

Abstract
Diazo compounds have been largely used as carbene precursors for carbene transfer reactions in a variety of functionalization reactions. However, the ease of carbene generation from the corresponding diazo compounds depends upon the electron donating/withdrawing substituents either side of the diazo functionality. These groups strongly impact the ease of N2 release. Recently, tris(pentafluorophenyl)borane [B(C6F5)3] has been shown to be an alternative transition metal‐free catalyst for carbene transfer reactions. Herein, a density functional theory (DFT) study on the generation of carbene species from α‐aryl α‐diazocarbonyl compounds using catalytic amounts of B(C6F5)3 is reported. The significant finding is that the efficiency of the catalyst depends directly on the nature of the substituents on both the aryl ring and the carbonyl group of the substrate. In some cases, the boron catalyst has negligible effect on the ease of the carbene formation, while in other cases there is a dramatic reduction in the activation energy of the reaction. This direct dependence is not commonly observed in catalysis and this finding opens the way for intelligent design of this and other similar catalytic reactions.
Keywords: carbene, density functional theory (DFT), diazo compounds, Lewis acid catalysis, mechanistic study, tris(pentafluorophenyl)borane
Extensive DFT calculations were carried out to interpret electronic effects of the substituents attached (electron donating/withdrawing substituents) to either side of a diazo functionality towards the ease of carbene generation from the corresponding diazo compounds in presence or absence of catalytic amounts of tris(pentafluorophenyl)borane [B(C6F5)3].

Introduction
Diazo compounds have been extensively used as carbene precursors and have been employed as reagents for a range of functionalization reactions of organic molecules. [1] The use of a precious transition metal catalyst is typically required for the generation of a metal carbenoid species. [2] However, metal‐free approaches towards carbene generation are highly desirable for drug discovery where trace amounts of metal impurities in the target molecules can cause an issue. [3] Recent studies have demonstrated that boranes can be used as an alternative to several transition metal catalysts to activate diazo compounds. [4] In this regard, borane catalyzed carbene transfer has been employed for a range of reactions such as C−H, [5] N−H, [6] O−H [7] insertion, C−C functionalization, [8] carbocycle formation,[ 5a , 9 ] and ring opening reactions. [5a] The efficacy of borane catalysts for carbene transfer reactions has raised our curiosity towards interpreting the mechanism for diazo‐activation. Many of the reports to date have included theoretical studies in which the mechanism for the borane catalyzed carbene transfer reaction has been proposed.[ 5a , 10 ] However, to date, the reactivity pattern between the Lewis acidic boranes and different donor‐acceptor diazo compounds has not yet been established. Theoretical calculations can provide a clear understanding on the stabilities and reactivities of diazo compounds, and their ease of carbene formation. [11] With B(C6F5)3, initial DFT studies by Stephan et al. demonstrated an interaction between the Lewis acidic borane and diphenyldiazomethane [12] which led to the formation of a reactive diazo‐borane adduct. Decomposition of the Ph2CN2 ⋅ B(C6F5)3 adduct subsequently led to the formation of a proposed carbene‐borane adduct [Ph2C ⋅ B(C6F5)3], which is exergonic by about 53 kcal/mol. In 2017, Wu et al. executed a theoretical calculation to rationalize the mode of activation of α‐aryl α‐diazocarbonyls using catalytic amounts of a Lewis acidic borane. [10]
DFT calculations showed that B(C6F5)3 can bind with the α‐aryl α‐diazocarbonyl compound in three possible ways; as a B−N adduct, a B−C adduct, or through B−O adduct formation. The results showed that boron preferred to bind to the carbonyl functionality, promoting N2 release (Scheme 1). The resulting carbene is generated as the B−O adduct, exists as a conjugated system enhancing the electrophilic character of the carbene carbon due to the electron‐withdrawing effect of B(C6F5)3. However, the formation of the B−O adduct, barrier to N2 release, and the electrophilicity of the carbene center can also be directly influenced by the substituents attached to the either side of the aryl ring or carbonyl functionality. Indeed, in our previous studies, we have observed differing rates of reactivity when using different diazo compounds.[ 5a , 5b , 5c ]
Scheme 1.
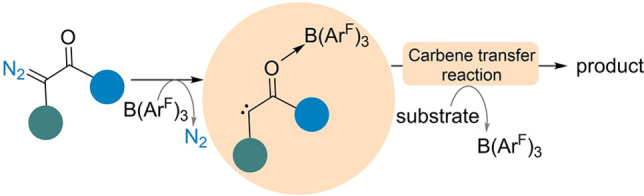
General mechanism for B(ArF)3‐catalyzed carbene transfer reaction.
Therefore, we decided to undertake a DFT study at the SMD/M06‐2X/def2‐TZVP//SMD/M06‐2X/6‐31G(d) level of theory in dichloromethane (see Supporting Information) to establish the energy barriers for B(C6F5)3‐catalyzed carbene formation from the corresponding α‐aryl α‐diazocarbonyl compounds when varying the electronic effects on the diazo compound. The primary focus of this research is on the specific influence of substituents on diazo substrates on catalyst efficiency, rather than on the mechanism of the reaction, which has already been established.[ 5a , 10 ]
Results and Discussion
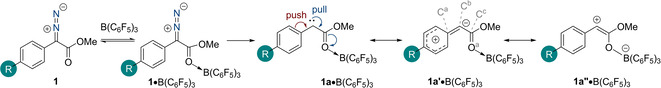
We began our investigations with an examination of how readily a free carbene can be generated from an α‐aryl α‐diazocarbonyl precursor in the absence of the boron Lewis acid catalyst (Scheme 2a). Different R substitutions on the aryl ring were introduced when R′=OMe and their influence on the generation of a free carbene was calculated (Table 1). As shown in Table 1, the reaction free energy for carbene formation (ΔG1) was found to be highly dependent upon the electronic nature of the R group. An upward shift in the ΔG1 value was observed going from electron donating groups (NMe2, NH2, OMe, and Me; Table 1; entries 1–4) to electron withdrawing groups (CF3, CN, NO2; Table 1; entries 8–10).
Scheme 2.

a) Carbene generation without using B(C6F5)3; b) Carbene generation in presence of B(C6F5)3; and c) Generic representation for carbene formation and the free energies associated with each step using catalytic B(C6F5)3.
Table 1.
Calculated free energies for the uncatalyzed and B(C6F5)3 catalyzed carbene formation from α‐aryl α‐diazocarbonyl with different R groups. Free energies are given in kcal/mol. The distance (rC a –C b) and WBI for the Ca−Cb bond in 1 a (Scheme 2a). The B−O bond distance (rB−O) in 1 a ⋅ B(C6F5)3. Hammett σp values for different R groups
|
| |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
Entry |
R |
ΔG≠ 1 |
ΔG1 |
rC a−C b [Å] |
σp |
WBI Ca−Cb |
ΔG≠ 2 |
ΔG2 |
rB−O [Å] |
ΔΔG≠ 1=ΔG≠ 1−ΔG≠ 2 |
ΔΔG1=ΔG1−ΔG2 |
|
Uncatalyzed |
B(C6F5)3 catalyzed |
Catalyst efficiency [x] |
|||||||||
|
1 |
NMe2 |
26.7 |
−0.1 |
1.388 |
−0.83 |
1.460 |
15.4 |
−13.8 |
1.516 |
11.3 |
13.7 |
|
2 |
NH2 |
26.8 |
0.3 |
1.389 |
−0.66 |
1.457 |
16.5 |
−11.9 |
1.517 |
10.3 |
12.2 |
|
3 |
OMe |
29.1 |
6.6 |
1.403 |
−0.27 |
1.369 |
21.1 |
−1.9 |
1.519 |
8.0 |
8.5 |
|
4 |
Me |
30.7 |
11.2 |
1.415 |
−0.17 |
1.310 |
25.1 |
5.9 |
1.539 |
5.6 |
5.3 |
|
5 |
F |
31.1 |
11.3 |
1.415 |
0.06 |
1.305 |
26.3 |
6.1 |
1.529 |
4.8 |
5.2 |
|
6 |
H |
32.0 |
13.1 |
1.421 |
0.0 |
1.282 |
25.9 |
6.5 |
1.533 |
6.1 |
6.6 |
|
7 |
Cl |
32.1 |
13.8 |
1.421 |
0.23 |
1.277 |
27.7 |
9.8 |
1.546 |
4.4 |
4.0 |
|
8 |
CF3 |
33.0 |
16.1 |
1.430 |
0.54 |
1.243 |
30.5 |
14.2 |
1.542 |
2.5 |
1.9 |
|
9 |
CN |
34.6 |
18.3 |
1.432 |
0.66 |
1.233 |
32.2 |
14.9 |
1.548 |
2.4 |
3.4 |
|
10 |
NO2 |
35.1 |
19.1 |
1.435 |
0.78 |
1.219 |
34.3 |
16.7 |
1.551 |
0.8 |
2.4 |
[x] Ability of the catalyst to reduce the activation free energies. The large values for ΔΔG≠ 1/ΔΔG1 indicate a high efficiency of the catalyst while the small values indicate a low efficiency of the catalyst; All the σp values for different R groups reported in the Table 1 were taken from the literature report (C. Hansch, A. Leo, R. W. Taft, Chem. Rev. 1991, 91, 165–195).
The same trend was observed with the activation barrier for the reaction (ΔG≠ 1) which increased when moving from electron donating groups (NMe2: 26.7 kcal/mol) to electron withdrawing groups (NO2: 35.1 kcal/mol). These results indicate that the stability of the formed carbene determines the activation barrier for the reaction. Thus, more stabilized carbenes are formed through a lower energy transition structure. Consistent with Hammond's postulate, [13] the more thermodynamically favorable the carbene formation, the lower the barrier to the N2 release. This finding is supported by the strong correlation between ΔG≠ 1 and ΔG1 with an R2 (the squared correlation coefficient) value of 0.99 (Figure 1a). The stability of the formed carbene 1 a bearing an electron donating group (NMe2/NH2/OMe) is a result of the π‐donation from the aromatic ring to the empty p orbital on the Cb atom and is enhanced by increasing the contribution of resonance structure 1 a′ (Scheme 2a).
Figure 1.
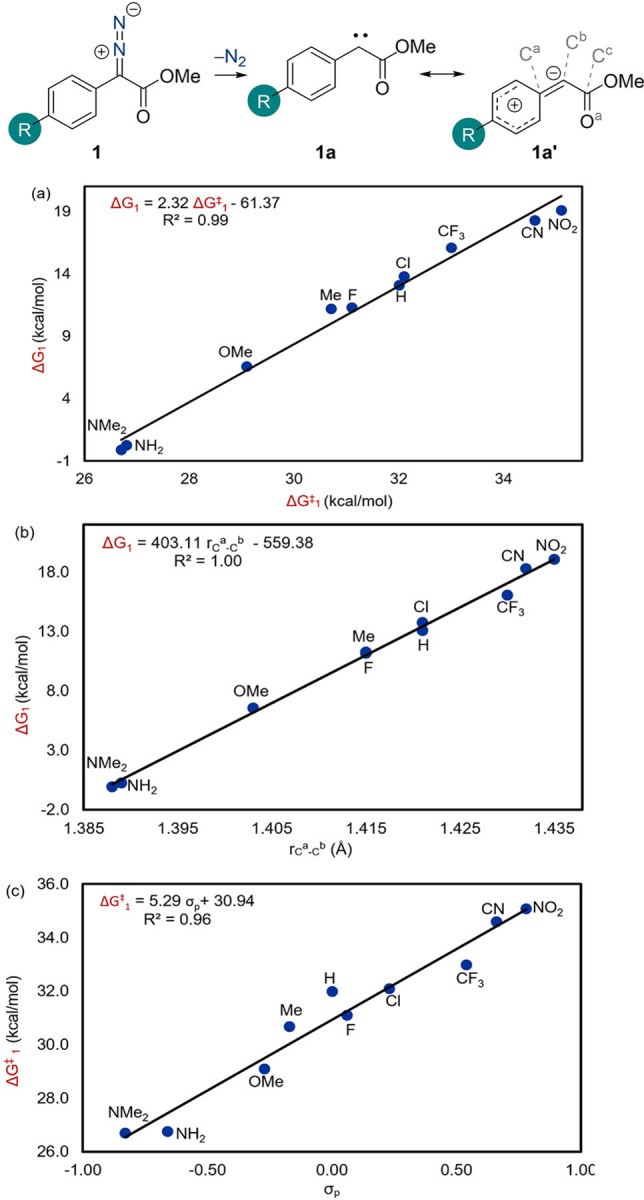
Correlation plots for uncatalyzed carbene formation. a) ΔG≠ 1 versus ΔG1; b) ΔG1 versus rC a−C b, and c) ΔG≠ 1 versus σp.
The degree of this stabilization can be ascertained through analysis of the Ca−Cb bond lengths in 1 a, with shorter bond lengths indicative of increased stabilization of the carbene. This was supported by a strong correlation between the Ca−Cb bond length in 1 a and the Gibbs free energy for carbene formation (ΔG1) (Table 1, Figure 1b). Shorter bond distances are observed when R is a π‐donating group, implying that the π‐bond character between the Ca and Cb atoms is increased, and hence structure 1 a′ has a greater contribution (Scheme 2a) This is reiterated by the Wiberg bond indices (WBI) analysis, [14] which showed the largest WBI value for R=NMe2 (1.460) and the smallest for R=NO2 (1.219, Table 1).
The importance of the electronic nature of the R group with respect to the carbene stabilization is evidenced by the direct correlation between the Hammett σp value and the activation barrier to N2 release (ΔG≠ 1) (Figure 1c).
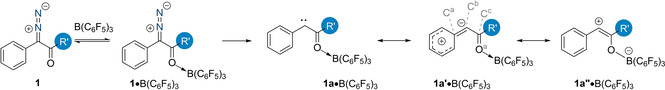
We next turned our attention to understand the effect of the B(C6F5)3 catalyst on the activation energy for N2 release from the α‐aryl α‐diazocarbonyl compound (Scheme 2b). The binding of the Lewis acidic borane to the carbonyl functionality (O→B adduct) facilitates this process both kinetically and thermodynamically (Table 1). [5a] However, this facilitating effect is primarily dependent on the identity of the R group. The magnitude of ΔΔG≠ 1 determines the extent of this effect on the kinetic property (Table 1); ΔΔG≠ 1 is the difference in the activation energies between the uncatalyzed and catalyzed reactions, i. e., ΔG≠ 1‐ΔG≠ 2. Accordingly, the maximum effect is found for the systems bearing an electron‐donating R group and the minimum effect for the systems where the R group is highly electron withdrawing. This means that the nature of the R group influences the efficiency of the catalyst. For example, for R=NMe2 with ΔΔG≠ 1=11.3 kcal/mol (Table 1, entry 1), the activation barrier (ΔG≠ 2) for the catalyzed N2 release is calculated to be 15.4 kcal/mol, whereas the uncatalyzed reaction using the same functional group requires an activation energy of 26.7 kcal/mol (ΔG≠ 1). No significant energy difference was observed for electron‐withdrawing substituents such as the NO2 functionality; here, ΔΔG≠ 1 was found to be only 0.8 kcal/mol (Table 1, entry 10). These findings are consistent with our previous experimental study, [15] in which we clearly demonstrated that the diazo compound bearing a strong electron‐withdrawing substituent (CF3) required a strong Lewis acidic borane [16] and high temperature to generate the corresponding carbene intermediate.
Our DFT studies disclosed the same trend for ΔΔG1 which reveals the difference between the free energies of the uncatalyzed and catalyzed reactions (ΔG1‐ΔG2, Table 1, Scheme 2c). The magnitude of ΔΔG1 indicates how strongly the borane catalyst binds to the carbene species (1 a). Our results showed that the diazo compounds bearing a strongly electron donating R group exhibit strong coordination between the borane catalyst and the formed carbene. Likewise, a weak coordination is observed for diazo compounds bearing electron withdrawing R groups. For example, when R=NMe2, ΔΔG1 is found to be 13.7 kcal/mol, but when R is NO2, ΔΔG1 is only 2.4 kcal/mol (Table 1). Interestingly, we found a strong positive correlation between ΔΔG1 and ΔΔG≠ 1 with R2=0.95 (Figure 2a). This strong correlation indicates that the strength of the O−B bond in carbene 1 a ⋅ B(C6F5)3 governs catalyst efficiency; the more electron donating the R group, the stronger the borane coordination to the carbene, the greater the ΔΔG≠ 1 value, and the more efficient the catalyst. This finding can serve as a guideline for determining how to alter the R group in a diazo compound in order to maximize catalyst efficiency. [16]
Figure 2.

Correlation plots for B(C6F5)3 catalyzed carbene formation. a) ΔΔG1 versus ΔΔG≠ 1; b) WBI for 1 a and 1 a ⋅ B(C6F5)3 with R=NMe2 and NO2; c) ΔG≠ 2 versus ΔG2, and d) ΔG≠ 2 versus σp.
In line with the above discussion, an electron donating R group is expected to increase the contribution of the resonance structure 1 a′ ⋅ B(C6F5)3 (Table 1) which increases the electron density on the Cb atom. The borane bonded to the carbonyl group in 1 a ⋅ B(C6F5)3 receives some of this electron density by involving resonance structure 1 a′′ ⋅ B(C6F5)3. Owing to the existence of a kind of push‐pull interaction in 1 a ⋅ B(C6F5)3 (Table 1), the contribution of these two resonance structures is expected to be increased by increasing the electron donating ability of the R group. An increase in the contribution of these two resonance structures raises the π‐bond character between the Ca and Cb atoms and between the Cb and Cc atoms while decreasing the π‐bond character between the Cc and Oa atoms. This claim is supported by the WBI analysis for 1 a and 1 a ⋅ B(C6F5)3 with R=NO2 and NMe2 (Figure 2b). Accordingly, 1 a ⋅ B(C6F5)3 with R=NMe2 has the highest WBI values for the Ca−Cb and Cb−Cc bonds and the lowest one for the Cc−O bond, supporting the π‐bond character argument.
An increase in the contribution of these two resonance structures causes the boron catalyst to bind more strongly to the carbene, corroborated by the shortening of the B−O bond distance in 1 a ⋅ B(C6F5)3 upon moving from R=NO2 to NMe2 (Table 1).
Figure 2c shows a plot with strong correlation between ΔG2 and ΔG≠ 2 (R2=0.98). This means that regardless of whether the borane is involved in the N2 release process, the thermodynamic favourability of carbene formation influences its activation barrier. [17] The formula in Figure 2d, which is based on the strong correlation between the Hammett σp value and ΔG≠ 2, will allow researchers to estimate the barrier to N2 release from a specific α‐diazo‐carbonyl compound using the σp value.
Next, attention was turned to investigating the effect of changing the R′ substituent on the N2 release process while keeping R=H (Table 2). Our calculations revealed that in the absence of the boron‐catalyst, changing R′ plays no significant role in the ease of the N2 release process compared to changing R. This can be attributed to the inability of this group to stabilize the formed carbene. Specifically, the activation free energy (ΔG≠ 3) for N2 release occurs over a very narrow range of ∼30–32 kcal/mol for all R′ groups. The same is true for the reaction energy (ΔG3, Table 2, Scheme 2a).
Table 2.
Calculated free energies for uncatalyzed carbene formation with different R′ groups, free energy difference between B(C6F5)3 catalyzed and uncatalyzed carbene formation, and free energy values for B(C6F5)3 catalyzed carbene formation. Free energies are given in kcal/mol. The B−O bond distance (rB−O) in 1 a ⋅ B(C6F5)3 .
|
| ||||||||
|---|---|---|---|---|---|---|---|---|
|
Entry |
R′ |
ΔG≠ 3 |
ΔG3 |
ΔG≠ 4 |
ΔG4 |
rB−O [Å] |
ΔΔG≠ 2=ΔG≠ 3–ΔG≠ 4 |
ΔΔG2=ΔG3–ΔG4 |
|
|
|
Uncatalyzed |
B(C6F5)3 catalyzed |
Catalyst efficiency [a] |
||||
|
1 |
Me |
30.4 |
10.4 |
9.5 |
−6.4 |
1.509 |
20.9 |
16.8 |
|
2 |
H |
31.3 |
13.4 |
10.5 |
−6.8 |
1.512 |
20.8 |
20.2 |
|
3 |
Ph |
31.2 |
9.3 |
11.4 |
−6.1 |
1.518 |
19.8 |
15.4 |
|
4 |
OMe |
32.0 |
13.1 |
25.9 |
6.5 |
1.533 |
6.1 |
6.6 |
|
5 |
OH |
30.6 |
12.1 |
26.1 |
7.8 |
1.534 |
4.5 |
4.3 |
|
6 |
F |
30.1 |
11.1 |
27.3 |
9.0 |
1.543 |
2.8 |
2.1 |
[a] Ability of the catalyst to reduce the activation free energies. The large values for ΔΔG≠ 2/ΔΔG2 indicate a high efficiency of the catalyst while the small values indicate a low efficiency of the catalyst.
Finally, attention was turned towards the influence of R′ on the ease of the N2 release process in the presence of the boron‐catalyst, while keeping R=H (Table 2, Scheme 2b, ΔG4 and ΔG≠ 4). Interestingly, in this case, the electronic nature of R′ has a profound impact on the N2 release process, with R′ substituents lacking a lone pair (R′=Me, H and Ph) favoring this process.
Again, we discovered that the activation barrier to the N2 liberation can be determined by how strongly the borane binds to the carbene in 1 a ⋅ B(C6F5)3. A plot between ΔΔG2 and ΔΔG≠ 2 was found to be a straight line with R2=0.96 (Figure 3a) where ΔΔG2=ΔG3−ΔG4 and ΔΔG≠ 2=ΔG≠ 3−ΔG≠ 4 (Scheme 2c).
Figure 3.

B(C6F5)3 catalyzed correlation plots for carbene formation with different R′ groups. a) ΔΔG≠ 2 versus ΔΔG2, and b) WBI for 1 a ⋅ B(C6F5)3 with R′=H and F.
This result once again demonstrates that the stronger the borane coordination to the formed carbene, the lower the activation barrier to N2 liberation, the greater the efficiency of the catalyst.
The resonance contributor 1 a’′ ⋅ B(C6F5)3 (Table 2) is favored when R′=Me, H, Ph, whereas lone pair bearing donor R′ groups, such as OMe, OH, and F, significantly reduce this contribution. Indeed, the presence of a lone pair on R′ does not allow the carbene lone pair to effectively interact with the carbonyl functional group, resulting in the contribution of 1 a′′ ⋅ B(C6F5)3 being decreased.
The above assumption is supported by a comparison of the WBI values for 1 a ⋅ B(C6F5)3 with R′=H and F (Figure 3b). The WBI for the bond between the Cb and Cc atoms is greater for R′=H than for R′=F, whereas WBI values between the Cc and Oa atoms has the opposite order. This indicates that, for R′=H, the π‐bond character between the Cb and Cc atoms is greater, while a reverse order is observed for the bond between the Cc and Oa atoms. This analysis demonstrates a greater contribution of 1 a′′ ⋅ B(C6F5)3 for the carbene with R′=H. The greater contribution of 1 a′′ ⋅ B(C6F5)3 leads to a stronger coordination of the borane to carbene, evidenced by a shorter B−O bond distance for the carbene with R′=Me, H and Ph (Table 2). We also found an excellent correlation between the ΔG4 and ΔG≠ 4 with R2=0.99, reconfirming the strong relationship between the stability of the formed carbene and the ease of N2 liberation (Figure S2). [18]
Conclusion
In conclusion, we have demonstrated computationally the influence of different substituents on the aryl ring and carbonyl functionality of α‐aryl α‐diazocarbonyls towards the formation of carbene species in the presence and absence of a B(C6F5)3 catalyst. We have several important findings, which are detailed below. Based on these DFT calculations, we conclude that there is a strong correlation between the activation barrier to the N2 liberation and the reaction free energy of the carbene formation. In general, the more stable the carbene generated, the lower the activation barrier to the process. In the absence of any Lewis acidic borane catalyst, a change in the electronic nature of the R group attached to the aryl ring has a considerable impact upon the ease of the N2 release, whilst the identity of the R′ group is unimportant. The activation barrier is reduced as the π‐donor ability of the R group is enhanced. This is because the carbene becomes more stable as the π‐donor ability of the R group increases. The most important finding from this study is that when the borane catalyst is present, both the R and R′ substituents have a direct influence on the stability of the carbene and, as a result, on the efficiency of the catalyst. The catalyst efficiency is determined by how strongly the borane is bonded to the formed carbene and we found that it is the greatest if the R group is a strong π‐donor and the R′ group is a weak π‐donor. In other words, the nature of the diazo substrate affects the catalyst efficiency. In short, the stronger the coordination of the borane to the generated carbene, the more thermodynamically favorable is the carbene formation, the lower the activation barrier to the N2 liberation, and the more efficient the catalyst. These computational calculations will aid the further exploration of B(C6F5)3 as a catalyst for carbene transfer reactions and will help to understand both the reactivity of diazo substrates with other nucleophiles as well as the relative rates of reaction.
Experimental Section
Gaussian 16 [19] was used to fully optimize all the structures at the M06‐2X level [20] of theory using the SMD solvation model [21] in dichloromethane. The 6‐31G(d) basis set [22] was chosen for all atoms. Frequency calculations were carried out at the same level of theory as those for the structural optimization. Transition structures were located using the Berny algorithm and intrinsic reaction coordinate (IRC) calculations [23] were employed to confirm the connectivity between transition structures and minima.
To further refine the energies obtained from the SMD/M06‐2X/6‐31G(d) calculations, single‐point energy calculations using the M06‐2X functional method were carried out for all of the structures with a larger basis set def2‐TZVP [24] and the SMD solvation model in dichloromethane. All thermodynamic data were calculated in the standard state (298.15 K and 1 atm). An additional correction for compression of 1 mol of an ideal gas from 1 atm to the 1 M solution phase standard state (1.89 kcal/mol) was applied. [25] Wiberg Bond Index (WBI) calculations were determined by the NBO6 program. [26]
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
RB, BFY, CJH, and AA thank the Australian Research Council (ARC) for project funding (DP180100904), and the Australian National Computational Infrastructure and Digital Research Services, IT Services at the University of Tasmania for the generous allocation of computing time. AD and RLM would like to acknowledge the EPSRC for an Early Career Fellowship for funding (EP/R026912/1).
R. Babaahmadi, A. Dasgupta, C. J. T. Hyland, B. F. Yates, R. L. Melen, A. Ariafard, Chem. Eur. J. 2022, 28, e202104376.
Contributor Information
Prof. Rebecca L. Melen, Email: melenr@cardiff.ac.
Prof. Alireza Ariafard, Email: alireza.ariafard@utas.edu.au.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.For selected reviews see:
- 1a. Hu D., Chen L., Fan H., Yao Q., Zhu S., Chem. Soc. Rev. 2020, 49, 908 – 950; [DOI] [PubMed] [Google Scholar]
- 1b. Xiang Y., Wang C., Ding Q., Peng Y., Adv. Synth. Catal. 2019, 361, 919 – 944; [Google Scholar]
- 1c. Ciszewski Ł. W., Rybicka-Jasińska K., Gryko D., Org. Biomol. Chem. 2019, 17, 432 – 448; [DOI] [PubMed] [Google Scholar]
- 1d. Xia Y., Qiu D., Wang J., Chem. Rev. 2017, 117, 13810 – 13889; [DOI] [PubMed] [Google Scholar]
- 1e. Mix K. A., Aronoff M. R., Raines R. T., ACS Chem. Biol. 2016, 11, 3233 – 3244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.For selected reviews see:
- 2a. Davies H. M. L., Liao K., Nat. Chem. Rev. 2019, 3, 347 – 360; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Gillingham D., Fei N., Chem. Soc. Rev. 2013, 42, 4918 – 4931; [DOI] [PubMed] [Google Scholar]
- 2c. Davies H. M. L., Manning J. R., Nature 2008, 451, 417 – 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Tchounwou P. B., Yedjou C. G., Patlolla A. K., Sutton D. J., in: Molecular, Clinical and Environmental Toxicology, Volume 3: Environmental Toxicology (Ed.: Luch A.), Springer Basel, Basel, 2012, pp. 133 –164; [Google Scholar]
- 3b. Egorova K. S., Ananikov V. P., Organometallics 2017, 36, 4071 – 4090. [Google Scholar]
- 4.For selected examples see:
- 4a. Dasgupta A., Richards E., R L Melen, ACS Catal. 2022, 12, 442 – 452; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Dasgupta A., Pahar S., Gierlichs L., Babaahmadi R., Yates B. F., Ariafard R L Melen A., Adv. Synth. Catal. 2021, 10.1002/adsc.202101312; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Rao S., Ashwathappa P. K. S., Prabhu K. R., Asian J. Org. Chem. 2019, 8, 320 – 323; [Google Scholar]
- 4d. Melen R. L., Angew. Chem. Int. Ed. 2018, 57, 880 – 882; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 890–892; [Google Scholar]
- 4e. Geri J. B., Shanahan J. P., Szymczak N. K., J. Am. Chem. Soc. 2017, 139, 5952 – 5956; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4f. Simonneau A., Turrel R., Vendier L., Etienne M., Angew. Chem. Int. Ed. 2017, 56, 12268 – 12272; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 12436–12440. [Google Scholar]
- 5.
- 5a. Dasgupta A., Babaahmadi R., Slater B., Yates B. F., Ariafard A., Melen R. L., Chem 2020, 6, 2364 – 2381; [Google Scholar]
- 5b. Dasgupta A., Stefkova K., Babaahmadi R., Gierlichs L., Ariafard A., Melen R. L., Angew. Chem. Int. Ed. 2020, 59, 15492 – 15496; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 15621–15626; [Google Scholar]
- 5c. Nori V., Dasgupta A., Babaahmadi R., Carlone A., Ariafard A., Melen R. L., Catal. Sci. Technol. 2020, 10, 7523 – 7530; [Google Scholar]
- 5d. San H. H., Wang C.-Y., Zeng H.-P., Fu S.-T., Jiang M., Tang X.-Y., J. Org. Chem. 2019, 84, 4478 – 4485; [DOI] [PubMed] [Google Scholar]
- 5e. San H. H., Wang S.-J., Jiang M., Tang X.-Y., Org. Lett. 2018, 20, 4672 – 4676. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Zhao Y., Mandal D., Guo J., Wu Y., Stephan D. W., Chem. Commun. 2021, 57, 7758 – 7761; [DOI] [PubMed] [Google Scholar]
- 6b. He F., Koenigs R. M., Org. Lett. 2021, 23, 5831 – 5835. [DOI] [PubMed] [Google Scholar]
- 7. Yu Z., Li Y., Shi J., Ma B., Liu L., Zhang J., Angew. Chem. Int. Ed. 2016, 55, 14807 – 14811; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 15027–15031. [Google Scholar]
- 8. Rao S., Kapanaiah R., Prabhu K. R., Adv. Synth. Catal. 2019, 361, 1301 – 1306. [Google Scholar]
- 9.
- 9a. Stefkova K., Heard M. J., Dasgupta A., Melen R. L., Chem. Commun. 2021, 57, 6736 – 6739; [DOI] [PubMed] [Google Scholar]
- 9b. Mancinelli J. P., Wilkerson-Hill S. M., ACS Catal. 2020, 10, 11171 – 11176. [Google Scholar]
- 10. Zhang Q., Zhang X.-F., Li M., Li C., Liu J.-Q., Jiang Y.-Y., Ji X., Liu L., Wu Y.-C., J. Org. Chem. 2019, 84, 14508 – 14519. [DOI] [PubMed] [Google Scholar]
- 11. Green S. P., Wheelhouse K. M., Payne A. D., Hallett J. P., Miller P. W., Bull J. A., Org. Process Res. Dev. 2020, 24, 67 – 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tang C., Liang Q., Jupp A. R., Johnstone T. C., Neu R. C., Song D., Grimme S., Stephan D. W., Angew. Chem. Int. Ed. 2017, 56, 16588 – 16592; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16815–16819. [Google Scholar]
- 13. Hammond G. S., J. Am. Chem. Soc. 1955, 77, 334 – 338. [Google Scholar]
- 14. Wiberg K. B., Tetrahedron 1968, 24, 1083 – 1096. [Google Scholar]
- 15. Santi M., Ould D. M. C., Wenz J., Soltani Y., Melen R. L., Wirth T., Angew. Chem. Int. Ed. 2019, 58, 7861 – 7865; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7943–7947. [Google Scholar]
- 16.The identity of the borane catalyst may also have an impact on its efficiency. Our calculations indicate that the N2 release process is facilitated more effectively if a borane with a higher acidity is used (Table S2).
- 17.The boron binds to the diazo substrate in an endergonic manner, implying that no catalyst poisoning is caused by the boron coordination (see Tables S1 and Figure S1).
- 18.For existence of any correlation between ΔG2 and rCa−Cb and between ΔG4 and rCa−Cb see the Supporting Information.
- 19.Gaussian 16, Revision C.01, M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C.; Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, D. J. Fox, Gaussian, Inc., Wallingford CT, 2016.
- 20. Zhao Y., Truhlar D. G., Acc. Chem. Res. 2008, 41, 157 – 167. [DOI] [PubMed] [Google Scholar]
- 21. Marenich A. V., Cramer C. J., Truhlar D. G., J. Phys. Chem. B. 2009, 113, 6378 – 6396. [DOI] [PubMed] [Google Scholar]
- 22. Hariharan P. C., Pople J., Theor. Chim. Acta 1973, 28, 213 – 222. [Google Scholar]
- 23.
- 23a. Fukui K., J. Phys. Chem. 1970, 74, 4161 – 4163; [Google Scholar]
- 23b. Fukui K., Acc. Chem. Res. 1981, 14, 363 – 368. [Google Scholar]
- 24. Weigend F., Furche F., Ahlrichs R., J. Chem. Phys. 2003, 119, 12753 – 12762. [Google Scholar]
- 25.J. W. Ochterski, Thermochemistry, Gaussian, Inc., Wallingford, CT, 2000.
- 26.E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013; http://nbo6.chem.wisc.edu/.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.