

Abstract
The genomic region surrounding the Tenascin‐XB gene ( TNXB ) is a complex and duplicated region, with several pseudogenes that predispose to high rates of homologous recombination. Classical‐like Ehlers–Danlos syndrome (clEDS) is the result of tenascin‐X deficiency due to biallelic loss of function variants in the TNXB gene. Here we present a patient with clEDS and spontaneous pneumothorax, a feature not previously reported to be associated with this condition. Two inherited pathogenic/likely pathogenic variants were identified; a previously reported deletion resulting in a TNXA/TNXB chimeric gene and a novel frameshift variant. The Tenascin‐XB gene is well described in the literature to be associated with collagen metabolism, stabilization of the fibrillar‐collagen matrix and is expressed abundantly in the extracellular matrix. We propose that tenascin‐X deficiency is directly related to pneumothorax predisposition. This case expands the phenotypic spectrum of clEDS and highlights the challenges with molecular analysis and diagnosis
Keywords: classical‐like Ehlers–Danlos syndrome, Ehlers–Danlos syndrome, pneumothorax, tenascin‐X deficiency
1. INTRODUCTION
The Tenascin‐XB gene (TNXB; OMIM 600985) contains 44 exons and codes for tenascin‐XB (TNXB‐protein), a glycoprotein of the extracellular matrix that is predominantly located in the outer reticular lamina of the basement membrane. The gene is located within the class III region of the HLA locus on human chromosome 6p21. This region of the genome is duplicated and contains many closely related and overlapping genes making molecular analysis challenging (Higashi et al., 1986). Notably, this region contains the genes for the fourth component of human complement (C4; OMIM 120810) as well as the CYP21A2[21B] gene (OMIM 613815), which specifically overlaps the 3′ region of TNXB and encodes the enzyme 21‐hydroxylase (Morel et al., 1989). Further complicating this region are pseudogenes TNXA, a truncated duplicate that is 97% identical to the 3′ end of the TNXB (exons 32–44) and CYP21A1P[21A], both of which predispose to high‐rate homologous recombination (Tee et al., 1995) (see Figure 1).
FIGURE 1.
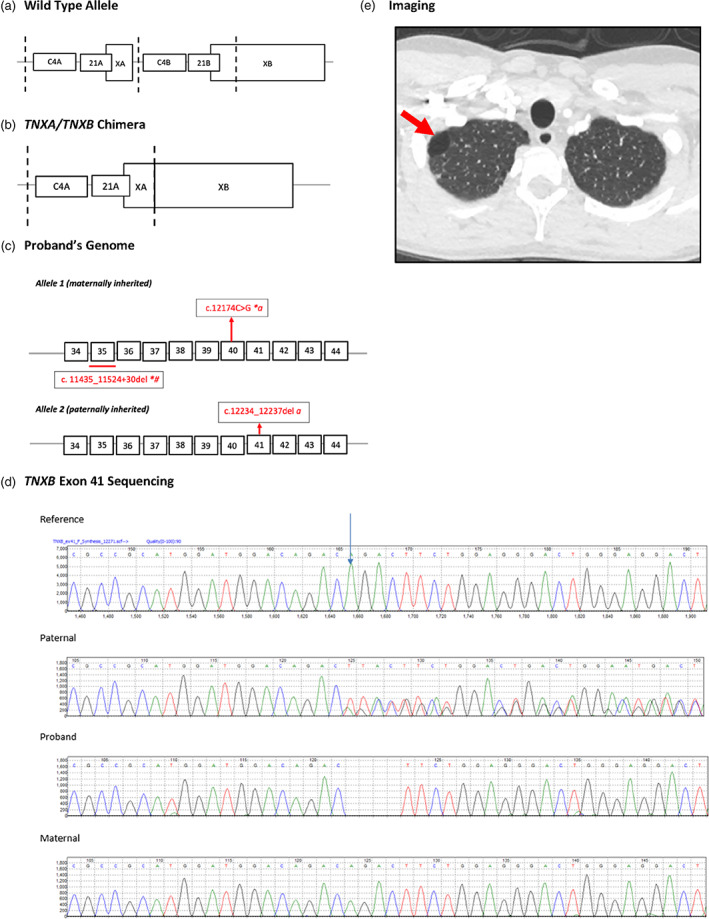
(a, b) Genetic map of the tenascin‐X locus for wild‐type and TNXA/TNXB chimera. Vertical dotted lines indicate duplication boundaries. Relevant coding region of TNXB denoted by white boxes and introns by intervening black horizontal lines. (c) Variants identified in the individual are represented in red. (d) TNXB exon 41 Sanger sequencing results for paternal, proband, and maternal samples. Start of TNXB (NM_019105):c.12234_12237del;p.(Asp4079Serfs*29) variant indicated by arrow on reference sequence. Variant was detected in paternal and proband samples. Variant was not present in maternal sample. Long range forward primer is located in region specific to TNXB exon 35. Only one allele amplified in proband and maternal samples as both carry TNXB exon 35 del. (e) High‐resolution CT chest of proband demonstrating persistent right apical bleb (red arrow). *, consistent with TNXB/TNXA fusion gene, α‐detected by Sanger sequencing; #, detected by Sanger sequencing and MLPA
21‐hydroxylase deficiency (OMIM 201910) is a form of congenial adrenal hyperplasia (CAH) resulting in defective cortisol synthesis, aldosterone deficiency and androgen excess leading to virilization in females (Speiser et al., 2018). Approximately 9% of patients with CAH due to 21‐hydroxylase deficiency have an associated connective tissue disorder due to a contiguous gene deletion affecting the TNXB gene and CYP21A2[21B] gene (Morissette et al., 2015). TNXA/TNXB chimeric genes, produced as a result of aberrant recombination, have been classified into three types (Ch‐1 to Ch‐3) based on the recombination site. CH‐1 is characterized by a 120‐bp deletion at the boundary of exon 35 and intron 35 of TNXB, CH‐2 by a c.12174c>G (p.Cys4058Trp) pseudogene variant in exon 40 and CH‐3 by a cluster of three pseudo gene variants: c.12218G>A (p.Arg4073His) in exon 41, and c.12514G>A (p.Asp4172Asn) and c.12524G>A (p.Ser4175Asn) in exon 43 (Chen et al., 2016; Miller & Merke, 2018).
Ehlers–Danlos syndrome (EDS) refers to a group of genetically heterogenous defects in the fibrillar‐collagen matrix and linked by the major criteria of joint hypermobility, skin and vascular fragility, and generalized connective tissue friability (Brady et al., 2017). There are currently 13 widely accepted subtypes (Malfait et al., 2017). Classical‐like EDS (clEDS: OMIM 606408), caused by deficiency of TNXB‐protein due to biallelic loss of function variants in TNXB, was first reported in 1997 (Burch et al., 1997). In contrast to classical‐EDS (cEDS: OMIM 618000), this subtype is not associated with atrophic skin scarring and has autosomal recessive inheritance (Egging et al., 2007; Schalkwijk et al., 2001). In addition to the well‐documented major criteria of clEDS, several other associated minor criteria as well as features not included in the diagnostic criteria have been reported. Within the gastrointestinal and genitourinary system, connective tissue fragility predisposes to diverticulosis, hollow viscous rupture, and rectal and vaginal prolapse. Cardiovascular abnormalities include spontaneous and provoked haematomas, arterial aneurysm, subconjunctival hemorrhages, and cardiac valvular pathology. Several musculoskeletal implications have been described including hand and foot deformities such as hallux valgus, pes planus, brachydactyly, clinodactyly, as well as neurological symptoms such as fatigue, muscle pain, paraesthesia, and weakness (Demirdas et al., 2017; Green et al., 2020).
Here we present a patient with clEDS, biallelic TNXB variants, including a novel, likely pathogenic variant, and spontaneous pneumothorax, a phenotypic feature not previously reported.
2. CLINICAL REPORT
The male patient is the only child of nonconsanguineous Caucasian parents with no relevant family history. He first came to the attention of a clinical geneticist at 10 years of age for investigation of recurrent shoulder dislocations, skin hyperextensibility and easy bruising. At this time, he was clinically suspected to have vascular EDS (vEDS; OMIM 130050), although subsequent type III collagen studies were normal and Sanger sequencing of COL3A1 (OMIM 120180), TGFBR1 (OMIM 190181) and TGFBR2 (OMIM 190182) genes did not identify a pathogenic variant. Thus, a specific molecular diagnosis was not made.
At age 34 years he presented for genetics review. History and examination revealed the following with regards to the major criterion for clEDS. His skin was translucent over his chest wall, smooth and velvety over his palms and hyperextensible with redundancy over his elbows. He had striae over his lateral hips and groin, however his skin was not fragile and healed without atrophic scarring. He reported easy and spontaneous bruising since childhood and there was a fading bruise on his left arm above his elbow as well as hemosiderin staining of both elbows. He had a normal coagulation profile. He first sustained a traumatic left shoulder dislocation at 6 years old, followed by recurrent spontaneous dislocations until 13 years of age. Recurrent spontaneous dislocations of his jaw and subluxation of his fingers and knees accompanied this. Beighton score for hypermobility was 8 out of a possible 9. He was also noted to have piezogenic papules on his feet, which is a minor criterion for clEDS. Additional features included deep set eyes, acrogeric facies, and pale blue sclera. He reported many years of daily fatigue and musculoskeletal pain. Echocardiogram at 33 years old detected mild aortic root dilatation at the sinus of Valsalva (3.9 cm; height 174.5 cm; weight 70 kg; BSA 1.84; z‐score 2.26), borderline mitral valve prolapse and trivial mitral regurgitation. Important negative findings included normal body proportions (no increase span/height ration, no reduction of upper/lower segment), absence of arachnodactyly and thumb and wrist sign. Additionally, he did not have any clinical or biochemical features to suggest 21‐hydroxylase deficiency (salt‐wasting crisis or electrolyte imbalance).
He first sustained a left‐sided spontaneous pneumothorax at 20 years old and a subsequent right‐sided spontaneous pneumothorax at 22 years old, both of which required hospital admission for insertion of an intercostal drain. A CT chest performed at 24 years old demonstrated an area of persistent para‐septal emphysema (right‐apical and left‐lateral) with no other parenchymal anomalies. At 29 years old he sustained a further, larger than previous, spontaneous right‐sided pneumothorax which again required insertion of an intercostal drain. High‐resolution CT scan of the chest, after resolution of the pneumothorax, demonstrated a persistent 2.4 cm bleb at the right lung apex (see Figure 1), a 1.3 cm superior right lower‐lobe bleb and multiple bilateral subpleural nodules in the context of apical scarring. He underwent right pleurodesis, pleurectomy and resection of the right apical lobe at 30 years old. The surgeon noted that the pleura was “very thin.” Macroscopically, within the resected right‐upper lobe, three cystic spaces were identified consistent with the known bleb and there were no other gross parenchymal changes. Microscopically there were multiple foci of subpleural fibroelastic scarring with neovascularization and patchy mild chronic inflammation. Para‐septal (distal acinar) emphysema was noted deep to the involved pleural. Normal parenchyma, with sparing of the surrounding acinar was seen elsewhere. Importantly, no eosinophilic pleurisy, granulomatous inflammation, hemorrhage, or malignancy was noted.
Given the presence of three major and one minor criterion for clEDS, further genetic testing was offered.
3. GENETIC RESULTS
A next generation sequencing (NGS) gene panel, with long‐range PCR (LRPCR) optimization of the TNXB gene, was arranged. The panel included 39 genes relevant to generalized connective tissue disorders, including nine genes with known association with pneumothorax (FBN1 (OMIM 134797), FLCN (OMIM 607273), TGFBR1 (OMIM 190181), TGFBR2 (OMIM 190182), TGFBR3 (OMIM 600742), TGFB2 (OMIM 190220), TGFB3 (OMIM 190230), SMAD3 (OMIM 603019), and COL3A1 (OMIM 120180)). The testing identified biallelic variants in the TNXB gene (NM_019105.6) (see Figure 1).
Proband TNXB testing initially used a combination of NGS and Sanger backfill of relevant regions. This detected the 120 bp deletion across the exon 35/intron 35 boundary that is associated with either the CH‐1 chimera or a gene conversion event. Subsequent testing by multiplex ligation‐dependent probe amplification (MLPA) that includes probes for CYP21A2 and CYP21A1P confirmed a heterozygous, previously published TNXB/TNXA fusion gene in the proband as indicated by a CYP21A2 gene deletion and the TNXA‐derived c.12234_11524+30del including exon 35. This is a known TNXA/TNXB chimera variant (CH‐1) that is reported to be pathogenic for clEDS. Additionally, the proband was heterozygous for the CH‐2 missense variant c.12174C>G (p.Cys4058Trp) carried by the CH‐1 allele, previously reported as part of a TNXA/TNXB fusion gene (Demirdas et al., 2017). A third novel heterozygous TNXB variant designated c.12234_12237del was also found. This is predicted to result in frameshift and premature protein termination (p.Asp4079Serfs*29) and is likely to be pathogenic. Long‐range primers were designed to take advantage of a 120 bp region that is specific to the TNXB gene, located in exon/intron 35 (Forward) and the 3′UTR (Reverse), to amplify exons 35–44. This could confirm the c.12234_12237del is in the functional gene, as the LRPCR does not amplify the fusion TNXA/TNXB gene. MLPA and Sanger sequencing of CYP21A2 was also undertaken to rule out biallelic variants, thereby confirming proband carrier status for 21‐hydroxylase deficiency.
Parental testing used a combination of long‐ranged and nested PCR, Sanger sequencing and MLPA to determine phase of the variants. Results confirmed the variants are in trans, the TNXA/TNXB fusion gene maternally inherited and the TNXB c.12234_12237del paternally inherited (see Table 1).
TABLE 1.
Variants detected in this family
| Variant | Proband | Maternal | Paternal |
|---|---|---|---|
| TNXB c.11435_11524+30del (sequencing) | Heterozygous a | Presumed heterozygous b | Not detected |
| CH‐1 Fusion gene detected by MLPA | Detected c | Detected c | Not detected |
| TNXB c.12234_12237del (exon 41) | Heterozygous a , b | Not detected | Heterozygous |
Variant was detected in the proband by biallelic Sanger sequencing.
LRPCR forward primer is located in exon 35 of TNXB and therefore does not amplify the fusion gene. Only the wild‐type allele was amplified by LR‐PCR.
Proband and maternal MLPA results showed CYP21A2 del and TNXB exon 35 del (also consistent with c.11435_11524+30del).
4. DISCUSSION
Classical‐like EDS (clEDS) is an autosomal recessive disorder resulting from biallelic loss of function TNXB variants. We report a case of clEDS with compound heterozygous (likely) pathogenic variants in the TNXB gene leading to TNXB‐protein deficiency and a clEDS phenotype. Specifically, our patient had a known pathogenic CH‐1 mutation and a previously unreported likely pathogenic variant c.12234_12237del that is predicted to result in premature protein termination. This report also documents a feature that has previously been unreported within the cl‐EDS clinical spectrum but is associated with vEDS – spontaneous recurrent pneumothorax.
A tissue's susceptibility for spontaneous rupture is dependent on the architectural structure of the tissue and the surrounding extracellular matrix. TNXB mRNA is most expressed in musculoskeletal, cardiac and dermal tissues, but also found at varying quantities in the tissues of the lung, kidney, mammary gland, blood vessels, testis, ovaries, and digestive tract (Bristow et al., 1993; Matsumoto et al., 1994; Valcourt et al., 2015). Specifically, the lung interstitial matrix is comprised of mostly collagen and elastin, which form a three‐dimensional framework and impart most of the mechanical attributes (Mereness & Mariani, 2021). TNXB‐protein has been shown to regulate collagen metabolism, specifically reducing the deposition of fibrils independent of collagen synthesis or fibrillogensis, as well as play a critical role in stabilization of fibrillar‐collagen matrix (Mao et al., 2002; Minamitani et al., 2004; Veit et al., 2006). In addition to its architectural function, TNXB‐protein is dynamically expressed within the extra‐cellular matrix and plays a crucial role in the regulation of cell adhesion‐de‐adhesion (Valcourt et al., 2015). TNXB‐protein is therefore integral to the tensile strength of lung tissue and under significant positive transmural pressures, a deficiency can result in visceral pleural rupture.
Spontaneous pneumothoraces are a known phenotypic feature of generalized connective tissue disorders such as vEDS and Marfan syndrome (MS). vEDS, caused by pathogenic variants in the COL3A1 gene, is associated with spontaneous pneumothorax in 16%–40% of cases (Oderich et al., 2005). In vEDS, fragility of the alveolar wall due to vascular injury predisposes to rupture. Unsurprisingly, histology of the pleuropulmonary tissue of vEDS patients characteristically demonstrates haematomas, intraluminal and interstitial hemosiderin‐containing macrophages, as well as subpleural fibrosis, nodules and other emphysematous changes (Kawabata et al., 2010; Shimaoka et al., 2010). MS is caused by pathogenic variants in the FBN1 gene encoding the connective tissue protein fibrillin‐1 (Boileau et al., 2005). Contrastingly, pneumothorax in this patient population is commonly related to rupture of a subpleural bulla due to abnormal lung connective tissue parenchyma, most often occurring in the lung apices, with normal alveolar vasculature (Karpman et al., 2011). The most consistent histological finding in MS is distal acinar emphysema with sparing of surrounding parenchyma. Additional nonspecific changes can include focal pneumonia or bronchiectasis, congenital pulmonary malformations, congestion, fibrous pleural adhesions, and pleuritis (Dyhdalo & Farver, 2011). As described above, our patient had apical pneumothoraces, bullae, and key histological changes in the pulmonary pleura—specifically, distal acinar emphysema and pleural scarring. Importantly there was no evidence of pulmonary hemorrhage. Therefore, we propose that the pathology of spontaneous pneumothorax in clEDS more likely resembles that of MS than vEDS.
clEDS shares many of the same clinical features that characterize other subtypes along the EDS phenotypic spectrum and together with the challenges of TNXB mutational analysis, specifically the presence of a pseudogene, it is likely that clEDS is underreported and under diagnosed. We postulate that spontaneous pneumothorax is directly related to TNXB‐protein deficiency and shares similar imaging and histology findings with other related connective tissue disorders.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
ACKNOWLEDGMENT
Open access publishing facilitated by The University of Adelaide, as part of the Wiley ‐ The University of Adelaide agreement via the Council of Australian University Librarians.
Santoreneos, R. , Vakulin, C. , Ellul, M. , Rawlings, L. , Hardy, T. , & Poplawski, N. (2022). Recurrent pneumothorax in a case of tenascin‐X deficient Ehlers–Danlos syndrome: Broadening the phenotypic spectrum. American Journal of Medical Genetics Part A, 188A:1583–1588. 10.1002/ajmg.a.62674
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- Boileau, C. , Jondeau, G. , Mizuguchi, T. , & Matsumoto, N. (2005). Molecular genetics of Marfan syndrome. Current Opinion in Cardiology, 20(3), 194–200. [DOI] [PubMed] [Google Scholar]
- Brady, A. F. , Demirdas, S. , Fournel‐Gigleux, S. , Ghali, N. , Giunta, C. , Kapferer‐Seebacher, I. , Kosho, T. , Mendoza‐Londono, R. , Pope, M. F. , Rohrbach, M. , Van Damme, T. , Vandersteen, A. , van Mourik, C. , Voermans, N. , Zschocke, J. , & Malfait, F. (2017). The Ehlers–Danlos syndromes, rare types. American Journal of Medical Genetics. Part C, Seminars in Medical Genetics, 175(1), 70–115. [DOI] [PubMed] [Google Scholar]
- Bristow, J. , Tee, M. K. , Gitelman, S. E. , Mellon, S. H. , & Miller, W. L. (1993). Tenascin‐X: A novel extracellular matrix protein encoded by the human XB gene overlapping P450c21B. Journal of Cell Biology, 122(1), 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch, G. H. , Gong, Y. , Liu, W. , Dettman, R. W. , Curry, C. J. , Smith, L. , Miller, W. L. , & Bristow, J. (1997). Tenascin‐X deficiency is associated with Ehlers–Danlos syndrome. Nature Genetics, 17(1), 104–108. [DOI] [PubMed] [Google Scholar]
- Chen, W. , Perritt, A. F. , Morissette, R. , Dreiling, J. L. , Bohn, M. F. , Mallappa, A. , Xu, Z. , Quezado, M. , & Merke, D. P. (2016). Ehlers–Danlos syndrome caused by biallelic TNXB variants in patients with congenital adrenal hyperplasia. Human Mutation, 37(9), 893–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demirdas, S. , Dulfer, E. , Robert, L. , Kempers, M. , van Beek, D. , Micha, D. , van Engelen, B. G. , Hamel, B. , Schalkwijk, J. , Loeys, B. , Maugeri, A. , & Voermans, N. C. (2017). Recognizing the tenascin‐X deficient type of Ehlers–Danlos syndrome: A cross‐sectional study in 17 patients. Clinical Genetics, 91(3), 411–425. [DOI] [PubMed] [Google Scholar]
- Dyhdalo, K. , & Farver, C. (2011). Pulmonary histologic changes in Marfan syndrome: A case series and literature review. American Journal of Clinical Pathology, 136(6), 857–863. [DOI] [PubMed] [Google Scholar]
- Egging, D. , van Vlijmen‐Willems, I. , van Tongeren, T. , Schalkwijk, J. , & Peeters, A. (2007). Wound healing in tenascin‐X deficient mice suggests that tenascin‐X is involved in matrix maturation rather than matrix deposition. Connective Tissue Research, 48(2), 93–98. [DOI] [PubMed] [Google Scholar]
- Green, C. , Ghali, N. , Akilapa, R. , Angwin, C. , Baker, D. , Bartlett, M. , Bowen, J. , Brady, A. F. , Brock, J. , Chamberlain, E. , Cheema, H. , McConnell, V. , Crookes, R. , Kazkaz, H. , Johnson, D. , Pope, F. M. , Vandersteen, A. , Sobey, G. , & van Dijk, F. S. (2020). Classical‐like Ehlers–Danlos syndrome: A clinical description of 20 newly identified individuals with evidence of tissue fragility. Genetics in Medicine, 22(10), 1576–1582. [DOI] [PubMed] [Google Scholar]
- Higashi, Y. , Yoshioka, H. , Yamane, M. , Gotoh, O. , & Fujii‐Kuriyama, Y. (1986). Complete nucleotide sequence of two steroid 21‐hydroxylase genes tandemly arranged in human chromosome: A pseudogene and a genuine gene. Proceedings of the National Academy of Sciences of the United States of America, 83(9), 2841–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpman, C. , Aughenbaugh, G. L. , & Ryu, J. H. (2011). Pneumothorax and bullae in Marfan syndrome. Respiration, 82(3), 219–224. [DOI] [PubMed] [Google Scholar]
- Kawabata, Y. , Watanabe, A. , Yamaguchi, S. , Aoshima, M. , Shiraki, A. , Hatamochi, A. , Kawamura, T. , Uchiyama, T. , Watanabe, A. , & Fukuda, Y. (2010). Pleuropulmonary pathology of vascular Ehlers–Danlos syndrome: Spontaneous laceration, haematoma and fibrous nodules. Histopathology, 56(7), 944–950. [DOI] [PubMed] [Google Scholar]
- Malfait, F. , Francomano, C. , Byers, P. , Belmont, J. , Berglund, B. , Black, J. , Bloom, L. , Bowen, J. M. , Brady, A. F. , Burrows, N. P. , Castori, M. , Cohen, H. , Colombi, M. , Demirdas, S. , De Backer, J. , De Paepe, A. , Fournel‐Gigleux, S. , Frank, M. , Ghali, N. , … Tinkle, B. (2017). The 2017 international classification of the Ehlers‐Danlos syndromes. American Journal of Medical Genetics Part C, Seminars in Medical Genetics, 175(1), 8–26. [DOI] [PubMed] [Google Scholar]
- Mao, J. R. , Taylor, G. , Dean, W. B. , Wagner, D. R. , Afzal, V. , Lotz, J. C. , Rubin, E. M. , & Bristow, J. (2002). Tenascin‐X deficiency mimics Ehlers–Danlos syndrome in mice through alteration of collagen deposition. Nature Genetics, 30(4), 421–425. [DOI] [PubMed] [Google Scholar]
- Matsumoto, K. , Saga, Y. , Ikemura, T. , Sakakura, T. , & Chiquet‐Ehrismann, R. (1994). The distribution of tenascin‐X is distinct and often reciprocal to that of tenascin‐C. Journal of Cell Biology, 125(2), 483–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereness, J. A. , & Mariani, T. J. (2021). The critical role of collagen VI in lung development and chronic disease. Matrix Biology Plus, 10, 100058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, W. L. , & Merke, D. P. (2018). Tenascin‐X, congenital adrenal hyperplasia, and the CAH‐X syndrome. Hormone Research in Pædiatrics, 89(5), 352–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamitani, T. , Ariga, H. , & Matsumoto, K. (2004). Deficiency of tenascin‐X causes a decrease in the level of expression of type VI collagen. Experimental Cell Research, 297(1), 49–60. [DOI] [PubMed] [Google Scholar]
- Morel, Y. , Bristow, J. , Gitelman, S. E. , & Miller, W. L. (1989). Transcript encoded on the opposite strand of the human steroid 21‐hydroxylase/complement component C4 gene locus. Proceedings of the National Academy of Sciences of the United States of America, 86(17), 6582–6586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette, R. , Chen, W. , Perritt, A. F. , Dreiling, J. L. , Arai, A. E. , Sachdev, V. , Hannoush, H. , Mallappa, A. , Xu, Z. , McDonnell, N. B. , Quezado, M. , & Merke, D. P. (2015). Broadening the spectrum of Ehlers–Danlos syndrome in patients with congenital adrenal hyperplasia. Journal of Clinical Endocrinology and Metabolism, 100(8), E1143–E1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oderich, G. , Panneton, G. , Bower, T. , Lindor, N. , Cherry, T. , & Noel, A. (2005). The spectrum, management and clinical outcome of Ehlers–Danlos syndrome type IV: A 30‐year experience. Journal of Vascular Surgery, 42, 98–106. [DOI] [PubMed] [Google Scholar]
- Schalkwijk, J. , Zweers, M. C. , Steijlen, P. M. , Dean, W. B. , Taylor, G. , van Vlijmen, I. M. , van Haren, B. , Miller, W. L. , & Bristow, J. (2001). A recessive form of the Ehlers–Danlos syndrome caused by tenascin‐X deficiency. New England Journal of Medicine, 345(16), 1167–1175. [DOI] [PubMed] [Google Scholar]
- Shimaoka, Y. , Kosho, T. , Wataya‐Kaneda, M. , Funakoshi, M. , Suzuki, T. , Hayashi, S. , Mitsuhashi, Y. , Isei, T. , Aoki, Y. , Yamazaki, K. , Ono, M. , Makino, K. , Tanaka, T. , Kunii, E. , & Hatamochi, A. (2010). Clinical and genetic features of 20 Japanese patients with vascular‐type Ehlers–Danlos syndrome. British Journal of Dermatology, 163, 704–710. [DOI] [PubMed] [Google Scholar]
- Speiser, P. W. , Arlt, W. , Auchus, R. J. , Baskin, L. S. , Conway, G. S. , Merke, D. P. , Meyer‐Bahlburg, H. , Miller, W. L. , Murad, M. H. , Oberfield, S. E. , & White, P. C. (2018). Congenital adrenal hyperplasia due to steroid 21‐hydroxylase deficiency: An Endocrine Society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism, 103(11), 4043–4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee, M. K. , Thomson, A. A. , Bristow, J. , & Miller, W. L. (1995). Sequences promoting the transcription of the human XA gene overlapping P450c21A correctly predict the presence of a novel, adrenal‐specific, truncated form of tenascin‐X. Genomics, 28(2), 171–178. [DOI] [PubMed] [Google Scholar]
- Valcourt, U. , Alcaraz, L. B. , Exposito, J. Y. , Lethias, C. , & Bartholin, L. (2015). Tenascin‐X: Beyond the architectural function. Cell Adhesion & Migration, 9(1–2), 154–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veit, G. , Hansen, U. , Keene, D. R. , Bruckner, P. , Chiquet‐Ehrismann, R. , Chiquet, M. , & Koch, M. (2006). Collagen XII interacts with avian tenascin‐X through its NC3 domain. Journal of Biological Chemistry, 281(37), 27461–27470. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.