long JM, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR inhibitors: rationale and importance to inhibiting these pathways in human health. Oncotarget. 2011;2(3):135‐164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Liu Y, Sheikh MS. Melanoma: molecular pathogenesis and therapeutic management. Mol Cell Pharmacol. 2014;6(3):228. [PMC free article] [PubMed] [Google Scholar]
- 89. Fedorenko IV, Gibney GT, Smalley KSM. NRAS mutant melanoma: biological behavior and future strategies for therapeutic management. Oncogene. 2013;32(25):3009‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rubinstein JC, Sznol M, Pavlick AC, et al. Incidence of the V600K mutation among melanoma patients with BRAF mutations, and potential therapeutic response to the specific BRAF inhibitor PLX4032. J Transl Med. 2010;8:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Lovly CM, Dahlman KB, Fohn LE, et al. Routine multiplex mutational profiling of melanomas enables enrollment in genotype‐driven therapeutic trials. PLOS One. 2012;7(4):e35309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Davies H, Bignell GR, Cox C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417(6892):949‐954. [DOI] [PubMed] [Google Scholar]
- 93. Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33(1):19‐20. [DOI] [PubMed] [Google Scholar]
- 94. Dong J, Phelps RG, Qiao R, et al. BRAF oncogenic mutations correlate with progression rather than initiation of human melanoma. Cancer Res. 2003;63(14):3883‐3885. [PubMed] [Google Scholar]
- 95. Oliveira Júnior R, Ferraz C, Silva M, et al. Flavonoids: promising natural products for treatment of skin cancer (melanoma). In: Koehn FE, ed. Natural Products and Cancer Drug Discovery. InTechOpen Limited; 2017:161‐210. [Google Scholar]
- 96. Houben R, Becker JC, Kappel A, et al. Constitutive activation of the Ras‐Raf signaling pathway in metastatic melanoma is associated with poor prognosis. J Carcinog. 2004;3:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Platz A, Egyhazi S, Ringborg U, Hansson J. Human cutaneous melanoma: a review of NRAS and BRAF mutation frequencies in relation to histogenetic subclass and body site. Mol Oncol. 2008;1(4):395‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Roskoski RJ. Structure and regulation of Kit protein‐tyrosine kinase – the stem cell factor receptor. Biochem Biophys Res Commun. 2005;338(3):1307‐1315. [DOI] [PubMed] [Google Scholar]
- 99. Nissan MH, Pratilas CA, Jones AM, et al. Loss of NF1 in cutaneous melanoma is associated with RAS activation and MEK dependence. Cancer Res. 2014;74(8):2340‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Handolias D, Salemi R, Murray W, et al. Mutations in KIT occur at low frequency in melanomas arising from anatomical sites associated with chronic and intermittent sun exposure. Pigment Cell Melanoma Res. 2010;23(2):210‐215. [DOI] [PubMed] [Google Scholar]
- 101. Shi H, Hugo W, Kong X, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4(12):988‐1004. [DOI] [PubMed] [Google Scholar]
- 103. Mirmohammadsadegh A, Marini A, Nambiar S, et al. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66(13):6546‐6552. [DOI] [PubMed] [Google Scholar]
- 104. Liu L, Dilworth D, Gao L, et al. Mutation of the CDKN2A 5' UTR creates an aberrant initiation codon and predisposes to melanoma. Nat Genet. 1999;21(1):128‐132. [DOI] [PubMed] [Google Scholar]
- 105. Shain AH, Yeh I, Kovalyshyn I, et al. The genetic evolution of melanoma from precursor lesions. N Engl J Med. 2015;373(20):1926‐1936. [DOI] [PubMed] [Google Scholar]
- 106. Massagué J. G1 cell‐cycle control and cancer. Nature. 2004;432(7015):298‐306. [DOI] [PubMed] [Google Scholar]
- 107. McNeal AS, Liu K, Nakhate V, et al. CDKN2B loss promotes progression from benign melanocytic nevus to melanoma. Cancer Discov. 2015;5(10):1072‐1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Garbe C, Amaral T, Peris K, et al. European consensus‐based interdisciplinary guideline for melanoma. Part 2: treatment – update 2019. Eur J Cancer. 2020;126:141‐177. [DOI] [PubMed] [Google Scholar]
- 109. Martinez JC, Otley CC. The management of melanoma and nonmelanoma skin cancer: a review for the primary care physician. Mayo Clin Proc. 2001;76(12):1253‐1265. [DOI] [PubMed] [Google Scholar]
- 110. Joyce KM. Surgical management of melanoma. In: Ward WH, Farma JM, eds. Cutaneous Melanoma: Etiology and Therapy. Codon Publications; 2017:91‐101. [PubMed] [Google Scholar]
- 111. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87‐108. [DOI] [PubMed] [Google Scholar]
- 112. Garbe C, Peris K, Hauschild A, et al. Diagnosis and treatment of melanoma. European consensus‐based interdisciplinary guideline – update 2012. Eur J Cancer. 2012;48(15):2375‐2390. [DOI] [PubMed] [Google Scholar]
- 113. Harries M, Malvehy J, Lebbe C, et al. Treatment patterns of advanced malignant melanoma (stage III‐IV) – A review of current standards in Europe. Eur J Cancer. 2016;60:179‐189. [DOI] [PubMed] [Google Scholar]
- 114. Bhatia S, Tykodi SS, Thompson JA. Treatment of metastatic melanoma: an overview. Oncology. 2009;23(6):488‐496. [PMC free article] [PubMed] [Google Scholar]
- 115. Bilir SP, Ma Q, Zhao Z, Wehler E, Munakata J, Barber B. Economic burden of toxicities associated with treating metastatic melanoma in the United States. Am Health Drug Benefits. 2016;9(4):203‐213. [PMC free article] [PubMed] [Google Scholar]
- 116. Kim T, Amaria RN, Spencer C, Reuben A, Cooper ZA, Wargo JA. Combining targeted therapy and immune checkpoint inhibitors in the treatment of metastatic melanoma. Cancer Biol Med. 2014;11(4):237‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Millet A, Martin AR, Ronco C, Rocchi S, Benhida R. Metastatic melanoma: insights into the evolution of the treatments and future challenges. Med Res Rev. 2017;37(1):98‐148. [DOI] [PubMed] [Google Scholar]
- 118. Bajetta E, Del Vecchio M, Bernard‐Marty C, et al. Metastatic melanoma: chemotherapy. Semin Oncol. 2002;29(5):427‐445. [DOI] [PubMed] [Google Scholar]
- 119. Su MY, Fisher DE. Immunotherapy in the precision medicine era: melanoma and beyond. PLoS Med. 2016;13(12):e1002196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Tang T, Eldabaje R, Yang L. Current status of biological therapies for the treatment of metastatic melanoma. Anticancer Res. 2016;36(7):3229‐3241. [PubMed] [Google Scholar]
- 121. Singh BP, Salama AKS. Updates in therapy for advanced melanoma. Cancers. 2016;8(1):17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Ascierto PA, McArthur GA, Dréno B, et al. Cobimetinib combined with vemurafenib in advanced BRAF(V600)‐mutant melanoma (coBRIM): updated efficacy results from a randomised, double‐blind, phase 3 trial. Lancet Oncol. 2016;17(9):1248‐1260. [DOI] [PubMed] [Google Scholar]
- 123. Chae HS, Xu R, Won JY, Chin YW, Yim H. Molecular targets of genistein and its related flavonoids to exert anticancer effects. Int J Mol Sci. 2019;20(10):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Liskova A, Koklesova L, Samec M, et al. Flavonoids in cancer metastasis. Cancers. 2020;12(6):1‐29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Scheau C, Mihai LG, Bădărău IA, Căruntu C. Emerging applications of some important natural compounds in the field of oncology. Farmacia. 2020;68(6):992‐998. [Google Scholar]
- 126. Kanadaswami C, Lee LT, Lee PPH, et al. The antitumor activities of flavonoids. In Vivo. 2005;19(5):895‐910. [PubMed] [Google Scholar]
- 127. Lotha R, Sivasubramanian A. Flavonoids nutraceuticals in prevention and treatment of cancer: a review. Asian J Pharm Clin Res. 2018;11(1):42‐47. [Google Scholar]
- 128. Chirumbolo S, Bjørklund G, Lysiuk R, Vella A, Lenchyk L, Upyr T. Targeting cancer with phytochemicals via their fine tuning of the cell survival signaling pathways. Int J Mol Sci. 2018;19(11):3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Ombra MN, Paliogiannis P, Stucci LS, et al. Dietary compounds and cutaneous malignant melanoma: recent advances from a biological perspective. Nutr Metab. 2019;16(1):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Momtaz S, Niaz K, Maqbool F, Abdollahi M, Rastrelli L, Nabavi SM. STAT3 targeting by polyphenols: novel therapeutic strategy for melanoma. Biofactors. 2017;43(3):347‐370. [DOI] [PubMed] [Google Scholar]
- 131. Seelinger G, Merfort I, Wölfle U, Schempp CM. Anti‐carcinogenic effects of the flavonoid luteolin. Molecules. 2008;13(10):2628‐2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. González‐Vallinas M, González‐Castejón M, Rodríguez‐Casado A, Ramírez, de Molina A. Dietary phytochemicals in cancer prevention and therapy: a complementary approach with promising perspectives. Nutr Rev. 2013;71(9):585‐599. [DOI] [PubMed] [Google Scholar]
- 133. Pan M‐H, Lai C‐S, Wu J‐C, Ho C‐T. Epigenetic and disease targets by polyphenols. Curr Pharm Des. 2013;19(34):6156‐6185. [DOI] [PubMed] [Google Scholar]
- 134. Ganai SA, Sheikh FA, Baba ZA, Mir MA, Mantoo MA, Yatoo MA. Anticancer activity of the plant flavonoid luteolin against preclinical models of various cancers and insights on different signalling mechanisms modulated. Phytother Res. 2021;35(7):3509‐3532. [DOI] [PubMed] [Google Scholar]
- 135. Islam SU, Ahmed MB, Ahsan H, et al. An update on the role of dietary phytochemicals in human skin cancer: new insights into molecular mechanisms. Antioxidants. 2020;9(10):1‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. López‐Lázaro M. Distribution and biological activities of the flavonoid luteolin. Mini Rev Med Chem. 2009;9(1):31‐59. [DOI] [PubMed] [Google Scholar]
- 137. Horváthová K, Chalupa I, Šebová L, Tóthová D, Vachálková A. Protective effect of quercetin and luteolin in human melanoma HMB‐2 cells. Mutat Res – Genet Toxicol Environ Mutagen. 2005;565(2):105‐112. [DOI] [PubMed] [Google Scholar]
- 138. Seito LN, Ruiz ALTG, Vendramini‐Costa D, et al. Antiproliferative activity of three methoxylated flavonoids isolated from Zeyheria montana Mart. (Bignoniaceae) leaves. Phyther Res. 2011;25(10):1447‐1450. [DOI] [PubMed] [Google Scholar]
- 139. Said A, Tundis R, Hawas UW, et al. In vitro antioxidant and antiproliferative activities of flavonoids from Ailanthus excelsa (Roxb.) (Simaroubaceae) leaves. Z Naturforsch C. 2010;65(3–4):180‐186. [DOI] [PubMed] [Google Scholar]
- 140. Kawaii S, Tomono Y, Katase E, Ogawa K, Yano M. Antiproliferative activity of flavonoids on several cancer cell lines. Biosci Biotechnol Biochem. 1999;63(5):896‐899. [DOI] [PubMed] [Google Scholar]
- 141. Akihisa T, Kawashima K, Orido M, et al. Antioxidative and melanogenesis‐inhibitory activities of caffeoylquinic acids and other compounds from moxa. Chem Biodivers. 2013;10(3):313‐327. [DOI] [PubMed] [Google Scholar]
- 142. Nagao T, Abe F, Kinjo J, Okabe H. Antiproliferative constituents in plants 10. Flavones from the leaves of Lantana montevidensis Briq. and consideration of structure‐activity relationship. Biol Pharm Bull. 2002;25(7):875‐879. [DOI] [PubMed] [Google Scholar]
- 143. Meng TX, Irino N, Kondo R. Melanin biosynthesis inhibitory activity of a compound isolated from young green barley (Hordeum vulgare L.) in B16 melanoma cells. J Nat Med. 2015;69(3):427‐431. [DOI] [PubMed] [Google Scholar]
- 144. Cattaneo L, Cicconi R, Mignogna G, et al. Anti‐proliferative effect of Rosmarinus officinalis L. Extract on human melanoma A375 cells. PLOS One. 2015;10(7):1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Dar AA, Dangroo NA, Raina A, et al. Biologically active xanthones from Codonopsis ovata . Phytochemistry. 2016;132:102‐108. [DOI] [PubMed] [Google Scholar]
- 146. Jiang W, Xia T, Liu C, Li J, Zhang W, Sun C. Remodeling the epigenetic landscape of cancer‐application potential of flavonoids in the prevention and treatment of cancer. Front Oncol. 2021;11:705903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Arora I, Sharma M, Tollefsbol TO. Combinatorial epigenetics impact of polyphenols and phytochemicals in cancer prevention and therapy. Int J Mol Sci. 2019;20(18):4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Kang KA, Piao MJ, Hyun YJ, et al. Luteolin promotes apoptotic cell death via upregulation of Nrf2 expression by DNA demethylase and the interaction of Nrf2 with p53 in human colon cancer cells. Exp Mol Med. 2019;51(4):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Juszczak AM, Czarnomysy R, Strawa JW, Zovko Končić M, Bielawski K, Tomczyk M. In vitro anticancer potential of Jasione montana and its main components against human amelanotic melanoma cells. Int J Mol Sci. 2021;22(7):3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Yao X, Jiang W, Yu D, Yan Z. Luteolin inhibits proliferation and induces apoptosis of human melanoma cells: in vivo and in vitro by suppressing MMP‐2 and MMP‐9 through the PI3K/AKT pathway. Food Funct. 2019;10(2):703‐712. [DOI] [PubMed] [Google Scholar]
- 151. Schomberg J, Wang Z, Farhat A, et al. Luteolin inhibits melanoma growth in vitro and in vivo via regulating ECM and oncogenic pathways but not ROS. Biochem Pharmacol. 2020;177:114025. [DOI] [PubMed] [Google Scholar]
- 152. Byun S, Lee KW, Jung SK, et al. Luteolin inhibits protein kinase Cε and c‐Src activities and UVB‐induced skin cancer. Cancer Res. 2010;70(6):2415‐2423. [DOI] [PubMed] [Google Scholar]
- 153. Tian L, Wang S, Jiang S, et al. Luteolin as an adjuvant effectively enhances CTL anti‐tumor response in B16F10 mouse model. Int Immunopharmacol. 2021;94:107441. [DOI] [PubMed] [Google Scholar]
- 154. Ralph SJ, Rodríguez‐Enríquez S, Neuzil J, Saavedra E, Moreno‐Sánchez R. The causes of cancer revisited: “mitochondrial malignancy” and ROS‐induced oncogenic transformation – why mitochondria are targets for cancer therapy. Mol Aspects Med. 2010;31(2):145‐170. [DOI] [PubMed] [Google Scholar]
- 155. Kim JK, Kang KA, Ryu YS, et al. Induction of endoplasmic reticulum stress via reactive oxygen species mediated by luteolin in melanoma cells. Anticancer Res. 2016;36(5):2281‐2289. [PubMed] [Google Scholar]
- 156. Rodriguez J, Yáñez J, Vicente V, et al. Effects of several flavonoids on the growth of B16F10 and SK‐MEL‐1 melanoma cell lines: relationship between structure and activity. Melanoma Res. 2002;12(2):99‐107. [DOI] [PubMed] [Google Scholar]
- 157. Yáñez J, Vicente V, Alcaraz M, et al. Cytotoxicity and antiproliferative activities of several phenolic compounds against three melanocytes cell lines: relationship between structure and activity. Nutr Cancer. 2004;49(2):191‐199. [DOI] [PubMed] [Google Scholar]
- 158. Kushiro K, Chu RA, Verma A, Núñez NP. Adipocytes promote B16BL6 melanoma cell invasion and the epithelial‐to‐mesenchymal transition. Cancer Microenviron. 2012;5(1):73‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Hussain Y, Cui JH, Khan H, Aschner M, Batiha GES, Jeandet P. Luteolin and cancer metastasis suppression: focus on the role of epithelial to mesenchymal transition. Med Oncol. 2021;38(6):1‐12. [DOI] [PubMed] [Google Scholar]
- 160. Lin Y, Tsai P, Kandaswami CC, et al. Effects of dietary flavonoids, luteolin, and quercetin on the reversal of epithelial–mesenchymal transition in A431 epidermal cancer cells. Cancer Sci. 2011;102(10):1829‐1839. [DOI] [PubMed] [Google Scholar]
- 161. Ruan JS, Liu YP, Zhang L, et al. Luteolin reduces the invasive potential of malignant melanoma cells by targeting β3 integrin and the epithelial‐mesenchymal transition. Acta Pharmacol Sin. 2012;33(10):1325‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Huang YT, Hwang JJ, Lee PP, et al. Effects of luteolin and quercetin, inhibitors of tyrosine kinase, on cell growth and metastasis‐associated properties in A431 cells overexpressing epidermal growth factor receptor. Br J Pharmacol. 1999;128(5):999‐1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Li C, Wang Q, Shen S, Wei X, Li G. HIF‐1α/VEGF signaling‐mediated epithelial–mesenchymal transition and angiogenesis is critically involved in anti‐metastasis effect of luteolin in melanoma cells. Phyther Res. 2019;33(3):798‐807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Fan JJ, Hsu WH, Lee KH, et al. Dietary flavonoids luteolin and quercetin inhibit migration and invasion of squamous carcinoma through reduction of Src/Stat3/S100A7 signaling. Antioxidants. 2019;8(11):557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lin YC, Tsai PH, Lin CY, et al. Impact of flavonoids on matrix metalloproteinase secretion and invadopodia formation in highly invasive A431‐III cancer cells. PLOS One. 2013;8(8):e71903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Lee L‐T, Huang Y‐T, Hwang J‐J, et al. Transinactivation of the epidermal growth factor receptor tyrosine kinase and focal adhesion kinase phosphorylation by dietary flavonoids: effect on invasive potential of human carcinoma cells. Biochem Pharmacol. 2004;67(11):2103‐2114. [DOI] [PubMed] [Google Scholar]
- 167. Iwashita K, Kobori M, Yamaki K, Tsushida T. Flavonoids inhibit cell growth and induce apoptosis in B16 melanoma 4A5 cells. Biosci Biotechnol Biochem. 2000;64(9):1813‐1820. [DOI] [PubMed] [Google Scholar]
- 168. Gilchrest BA, Eller MS. DNA photodamage stimulates melanogenesis and other photoprotective responses. J Invest Dermatol. 1999;4(1):35‐40. [DOI] [PubMed] [Google Scholar]
- 169. Parvez S, Kang M, Chung H‐S, et al. Survey and mechanism of skin depigmenting and lightening agents. Phytother Res. 2006;20(11):921‐934. [DOI] [PubMed] [Google Scholar]
- 170. Wu QY, Wong ZCF, Wang C, et al. Isoorientin derived from Gentiana veitchiorum Hemsl. flowers inhibits melanogenesis by down‐regulating MITF‐induced tyrosinase expression. Phytomedicine. 2019;57:129‐136. [DOI] [PubMed] [Google Scholar]
- 171. An SM, Kim HJ, Kim J‐E, Boo YC. Flavonoids, taxifolin and luteolin attenuate cellular melanogenesis despite increasing tyrosinase protein levels. Phytother Res. 2008;22(9):1200‐1207. [DOI] [PubMed] [Google Scholar]
- 172. Choi MY, Song HS, Hur HS, Sim SS. Whitening activity of luteolin related to the inhibition of cAMP pathway in α‐MSH‐stimulated B16 melanoma cells. Arch Pharm Res. 2008;31(9):1166‐1171. [DOI] [PubMed] [Google Scholar]
- 173. Takekoshi S, Nagata H, Kitatani K. Flavonoids enhance melanogenesis in human melanoma cells. Tokai J Exp Clin Med. 2014;39(3):116‐121. [PubMed] [Google Scholar]
- 174. Yamauchi K, Fujieda A, Mitsunaga T. Selective synthesis of 7‐O‐substituted luteolin derivatives and their melanonenesis and proliferation inhibitory activity in B16 melanoma cells. Bioorganic Med Chem Lett. 2018;28(14):2518‐2522. [DOI] [PubMed] [Google Scholar]
- 175. Horibe I, Satoh Y, Shiota Y, et al. Induction of melanogenesis by 4′‐O‐methylated flavonoids in B16F10 melanoma cells. J Nat Med. 2013;67(4):705‐710. [DOI] [PubMed] [Google Scholar]
- 176. Shi R, Huang Q, Zhu X, et al. Luteolin sensitizes the anticancer effect of cisplatin via c‐Jun NH2‐terminal kinase‐mediated p53 phosphorylation and stabilization. Mol Cancer Ther. 2007;6(4):1338‐1347. [DOI] [PubMed] [Google Scholar]
- 177. Bu J, Ma PC, Chen ZQ, et al. Inhibition of MITF and tyrosinase by paeonol‐stimulated JNK/SAPK to reduction of phosphorylated CREB. Am J Chin Med. 2008;36(2):245‐263. [DOI] [PubMed] [Google Scholar]
- 178. Lee SW, Kim JH, Song H, Seok JK, Hong SS, Boo YC. Luteolin 7‐sulfate attenuates melanin synthesis through inhibition of CREB‐ and MITF‐mediated tyrosinase expression. Antioxidants. 2019;8(4):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Kwak JY, Seok JK, Suh H‐J, et al. Antimelanogenic effects of luteolin 7‐sulfate isolated from Phyllospadix iwatensis Makino. Br J Dermatol. 2016;175(3):501‐511. [DOI] [PubMed] [Google Scholar]
- 180. Martínez C, Yàñez J, Vicente V, et al. Effects of several polyhydroxylated flavonoids on the growth of B16F10 melanoma and Melan—a melanocyte cell lines: influence of the sequential oxidation state of the flavonoid skeleton. Melanoma Res. 2003;13(1):3‐9. [DOI] [PubMed] [Google Scholar]
- 181. Nakashima S, Matsuda H, Oda Y, Nakamura S, Xu F, Yoshikawa M. Melanogenesis inhibitors from the desert plant Anastatica hierochuntica in B16 melanoma cells. Bioorg Med Chem. 2010;18(6):2337‐2345. [DOI] [PubMed] [Google Scholar]
- 182. Dar AA, Rath SK, Qaudri A, et al. Isolation, cytotoxic evaluation, and simultaneous quantification of eight bioactive secondary metabolites from Cicer microphyllum by high‐performance thin‐layer chromatography. J Sep Sci. 2015;38(23):4021‐4028. [DOI] [PubMed] [Google Scholar]
- 183. Touil YS, Fellous A, Scherman D, Chabot GG. Flavonoid‐induced morphological modifications of endothelial cells through microtubule stabilization. Nutr Cancer. 2009;61(3):310‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Manthey JA, Guthrie N. Antiproliferative activities of citrus flavonoids against six human cancer cell lines. J Agr Food Chem. 2002;50(21):5837‐5843. [DOI] [PubMed] [Google Scholar]
- 185. Peng H, Xing Y, Gao L, Zhang L, Zhang G. Simultaneous separation of apigenin, luteolin and rosmarinic acid from the aerial parts of the copper‐tolerant plant Elsholtzia splendens . Environ Sci Pollut Res Int. 2014;21(13):8124‐8132. [DOI] [PubMed] [Google Scholar]
- 186. Chen KC, Hsu WH, Ho JY, et al. Flavonoids luteolin and quercetin inhibit RPS19 and contributes to metastasis of cancer cells through c‐Myc reduction. J Food Drug Anal. 2018;26(3):1180‐1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lin CW, Lai GM, Chen KC, et al. RPS12 increases the invasiveness in cervical cancer activated by c‐Myc and inhibited by the dietary flavonoids luteolin and quercetin. J Funct Foods. 2015;19:19236‐19247. [Google Scholar]
- 188. Gálvez M, Martín‐Cordero C, López‐Lázaro M, Cortés F, Ayuso MJ. Cytotoxic effect of Plantago spp. on cancer cell lines. J Ethnopharmacol. 2003;88(2–3):125‐130. [DOI] [PubMed] [Google Scholar]
- 189. Yamauchi K, Mitsunaga T, Itakura Y, Batubara I. Extracellular melanogenesis inhibitory activity and the structure‐activity relationships of ugonins from Helminthostachys zeylanica roots. Fitoterapia. 2015;104:10469‐10474. [DOI] [PubMed] [Google Scholar]
- 190. Choi J, Lee D‐H, Park S‐Y, Seol J‐W. Diosmetin inhibits tumor development and block tumor angiogenesis in skin cancer. Biomed Pharmacother. 2019;117:109091. [DOI] [PubMed] [Google Scholar]
- 191. Chaabane F, Mustapha N, Mokdad‐Bzeouich I, et al. In vitro and in vivo anti‐melanoma effects of Daphne gnidium aqueous extract via activation of the immune system. Tumour Biol. 2016;37(5):6511‐6517. [DOI] [PubMed] [Google Scholar]
- 192. Arung E, Kuspradini H, Kusuma I, et al. Effects of isolated compound from Sonneratia caseolaris leaf: a validation of traditional utilization by melanin biosynthesis and antioxidant assays. J Appl Pharm Sci. 2015;5(10):39‐43. [Google Scholar]
- 193. Apalla Z, Nashan D, Weller RB, Castellsagué X. Skin cancer: epidemiology, disease burden, pathophysiology, diagnosis, and therapeutic approaches. Dermatol Ther. 2017;7:5‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Olson AL, Gaffney CA, Starr P, Dietrich AJ. The impact of an appearance‐based educational intervention on adolescent intention to use sunscreen. Health Educ Res. 2008;23(5):763‐769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.National Cancer Institute (NIH). Skin Cancer Prevention (PDQ®)–Health Professional Version. Accessed July 13, 2021.
- 196. Demierre M‐F, Nathanson L. Chemoprevention of melanoma: an unexplored strategy. J Clin Oncol. 2003;21(1):158‐165. [DOI] [PubMed] [Google Scholar]
- 197. Lao CD, Demierre M‐F, Sondak VK. Targeting events in melanoma carcinogenesis for the prevention of melanoma. Expert Rev Anticancer Ther. 2006;6(11):1559‐1568. [DOI] [PubMed] [Google Scholar]
- 198. Lien EJ, Ren S, Bui HH, Wang R. Quantitative structure‐activity relationship analysis of phenolic antioxidants. Free Radic Biol Med. 1999;26(3–4):285‐294. [DOI] [PubMed] [Google Scholar]
- 199. Romanova D, Vachálkova A, Čipák L, Ovesná Z, Rauko P. Study of antioxidant effect of apigenin, luteolin and quercetin by DNA protective method. Neoplasma. 2001;48(2):104‐107. [PubMed] [Google Scholar]
- 200. Ross JA, Kasum CM. Dietary flavonoids: bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:2219‐2234. [DOI] [PubMed] [Google Scholar]
- 201. Horváthová K, Novotny L, Vachalkova A. The free radical scavenging activity of four flavonoids determined by the comet assay. Neoplasma. 2003;50(4):291‐295. [PubMed] [Google Scholar]
- 202. Shimoi K, Masuda S, Furugori M, Esaki S, Kinae N. Radioprotective effect of antioxidative flavonoids in gamma‐ray irradiated mice. Carcinogenesis. 1994;15(11):2669‐2672. [DOI] [PubMed] [Google Scholar]
- 203. Gendrisch F, Esser PR, Schempp CM, Wölfle U. Luteolin as a modulator of skin aging and inflammation. Biofactors. 2021;47(2):170‐180. [DOI] [PubMed] [Google Scholar]
- 204. Brown JE, Rice‐Evans CA. Luteolin‐rich artichoke extract protects low density lipoprotein from oxidation in vitro . Free Radic Res. 1998;29(3):247‐255. [DOI] [PubMed] [Google Scholar]
- 205. Wölfle U, Esser PR, Simon‐Haarhaus B, Martin SF, Lademann J, Schempp CM. UVB‐induced DNA damage, generation of reactive oxygen species, and inflammation are effectively attenuated by the flavonoid luteolin in vitro and in vivo . Free Radic Biol Med. 2011;50(9):1081‐1093. [DOI] [PubMed] [Google Scholar]
- 206. Ju W, Wang X, Shi H, Chen W, Belinsky SA, Lin Y. A critical role of luteolin‐induced reactive oxygen species in blockage of tumor necrosis factor‐activated nuclear factor‐κB pathway and sensitization of apoptosis in lung cancer cells. Mol Pharmacol. 2007;71(5):1381‐1388. [DOI] [PubMed] [Google Scholar]
- 207. Xian D, Guo M, Xu J, Yang Y, Zhao Y, Zhong J. Current evidence to support the therapeutic potential of flavonoids in oxidative stress‐related dermatoses. Redox Rep. 2021;26(1):134‐146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Xagorari A, Papapetropoulos A, Mauromatis A, Economou M, Fotsis T, Roussos C. Luteolin inhibits an endotoxin‐stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J Pharmacol Exp Ther. 2001;296(1):181‐187. [PubMed] [Google Scholar]
- 209. Kumazawa Y, Kawaguchi K, Takimoto H. Immunomodulating effects of flavonoids on acute and chronic inflammatory responses caused by tumor necrosis factor alpha. Curr Pharm Des. 2006;12(32):4271‐4279. [DOI] [PubMed] [Google Scholar]
- 210. Hayden MS, Ghosh S. Signaling to NF‐kappaB. Genes Dev. 2004;18(18):2195‐2224. [DOI] [PubMed] [Google Scholar]
- 211. Lim SH, Jung SK, Byun S, et al. Luteolin suppresses UVB‐induced photoageing by targeting JNK1 and p90RSK2. J Cell Mol Med. 2013;17(5):672‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Verschooten L, Smaers K, Van Kelst S, et al. The flavonoid luteolin increases the resistance of normal, but not malignant keratinocytes, against UVB‐induced apoptosis. J Invest Dermatol. 2010;130(9):2277‐2285. [DOI] [PubMed] [Google Scholar]
- 213. Szekalska M, Sosnowska K, Tomczykowa M, Winnicka K, Kasacka I, Tomczyk M. In vivo anti‐inflammatory and anti‐allergic activities of cynaroside evaluated by using hydrogel formulations. Biomed Pharmacother. 2020;121:109681. [DOI] [PubMed] [Google Scholar]
- 214. Yi YS. Regulatory roles of flavonoids on inflammasome activation during inflammatory responses. Mol Nutr Food Res. 2018;62(13):1‐45. [DOI] [PubMed] [Google Scholar]
- 215. Casagrande F, Darbon JM. Effects of structurally related flavonoids on cell cycle progression of human melanoma cells: regulation of cyclin‐dependent kinases CDK2 and CDK1. Biochem Pharmacol. 2001;61(10):1205‐1215. [DOI] [PubMed] [Google Scholar]
- 216. Jung S, Shin J, Oh J, et al. Cytotoxic and apoptotic potential of Phyllodium elegans extracts on human cancer cell lines. Bioengineered. 2019;10(1):501‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Ullah MF, Ahmad A, Khan HY, Zubair H, Sarkar FH, Hadi SM. The prooxidant action of dietary antioxidants leading to cellular DNA breakage and anticancer effects: implications for chemotherapeutic action against cancer. Cell Biochem Biophys. 2013;67(2):431‐438. [DOI] [PubMed] [Google Scholar]
- 218. Denault J‐B, Salvesen G. Caspases: keys in the ignition of cell death. Chem Rev. 2003;34:1024489‐1024500. [DOI] [PubMed] [Google Scholar]
- 219. Porter AG, Jänicke RU. Emerging roles of caspase‐3 in apoptosis. Cell Death Differ. 1999;6(2):99‐104. [DOI] [PubMed] [Google Scholar]
- 220. Danciu C, Zupko I, Bor A, et al. Botanical therapeutics: phytochemical screening and biological assessment of chamomile, parsley and celery extracts against A375 human melanoma and dendritic cells. Int J Mol Sci. 2018;19(11):1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Guruvayoorappan C, Kuttan G. Apoptotic effect of Biophytum sensitivum on B16F‐10 cells and its regulatory effects on nitric oxide and cytokine production on tumor‐associated macrophages. Integr Cancer Ther. 2007;6(4):373‐380. [DOI] [PubMed] [Google Scholar]
- 222. Guruvayoorappan C, Girija K. Biophytum sensitivum (L.) DC inhibits tumor cell invasion and metastasis through a mechanism involving regulation of MMPs, prolyl hydroxylase, lysyl oxidase, nm23, ERK‐1, ERK‐2, STAT‐1, and proinflammatory cytokine gene expression in metastatic lung tissue. Integr Cancer Ther. 2008;7(1):42‐50. [DOI] [PubMed] [Google Scholar]
- 223. Lin YL, Wang WY. Chemical constituents of Biophytum sensitivum . Chinese Pharm J. 2003;55:71‐75. [Google Scholar]
- 224. Di Petrillo A, González‐Paramás AM, Era B, et al. Tyrosinase inhibition and antioxidant properties of Asphodelus microcarpus extracts. BMC Complement Altern Med. 2016;16(1):453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Rauca V‐F, Vlase L, Casian T, et al. Biologically active Ajuga species extracts modulate supportive processes for cancer cell development. Front Pharmacol. 2019;10:334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Chao HC, Najjaa H, Villareal MO, et al. Arthrophytum scoparium inhibits melanogenesis through the down‐regulation of tyrosinase and melanogenic gene expressions in B16 melanoma cells. Exp Dermatol. 2013;22(2):131‐136. [DOI] [PubMed] [Google Scholar]
- 227. Said A, El‐Fiky NM, Rashed K, et al. Anticancer, anti HIV‐1 and antimicrobial potentials of methanol extract and non polar fractions of Citrus volkameriana leaves and phytochemical composition. Res J Med Plant. 2015;9(5):201‐214. [Google Scholar]
- 228. Shomirzoeva O, Li J, Numonov S, et al. Chemical components of Hyssopus seravshanicus: antioxidant activity, activations of melanogenesis and tyrosinase, and quantitative determination by UPLC‐DAD. Nat Prod Res. 2019;33(6):866‐870. [DOI] [PubMed] [Google Scholar]
- 229. Arun KP, Brindha P. Investigations into phenolic and alkaloid constituents of Jatropha tanjorensis by LC‐MS/MS and evaluating its bioactive property. Asian J Chem. 2015;27(9):3249‐3253. [Google Scholar]
- 230. Makowska‐Wąs J, Galanty A, Gdula‐Argasińska J, et al. Identification of predominant phytochemical compounds and cytotoxic activity of wild Olive leaves (Olea europaea L. ssp. sylvestris) harvested in South Portugal. Chem Biodivers. 2017;14(3):1‐10. [DOI] [PubMed] [Google Scholar]
- 231. Boruga M, Enatescu V, Pinzaru J, et al. Assessment of olive leaves extract ‐ cytotoxicity in vitro and angiogenesis in ovo . Farmacia. 2021;69(1):38‐43. [Google Scholar]
- 232. Jeong D, Lee J, Park SH, et al. Antiphotoaging and antimelanogenic effects of Penthorum chinense pursh ethanol extract due to antioxidant – and autophagy‐inducing properties. Oxid Med Cell Longev. 2019;2019:9679731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Yi J, Wang Z, Bai H, Yu X, Jing J, Zuo L. Optimization of purification, identification and evaluation of the in vitro antitumor activity of polyphenols from Pinus koraiensis pinecones. Molecules. 2015;20(6):10450‐10467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Sadhu SK, Ahmed F, Ohtsuki T, Ishibashi M. Flavonoids from Sonneratia caseolaris . J Nat Med. 2006;60(3):264‐265. [DOI] [PubMed] [Google Scholar]
- 235. Manzoor MF, Ahmad N, Ahmed Z, et al. Novel extraction techniques and pharmaceutical activities of luteolin and its derivatives. J Food Biochem. 2019;43(9):e12974. [DOI] [PubMed] [Google Scholar]
- 236. Sabir F, Barani M, Rahdar A, et al. How to face skin cancer with nanomaterials: a review. Biointerface Res Appl Chem. 2021;11(4):11931‐11955. [Google Scholar]
- 237. Padya BS, Pandey A, Pisay M, et al. Stimuli‐responsive and cellular targeted nanoplatforms for multimodal therapy of skin cancer. Eur J Pharmacol. 2021;890:173633. [DOI] [PubMed] [Google Scholar]
- 238. Dasari S, Yedjou CG, Brodell RT, Cruse AR, Tchounwou PB. Therapeutic strategies and potential implications of silver nanoparticles in the management of skin cancer. Nanotechnol Rev. 2020;9(1):1500‐1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Gupta S, Bansal R, Gupta S, Jindal N, Jindal A. Nanocarriers and nanoparticles for skin care and dermatological treatments. Indian Dermatol Online J. 2013;4(4):267‐272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Wu G, Li J, Yue J, Zhang S, Yunusi K. Liposome encapsulated luteolin showed enhanced antitumor efficacy to colorectal carcinoma. Mol Med Rep. 2018;17(2):2456‐2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Koudelka S, Turanek Knotigova P, Masek J, et al. Liposomal delivery systems for anti‐cancer analogues of vitamin E. J Control Release. 2015;207:59‐69. [DOI] [PubMed] [Google Scholar]
- 242. Dianzani C, Zara GP, Maina G, et al. Drug delivery nanoparticles in skin cancers. BioMed Res Int. 2014;2014:895986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Tsilioni I, Taliou A, Francis K, Theoharides TC. Children with autism spectrum disorders, who improved with a luteolin‐containing dietary formulation, show reduced serum levels of TNF and IL‐6. Transl Psychiatry. 2015;5(9):e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Karrasch T, Kim J‐S, Jang BI, Jobin C. The flavonoid luteolin worsens chemical‐induced colitis in NF‐κBEGFP transgenic mice through blockade of NF‐κB‐dependent protective molecules. PLOS One. 2007;2(7):1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.