

Summary
Background
Bimekizumab is a monoclonal antibody that selectively inhibits both interleukin (IL)‐17A and IL‐17F, which is currently under investigation for treatment of moderate‐to‐severe plaque psoriasis. Maintenance dosing every 4 weeks is well established with IL‐17 inhibitors for psoriasis.
Objectives
To investigate the possible dosing interval during bimekizumab maintenance therapy to maintain clear skin, to inform phase III studies.
Methods
Forty‐nine patients with moderate‐to‐severe plaque psoriasis received bimekizumab 320 mg at weeks 0/4, followed at week 16 by bimekizumab 320 mg (n = 17) or placebo (n = 32). Efficacy, safety, pharmacokinetics, immunogenicity and biopsy transcriptomic analyses were assessed to week 28.
Results
At week 8, 47% of patients achieved a 100% improvement from baseline in Psoriasis Area and Severity Index (PASI 100), increasing to 57% at week 12 (8 weeks after the second dose) before decreasing. In those who received bimekizumab at week 16, PASI 100 rate increased to comparable peak levels at week 20, but reduced by week 28 to 41% (12 weeks after the third dose). The week 8 transcriptional signature observed in lesional psoriatic skin rapidly normalized to levels consistent with nonlesional skin, resulting in molecular remission. Keratinocyte‐related gene products such as CXCL1 (C‐X‐C motif chemokine ligand 1), IL‐8 (encoded by the CXCL8 gene), CCL20 (C‐C motif chemokine 20), IL‐36γ and IL‐17C were profoundly normalized to levels associated with nonlesional skin.
Conclusions
Here, inhibition of IL‐17F in addition to IL‐17A resulted in rapid, deep clinical responses. Additionally, profound normalization of keratinocyte biology and the psoriatic transcriptome was observed, including normalization of both IL17 and IL23 gene expression by week 8. These data provide evidence to support evaluation of bimekizumab maintenance dosing both every 8 and every 4 weeks in phase III clinical trials.
What is already known about this topic?
Strong clinical responses have been demonstrated with biologics that inhibit interleukin (IL)‐17A, and inhibition of this cytokine has been shown to affect the transcriptome of patients with psoriasis.
Prolonged maintenance dosing intervals are preferred by patients, with sustainable clinical responses key to improvements in quality of life.
Maintenance dosing every 4 weeks is well established with IL‐17A inhibitors in psoriasis.
What does this study add?
A first assessment of the effects of a monoclonal antibody that selectively inhibits both IL‐17A and IL‐17F on the transcriptome of patients with moderate‐to‐severe plaque psoriasis shows rapid normalization of the transcriptional signature in lesional psoriatic skin after 8 weeks.
These data provide evidence to support further evaluation of bimekizumab maintenance dosing both every 8 weeks and every 4 weeks in psoriasis in phase III studies.
Linked Comment: S. Gerdes and J. Albrecht. Br J Dermatol 2022; 186:603–604.
Plain language summary available online
The introduction of biologics that target tumour necrosis factor (TNF) or interleukin (IL)‐12/23 has enabled the majority of patients with plaque psoriasis to achieve a 75% improvement from baseline in Psoriasis Area and Severity Index (PASI 75). 1 More recently, biologics targeting IL‐23 and IL‐17A have further raised treatment expectations, with PASI 90 becoming a realistic goal, and many patients also achieving PASI 100 (complete skin clearance). 2 , 3 , 4 , 5
IL‐17A, the signature cytokine of T helper (Th)17 cells, is a proinflammatory cytokine overexpressed in psoriatic tissue. 6 There is significant clinical evidence that IL‐17A inhibition leads to rapid clinical, histological and molecular resolution of psoriasis. IL‐17F is a close family member of IL‐17A, and while it has been shown to be around 100‐fold less potent than IL‐17A in inducing proinflammatory cytokines such as CXCL1 (C‐X‐C motif chemokine ligand 1), IL‐8 (encoded by the CXCL8 gene) and IL‐6, it is much more abundant in psoriatic skin. 7 , 8 Increasing evidence highlights the central role of both IL‐17A and IL‐17F in the underlying disease pathobiology of psoriasis, by driving aberrant keratinocyte biology. Both keratinocyte proliferation and release of factors such as IL‐36γ and IL‐17C amplify inflammation and plaque chronicity. 9 , 10 , 11
IL‐17A and IL‐17F are principally produced by activated Th17 cells and exist in dimeric isoforms (homodimers IL‐17A/A and IL‐17F/F, and heterodimer IL‐17A/F). 6 , 12 Each dimer binds an IL receptor complex, IL‐17RA/IL‐RC, and therefore can transduce similar signalling and activation of target genes. 12 It has been found that all IL‐17 isoforms are produced in active plaques of psoriasis vulgaris, and thus each has potential to stimulate ‘downstream’ activation of keratinocytes and other cell types that ultimately create visible psoriasis lesions. 7
A significant challenge in chronic diseases such as psoriasis is the need for patients to receive medication over long periods of time. Less frequent treatment administration may increase treatment adherence and quality of life. 13 , 14 , 15 The commonly used IL‐17A inhibitors, secukinumab and ixekizumab, 16 , 17 both require dosing every 4 weeks (Q4W) during maintenance treatment, and loss of response with these agents has driven efforts to optimize response with more frequent intervals, 5 , 18 as already used with the IL‐17 receptor inhibitor, brodalumab. 19 There remains an unmet need for a treatment that provides both high rates of complete skin clearance and less frequent dosing. 20
Bimekizumab is a monoclonal IgG1 antibody that selectively inhibits both IL‐17A and IL‐17F. 21 , 22 , 23 Preclinical studies in human in vitro assays have shown that dual inhibition of IL‐17A and IL‐17F with bimekizumab leads to a greater inhibitory effect than blockade of either cytokine alone. 11 Phase II studies in patients with moderate‐to‐severe plaque psoriasis demonstrated high rates of skin clearance with bimekizumab, which were well‐maintained with Q4W maintenance dosing. 24 , 25
Here, we present results from a phase II exploratory trial designed to investigate whether high rates of skin clearance can be achieved with a prolonged bimekizumab maintenance dosing interval, and whether bimekizumab treatment is associated with the molecular resolution of psoriasis and other inflammatory markers.
Patients and methods
Study design
This prospective, randomized, patient‐blinded, investigator‐blinded, parallel‐group phase IIa study (NCT03025542) was conducted in eight study sites in Australia, Canada, USA and Moldova. Patients were randomized 2 : 1 to two treatment groups, both of which received bimekizumab dosed at 320 mg via subcutaneous administration at weeks 0 and 4; at week 16 the first group received placebo [the bimekizumab plus placebo (BKZ+PBO) group] and the second group received a third dose of bimekizumab 320 mg (the BKZ group) (Figure 1). Further details of randomization and study blinding are described in Appendix S1 (see Supporting Information).
Figure 1.
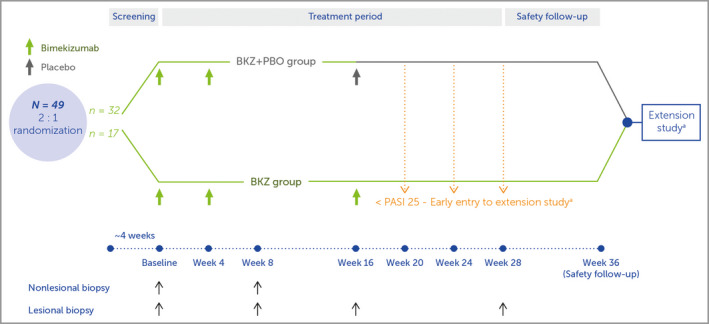
Study design. Study visits occurred at week 0 (baseline), and weeks 2, 4, 8, 12, 16, 20, 28 and 36 (safety follow‐up). The bimekizumab plus placebo (BKZ+PBO) group received two doses of bimekizumab 320 mg, at weeks 0 and 4, with placebo at week 16; the BKZ group received three doses of bimekizumab 320 mg, at weeks 0, 4 and 16. aOnly patients who achieved a 25% improvement from baseline Psoriasis Area and Severity Index score (PASI 25) in the first 16 weeks were eligible for entry to the extension study (NCT03230292) after the safety follow‐up visit; if these patients failed to achieve PASI 25 at weeks 20, 24 or 28 they were permitted to enter the extension study early, receiving the first dose of study drug at that visit.
The institutional review board or independent ethics committee at all the investigational sites approved the protocol, and the study was performed in accordance with the Declaration of Helsinki and Good Clinical Practice.
Patients
Eligible patients were aged between 18 and 70 years; they had plaque psoriasis for ≥ 6 months prior to screening, and moderate‐to‐severe plaque psoriasis [defined as PASI ≥ 12, body surface area (BSA) involvement ≥ 10% and Investigator’s Global Assessment (IGA) of ≥ 3 (5‐point scale)] at screening and baseline; and were candidates for systemic psoriasis therapy and/or phototherapy and/or chemophototherapy. Patients were excluded if they were taking medications that could influence treatment efficacy, including prior bimekizumab treatment, or had erythrodermic, guttate, pustular or drug‐induced psoriasis.
Endpoints and assessments
The primary efficacy endpoint was change from baseline in PASI at week 28 in each treatment group. The primary pharmacokinetic variable was plasma bimekizumab concentration over the course of the trial. The primary immunological variable was anti‐bimekizumab antibody detection prior to and following study treatment. The primary safety variable was incidence of adverse events (AEs). Secondary efficacy endpoints were the proportion of patients achieving PASI 75, PASI 90 and PASI 100, and IGA 0/1 [IGA score of 0 or 1 (clear or almost clear) with ≥ 2‐point improvement from baseline] at week 16.
Bioanalytical and transcriptome analyses
Blood samples for the purpose of measuring pharmacokinetics were collected at baseline (predose) and at all subsequent study visits. Samples for anti‐drug antibody (ADA) analysis were also taken at each visit, except week 2. Skin biopsies for transcriptome analysis were taken from lesional psoriatic plaques and paired nonlesional skin at baseline and week 8, and from lesional psoriatic plaques only at weeks 16 and 28 (Figure 1). Detailed methods for bioanalytical and transcriptome analyses can be found in Appendix S1.
Statistical analyses and sample size
The safety set consisted of all patients who received ≥ 1 dose of study drug, with the full analysis set (FAS) consisting of the subset of the safety set who had a valid PASI measurement at baseline.
Efficacy variables were based on the FAS. Results were combined across all patients for the first 16 weeks and separated by treatment group thereafter. Changes from baseline and responder variables were defined relative to the last available measurement prior to the first bimekizumab injection. Missing data were imputed as last observation carried forward for continuous variables and using nonresponder imputation for binary variables. Descriptive statistics were used.
All safety variables were performed using the safety set. Treatment‐emergent AEs (TEAEs) were defined as any AE occurring from the first dose of study drug up to 140 days after the final dose (i.e. up to the week‐36 safety follow‐up visit). AEs occurring after this date were not classified as TEAEs and were excluded. AEs were coded using MedDRA version 19.0 (https://www.meddra.org).
For pharmacokinetic data, actual blood sampling times for bimekizumab concentrations were obtained relative to the start time of the first injection. Individual concentrations were summarized by treatment group at each scheduled visit based on the pharmacokinetic per‐protocol set. ADA status (positive/negative) was summarized as a categorical endpoint (number and percentage of patients) by treatment group for all timepoints and overall, based on the safety set.
No formal statistical sample size estimation was performed owing to the exploratory nature of this study, which was not powered for any conclusive statistical analysis of efficacy, pharmacokinetics, safety or immunological variables. The sample size and 2 : 1 randomization were determined based on the number of patients required in the current study to provide reliable pharmacokinetic/pharmacodynamic model parameter estimates when combined with data from the phase IIb trial, BE ABLE 1. 25
Results
Patients and baseline demographics
Beginning 27 December 2016, 73 patients were screened and 49 patients were randomized: 32 to the BKZ+PBO group and 17 to the BKZ group (Figure 2). The last study visit occurred on 11 December 2017. Patient demographics and baseline characteristics were generally balanced (Table 1), with the exception of the higher mean BSA involvement in the BKZ group, attributed to two patients with BSA involvement > 60% (82·5% and 72·1%, respectively). Mean baseline PASI was 18·3 in the BKZ+PBO group and 23·4 in the BKZ group. The overall study population was comparable to that in other studies of bimekizumab in psoriasis. 25 , 26 , 27 , 28 , 29
Figure 2.
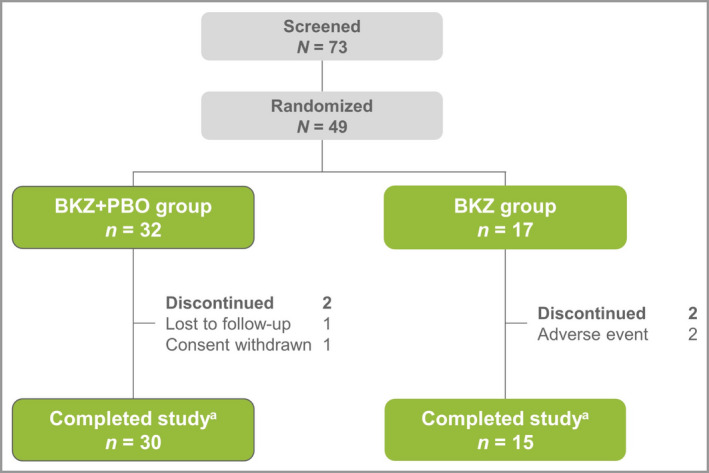
Patient disposition. The bimekizumab plus placebo (BKZ+PBO) group received two doses of bimekizumab 320 mg at weeks 0 and 4, with placebo at week 16; the BKZ group received three doses of bimekizumab 320 mg, at weeks 0, 4 and 16. aStudy completion was defined as completing week 28 or completing the visits at weeks 20, 24 or 28 and entering the extension study early.
Table 1.
Demographics and baseline characteristics
|
BKZ+PBO groupa (n = 32) |
BKZ groupa (n = 17) |
All patients (N = 49) |
|
|---|---|---|---|
| Demographics | |||
| Age (years), mean (SD) | 41·8 (14·1) | 45·9 (10·4) | 43·2 (13·0) |
| Male, n (%) | 15 (47) | 11 (65) | 26 (53) |
| White, n (%) | 30 (94) | 12 (71) | 42 (86) |
| Body mass index, kg m‐2, mean (SD) | 30·1 (5·6) | 31·0 (5·9) | 30·4 (5·7) |
| Baseline disease characteristics | |||
| Disease duration (years), mean (SD) | 13·5 (12·3) | 14·9 (10·5) | 14·0 (11·6) |
| PASI, mean ± SD | 18·3 (6·7) | 23·4 (10·1) | 20·1 (8·3) |
| Body surface area (%), mean (SD) | 22·0 (12·4) | 34·8 (21·5) | 26·4 (17·1) |
| Investigator’s Global Assessment, n (%) | |||
| 3: moderate | 28 (88) | 14 (82) | 42 (86) |
| 4: severe | 4 (13) | 3 (18) | 7 (14) |
| Prior biologic therapy, n (%) | 4 (13) | 0 (0) | 4 (8) |
PASI, Psoriasis Area and Severity Index. aBoth the BKZ+PBO and BKZ groups received bimekizumab 320 mg at weeks 0 and 4; at week 16 the BKZ+PBO group received placebo and the BKZ group received a third dose of bimekizumab 320 mg.
Clinical response
The primary efficacy endpoint in this trial was change in PASI from baseline at week 28. Absolute change from baseline at week 28 was –10·8 [95% confidence interval (CI) –13·5 to –8.0] in the BKZ+PBO group and –19·7 (95% CI –24·2 to –15·2) in the BKZ group. Substantial improvements in PASI response rate were observed after two bimekizumab doses, at weeks 0 and 4, reaching a maximum mean decrease across all patients of 94% at week 16 (12 weeks after the second dose).
Rapid responses were demonstrated by the proportion of patients achieving PASI 75, PASI 90 and PASI 100 responses during the first 8 weeks, with 16% achieving PASI 100 at week 4 (Figure 3). This response rate increased to 47% by week 8 (4 weeks after the second dose). PASI 75 and PASI 90 responder rates reached a maximum at week 16 (92% and 80%, respectively), whereas the maximal PASI 100 response rate of 57% was observed at week 12 (8 weeks after the second dose), before decreasing to 49% at week 16 (12 weeks after the second dose).
Figure 3.
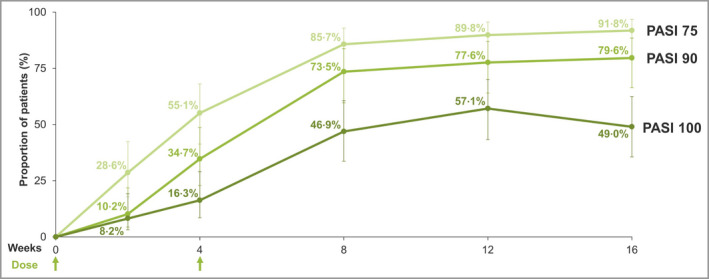
Psoriasis Area and Severity Index (PASI) response to week 16 for the combined treatment groups (nonresponder imputation). PASI 75/90/100, ≥ 75%/90%/100% improvement from baseline PASI.
Patients who received an additional bimekizumab dose at week 16 generally maintained their PASI 90 response at week 28 (12 weeks later), with a similar trend observed for IGA 0/1 response, while responses were diminished at week 28 in patients who received placebo at week 16 (Figure 4). In patients who received an additional dose at week 16, the PASI 100 response rate was increased at week 20, but a moderate reduction was observed at week 28, whereas a large reduction was observed at week 28 in patients who received no additional dose (week 28 response rates 41% vs. 13%, respectively).
Figure 4.
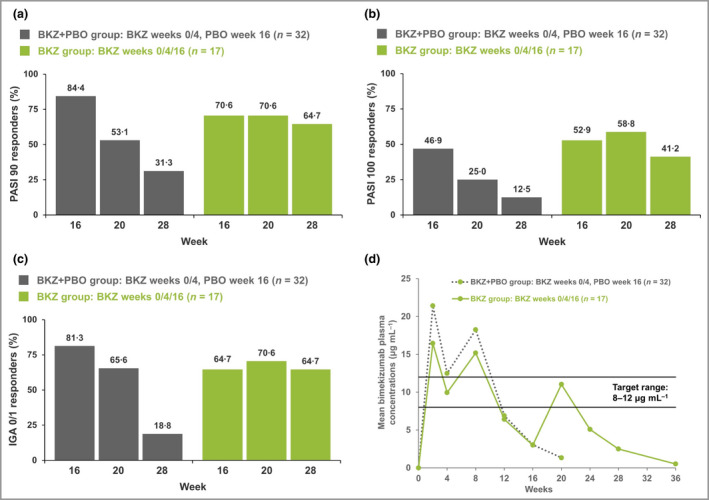
Psoriasis Area and Severity Index (PASI) score and Investigator’s Global Assessment (IGA) 0/1 responses at weeks 16, 20 and 28 (nonresponder imputation); and bimekizumab (BKZ) plasma concentration from baseline to week 36. The bimekizumab plus placebo (BKZ+PBO) group received two doses of BKZ 320 mg at weeks 0 and 4, with placebo at week 16; the BKZ group received three doses of bimekizumab 320 mg, at weeks 0, 4 and 16. (a) 90% improvement in PASI (PASI 90) responses. (b) PASI 100 responses (total clearance). (c) IGA 0/1 responses: score of 0 (clear) or 1 (almost clear) with ≥ 2‐category improvement relative to baseline IGA, scored on a 5‐point scale. (d) Bimekizumab plasma concentration. See Figure S1 (Supporting Information) for the bimekizumab plasma concentration target range rationale.
Safety
Overall, TEAEs were reported in 88% of patients at a similar incidence between treatment groups (Table 2). Among the TEAEs occurring in ≥ 5% of all patients, the two most common were upper respiratory tract infection (18%) and nasopharyngitis (12%).
Table 2.
Treatment‐emergent adverse events (TEAEs)
|
BKZ+PBO groupa (n = 32) |
BKZ groupa (n = 17) |
All patients (N = 49) |
|
|---|---|---|---|
| Any TEAEs | 28 (88) | 15 (88) | 43 (88) |
| Serious TEAEs | 2 (6) | 1 (6) | 3 (6) |
| Discontinuation due to TEAEs | 0 (0) | 2 (12) | 2 (4) |
| Treatment‐related TEAEs | 11 (34) | 9 (53) | 20 (41) |
| Severe TEAEs | 1 (3) | 3 (18) | 4 (8) |
| Deaths | 0 (0) | 0 (0) | 0 (0) |
| Most common TEAEs (≥ 5% of all patients) | |||
| Infections and infestationsb | |||
| Upper respiratory tract infection | 6 (19) | 3 (18) | 9 (18) |
| Nasopharyngitis | 2 (6) | 4 (24) | 6 (12) |
| Urinary tract infection | 4 (13) | 0 (0) | 4 (8) |
| Viral upper respiratory tract infection | 2 (6) | 2 (12) | 4 (8) |
| Gastrointestinal viral | 2 (6) | 1 (6) | 3 (6) |
| Investigations | |||
| Alanine aminotransferase increased | 2 (6) | 3 (18) | 5 (10) |
| Gamma‐glutamyltransferase increased | 2 (6) | 3 (18) | 5 (10) |
| White blood cell count decreased | 2 (6) | 3 (18) | 5 (10) |
| Aspartate aminotransferase increased | 2 (6) | 2 (12) | 4 (8) |
| Neutrophil count decreased | 1 (3) | 3 (18) | 4 (8) |
| Blood cholesterol increased | 1 (3) | 2 (12) | 3 (6) |
| Metabolism and nutrition disorders | |||
| Hyperkalaemia | 4 (13) | 1 (6) | 5 (10) |
| Nervous system disorders | |||
| Headache | 3 (9) | 1 (6) | 4 (8) |
All values are n (%). aBoth the BKZ+PBO and BKZ groups received bimekizumab 320 mg at weeks 0 and 4; at week 16 the BKZ+PBO group received placebo and the BKZ group received a third dose of bimekizumab 320 mg. bIn addition, two cases of oral candidiasis, an adverse event of special interest, were reported, in patients in the BKZ+PBO group (4% of all patients).
Three patients reported serious TEAEs: one case of nonrelated, severe peripheral sensorimotor neuropathy and one case of nonrelated, severe syncope (114 days after the final dose of study medication), in two patients in the BKZ+PBO group, and one case of nonrelated, acute cholecystitis and pancreatitis in a patient in the BKZ group. No serious TEAEs were considered related to the study drug, and all resolved with no sequelae. Two patients, both in the BKZ group, discontinued treatment due to TEAEs: one reported a nonserious TEAE of decreased lymphocyte count (29 days after study commencement) and the other reported one TEAE of increased alanine aminotransferase and two TEAEs of increased gamma‐glutamyltransferase (the first being 15 days after the last dose of study medication). There were no deaths.
Pharmacokinetics and anti‐drug antibodies
The plasma bimekizumab concentrations following subcutaneous administration were consistent with expectations and other bimekizumab studies, maintaining trough concentrations of around 8–12 µg mL–1 over the first 10–12 weeks from the start of treatment (Figure 4d) (Figure S1 shows plasma concentration target range rationale; see Supporting Information). In patients who received a third dose at week 16 (the BKZ group), an increase in plasma bimekizumab concentrations was observed, whereas an exponential decrease in levels was observed in patients who did not receive an additional dose, with the expected elimination rate associated with a T½ of ˜23 days.
The total incidence of anti‐bimekizumab antibodies over the 36‐week trial period, defined as a single postdose titre that was confirmed positive, was 34% (11 of 32) in the BKZ+PBO group and 47% (eight of 17) in the BKZ group (noting that a neutralizing antibody assay was not available at time of study). One patient in the BKZ+PBO group was confirmed positive at baseline and again at weeks 20, 24 and 28, and at the safety follow‐up visit, but with no increase in titre. Review of the individual pharmacokinetic and PASI time‐course data suggests there was no impact of anti‐bimekizumab antibodies on any of drug exposure, clinical response or safety across the 28‐week treatment period.
Transcriptome analyses: molecular resolution of the psoriasis transcriptome by bimekizumab
The molecular effect of bimekizumab was examined using changes in transcript abundance at week 8 vs. baseline. These changes were analysed relative to the psoriasis transcriptome, defined as the set of differentially expressed probesets (and unique mapped genes) in baseline lesional skin compared with paired nonlesional skin biopsies.
Bimekizumab treatment led to a rapid, deep normalization of the psoriasis transcriptome at week 8, following two doses. Probesets that were more highly expressed in psoriatic skin had a median percentage improvement of 97%, representing normalization to near nonlesional levels (Figure 5). Bimekizumab also normalized probesets that were downregulated in lesional skin by 84%. Overall, there was a strong negative correlation between gene expression changes at baseline and gene expression changes elicited by bimekizumab treatment (Pearson correlation coefficient r = –0·97, P < 0·001). Principal component analysis indicated a homogeneous and deep molecular response across patients, with lesional samples at week 8 closely resembling baseline nonlesional samples in latent space. A heatmap of the expression of probesets in the psoriasis transcriptome further illustrates the comprehensive scope of the molecular response to bimekizumab, both in terms of the breadth of disease genes whose expression was reversed towards nonlesional levels and the homogeneity of the molecular response among patients (Figure 5c).
Figure 5.
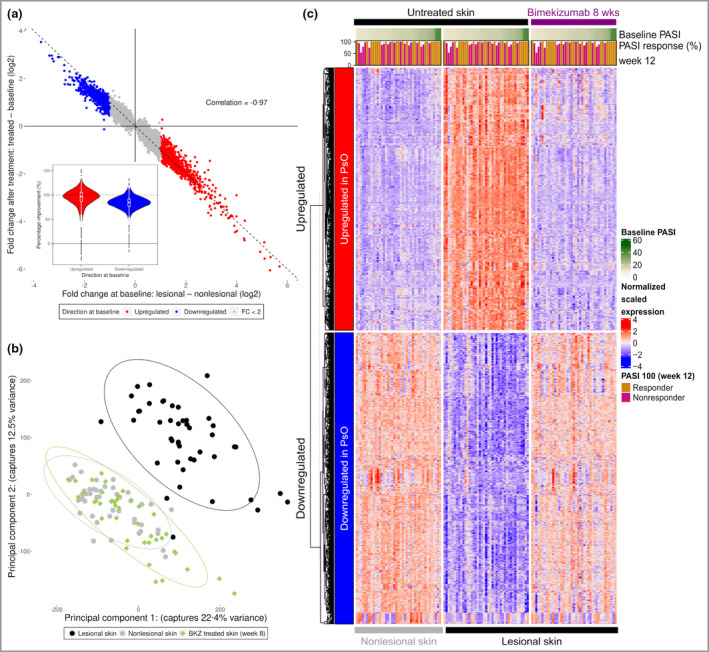
Molecular resolution of the psoriasis (PsO) transcriptome by bimekizumab (BKZ). (a) Scatter plot comparing the fold change (FC) at baseline and after treatment for each probeset significantly dysregulated at baseline. Probesets are coloured by their direction at baseline. Violin plot insert of percentage improvements of psoriasis probesets. (b) Results of principal component analysis used to project the sample transcriptomes onto two dimensions (together explaining 35% of global variance in the data). (c) Heat map of expression profiles for probesets in the psoriasis transcriptome at baseline and after treatment. Expression values are scaled by row, and probesets are split first by direction and then clustered using hierarchical clustering. Samples are arranged in ascending order of Psoriasis Area and Severity Index (PASI) scores at baseline. Bars signify the percentage PASI response at week 12, with PASI 100 responders (those with 100% improvement from baseline PASI) further highlighted in orange.
Of the 1082 probesets differentially upregulated in lesional skin at baseline [842 differentially expressed genes (DEGs)], 880 (81%) were also significantly reversed following bimekizumab treatment at week 8. Conversely, 716 (60%) of the 1201 downregulated probesets (835 DEGs) in the psoriasis transcriptome were significantly upregulated at week 8. By relaxing the treatment effect size threshold from ≥ 2·0 to ≥ 1·5, the number of reversed probesets rose to 1069 (99%) and 1182 (98%), respectively. Tables S1 and S2 (see Supporting Information) detail the top 50 probesets downregulated and upregulated by bimekizumab in lesional skin at week 8.
IL‐17‐responsive genes, induced by stimulation of cultured keratinocytes with supernatant from Th17 cells [Figure 6; and Figure S2 (see Supporting Information)], were among the more strongly normalized genes in the psoriasis transcriptome, with a median percentage improvement of 105% 4 weeks after the second dose. Selected marker genes that drive plaque formation were also deeply normalized by week 8 (Figures 7 and 8). These genes capture key upstream cytokines (IL17A, IL17F, and IL23A and IL12B); downstream effectors such as antimicrobial peptides (S100A7) and proinflammatory chemotactic factors [CXCL1, CXCL8, CCL20 (C‐C motif chemokine 20)]; markers of keratinocyte proliferation (KRT16); and feedforward cytokines (IL17C, IL36G, IL36A).
Figure 6.
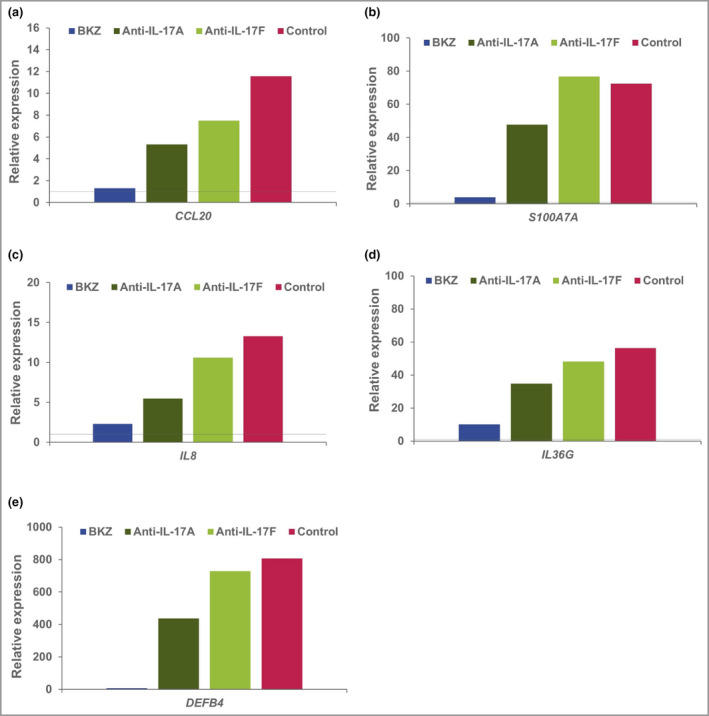
Relative expression of key genes in T helper (Th)17‐stimulated keratinocytes. Human keratinocytes were stimulated with Th17 supernatant, with anti‐interleukin (IL)‐16 blocking antibodies, or without (control). Expression relative to unstimulated cells (shown as a horizontal line) is reported. BKZ, bimekizumab.
Figure 7.
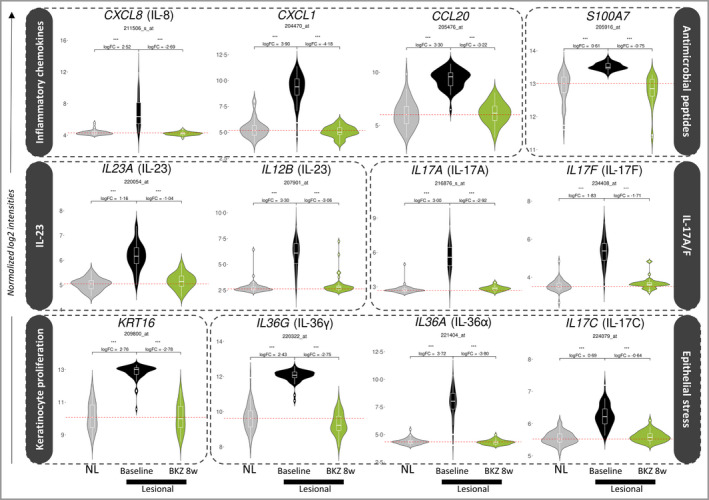
Expression of 12 key genes in treated lesional tissue at week 8 relative to baseline nonlesional (NL) and lesional tissue. Log2 normalized expression of probesets corresponding to specific marker genes that underpin psoriasis pathology, in baseline lesional and nonlesional samples and treated lesional samples at week 8. Hashed red horizontal lines correspond to the median baseline expression of a probeset in nonlesional tissue. Log fold change (FC) and false discovery rate (FDR)‐adjusted P‐values are calculated using the limma moderated t‐test. IL, interleukin. BKZ, bimekizumab; CXCL, C‐X‐C motif chemokine ligand 1; CCL, C‐C motif chemokine; KRT, keratin. ***FDR < 0·001.
Figure 8.
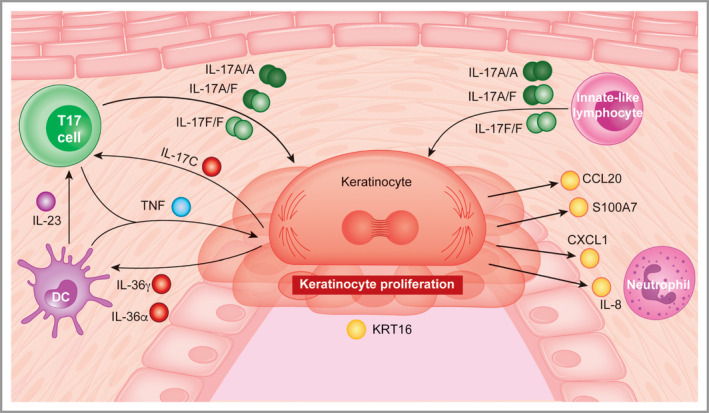
Drivers of plaque formation in psoriasis. Proinflammatory chemotactic factors: CXCL1 (C‐X‐C motif chemokine ligand 1), interleukin (IL)‐8 (encoded by CXCL8) and CCL20 (C‐C motif chemokine 20); upstream cytokines: IL‐17A, IL‐17F, IL‐23A and IL‐12B; markers of epithelial stress: IL‐19, IL‐36γ and IL‐36α; proliferation: KRT16; antimicrobial peptide: S100A7. TNF, tumour necrosis factor. The T17 cell represents IL‐17‐producing T cells (including cytotoxic T cells and T helper cells).
To evaluate response durability at a molecular level, median percentage improvement of the psoriasis transcriptome was assessed at weeks 8, 16 and 28 (Figure S3; see Supporting Information). Percentage improvement of upregulated genes reduced from a maximum of 97% at week 8 to 90% by week 16, 12 weeks after the second bimekizumab dose. At week 28, normalization was maintained in patients who received an additional dose at week 16 (94% improvement), but was substantially diminished if no extra dose was administered (60% improvement).
Discussion
Bimekizumab is a monoclonal antibody that selectively inhibits all IL‐17A and IL‐17F dimeric isoforms. Increasing evidence highlights the central role of both cytokines in psoriasis pathogenesis. Since the introduction of anti‐IL‐17 agents for psoriasis treatment, 16 , 17 , 19 patient expectations have shifted from achieving PASI 75 to maintenance of PASI 100, with infrequent dosing. 13 , 14 , 15
Analyses have shown that improvements in quality of life correlate with the achievement of complete skin clearance, 30 , 31 which can also be attained through longer dosing intervals. 31 Here, bimekizumab treatment resulted in a rapid response onset and achievement of high complete skin clearance rates in the first 16 weeks. While PASI 90 was maintained from week 8 to week 16, PASI 100 response was maximal at week 12 (8 weeks after the second bimekizumab dose) before decreasing. At week 16, patients received either placebo or a third bimekizumab dose. At week 28, 12 weeks later, substantial reductions in all efficacy endpoints were observed in patients who received placebo. In contrast, PASI 90 and IGA 0/1 were maintained in patients who received an additional bimekizumab dose, with a slight reduction in PASI 100. These observations suggest maintenance dosing every 12 weeks would be too infrequent for sustaining complete skin clearance, but this may be possible with dosing every 8 weeks (Q8W). These data helped provide an understanding of the basis for the dosing regimens selected for the phase III trials, which evaluated Q8W dosing, alongside Q4W dosing. 26 , 28 , 29
These data were combined with those from the phase IIb study BE ABLE 1 25 for pharmacokinetic/pharmacodynamic modelling and subsequent simulation (combined data not reported). It was concluded that an 8‐week dosing interval during maintenance treatment (post week 16) was an option for the maintenance of high skin clearance (PASI 90/100) in the majority of patients with moderate‐to‐severe psoriasis. Furthermore, less frequent dosing (e.g. every 12 weeks or intermittently) is likely to lead to loss of response in some patients.
Transcriptomic analysis of nonlesional and lesional biopsies showed a clear and distinct molecular profile. Dual inhibition of IL‐17A and IL‐17F with bimekizumab normalized the psoriatic transcriptome associated with aberrant keratinocyte biology in psoriatic skin, leading to molecular resolution. Specifically, expression of genes encoding the antimicrobial peptides β‐defensin 2 (DEFB4A), S100 family, neutrophil chemotactic factors such as CXCL1 and IL‐8 (CXCL8), and the chemotactic factor for Th17 cells, CCL20, was profoundly normalized. Other keratinocyte‐derived factors such as IL‐17C and IL‐36γ, which are thought to amplify the IL‐23/IL‐17 axis in driving lesion chronicity, were also normalized to levels seen in nonlesional skin. Furthermore, expression of IL23 transcripts, along with IL17A and IL17F transcripts themselves, was normalized to nonlesional levels. The affected pathways are consistent with data reported for other IL‐17 therapies such as secukinumab and brodalumab, 32 , 33 and in this study bimekizumab induced a 97% median normalization of the psoriasis transcriptome by week 8.
Therapies that directly target IL‐23, such as risankizumab and guselkumab, demonstrate durable clinical responses and require maintenance dosing less frequent than Q4W. 2 , 3 The near normalization of IL23 transcript expression at 8 weeks after two bimekizumab doses, along with the almost complete normalization of the psoriatic transcriptome (including IL17A and IL17F expression), may in part explain the prolonged efficacy observed within the withdrawal arm of the study, and further supported an 8‐week dosing interval with bimekizumab in phase III trials. 26 , 28 , 29 Similar observations of prolonged efficacy were subsequently confirmed across the phase III studies. 26 , 28 , 29
In this study, we observed that bimekizumab, the only monoclonal antibody under investigation designed to selectively inhibit both IL‐17A and IL‐17F, demonstrates rapid and deep clinical responses along with a profound normalization of the psoriatic transcriptome, providing strong evidence that both IL‐17A and IL‐17F are key inflammatory entities underpinning psoriasis pathogenesis. In addition, unlike IL‐17A therapeutics that require Q4W (or more frequent) maintenance dosing, these data supported the decision to evaluate Q8W maintenance dosing alongside Q4W dosing with bimekizumab in subsequent phase III psoriasis studies. 26 , 28 , 29
Author Contribution
Ruth Oliver: Conceptualization (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). James G. Krueger: Data curation (equal); Investigation (equal); Methodology (equal); Writing‐review & editing (equal). Sophie Glatt: Conceptualization (equal); Visualization (equal); Writing‐review & editing (equal). Pavan Vajjah: Conceptualization (equal); Data curation (equal); Formal analysis (equal); Visualization (equal); Writing‐review & editing (equal). Chetan Mistry: Visualization (equal); Writing‐review & editing (equal). Matthew Page: Data curation (equal); Formal analysis (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Hannah Edwards: Data curation (equal); Formal analysis (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Sandra Garcet: Investigation (equal); Writing‐review & editing (equal). Xuan Li: Investigation (equal); Writing‐review & editing (equal). Benjamin Dizier: Visualization (equal); Writing‐review & editing (equal). Ash Maroof: Data curation (equal); Formal analysis (equal); Methodology (equal); Software (equal); Supervision (equal); Validation (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal). Mark Watling: Conceptualization (equal); Methodology (equal); Project administration (equal); Supervision (equal); Visualization (equal); Writing‐review & editing (equal). Assem el Baghdady: Visualization (equal); Writing‐review & editing (equal). Dominique Baeten: Visualization (equal); Writing‐review & editing (equal). Lucian Ionescu: Conceptualization (equal); Visualization (equal); Writing‐review & editing (equal). Stevan Shaw: Conceptualization (equal); Visualization (equal); Writing‐original draft (equal); Writing‐review & editing (equal).
Supporting information
Appendix S1 Supplementary methods.
Figure S1 Target plasma concentration range.
Figure S2 Molecular resolution of interleukin‐17 pathway genes.
Figure S3 Time course of molecular resolution of the psoriasis transcriptome by bimekizumab.
Table S1 Top 50 genes downregulated by bimekizumab in lesional skin at week 8.
Table S2 Top 50 genes upregulated by bimekizumab in lesional skin at week 8.
Acknowledgments
The authors thank the patients, the investigators and their teams who took part in this study. The authors also acknowledge Susanne Wiegratz, MSc, UCB Pharma, Monheim, Germany, for publication coordination; Eva Cullen, PhD, UCB Pharma, Brussels, Belgium, for critical review; and Joe Dixon, PhD, from Costello Medical, UK, for medical writing and editorial assistance based on the authors’ input and direction. This study was funded by UCB Pharma.
Funding sources This study was funded by UCB Pharma. Support for third‐party writing assistance for this article, provided by Joe Dixon, PhD, Costello Medical, UK, was funded by UCB in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org/gpp3).
Conflicts of interest R.O., S. Glatt, P.V., M.P., H.E., B.D., A.M., M.W., D.B., L.I. and S.S. are employees of UCB Pharma. J.G.K. has received grants paid to his institution from AbbVie, Akros, Allergan, Amgen, Avillion, Biogen MA Inc., Boehringer Ingelheim, Botanix Pharmaceuticals, Bristol Myers Squibb, Celgene, Eli Lilly, Exicure, Inc., Incyte Corporation, Innovaderm Research Inc., Janssen Pharmaceuticals, LEO Pharma, Novan, Novartis, Parexel, Pfizer, Regeneron Pharmaceuticals, Sienna Biopharmaceuticals, UCB and Vitae Pharmaceuticals; and he has received personal fees from AbbVie, Aclaris Therapeutics Inc., Allergan, Almirall, Amgen, Arena Pharmaceuticals Ltd, Aristea Therapeutics, Asana BioSciences, Aurigene, Biogen Idec, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Eli Lilly, Escalier Biosciences, Galapagos NV, LEO Pharma, Menlo Therapeutics, Nimbus Therapeutics, Novartis, Pfizer, Sanofi, Sienna, Sun Pharmaceutical Industries Ltd, UCB, Valeant and Ventyx Biosciences. C.M. is a Veramed statistical consultant for UCB Pharma. A.e.B. is an external consultant contracted by UCB, and is a visiting senior lecturer at the Institute of Pharmaceutical Science, King’s College London. S. Garcet and X.L. declare they have no conflicts of interest.
R.O. and J.G.K. are joint primary authors.
Data availability statement Underlying data from this manuscript may be requested by qualified researchers 6 months after product approval in the USA and/or Europe, or global development is discontinued, and 18 months after trial completion. Investigators may request access to anonymized individual patient‐level data and redacted trial documents, which may include: analysis‐ready datasets, study protocol, annotated case report form, statistical analysis plan, dataset specifications and clinical study report. Prior to use of the data, proposals need to be approved by an independent review panel at Vivli (vivli.org) and a signed data sharing agreement will need to be executed. All documents are available in English only, for a prespecified time, typically 12 months, on a password protected portal.
Plain language summary available online
References
- 1. Sbidian E, Chaimani A, Afach S et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta‐analysis. Cochrane Database Syst Rev 2020; 12:CD011535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gordon KB, Strober B, Lebwohl M et al. Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet 2018; 392:650–61. [DOI] [PubMed] [Google Scholar]
- 3. Reich K, Armstrong AW, Langley RG et al. Guselkumab versus secukinumab for the treatment of moderate‐to‐severe psoriasis (ECLIPSE): results from a phase 3, randomised controlled trial. Lancet 2019; 394:831–9. [DOI] [PubMed] [Google Scholar]
- 4. Blauvelt A, Reich K, Tsai T‐F et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate‐to‐severe plaque psoriasis up to 1 year: results from the CLEAR study. J Am Acad Dermatol 2017; 76:60–9.e9. [DOI] [PubMed] [Google Scholar]
- 5. Langley R, Papp K, Gooderham M et al. Efficacy and safety of continuous every‐2‐week dosing of ixekizumab over 52 weeks in patients with moderate‐to‐severe plaque psoriasis in a randomized phase III trial (IXORA‐P). Br J Dermatol 2018; 178:1315–23. [DOI] [PubMed] [Google Scholar]
- 6. Johansen C, Usher P, Kjellerup R et al. Characterization of the interleukin‐17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 2009; 160:319–24. [DOI] [PubMed] [Google Scholar]
- 7. Kolbinger F, Loesche C, Valentin M‐A et al. β‐Defensin 2 is a responsive biomarker of IL‐17A–driven skin pathology in patients with psoriasis. J Allergy Clin Immunol 2017; 139:923–32.e8. [DOI] [PubMed] [Google Scholar]
- 8. Wright JF, Guo Y, Quazi A et al. Identification of an interleukin 17F/17A heterodimer in activated human CD4+ T cells. J Biol Chem 2007; 282:13447–55. [DOI] [PubMed] [Google Scholar]
- 9. Blauvelt A, Chiricozzi A. The immunologic role of IL‐17 in psoriasis and psoriatic arthritis pathogenesis. Clin Rev Allergy Immunol 2018; 55:379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Johnston A, Fritz Y, Dawes SM et al. Keratinocyte overexpression of IL‐17C promotes psoriasiform skin inflammation. J Immunol 2013; 190:2252–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maroof A, Baeten D, Archer S et al. IL‐17F contributes to human chronic inflammation in synovial tissue: Preclinical evidence with dual IL‐17A and IL‐17F inhibition with bimekizumab in psoriatic arthritis. Ann Rheum Dis 2017; 76 (Suppl. 1):A13. Poster 02.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wright JF, Bennett F, Li B et al. The human IL‐17F/IL‐17A heterodimeric cytokine signals through the IL‐17RA/IL‐17RC receptor complex. J Immunol 2008; 181:2799–805. [DOI] [PubMed] [Google Scholar]
- 13. Bhoi P, Bessette L, Bell MJ et al. Adherence and dosing interval of subcutaneous antitumour necrosis factor biologics among patients with inflammatory arthritis: analysis from a Canadian administrative database. BMJ Open 2017; 7:e015872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Coleman CI, Limone B, Sobieraj DM et al. Dosing frequency and medication adherence in chronic disease. J Manag Care Pharm 2012; 18:527–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Reginster JY, Rabenda V, Neuprez A. Adherence, patient preference and dosing frequency: understanding the relationship. Bone 2006; 38:S2–S6. [DOI] [PubMed] [Google Scholar]
- 16. Pariser D, Frankel E, Schlessinger J et al. Efficacy of secukinumab in the treatment of moderate to severe plaque psoriasis in the North American subgroup of patients: pooled analysis of four phase 3 studies. Dermatol Ther (Heidelb) 2018; 8:17–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mease PJ, Smolen JS, Behrens F et al. A head‐to‐head comparison of the efficacy and safety of ixekizumab and adalimumab in biological‐naïve patients with active psoriatic arthritis: 24‐week results of a randomised, open‐label, blinded‐assessor trial. Ann Rheum Dis 2020; 79:123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phung M, Georgakopoulos JR, Ighani A et al. Secukinumab dose optimization in adult psoriasis patients: a retrospective, multicenter case series. JAAD Case Rep 2018; 4:310–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farahnik B, Beroukhim K, Abrouk M et al. Brodalumab for the treatment of psoriasis: a review of phase III trials. Dermatol Ther (Heidelb) 2016; 6:111–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brezinski EA, Armstrong AW. Off‐label biologic regimens in psoriasis: a systematic review of efficacy and safety of dose escalation, reduction, and interrupted biologic therapy. PLoS One 2012; 7:e33486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Adams R, Maroof A, Baker T et al. Bimekizumab, a novel humanized IgG1 antibody that neutralizes both IL‐17A and IL‐17F. Front Immunol 2020; 11:1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Glatt S, Baeten D, Baker T et al. Dual IL‐17A and IL‐17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo‐controlled clinical trial that IL‐17F contributes to human chronic tissue inflammation. Ann Rheum Dis 2018; 77:523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Glatt S, Helmer E, Haier B et al. First‐in‐human randomized study of bimekizumab, a humanized monoclonal antibody and selective dual inhibitor of IL‐17A and IL‐17F, in mild psoriasis. Br J Clin Pharmacol 2017; 83:991–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Blauvelt A, Papp KA, Merola JF et al. Bimekizumab for patients with moderate‐to‐severe plaque psoriasis: 60‐week results from BE ABLE 2, a randomized, double‐blinded, placebo‐controlled phase 2b extension study. J Am Acad Dermatol 2020; 83:1367–74. [DOI] [PubMed] [Google Scholar]
- 25. Papp KA, Merola JF, Gottlieb AB et al. Dual neutralization of both interleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: results from BE ABLE 1, a 12‐week randomized, double‐blinded, placebo‐controlled phase 2b trial. J Am Acad Dermatol 2018; 79:277–86.e10. [DOI] [PubMed] [Google Scholar]
- 26. Gordon KB, Foley P, Krueger JG et al. Bimekizumab efficacy and safety in moderate to severe plaque psoriasis (BE READY): a multicentre, double‐blind, placebo‐controlled, randomised withdrawal phase 3 trial. Lancet 2021; 397:475–86. [DOI] [PubMed] [Google Scholar]
- 27. Reich K, Papp KA, Blauvelt A et al. Bimekizumab versus ustekinumab for the treatment of moderate to severe plaque psoriasis (BE VIVID): efficacy and safety from a 52‐week, multicentre, double‐blind, active comparator and placebo controlled phase 3 trial. Lancet 2021; 397:487–98. [DOI] [PubMed] [Google Scholar]
- 28. Reich K, Warren RB, Lebwohl M et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med 2021; 385:142–52. [DOI] [PubMed] [Google Scholar]
- 29. Warren RB, Blauvelt A, Bagel J et al. Bimekizumab versus adalimumab in plaque psoriasis. N Engl J Med 2021; 385:130–41. [DOI] [PubMed] [Google Scholar]
- 30. Strober B, Papp KA, Lebwohl M et al. Clinical meaningfulness of complete skin clearance in psoriasis. J Am Acad Dermatol 2016; 75:77–82.e7. [DOI] [PubMed] [Google Scholar]
- 31. Puig L, Thom H, Mollon P et al. Clear or almost clear skin improves the quality of life in patients with moderate‐to‐severe psoriasis: a systematic review and meta‐analysis. J Eur Acad Dermatol Venereol 2017; 31:213–20. [DOI] [PubMed] [Google Scholar]
- 32. Krueger JG, Wharton KA Jr, Schlitt T et al. IL‐17A inhibition by secukinumab induces early clinical, histopathologic, and molecular resolution of psoriasis. J Allergy Clin Immunol 2019; 144:750–63. [DOI] [PubMed] [Google Scholar]
- 33. Russell CB, Rand H, Bigler J et al. Gene expression profiles normalized in psoriatic skin by treatment with brodalumab, a human ant‐IL‐17 receptor monoclonal antibody. J Immunol 2014; 192:3828–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Supplementary methods.
Figure S1 Target plasma concentration range.
Figure S2 Molecular resolution of interleukin‐17 pathway genes.
Figure S3 Time course of molecular resolution of the psoriasis transcriptome by bimekizumab.
Table S1 Top 50 genes downregulated by bimekizumab in lesional skin at week 8.
Table S2 Top 50 genes upregulated by bimekizumab in lesional skin at week 8.