

Summary
The multiple teats trait is common in many species of mammals and is considered related to lactation ability in swine. However, in Hu sheep, related gene research is still relatively limited. In this study, a genome‐wide association study was used to identify genetic markers and genes related to the number of teats in the Hu sheep population, a native Chinese sheep breed. A single marker method and several multi‐locus methods were utilized. A total of 61 SNPs were found to be related to the number of teats. Among these, 11 SNPs and one SNP were consistently detected by two and three multi‐locus models respectively. Four SNPs were concordantly identified between the single marker and multi‐locus methods. We also performed quantitative real‐time PCR testing of these identified candidate genes, identifying three genes with significantly different expression. Our study suggested that the LHFP, DPYSL2, and TDP‐43 genes may be related to the number of teats in sheep. The combination of single and multi‐locus GWAS detected additional SNPs not found with only one model. Our results provide new and important insights into the genetic mechanisms of the mammalian multiparous teat phenotype. These findings may be useful for future breeding and understanding the genetics of sheep and other livestock.
Keywords: GWAS, multi‐locus, sheep, SNP, teat number
INTRODUCTION
The mammary gland is comprised of several ductal systems and acini and is an important structure that distinguishes mammals from other animals. Mammary gland development and function are tightly orchestrated, and its main function is to produce and deliver milk to offspring (Plante et al., 2011). Teats (or nipples), epidermal appendages on the udders or breasts of mammals (Pumfrey et al., 1980), play an important role in mammalian reproduction and offspring growth.
In addition to normal teats, some mammals, such as swine, cows, and sheep, also have supernumerary teats. In pigs, teat number is a reproductive trait that directly affects the lactation rate of sows and the survival rate of piglets. Previous studies have reported that teat number is increased due to the strong effect of an insertion mutation in the Vertebrae Development Associated (VRTN) gene on Sus scrofa chromosome 7 (SSC7) in Landrace and Korean pigs (Lee et al., 2014), Duroc pigs (Arakawa et al., 2015), Erhualian pigs (Wang et al., 2017a), and Large White pigs (Duijvesteijn et al., 2014). Different genes, SPRED2, MKX, TMSB4X, and ESR1, are involved in this trait in Chinese Sushan pigs (Zhou et al., 2019). In cattle, some interesting genes related to teat number have been identified. For instance, the inheritance of supernumerary teats in Holstein cattle depends on a QTL on chromosome 20 and a polygenic part (Joerg et al., 2014). Another study found that the gene LGR5 on chromosome 5 was a candidate for the presence of supernumerary teats (Butty et al., 2017). In contrast to numerous studies in pigs and cattle, a study showed that BBX and CD47 on chromosome 1 were commonly identified as significant by genome‐wide association study (GWAS) in Wadi sheep (Peng et al., 2017). However, genetic variants associated with teat number in Hu sheep are not known.
Teat number is a typical polygenic quantitative trait. Currently, use of the Bonferroni correction not only controls the false‐positive rate for single‐locus GWAS but also excludes some important loci with small effects (Zhang et al., 2019). Population structure and genetic correlations have been widely analysed by mixed linear models (Sul et al., 2018). Several recently developed multi‐locus models, including the fast multi‐locus random‐SNP‐effect EMMA (FASTmrMLM) (Tamba & Zhang, 2018), polygenic‐background‐control‐based least angle regression plus empirical Bayes (pLARmEB) (Zhang et al., 2017), polygenic‐background‐control‐based Kruskal–Wallis test plus empirical Bayes (pKWmEB) (Ren et al., 2018) and Iterative Sure Independence Screening (ISIS) EM–Bayesian LASSO (Tamba et al., 2017) have been shown to effectively resolve this issue.
Hu sheep, a descendant of Mongolian sheep, is a famous lambing breed in China. In ewes of the Hu sheep, TT (individuals with two normal teats) and MT (individuals with two normal teats and one or two supernumerary teats) account for 76–62% and 38–24% respectively. For MT ewes, the supernumerary teats are smaller than the normal teats, but some can produce milk. In the current study, we analyzed the complex genetic mechanism of differences in teat number using single‐locus and multi‐locus GWAS in a total of 160 Hu sheep. Our results provide new insight into the genetic mechanisms of teat number traits in sheep and will be useful for sheep breeding in the future. The aim of the present study was to identify SNPs and candidate genes associated with teat number traits in Hu sheep genotyped with a dense SNP array.
MATERIALS AND METHODS
Ethics statement
All animals used in this study met the guidelines for the care and use of experimental animals established by the Ministry of Agriculture of China. The whole study was performed according to protocols and guidelines approved by the Institutional Animal Care and Use Committee of the Beijing Academy of Agricultural Sciences (IAS2019–57). The experimental animals were not anesthetized or euthanized in order to conduct this study.
Sample collection and phenotyping
In the current study, experiment animals were raised in Inner Mongolia Golden Grassland Ecological Technology Group Co. Ltd (Bayannaoer, China). In brief, a total of 160 Hu sheep ewes (77 TT and 83 MT sheep) born within a month were analyzed. All sheep were subjected to the same growth and feeding condition. Ear marginal tissues were collected and stored in 1.5‐ml microcentrifuge tubes containing 75% ethanol. In all the cases, particular efforts were made, based on both pedigree information and the knowledge of local herdsmen, to ensure that the animals were as distantly related as possible.
Genotyping and quality control
The genomic DNA of each sheep from ear tissue was isolated following the standard phenol/chloroform method, quantified, and diluted to 50 ng/μl (Ozsensoy & Sahin, 2016). Genotyping was performed using the Affymetrix GeneSeek Genomic Profiler OvineHD (GGP OvineHD), and it included 566 129 SNPs across 27 autosomes. Quality control was performed using PLINK v1.90 (Purcell et al., 2007). The filtering criteria were as follows: (i) SNPs without chromosomal and physical locations; (ii) SNPs with missing genotypes >0.1; (iii) SNPs of minor allele frequency <0.01; (iv) individuals with a genotyping rate <90%; and (v) a p‐value of Fisher's exact test (Raymond & Rousset, 1995a,b) for Hardy–Weinberg equilibrium <0.00001; and (vi) SNPs that located on the sex chromosomes. We removed the SNPs and individuals who met any of these criteria. Moreover, pairwise relatedness among individuals was examined using the King v1.4 program (Raymond & Rousset, 1995b), and three of the pairwise individuals with a kinship coefficient Φ > 0.25 (e.g. parent‐offspring or full–sibs from two unrelated parents) were removed from further analyses. After filtering, 489 958 SNPs and 157 individuals (76 TT and 81 MT sheep) were retained in the dataset for within–population stratification analysis.
Population structure and linkage disequilibrium estimation
Principal component analysis (PCA) and linkage disequilibrium (LD) analysis were performed using the quantified SNPs to investigate the population structure among the TT and MT sheep. PCA was performed with GCTA (Yang et al., 2011). LD among SNPs were estimated as the squared correlation (r 2) of alleles with a window size of 5000. The average LD decay distance (r 2 = 0.05) was calculated by PLINK v1.90 (Purcell et al., 2007) for the whole genome of the Hu sheep. Moreover, Weir & Cockerham Fst analysis was performed with the filtered SNPs to estimate Within–breed genetic differentiation using VCFtools (Danecek et al., 2011).
Single–locus GWAS
In the present study, GEMMA software (Zhou & Stephens, 2012) was used to implement LMM for single–locus GWAS of teat number. GEMMA calculated the genomic relatedness matrix between TT and MT individuals within each population to account for population structure. The first two principal components calculated by GCTA tool (Yang et al., 2011) were embedded as covariates in the association analysis model to eliminate the confounding effect of population structure (Price et al., 2006). The model that tested the allelic effect of each SNP on teat number invoked by GEMMA was as follows:
where y is the vector of teat number in Hu sheep; W is the incidence matrix of covariates (fixed effects) including the top two eigenvectors of PCA; α is a vector of the corresponding coefficients including the intercept; X is the vector of all marker genotypes; β is the effect size of the marker and is an estimate of the marker/SNP additive effect; u is the vector of random effects; ε is the vector of errors; is the variance of the residual errors; is the ratio between the two variance components; K is a known relatedness matrix; is the identity matrix; denotes the n–dimensional multivariate normal distribution; and n refers to the number of sheep.
Given that Bonferroni correction is a stringent criterion, false discovery rate (FDR) was used to determine the threshold p values of single–locus GWAS (Wang et al., 2017b). In the present study, FDR was set as 0.005, and the threshold p value was defined as p = FDR × N/M, where N represents the number of SNPs with p value <0.005 in the results of GWAS and M refers to the total number of qualified SNPs for teat number in Hu sheep. The adjusted p value that limited the FDR was calculated using the Benjamini–Hochberg method (Benjamini & Hochberg, 1995).
Multi‐locus GWAS
Multi‐locus GWAS was performed with four models, including FASTmrMLM (Tamba & Zhang, 2018), pLARmEB (Zhang et al., 2017), pKWmEB (Ren et al., 2018), and ISIS EM–BLASSO (Tamba et al., 2017). All four multi–locus models were implemented in the R package mrMLM (Wang et al., 2016) to detect SNPs associated with teat number in Hu sheep. Q (population genetic structure) matrix was the same as that used in single‐locus GWAS and K (genomic relatedness) matrix was calculated using R package mrMLM. All SNPs were treated as random effects in the first stage of these five methods, in which the main purpose is to select all potentially relevant SNPs (Pan et al., 2018). In the second stage, the selected SNPs were fitted into the multi‐locus models and the markers with largest effects that surpassed the threshold of LOD values were regarded as promising trait associated SNPs (Wang et al., 2016). The critical p value parameters were set at default values in the first step. The critical threshold of LOD score was set to 3 for SNPs at final stage.
Annotation of candidate genes
Based on the LD decay distances of the Hu sheep, The sheep genome assembly 4.0 named Oar_v4.0 (https://www.ncbi.nlm.nih.gov/assembly/GCA_000298735.2/) were retrieved to characterize candidate genes in targeted regions. The search to for positional candidate genes was extended 200 kb up‐ and downstream from the significant SNPs.
Quantitative real‐time PCR
Six Hu sheep from the Inner Mongolia Golden Grassland Ecological Technology Group Co. Ltd were randomly selected, including three TT sheep and three MT sheep. The teats of the TT sheep and the MT sheep were collected and immediately stored in RNAlater for RNA extraction. Total RNA of each sample was isolated using RNAprep pure Tissue Kit (TIANGEN, China) according to the supplied manufacturer's instructions. For each sample, 1 μg of RNA was used for cDNA synthesis using PrimeScript™ RT reagent Kit (cat. # RR047A; Takara) according to the supplied protocol. The primer sequences were designed using Primer Premier 5.0 (Premier Biosoft International, San Francisco, CA, USA) and listed in Table S1. Quantitative real‐time PCR (qRT‐PCR) was performed on a 384 Real‐Time System (ABI Q7 Flex; Singapore) with a 10 μL reaction system using TB Green® Premix Ex Taq™ II (cat. # RR820A; Takara) according to the supplied protocol to examine the mRNA expression levels of LHFP, TDP‐43, DPYSL2, and TWIST1. Each sample had three technical duplicates, and GAPDH and ACTB were used as reference genes to normalize target gene expression. The thermal cycling process was as follows: 95°C for 15 min, followed by 45 cycles of 95°C for 20 s, 56°C for 20 s, and 72°C for 20 s. Relative gene expression was calculated using the 2–ΔΔCt method (Arocho et al., 2006).
RESULTS
SNP genotyping
The extracted DNA from each collected sample was genotyped using the Thermo Fisher/Affymetrix Genomic Geneseek Profilor Ovine HD 630K at Niuqin Company (Beijing, China). The quality of genotyping of the 157 Hu sheep was examined using PLINK v1.90 (Purcell et al., 2007). The characteristics of the SNPs in Hu sheep are summarized in Table S2, and Figure S1. These SNPs were roughly proportionally distributed on all 26 chromosomes of sheep, with the longest chromosome having the largest number of SNPs. The average marker density was approximately 202 71 SNPs per Mb in Hu sheep.
Population structure and LD decay
PCA was used to assess the population structure among individuals with different teat numbers, as is shown in Figure S2. To correct the population stratification, the first two principal components were embedded as covariates in the association analysis model. In addition, Q‐Q plots with genomic inflation factors (λgc) were generated to assess the influence of the population structure on the single‐locus GWAS (Figure 2b). Systematic inflation of the test statistics was not observed for the GWAS of either the TT or the MT sheep. The average LD decay distance of the Hu sheep was approximately 200 kb, where r 2 dropped to 0.05 (Figure 1). Furthermore, the pairwise Weir & Cockerham Fst value was 0.0004 between TT and MT individuals, implying little to moderate genetic differentiation (Gorssen et al., 2020).
FIGURE 2.
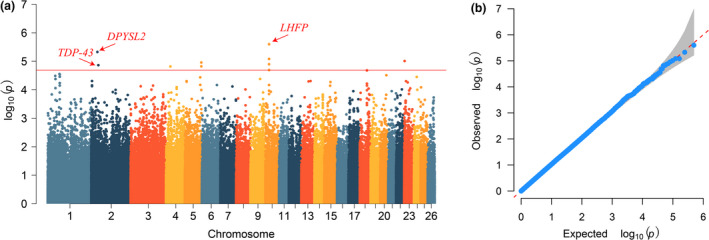
(a) Manhattan plots of the single–locus genome‐wide association study for teat number in Hu sheep. The x‐axis represents the chromosomes, and the y‐axis represents the –log10(p‐value). The dashed lines indicate the thresholds for teat number in Hu sheep (–log10(p) = 4.69). (b) Quantile–quantile (Q–Q) plots of single–locus genome‐wide association study for teat number in Hu sheep. Q–Q plots show the observed vs. expected negative log10 p values.
FIGURE 1.
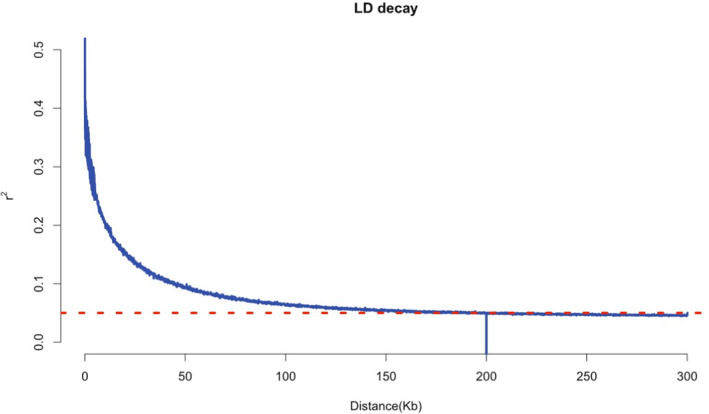
Linkage disequilibrium (LD) decay across the whole genome of the association panel. The red dotted line represents the LD threshold for the association panel (r 2 = 0.05)
Single‐locus GWAS for teat number
Significant SNPs detected by single‐locus GWAS (LMM) for the teat number of Hu sheep are shown in Table 1 and Figure 2a. Four SNPs in Chr10, two SNPs in Chr2, two SNPs in Chr5, one SNP in Chr23, and one SNP in Chr4 surpassed the threshold (–log10(p) = 4.69) with the FDR controlled at 0.005.
TABLE 1.
Significant SNPs associated with teat number in Hu sheep
| Chr. a | SNP | Position (bp) b | p‐value | Gene |
|---|---|---|---|---|
| 10 | AX–185209072 | 23384931 | 2.52E–06 | NHLRC3, PROSER1, LHFP, STOML3, FREM2 |
| 2 | AX–123201499 | 38790917 | 4.71E–06 | DPYSL2, PNMA2, ADRA1A |
| 10 | AX–185208896 | 23353832 | 8.27E–06 | NHLRC3, PROSER1, LHFP, STOML3, FREM2 |
| 10 | AX–272337568 | 23357343 | 8.27E–06 | NHLRC3, PROSER1, LHFP, STOML3, FREM2 |
| 23 | AX–272333176 | 3298777 | 9.79E–06 | TSHZ1, ZADH2 |
| 5 | AX–272369639 | 1.04E+08 | 1.10E–05 | FBXL17 |
| 10 | AX–272304405 | 23359639 | 1.27E–05 | NHLRC3, PROSER1, LHFP, STOML3, FREM2 |
| 2 | AX–272556355 | 44638685 | 1.37E–05 | TDP–43 |
| 5 | AX–272528738 | 1.04E+08 | 1.51E–05 | FBXL17 |
| 4 | AX–123228810 | 27722618 | 1.52E–05 | TWIST1, FERD3L |
Oar_v4.0 chromosome.
SNP position in NCBI.
Multi‐locus GWAS for teat number
Next, we performed multi‐locus GWAS using several methods including FASTmrMLM, pLARmEB, pKWmEB, and ISIS EM‐BLASSO. The four multi‐locus GWAS identified 55 teat number‐associated SNPs with LOD scores >3 (Table S3; Figure 3). Among these SNPs, FASTmrMLM and pLARmEB detected the highest number of SNPs (19), followed by pKWmEB (18) and ISIS EM‐BLASSO (18). Eleven and one were detected by two and three multi‐locus models respectively. Moreover, one SNP on Chr2 and one SNP on Chr4 detected by single‐locus LMM were also identified by multi‐locus models. Venn diagrams show the distribution of the SNPs from the four multi‐locus methods and highlight the concordance between the single‐locus method and the different multi‐locus methods (Figure 4). Two SNPs were concordantly identified between the single marker and the multi‐locus methods.
FIGURE 3.
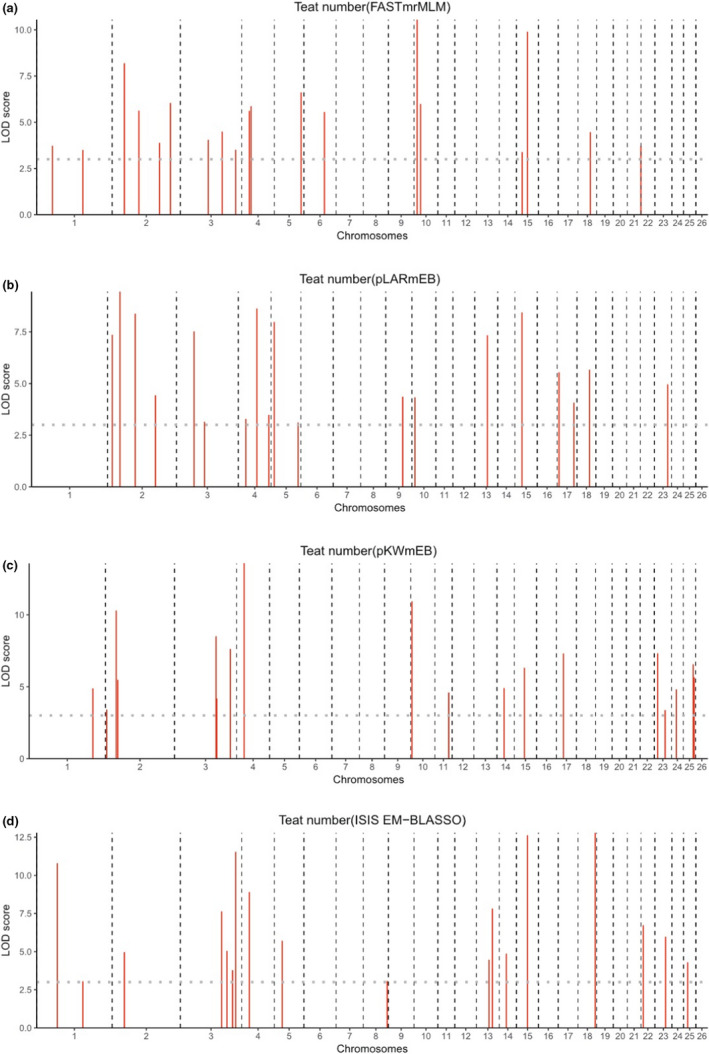
Manhattan plots of the four multi‐locus GWAS for teat number in Hu sheep. For (a–d), the Manhattan plots indicate LOD scores for genome‐wide SNPs (y‐axis) plotted against their respective positions on each chromosome (x‐axis), and the horizontal lines indicate the thresholds for significance (LOD score = 3)
FIGURE 4.
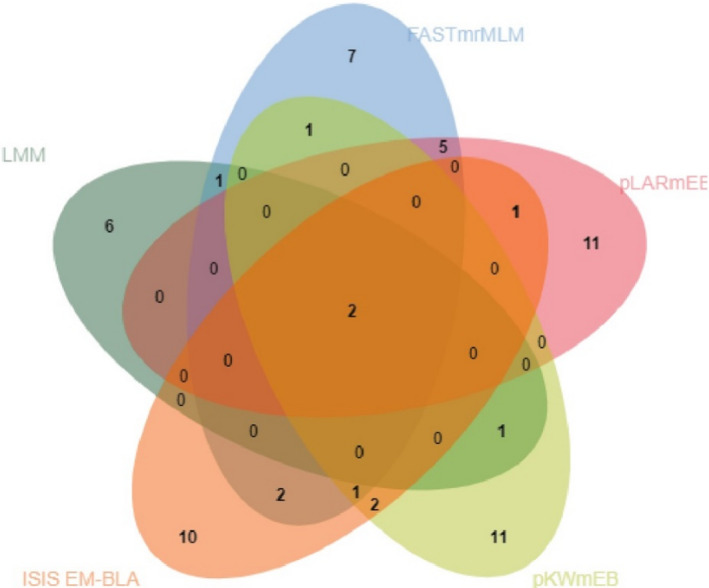
Venn diagrams show the distribution of SNPs from the four multi‐locus methods and also highlight the concordance between single marker method and different multi‐locus methods
Candidate genes search and functional annotation
Considering the genome‐wide LD decay distance of the Hu sheep used in the present study, genomic regions within 200 kb on either side of the 61 SNPs were used to mine candidate genes for teat number. To further understand the functions of the genes implicated by the GWAS, a final set of 208 genes within the LD regions of these SNPs were functionally annotated (Table 1; Table S3). Among them, TAR DNA‐binding protein 43‐like (TDP‐43) and twist family bHLH transcription Factor 1 (TWIST1) genes were identified simultaneously by the single‐locus LMM model and by all four multi‐locus models. LHFPL tetraspan subfamily member 6 (LHFP) was recognized by single‐locus LMM model and FASTmrMLM model. Meanwhile, dihydropyrimidinase like 2 (DPYSL2) was recognized by the single‐locus LMM model and the pKWmEB model. Therefore, we preliminarily identified these four genes as candidate genes and verified them by qRT‐PCR. The expression divergences of these four genes between TT and MT sheep were validated by qRT‐PCR as shown in Figure 5. From Figure 5, we can see that the expression levels of the LHFP, DPYSL2, and TDP‐43 genes in MT individuals were significantly higher than those in TT individuals, and the expression levels in MT individuals were more than twice as high as those in TT individuals. However, we can also see that the expression of the TWIST1 gene in individuals with MT is higher than that in individuals with TT, but it does not reach a significant level. Therefore, three genes including LHFP, DPYSL2, and TDP‐43 were further highlighted as promising candidates for teat number in Hu sheep.
FIGURE 5.
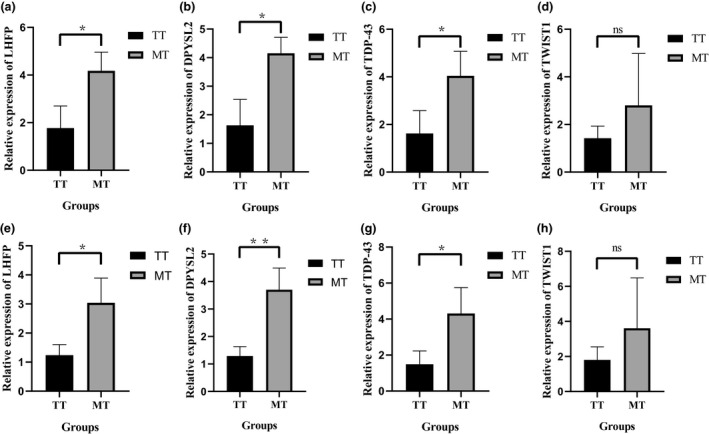
The relative expression of LHFP (a), DPYSL2 (b), TDP‐43 (c), and TWIST1 (d) between TT and MT Hu sheep with GAPDH as reference gene and the relative expression of LHFP (e), DPYSL2 (f), TDP‐43 (g), and TWIST1 (h) between TT and MT Hu sheep with ACTB as reference gene. Panels were plotted based on the results of quantitative real‐time PCR. *means the values of gene relative expression levels are significantly different between the TT and MT groups at the p < 0.05 level. **means the values of gene relative expression levels are significantly different between the TT and MT groups at the p < 0.01 level. (a) p = 0.026; (b) p = 0.015; (c) p = 0.041; (d) p = 0.34; (e) p = 0.028; (f) p = 0.008; (g) p = 0.039; (h) p = 0.35
DISCUSSION
In this study, we carried out a GWAS of teat number with a panel of 160 Hu sheep using one single‐locus model (LMM) and four multi‐locus models (FASTmrMLM, pLARmEB, pKWmEB, and ISIS EM‐BLASSO). The combination of the single‐locus and multi‐locus models significantly increased the power of GWAS and detected 61 significant SNPs. According to the results of the GWAS, four candidate genes were annotated using a series of functional annotations and qRT‐PCR. The findings provide new insight into further deciphering the genetic architecture of teat number in sheep.
Previous studies demonstrated that complex traits were focused only on single‐locus GWAS models by using general linear models and mixed linear models (MLMs) (He et al., 2018; Price et al., 2006; Sant'Ana et al., 2018). Nevertheless, the multi‐locus GWAS model has higher statistical ability and lower FPR and is better than the single‐locus GWAS model (Segura et al., 2012; Wang et al., 2016). For instance, Cui et al. performed a GWAS to detect loci related to rice salt tolerance at the seed germination stage using six multi‐locus GWAS methods (Cui et al., 2018). The six models detected 371 significant SNPs and 56 QTNs co‐detected by at least three methods.
In the present study, the SNPs detected by the single‐locus GWAS model did not exceed the threshold line significantly. To improve the efficiency of the study, we performed multi‐locus GWAS methods on the teat numbers of Hu sheep. The SNPs detected by the single‐locus and multi‐locus GWAS models were considered to be significantly correlated with teat number. In this study, the single‐locus method had less power for detecting SNPs with minor effects and it ignored the presence of additional QTL on quantitative traits. However, multi‐locus models consider multiple QTLs and treat them as random effects (Bu et al., 2020), which is close to the real genetic model of animals and plants.
In this study, we regarded LHFP, DPYSL2, and TDP‐43 as functional genes associated with teat number in Hu sheep by single‐locus method, multi‐locus models, and qRT‐PCR. Notably, TDP‐43, a highly conserved RNA‐ and DNA‐binding protein, can be cleaved by caspase3 to generate 25/35 kDa C‐terminal fragments (Zhang et al., 2007) and it induces apoptosis through a toxic gain‐of‐function (Zhang et al., 2009). According to a previous study, the 35 kDa fragment of TDP‐43 may serve as a potential therapeutic target to cure breast cancer (Nan et al., 2018). Interestingly, Zhao et al. (202) conducted research and found that the expression levels of TDP‐43 were positively correlated with higher milk output by using human milk samples from lactating women. Mechanistically, TDP‐43 is involved in posttranscriptional regulation of Btn1a1 and Xdh mRNA stability, which are required for the secretion of lipid droplets from epithelial cells into the lumen (Zhao et al., 2020). All of this evidence suggests that TDP‐43 plays an important role in the breast. In the present study, the expression levels of TDP‐43 in MT individuals were significantly higher than those in TT individuals. Therefore, we inferred that MT sheep might have higher milk output than TT sheep.
LHFP, as a common competing endogenous RNA hub in breast cancer is associated with mesenchymal differentiation in glioma (Nagaishi et al., 2012). Another study found that LHFP may be involved in the mechanisms that lead to the transformation or progression of the original tumor in human breast cancer (Levy, 2021), as well as lymph node metastasis. An earlier study found that DPYSLs are a family of proteins developmentally regulated during maturation of the nervous system, and DPYSL1, DPYSL2, and DPYSL3 may be prognostic markers in breast cancer (Levy, 2021; Zottel et al., 2020). The current findings, although in need of extensive validation, are expected to promote our understanding of the underlying regulatory mechanism of the teat number trait in sheep.
Our results revealed that teat number in sheep is a complex microeffect multigene trait. However, a number of the genes associated with teat number in cattle and pigs did not match our results (Arakawa et al., 2015; Butty et al., 2017; Duijvesteijn et al., 2014; Joerg et al., 2014; Lee et al., 2014; Wang et al., 2017a; Zhou et al., 2019). In our GWAS, we treated teat number as a disease status, which may differ from teat number in cattle and pigs. In addition, different sample sizes may identify many different candidate genes associated with teat number trait.
CONCLUSIONS
In conclusion, 10 and 55 SNPs were found to be associated with teat number by using single‐locus and multi‐locus methods respectively. The integration of single‐locus and multi‐locus GWAS detected additional SNPs in comparison with that using only one model. Based on the result of GWAS and qRT‐PCR, three novel candidate genes, LHFP, DPYSL2, and TDP‐43, were found. Our findings may provide the information resource to further understanding the complexity of the genetic mechanism of teat number trait, and a reference to optimize breeding program for improvement of reproductive performances in sheep.
CONFLICT OF INTEREST
The authors have declared that no competing interest exists.
Supporting information
Figure S1‐S2‐Table S1‐S2
Table S3
ACKNOWLEDGEMENTS
This work was supported by the National Natural Science Foundation of China (Nos. 31961143021), the earmarked fund for Modern Agro‐industry Technology Research System (CARS‐39‐01) and the Agricultural Science and Technology Innovation Program of China (ASTIP‐IAS01).
Zhao, Y. , Pu, Y. , Liang, B. , Bai, T. , Liu, Y. , Jiang, L. & et al (2022) A study using single‐locus and multi‐locus genome‐wide association study to identify genes associated with teat number in Hu sheep. Animal Genetics, 53, e13169. Available from: 10.1111/age.13169
Contributor Information
Lin Jiang, Email: jianglin@caas.cn.
Yuehui Ma, Email: mayuehui@caas.cn.
DATA AVAILABILITY STATEMENT
Phenotypes are available at: https://figshare.com/s/0b326ab8632bbd1c82d6. Genotypes are available at: https://figshare.com/s/197c20e3229490ccb5d6.
REFERENCES
- Arakawa, A. , Okumura, N. , Taniguchi, M. , Hayashi, T. , Hirose, K. , Fukawa, K. et al. (2015) Genome‐wide association QTL mapping for teat number in a purebred population of Duroc pigs. Animal Genetics, 46, 571–575. [DOI] [PubMed] [Google Scholar]
- Arocho, A. , Chen, B. , Ladanyi, M. & Pan, Q. (2006) Validation of the 2‐DeltaDeltaCt calculation as an alternate method of data analysis for quantitative PCR of BCR‐ABL P210 transcripts. Diagnostic Molecular Pathology, 15, 56–61. [DOI] [PubMed] [Google Scholar]
- Benjamini, Y. & Hochberg, Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Series B: Methodological, 57, 289–300. [Google Scholar]
- Bu, S. , Wu, W. & Zhang, Y.M. (2020) A multi‐locus association model framework for nested association mapping with discriminating QTL effects in various subpopulations. Front Genet, 11, 590012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butty, A.M. , Frischknecht, M. , Gredler, B. , Neuenschwander, S. , Moll, J. , Bieber, A. et al. (2017) Genetic and genomic analysis of hyperthelia in Brown Swiss cattle. Journal of Dairy Science, 100, 402–411. [DOI] [PubMed] [Google Scholar]
- Cui, Y. , Zhang, F. & Zhou, Y. (2018) The application of multi‐locus GWAS for the detection of salt‐tolerance loci in rice. Frontiers in Plant Science, 9, 1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C.A. , Banks, E. , DePristo, M.A. et al. (2011) The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duijvesteijn, N. , Veltmaat, J.M. , Knol, E.F. & Harlizius, B. (2014) High‐resolution association mapping of number of teats in pigs reveals regions controlling vertebral development. BMC Genomics, 15, 542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorssen, W. , Meyermans, R. , Buys, N. & Janssens, S. (2020) SNP genotypes reveal breed substructure, selection signatures and highly inbred regions in Piétrain pigs. Animal Genetics, 51, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He, L. , Xiao, J. , Rashid, K.Y. , Yao, Z. , Li, P. , Jia, G. et al. (2018) Genome‐wide association studies for pasmo resistance in flax (Linum usitatissimum L.). Frontiers Plant Science, 9, 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joerg, H. , Meili, C. , Ruprecht, O. , Bangerter, E. , Burren, A. & Bigler, A. (2014) A genome‐wide association study reveals a QTL influencing caudal supernumerary teats in Holstein cattle. Animal Genetics, 45, 871–873. [DOI] [PubMed] [Google Scholar]
- Lee, J.B. , Jung, E.J. , Park, H.B. , Jin, S. , Seo, D.W. , Ko, M.S. et al. (2014) Genome‐wide association analysis to identify SNP markers affecting teat numbers in an F2 intercross population between Landrace and Korean native pigs. Molecular Biology Reports, 41, 7167–7173. [DOI] [PubMed] [Google Scholar]
- Levy, W.A. (2021) Lipoma HMGIC fusion partner expression differs in primary tumors and lymph node metastases in malignancy. 10.21203/rs.3.rs-591947/v1 [DOI]
- Nagaishi, M. , Kim, Y.H. , Mittelbronn, M. , Giangaspero, F. , Paulus, W. , Brokinkel, B. et al. (2012) Amplification of the STOML3, FREM2, and LHFP genes is associated with mesenchymal differentiation in gliosarcoma. American Journal of Pathology, 180, 1816–1823. [DOI] [PubMed] [Google Scholar]
- Nan, Y. , Wang, S. & Jia, W. (2018) Caspase independent cleavages of TDP‐43 generates 35kD fragment that cause apoptosis of breast cancer cells. Biochemical and Biophysical Research Communications, 497, 51–57. [DOI] [PubMed] [Google Scholar]
- Ozsensoy, Y. & Sahin, S. (2016) Comparison of different DNA isolation methods and use of dodecyle trimethyl ammonium bromide (DTAB) for the isolation of DNA from meat products. Journal of Advanced Veterinary and Animal Research, 3(4), 368. [Google Scholar]
- Pan, L. , He, J. , Zhao, T. , Xing, G. , Wang, Y. , Yu, D. et al. (2018) Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two‐stage multi‐locus GWAS procedure. TAG. Theoretical and Applied Genetics, 131, 2581–2599. [DOI] [PubMed] [Google Scholar]
- Peng, W.F. , Xu, S.S. , Ren, X. , Lv, F.H. , Xie, X.L. , Zhao, Y.X. et al. (2017) A genome‐wide association study reveals candidate genes for the supernumerary nipple phenotype in sheep (Ovis aries). Animal Genetics, 48, 570–579. [DOI] [PubMed] [Google Scholar]
- Plante, I. , Stewart, M.K. & Laird, D.W. (2011) Evaluation of mammary gland development and function in mouse models. Journal of Visualized Experiments, (53), e2828. 10.3791/2828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, A.L. , Patterson, N.J. , Plenge, R.M. , Weinblatt, M.E. , Shadick, N.A. & Reich, D. (2006) Principal components analysis corrects for stratification in genome‐wide association studies. Nature Genetics, 38, 904–909. [DOI] [PubMed] [Google Scholar]
- Pumfrey, R.A. , Johnson, R.K. , Cunningham, P.J. & Zimmerman, D.R. (1980) Inheritance of teat number and its relationship to maternal traits in swine. Journal of Animal Science, 50, 1057–1060. [DOI] [PubMed] [Google Scholar]
- Purcell, S. , Neale, B. , Todd‐Brown, K. , Thomas, L. , Ferreira, M.A. , Bender, D. et al. (2007) PLINK: a tool set for whole‐genome association and population‐based linkage analyses. American Journal of Human Genetics, 81, 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond, M. & Rousset, F. (1995a) An exact test for population differentiation. Evolution, 49, 1280–1283. [DOI] [PubMed] [Google Scholar]
- Raymond, M. & Rousset, F. (1995b) GENEPOP (Version 1.2): population genetics software for exact tests and ecumenicism. Journal of Heredity, 68, 248–249. [Google Scholar]
- Ren, W.L. , Wen, Y.J. , Dunwell, J.M. & Zhang, Y.M. (2018) pKWmEB: integration of Kruskal‐Wallis test with empirical Bayes under polygenic background control for multi‐locus genome‐wide association study. Heredity, 120, 208–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sant'Ana, G.C. , Pereira, L.F.P. , Pot, D. , Ivamoto, S.T. , Domingues, D.S. , Ferreira, R.V. et al. (2018) Genome‐wide association study reveals candidate genes influencing lipids and diterpenes contents in Coffea arabica L. Scientific Reports, 8, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segura, V. , Vilhjálmsson, B.J. , Platt, A. , Korte, A. , Seren, Ü. , Long, Q. et al. (2012) An efficient multi‐locus mixed‐model approach for genome‐wide association studies in structured populations. Nature Genetics, 44, 825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sul, J.H. , Martin, L.S. & Eskin, E. (2018) Population structure in genetic studies: Confounding factors and mixed models. PLoS Genetics, 14, e1007309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamba, C.L. , Ni, Y.L. & Zhang, Y.M. (2017) Iterative sure independence screening EM‐Bayesian LASSO algorithm for multi‐locus genome‐wide association studies. PLoS Computational Biology, 13, e1005357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamba, C.L. & Zhang, Y.M. (2018). A fast mrMLM algorithm for multi‐locus genome‐wide association studies. 10.1101/341784 [DOI] [PMC free article] [PubMed]
- Wang, L. , Zhang, Y. , Zhang, T. , Zhang, L. , Yan, H. , Liu, X. et al. (2017a) Genotyping by sequencing reveals a new locus for pig teat number. Animal Genetics, 48, 470–472. [DOI] [PubMed] [Google Scholar]
- Wang, S.B. , Feng, J.Y. , Ren, W.L. , Huang, B. , Zhou, L. , Wen, Y.J. et al. (2016) Improving power and accuracy of genome‐wide association studies via a multi‐locus mixed linear model methodology. Scientific Reports, 6, 19444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Ding, X. , Tan, Z. , Ning, C. , Xing, K. , Yang, T. et al. (2017b) Genome‐wide association study of piglet uniformity and farrowing interval. Frontiers in Genetics, 8, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Lee, S.H. , Goddard, M.E. & Visscher, P.M. (2011) GCTA: a tool for genome‐wide complex trait analysis. American Journal of Human Genetics, 88, 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Feng, J.Y. , Ni, Y.L. , Wen, Y.J. , Niu, Y. , Tamba, C.L. et al. (2017) pLARmEB: integration of least angle regression with empirical Bayes for multilocus genome‐wide association studies. Heredity, 118, 517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.M. , Jia, Z. & Dunwell, J.M. (2019) Editorial: The applications of new multi‐locus GWAS methodologies in the genetic dissection of complex traits. Frontiers in Plant Science, 10, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.J. , Xu, Y.F. , Dickey, C.A. , Buratti, E. , Baralle, F. , Bailey, R. et al. (2007) Progranulin mediates caspase‐dependent cleavage of TAR DNA binding protein‐43. Journal of Neuroscience, 27, 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y.J. , Xu, Y.F. , Cook, C. , Gendron, T.F. , Roettges, P. , Link, C.D. et al. (2009) Aberrant cleavage of TDP‐43 enhances aggregation and cellular toxicity. Proceedings of the National Academy of Sciences, USA, 106, 7607–7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, L. , Ke, H. , Xu, H. , Wang, G.D. , Zhang, H. , Zou, L. et al. (2020) TDP‐43 facilitates milk lipid secretion by post‐transcriptional regulation of Btn1a1 and Xdh. Nature Communications, 11, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, L. , Zhao, W. , Fu, Y. , Fang, X. , Ren, S. & Ren, J. (2019) Genome‐wide detection of genetic loci and candidate genes for teat number and body conformation traits at birth in Chinese Sushan pigs. Animal Genetics, 50, 753–756. [DOI] [PubMed] [Google Scholar]
- Zhou, X. & Stephens, M. (2012) Genome‐wide efficient mixed‐model analysis for association studies. Nature Genetics, 44, 821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zottel, A. , Jovčevska, I. , Šamec, N. , Mlakar, J. , Šribar, J. , Križaj, I. et al. (2020) Anti‐vimentin, anti‐TUFM, anti‐NAP1L1 and anti‐DPYSL2 nanobodies display cytotoxic effect and reduce glioblastoma cell migration. Therapeutic Advances in Medical Oncology, 12, 1758835920915302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1‐S2‐Table S1‐S2
Table S3
Data Availability Statement
Phenotypes are available at: https://figshare.com/s/0b326ab8632bbd1c82d6. Genotypes are available at: https://figshare.com/s/197c20e3229490ccb5d6.