

Abstract
4‐Hydroxyphenylacetate 3‐hydroxylase (4HPA3H), a flavin‐dependent monooxygenase from E. coli that catalyzes the hydroxylation of monophenols to catechols, was modified by rational redesign to convert also more bulky substrates, especially phenolic natural products like phenylpropanoids, flavones or coumarins. Selected amino acid positions in the binding pocket of 4HPA3H were exchanged with residues from the homologous protein from Pseudomonas aeruginosa, yielding variants with improved conversion of spacious substrates such as the flavonoid naringenin or the alkaloid mimetic 2‐hydroxycarbazole. Reactions were followed by an adapted Fe(III)‐catechol chromogenic assay selective for the products. Especially substitution of the residue Y301 facilitated modulation of substrate specificity: introduction of nonaromatic but hydrophobic (iso)leucine resulted in the preference of the substrate ferulic acid (having a guaiacyl (guajacyl) moiety, part of the vanilloid motif) over unsubstituted monophenols. The in vivo (whole‐cell biocatalysts) and in vitro (three‐enzyme cascade) transformations of substrates by 4HPA3H and its optimized variants was strictly regiospecific and proceeded without generation of byproducts.
Keywords: biocatalysis, catalysis, flavoprotein monooxygenases, hydroxylation, whole-cell biotransformation
The ability to perform regiospecific hydroxylations of sterically challenging monophenols was engineered into a flavin‐dependent monooxygenase. Its variants are valuable tools for production of the corresponding catechols for in vitro or live whole‐cell conversions.

Introduction
Direct selective oxygenations of organic molecules with molecular oxygen (air) are among the most challenging reactions in organic chemistry. As such, they introduce a hydroxyl (or rarely a keto) group by substitution of a hydrogen atom or – less commonly – of another functionality. Although these reactions are an essential part of synthetic routes to most natural and other complex organic molecules, the selective introduction of a hydroxyl group into alkyl or, in particular, aryl moieties is demanding and often requires multiple steps if classical chemical synthesis is concerned. Hence, the number of methods for direct (aromatic) hydroxylation is still limited.[ 1 , 2 , 3 ] In contrast to these chemosynthetic approaches, enzyme‐mediated hydroxylation often proved to be more effective due to high regio‐ or stereoselectivity, and is increasingly favored. It usually also is more compatible environmentally (due to omission of problematic reagents or catalysts, e. g. chromium salts, and the potential to reduce consumption of organic solvents). [4]
Indeed, biocatalytic oxidative transformations have been used for hydroxylations of structurally divergent compounds such as steroids,[ 5 , 6 , 7 ] alkaloids, [5] terpenoids, [8] fatty acids or simple alkanes. [5] In biosynthetic pathways, selective oxidative functionalizations often rely on cytochrome P450‐type enzymes, which are the predominant type of hydroxylating proteins in all organisms. [9] Thus, a multitude of cytochromes have been produced recombinantly, paving the way for an in‐depth characterization with respect to substrate scope, as exemplified for the well‐known enzyme from Bacillus megaterium and its variants, including the “workhorse” of these biocatalysts BM3.[ 5 , 6 , 10 ] However, BM3 is a positive exception as most cytochrome enzymes proved to be incompatible to many in vitro or industrial processes. To name just a few drawbacks, they are sensitive (Fe‐complexing agents, H2O2 produced by shunt mechanisms) and many are membrane‐bound and cannot be produced in standard expression hosts such as E. coli, etc.
Alternatively, since a few years flavin‐dependent monooxygenases are increasingly used in oxidative biotransformations, including hydroxylation reactions.[ 10 , 11 , 12 , 13 , 14 , 15 ] These monooxygenases belong to a superfamily of enzymes that is involved in key metabolic processes in both prokaryotic and eukaryotic cells, such as the biosynthesis of polyketides, coenzymes and siderophores or the degradation of a variety of aromatic natural products.[ 12 , 16 , 17 , 18 ] The classification of flavin adenine dinucleotide (FAD)‐dependent monooxygenases is based on structural features as well as on the type of electron donor and oxygen transfer.[ 11 , 14 ] The prototypic enzyme 4‐hydroxy‐phenylacetate 3‐hydroxylase (4HPA3H or HpaB, EC 1.14.14.9) catalyzes the reaction of 4‐hydroxyphenylacetate with oxygen to 3,4‐dihydroxyphenylacetate. During the reaction, water is formed by consumption of the cofactor FADH2, which is produced from FAD by an additional reductase component (HpaC, E.C. 1.5.1.36). [20] Its highly specific ortho‐hydroxylation is not restricted to the natural substrate 4‐hydroxyphenylacetate. Therefore 4HPA3H is an attractive biocatalyst for challenging oxidations of monophenols to σ‐diphenols (catechols).[ 12 , 21 , 22 ] Coloumbel and co‐workers published a seminal study on the transformation of 4‐halophenols to 4‐halocatechols by 4HPA3H from Escherichia coli with live whole‐cell biocatalysts. [23]
The same enzyme was used for the quantitative oxidation of p‐coumaric acid to caffeic acid as part of a reconstructed plant biosynthetic pathway by Lin and Yan, [24] especially because all the corresponding enzymes from plant sources are cytochrome P450‐dependent monooxygenases which proved to be difficult to express functionally in E. coli. Notably, in this case 4HPA3H‐mediated hydroxylation proceeds without side reactions and thus no undesired byproducts were reported.
Other biotransformations include the use of wildtype 4HPA3H in the hydroxylation of L‐tyrosine to DOPA [25] or the low‐yield conversion of the basal flavonoid naringenin into eriodictyol. [26]
Despite its efficiency and reaction specificity, the applicability of 4HPA3H in biotechnology is still limited because a toolbox of enzymes with a substrate scope expanding beyond simple monophenols or flavonoids, e. g. natural products such as phenolic alkaloids, is not yet available. Encouraged by a recent report on the engineering of the enzyme's product specificity by site‐directed exchange of an active site moiety, which enabled the sequential conversion of caffeic acid into 3,4,5‐trihydroxycinnamate in modified bacterial cells, [27] we performed a detailed rational optimization of 4HPA3H from E. coli. Herein, we report on enzyme variants suitable for the conversion of a broad range of monophenols. In contrast to other reports on 4HPA3H, which were solely focused on its application in cellular systems, our improved enzymes could also advantageously applied in an optimized in in vitro reaction systems for producing the corresponding catechols cell‐free.
Results and Discussion
Substrate spectrum of wild‐type 4HPA3H
As mentioned above, bacteria producing recombinant 4HPA3H proved to be well suited for conducting hydroxylation reactions.[ 23 , 24 , 28 ] In this study, we first used these simple whole‐cell biocatalysts for probing the substrate requirements of 3HPA3H from E. coli. We applied the expression strain BL21(DE3) (producing high levels of the recombinant monooxygenase (up to 246 mg l−1 of culture) under auto‐inducing conditions, but in contrast to previous studies[ 21 , 22 , 23 , 24 ] the cells were not co‐transformed with a plasmid harboring the gene for a flavin reductase component as the endogenous reductase activity proved to be sufficient to enable in vivo transformation of substrates. Suspensions of the cells were tested for hydroxylation of a set of eight phenolic compounds (Figure 1) which belong to different classes of natural products: we included (I) phenolic acids / ketones such as p‐coumaric acid (1), ferulic acid (3), rheosmin (15) and p‐hydroxylbenzoic acid (9), and (II) polycyclic compounds such as the hydroxylcoumarin umbelliferone (13), the stilbene resveratrol (5) and the flavonoid naringenin (7). Additionally, 2‐hydroxylcarbazole (11) was included, a readily available alkaloid‐like substrate resembling carbazoles from plant (e. g. Clauralia alkaloids) or bacterial origin (carazostatin, carbazomadurins). [29] Similar to its known substrates 1, 5 and 13[ 17 , 19 ] the cells also converted rheosmin 15 with high efficiency (100 %) (Figure 2A). Remarkably, the biocatalysts were also capable of hydroxylation of phenols 9 and 11, albeit lower yields of catechol products (39 % and 9 %, resp.) were obtained. A potential involvement of other E. coli monooxygenases in these conversions was ruled out by control reactions (i. e. application of cells harboring the empty vector) which showed no conversion. In contrast to the substrates mentioned above, ferulic acid (3) and naringenin (7) were not accepted at a detectable level, most probably because of their bulky methoxy (3) or chromanone substituent (7). A low conversion of naringenin (7) by the 4HPA3H was reported in two previous papers,[ 26 , 30 ] which can be improved by a change in the cultivation conditions. [30] Certainly, the used E. coli strain might influence the transformation efficiency, which is maybe the reason for the lack of conversion seen in our experiments.
Figure 1.

Substrates (odd numbers) used in hydroxylation reactions and their corresponding catechol products (even numbers). The hydroxyl groups introduced into the monophenols by 4HPA3H‐mediated oxidation are depicted in dark grey and marked by dashed circles. Substrates: p‐coumaric acid (1), caffeic acid (2), ferulic acid (3), 5‐hydroxyferulic acid (4), resveratrol (5), piceatannol (6), naringenin (7), eriodictyol (8), p‐hydroxybenzoic acid (9), 3,4‐dihydroxybenzoic acid (10), 2‐hydroxycarbazole (11), 2,3‐dihydroxy‐carbazole (12), umbelliferone (13), esculetin (14), rheosmin (15), 4‐(3,4‐dihydroxyphenyl)‐butan‐2‐one (16).
Figure 2.

Hydroxylation with live whole‐cell biocatalysts. Substrates (structures are depicted in Figure 1) were added to suspensions of bacterial cells expressing 4HPA3H or its single (A) and double variants (B) in a final concentration of 200 μM. After incubation for 16 hours, catechols and residual substrates were extracted and analyzed by HPLC as described in the supporting methods (see Supporting Information).
We also tested 4HPA3H for conversion of 4‐methoxycinnamic acid and 4‐aminocinnamic acid. These two analogues of p‐coumaric acid (1) were no substrates for the enzyme (data not shown). Hence, in accordance with a previous study (which showed that 4HPA3H enzymes are unable to hydroxylate cinnamic acid), [21] the presence of a phenolic hydroxyl group in the substrate seems to be mandatory for enzymatic turnover.
Optimization of 4HPA3H for hydroxylation of bulky substrates
Unlike the 4HPA3H enzyme used in this study, the homologous protein from Pseudomonas aeruginosa (homology: 85 %, similarity: 73 %) was reported to readily hydroxylate ferulic acid (3). [21] Thus, we speculated whether comparison of both enzymes might yield structural features responsible for this altered substrate scope ‐ which are prerequisites for a rational engineering of 4HPA3H specificity.
In order to gain insight into the architecture of the individual active sites, the three‐dimensional structures of the proteins were modeled based on crystallographic data of the related enzyme from Thermus thermophilus (PDB ID: 2YYJ, (homology: 49 %, similarity: 30 %). [28] Both models were aligned to the template (RSMD values: 0.78 and 1.14 Å for the E. coli and pseudomonad enzyme), which also contains the substrate 4‐hydroxy‐phenylacetate and FAD (Figure 3). We inspected the active sites for residues which are proximate to the aromatic ring of 4‐hydroxyphenylacetate, and identified three positions which differ in the enzymes from E. coli (M293, Y301, S462) and Pseudomonas aeruginosa (P293, F301, A462). Remarkably, all these positions are occupied by less voluminous residues in the latter enzyme, resulting in a more spacious active site. Thus, we substituted those residues in the enzyme from E. coli to shift the substrate scope in favor of more bulky substrates, yielding the variants M293P, Y301F and S462A. For similar reasons, isoleucine 157 which seems to directly interact with the phenolic moiety of 4‐hydroxyphenylacetate (Figure 3) was exchanged against valine. Notably, the exchanges Y301F and S462A also lead to an altered hydrophobicity of the active site (as a hydroxylated side chain is substituted by a smaller but more hydrophobic residue), which might also contribute to a putative alteration in substrate scope.
Figure 3.

Alignment of crystal structure of 4HPA3H from Thermus thermophilus (PDB ID: 2YYJ) (green) with the models of the enzymes from E. coli (pale yellow) and Pseudomonas aeruginosa (light blue). The models were generated with the tool Phyre2 as described in the Experimental Section, and aligned by the software MOE (version 2019.0101). The amino acid residues in close vicinity to the substrate 4‐hydroxyphenylacetate (orange) which were subjected to mutagenesis are shown as labelled stick representations. Gray/magenta: riboflavin/diphosphate moiety of the FAD cofactor.
Similar to the wild‐type enzyme, the variants were applied for in vivo hydroxylation reactions after appropriate expression was confirmed by SDS‐PAGE (see Figure S1 in Supporting Information). Remarkably, the enzymes 4HPA3H‐Y301F and ‐S462A were comparably active towards substrates which were also well‐accepted by the wild‐type enzyme (1, 13, 15), whereas the variants M293P and, in particular, I157V showed reduced productivity (Figure 2A). As expected, the variants are characterized by an altered substrate specificity. For example, the enzyme Y301F proved to be superior in conversion of the substrates 5, 9 and 11. Strikingly, most variants were also capable of a detectable oxidation of naringenin (7) and, to a minor extend, of ferulic acid (3). This effect was even more pronounced in some double variants, e. g. the enzyme Y301F/S462A, which produced significantly more catechol product from the bulky substrate 2‐hydroxycarbazole 11 (Figure 2B) than its parent variant Y301F (Figure 2A).
Since the substitution Y301F had the greatest impact on the acceptance of challenging compounds, we decided to also introduce a leucine or isoleucine into this position. Both amino acids are similar to phenylalanine with respect to hydrophobicity, but are smaller in size. [31] Strikingly, the preferred substrate of the resulting mutant enzymes was ferulic acid (3) (which was converted up to 45 % in particular by 4HPA3H‐Y301I), excelling well‐known substrates such as p‐coumaric acid (1) (Figure 2A). Obviously, subtle changes in size and hydrophobicity of the active site – e. g. the formal replacement of a hydroxyl group by a hydrogen atom in the variants Y301F and S462A only – are sufficient to change the substrate scope of 4HPA3H enzymes.
A similar sensitivity to single amino acid substitutions was observed in the exchange of active site and particular second‐shell residues in 4HPA3H. The latter residues are not in direct contact with the substrates, but are supporting the structural integrity of the active site. A recent study carried out by Deng and co‐workers [32] revealed that the exchange of the active site positions V158, I157 and S210 either had no significant effect on the hydroxylation of a set of cinnamate and mandelate substrates, or strongly reduced activity. Likewise, substitution of the second‐shell arginine R379 in 4HPA3H by serine, cysteine or glycine was used to probe the enzymes product selectivity, yielding variants which were not able to convert the two substrates L‐tyrosine and 3‐hydroxyphenyl‐acetate accepted by the wildtype enzyme. [24] Dhammaraj and co‐authors engineered 4HPA3H for the aforementioned conversion of caffeic acid (2) [20] by exchange of the second‐shell tyrosine Y398. Taken together, these altered substrate preferences caused by exchange of second‐shell positions seem to influence the enzyme activity rather indirectly by slightly shifted positioning of active site residues.
Product specificity of 4HPA3H and variants
To gain structural information on the hydroxylation products, we performed whole‐cell biotransformations in a larger scale (500 mL of cell suspension). Each substrate was converted by the enzyme variant that showed highest conversion in the analytic in vivo reactions (see above). Similar to those transformations, the catechol products were extracted from the reaction mixture after 16 hours of incubation. The compounds, which were purified by preparative reserve‐phase HPLC, were identified as the catechols shown in Figure 1 by 1H‐NMR, MS and MS2 analysis (see Experimental Section). Noteworthy, 4HPA3H and its variants demonstrated strict regio‐ and product specificity. In contrast to the enzyme from Pseudomonas aeruginosa, which performs sequential multiple hydroxylations, [16] no further conversion of the formed catechols into pyrogallol‐like polyphenols was observed for both the wild‐type enzyme and its variants.
Activity of 4HPA3H and variants
In order to characterize 4HPA3H and its variants with respect to their specific activity, the development of a reliable in vitro assay was necessary. As the enzymes rely on a reductase component which provides the cofactor FADH2 (Figure 4), we focused on the screening of a suitable reductase enzyme in the first step. The flavin reductase HpaC from E. coli – the endogenous partner of 4HPA3H – could not be obtained as purified enzyme due to formation of mainly insoluble protein under a variety of expression conditions (data not shown). In the search for homologous proteins, PrnF from Pseudomonas protegens (NCBI accession AAY91318.1) which is a part of the pseudomonad two‐component arylamine oxygenase system[ 34 , 35 ] showed to have high sequence identity (99 %) to HpaC. Recombinant PrnF proved to be well expressed in E. coli (in a yield of 175 mg l−1 of culture). Similar to literature data, [36] this purified protein (see Figure S2 in Supporting Information) was highly active in the reduction of FAD with NADH (16,660±1,600 nmol min−1 mg−1).
Figure 4.
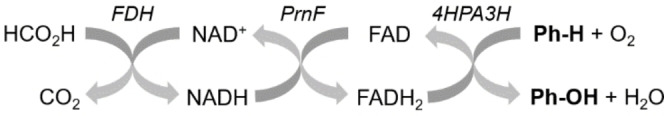
Scheme of enzyme cascade for in vitro hydroxylation. For recycling of the cofactors NADH and FAD used in the aromatic hydroxylation step (boldface), FDH from Candida boidinii and the flavin reductase PrnF from Pseudomonas protegens were applied. Enzymes are shown in italics.
For assaying 4HPA3H in microplates, 4HPA3H was incubated with PrnF, the substrate p‐coumaric acid (1) and formate dehydrogenase (FDH) from Candida boidinii. We introduced this enzyme for regeneration of the costly cofactor NADH: the NAD+ formed during the reaction is recycled with sodium formate, yielding a sequential three‐enzyme cascade for the transfer of redox equivalents / oxygen (Figure 4). The conversion of p‐coumaric acid (1) was measured by complexation of the produced caffeic acid with Fe3+ ions. The colored complex formed during this reaction was quantitatively assayed via spectrophotometry (see Figure S3 in Supporting Information). We assayed 4HPA3H under optimized conditions (see next section), yielding a specific activity (6.2±0.5 nmol min−1 mg−1) that is comparable to literature values. [24] The variant Y301F/S462A – which was also produced recombinantly (in a yield of 147 mg l−1 of culture) and applied as purified enzyme (see Figure S2 in Supporting Information) – showed reduced activity (1.6±0.3 nmol min−1 mg‐1) towards p‐coumaric acid (1). However, similar to the data obtained from whole‐cell biotransformations (see above), this variant was beneficial in the conversion of ferulic acid (3), which is also reflected by a comparably high conversion rate (1.1±0.3 nmol min−1 mg−1).
In addition, the developed spectrophotometric assay was used to determine how much of the shunt product H2O2 is formed by the 4HPA3H enzyme. Therefore, H2O2 was quantified during the hydroxylation of p‐coumaric acid (1) by an adapted peroxidase assay. [37] As common for flavin‐dependent enzymes, wild‐type 4HPA3H showed a significant uncoupling, which converted an additional amounts of NADH (42.0±0.1 % of the consumption caused by the hydroxylation reaction) into shunt H2O2. Noteworthy, the variant Y301F/S462A did not show a higher tendency for uncoupling than the wild type (see Table S1 in the Supporting Information).
In vitro production of catechols by 4HPA3H/PrnF
In a multitude of biocatalytic conversions, cell‐free (in vitro) systems proved to be superior to reactions with whole‐cell biocatalysts, especially with emphasis on substrate depletion by and toxicity to the used microorganism by involved compounds, side reactions, and for a more facile product purification. [38] In order to develop an effective and low‐cost enzyme system for in vitro hydroxylation by 4HPA3H, we optimized several parameters of the enzyme reaction with respect to maximum reaction rate. By means of our spectrophotometric assay (see section above), we screened different buffer systems and optimized the concentration of reagents (p‐coumaric acid (1), FAD, NADH and methanol), the incubation temperature and oxygen transfer, e. g. by variation of the shaking frequency (optimum conditions for best reaction rates are given in the Experimental Section). In particular, this fine‐tuning yielded a reduction in the content of NADH from 200 μM to 25 μM without loss of activity (see Figure S4 in Supporting Information). Advantageously, HPA3H showed tolerance to high concentrations of substrate (1 mM) and of the co‐solvent methanol (10 %, v/v), which is beneficial due to enhanced substrate solubility. The redox system was susceptible to high concentrations of FAD which proved to be inhibitory (see Figure S4 in Supporting Information). In contrast to the cofactors, the accompanying redox enzymes PrnF and FDH were used in excess to ensure an unrestrained coupling.
Subsequently, we applied the optimized reaction conditions to the in vitro oxidation of the substrates shown in Figure 1 (without further optimization for a specific substrate). In accordance with the substrate specificity of the enzyme in whole‐cell transformation experiments (Figure 2A), product yields in the conversion by the wild‐type 4HPA3H (Figure 5A) ranged from high (1, 13, 15) or moderate (5) to low (9, 11) (see footnote 1 ), whereas ferulic acid 3 was not oxidized. In addition, we selectively tested the mutant enzymes Y301F/S462A and Y301I from the initial screen (Figure 5B). Although a direct comparison between the in vivo and cell‐free system is not possible due to e. g. the different substrate concentration, the variants again proved to be optimal for transformation of the more challenging substrates such as naringenin (7) and ferulic acid (3), respectively (see footnotes1, 2 ). However, in contrast to the in vivo experiments 2‐hydroxycarbazole (11) was not accepted by the double variant Y301F/S462A. Obviously, the high concentration of the aromatic substrate 11 seems to trigger enzyme inhibition in this case, which was confirmed by detection of activity at a lower substrate concentration (see footnote 3 ). This effect has previously been observed in transformations with other 4HPA3H enzymes. [21]
Figure 5.

In vitro hydroxylation by the 4HPA3H/PrnF/FDH system. Purified 4HPA3H (A) or variants (B, Y301I depicted in white and Y301F/S462A depicted in black) were used for conversion of substrates (1 mM, structures with numbers are shown in Figure 1) under optimized conditions (10 μM FAD, 200 μM NADH, 10 % (v/v) MeOH) as specified in the supporting methods (see Supporting Information); b.d.: below detection.
Conclusions
This paper is the first report on rational optimization of the active site of the flavin‐dependent monooxygenase 4HPA3H towards a higher activity on non‐natural substrates. We showed that single substitutions are sufficient to engineer new functionalities into 4HPA3H, yielding enzymes which are optimal for conversion of e. g. sterically challenging substrates. This strategy affords robust biocatalysts which provide an alternative to other enzymatic hydroxylation systems (e. g. cytochromes), especially due to high yield, strict regio‐ and product specificity. In addition, the applicability of the enzyme not only in whole‐cell hydroxylation reactions but also in in vitro production systems will pave the way for its use in e. g. multienzyme and microscale reaction systems.
Experimental Section
Enzyme production and quantification
The 4HPA3H gene from Escherichia coli BL21(DE3) and the PrnF gene from Pseudomonas protegens were introduced into a modified pET28a(+)vector by BsaI‐directed ligation. Enzyme variants were constructed by mutagenesis of pET28a(+)‐4HPA3H by the Quik Change II Site‐Directed Mutagenesis Kit (Agilent, Santa Clara, USA). The recombinant enzymes, which carried a hexahistidine tag at their N‐terminal, were produced in E. coli BL21(DE3) (see Supporting Information) and purified by immobilized Co2+ ion affinity chromatography as described previously. [39] Purified proteins were checked for homogeneity by sodium dodecyl sulfate polyacrylamide gel electrophoresis, and their concentration was determined by the method of Bradford. [40]
Whole‐cell bioconversions and product identification
Reactions (10 mL) were performed in duplicates in auto‐inducing medium [41] containing 200 μM hydroxylase substrate. Cultures were inoculated with E. coli BL21(DE3) harboring either the plasmids for production of 4HPA3H or its variants (hydroxylation reactions) or the empty vector (controls). After 16 hours of incubation at 37 °C, samples of the cell suspension (in case of 2‐hydroxycarbazole) or of the supernatants from centrifugation (all other substrates) were acidified and extracted with ethyl acetate. The extracts were subjected to HPLC analysis (see Supporting Information). For product identification, preparative transformations were performed in larger scale (500 mL). Products extracted with ethyl acetate were purified by preparative HPLC and analyzed by 1H NMR and ESI‐FTMS (see Supporting Information).
Homology modeling
The three‐dimensional structures of 4HPA3H from E. coli and P. aeruginosa were modeled by means of the online protein fold recognition server Phyre2[42] The model with the highest score, which is based on crystallographic data of the 4HPA3H enzyme from Thermus thermophilus (PDB ID: 2YYJ) [19] was used for alignments and active site studies. The determination of RMSD values was conducted using the software Molecular Operating Environment (Chemical Computing Group ULC 2019).
Spectrophotometric activity assay for 4HPA3H
The reactions (80 μL) were performed in a microplate well that contained substrate (2 mM p‐coumaric acid or ferulic acid), cofactors (10–1000 μM FAD, 25–200 μM NADH) and a cofactor regeneration system (see Supporting Information). Reactions were started by addition of PrnF (566 mU mL−1, activity determined spectrophotometrically as described in the Supporting Information) and hydroxylase (1 mg mL−1). The reactions were stopped by mixing with 40 μL of catechol reagent to generate a colored Fe3+/product complex. From the absorbance at 595 nm, the concentration of formed product was calculated by means of a standard curve (Figure S3). The same assay was used to determine uncoupling in the 4HPA3H reaction. Therefore, quantification of formed caffeic acid was coupled to the determination of H2O2 using a modification of the method described by Dippe and Ulbrich‐Hofmann. [43]
In vitro conversion of monophenols
The reactions (200 μL) were performed in duplicates under optimized conditions (see Supporting Information). After 16 hours of incubation, the samples were centrifuged and the acidified supernatant was extracted with ethyl acetate. The extracts were analyzed by HPLC analysis (see Supporting Information).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We are thankful to Dr. Jürgen Schmidt for mass spectrum analysis. Moreover, we gratefully thank Anja Ehrlich and Martina Lerbs for excellent technical assistance, Gudrun Hahn, Andrea Porzel and Hidayat Hussain for help in NMR measurements, and Andrej Frolov for help in MS assays (all IPB Halle). The research leading to these results has received funding from the European Union's Seventh Framework Program FP7/2007‐2013 under grant agreement no 266025 (BIONEXGEN). This work was also funded by ScienceCampus Halle ‐ Plant‐Based Bio‐Economy. Open Access funding enabled and organized by Projekt DEAL.
S. Herrmann, M. Dippe, P. Pecher, E. Funke, M. Pietzsch, L. A. Wessjohann, ChemBioChem 2022, 23, e202100480.
Footnotes
HPLC analysis of the reaction products confirmed product specificity. All conversions – except for naringenin (7) where 4HPA3H‐Y301I was used – yielded the previously identified catechols (Figure 1) only.
In contrast to the experiments with live biocatalysts (Figure 2A), some low‐level conversion of naringenin (7) was observed by wild‐type 4HPA3H under in vitro conditions (25 % yield). This effect might be explained by a better substrate availability due to the higher naringenin concentration in vitro. This is accordance with the high KM value of wild‐type 4HPA3H for this substrate. [26]
The double variant Y301F/S462A showed high conversion at substrate concentrations of 200 μM and 500 μM. To a lesser extent, the single variant Y301I was capable of hydroxylating 2‐hydroxycarbazole (11) under in vitro conditions, i. e. at a concentration of 200 μM only (see Table S2 in the Supporting Information). This differential effect indicates a general inhibition by this substrate, which in addition seems to be dependent on the architecture of the active site.
References
- 1. Ullrich R., Hofrichter M., Cell. Mol. Life Sci. 2007, 64, 271–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dong J., Fernández-Fueyo E., Hollmann F., Paul C. E., Pesic M., Schmidt S., Wang Y., Younes S., Zhang W., Angew. Chem. Int. Ed. 2018, 57, 9238–9261; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 9380–9404. [Google Scholar]
- 3. Chakrabarty S., Wang Y., Perkins J. C., Narayan A. R. H., Chem. Soc. Rev. 2020, 49, 8137–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Woodley J. M., Trends Biotechnol. 2008, 26, 321–327. [DOI] [PubMed] [Google Scholar]
- 5. Urlacher V. B., Girhard M., Trends Biotechnol. 2012, 30, 26–36. [DOI] [PubMed] [Google Scholar]
- 6. Fasan R., ACS Catal. 2012, 2, 647–666. [Google Scholar]
- 7. Cha G. S., Rju S. H., Ahn T., Yun C.-H., Biotechnol. Lett. 2014, 36, 2501–2506. [DOI] [PubMed] [Google Scholar]
- 8. Bogazkaya A. M., von Bühler C. J., Kriening S., Busch A., Seifert A., Pleiss J., Laschat S., Urlacher V. B., Beilstein J. Org. Chem. 2014, 10, 1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rasool S., Mohamed R., Protoplasma 2016, 253, 1197–1209. [DOI] [PubMed] [Google Scholar]
- 10. Neufeld K., Marienhagen J., Schwaneberg U., Pietruszka J., Green Chem. 2013, 15, 2408–2421. [Google Scholar]
- 11. Sucharitakul J., Tinikul R., Chaiyen P., Arch. Biochem. Biophys. 2014, 555–556, 33–46. [DOI] [PubMed] [Google Scholar]
- 12. Ceccoli R. D., Bianchi D. A., Rial D. V., Front. Microbiol. 2014, 5 : 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mügge C., Heine T., Gomez Baraibar A., van Berkel W. J. H., Paul C. E., Tischler D., Appl. Microbiol. Biotechnol. 2020, 104, 6481–6499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Heine T., van Berkel W. J. H., Gassner G., van Pée K.-H., Tischler D., Biology 2018, 7, 42–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chenprakhon P., Wongnate T., Chaiyen P., Protein Sci. 2019, 28, 8–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Huijbers M. M. E., Montersino S., Westphal A. H., Tischler D., van Berkel W. J. H., Arch. Biochem. Biophys. 2014, 544, 2–17. [DOI] [PubMed] [Google Scholar]
- 17. Davison J., Fahad A., Cai M., Song Z., Yehia S. Y., Lazarus C. M., Bailey A. M., Simpson T. J., Cox R. J., Proc. Natl. Acad. Sci. USA 2016, 109, 7642–7647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Bosello M., Mielcarek A., Giessen T. W., Marahiel M. A., Biochemistry 2012, 51, 3059–3066; [DOI] [PubMed] [Google Scholar]
- 18b. Michiel M., Perchat N., Perret A., Tricot S., Papeil A., Besnard M., de Berardinis V., Salanoubat M., Fischer C., Environ. Microbiol. Rep. 2012, 6, 642–647; [DOI] [PubMed] [Google Scholar]
- 18c. Tang H., Yao Y., Zhang D., Meng X., Wang L., Yu H., Ma L., Xu P., J. Biol. Chem. 2012, 286, 9179–39187; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18d. Volkers G., Damas J. M., Palm G. J., Panjikar S., Soares C. M., Hinrichs W., Acta Crystallogr. Sect. D 2013, 69, 1758–1767; [DOI] [PubMed] [Google Scholar]
- 18e. Trivedi V. D., Majhi P., Phale P. S., Appl. Biochem. Biotechnol. 2014, 172, 3964–3977; [DOI] [PubMed] [Google Scholar]
- 18f. Ismail A., Leroux V., Smadja M., Gonzalez L., Lombard M., Pierrel F., Mellot-Draznieks C., Fontecave M., PLoS Comput. Biol. 2016, 12, e1004690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18g. Pimviriyakul P., Thotsaporn K., Sucharitakul J., Chaiyen P., J. Biol. Chem. 2017, 292, 4818–4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van Berkel W. J. H., Kamerbeek N. M., Fraaije M. W., J. Biotechnol. 2006, 124, 670–689. [DOI] [PubMed] [Google Scholar]
- 20. Galán B., Díaz E., Prieto M. A., García J. L., J. Bacteriol. 2000, 182, 627–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Furuya T., Kino K., Appl. Microbiol. Biotechnol. 2014, 98, 1145–1154. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a.Y. Yan, Y. Lin, Patent US 20140363858 A1, 2014;
- 22b.S. Herrmann, M. Dippe, L. A. Wessjohann, T. Geißler, K. Geißler, J. Ley, Patent application EP 15188136.4, 2015.
- 23. Coulombel L., Nolan L. C., Nicodinovic J., Doyle E. M., O‘Connor K. E., Appl. Microbiol. Biotechnol. 2010, 89, 1867–1875. [DOI] [PubMed] [Google Scholar]
- 24. Lin Y., Yan Y., Microb. Cell Fact. 2012, 11 : 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nakagawa A., Nakamura S., Matsumura E., Yashima Y., Takao M., Aburatani S., Yaoi K., Katayama T., Minami H., Appl. Microbiol. Biotechnol. 2021, 105, 5433–5447. [DOI] [PubMed] [Google Scholar]
- 26. Shen X., Zhou D., Lin Y., Wang J., Gao S., Kandavelu P., Zhang H., Zhang R., Wang B.-C., Rose J., Yuan Q., Yan Y., Sci. Rep. 2019, 9, 7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Dhammaraj T., Phintha A., Pinthong C., Medhanavyn D., Tinikul R., Chenprakhon P., Sucharitakul J., Vardhanabhuti N., Jiarpinitnun C., Chaiyen P., ACS Catal. 2015, 5, 4492–4502. [Google Scholar]
- 28. Kim S.-H., Hisano T., Takeda K., Iwasaki W., Ebihara A., Miki K., J. Biol. Chem. 2007, 282, 33107–33117. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Songsiang U., Thongthoom T., Boonyarat C., Yenjai C., J. Nat. Prod. 2011, 74, 208–212; [DOI] [PubMed] [Google Scholar]
- 29b. Hieda Y., Anraku M., Choshi T., Tomida H., Fujioka H., Hatae N., Hori O., Hirose J., Hibino S., Bioorg. Med. Chem. Lett. 2014, 24, 3530–3533. [DOI] [PubMed] [Google Scholar]
- 30. Jones J. A., Collins S. M., Vernacchio V. R., Lachance D. M., Koffas M. A. G., Biotechnol. Prog. 2016, 32, 21–25. [DOI] [PubMed] [Google Scholar]
- 31. Voet D., Voet J. G., in Biochemistry, Vol. 3, Wiley-VCH, Weinheim, 2004, pp. 66–67. [Google Scholar]
- 32. Deng Y., Faivre B., Back O., Lombard M., Pecqueur L., Fontecave M., ChemBioChem 2020, 21, 163–170. [DOI] [PubMed] [Google Scholar]
- 33. Kim H., Kim S., Kim D., Yoon S. H., BMC Microbiol. 2020, 20, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jensen C. N., Mielke T., Farrugia J. E., Frank A., Man H., Hart S., Turkenburg J. P., Grogan G., ChemBioChem 2015, 16, 968–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee J.-K., Zhao H., J. Bacteriol. 2007, 189, 8556–8563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tiwari M. K., Singh R. K., Lee J.-K., Zhao H., Bioorg. Med. Chem. Lett. 2012, 22, 1344–1347. [DOI] [PubMed] [Google Scholar]
- 37. Dippe M., Ulbrich-Hofmann R., J. Am. Oil Chem. Soc. 2010, 87, 1005–1011. [Google Scholar]
- 38.
- 38a. You C., Zhang Y.-H. P., Adv. Biochem. Eng./Biotechnol. 2012, 131, 89–119; [DOI] [PubMed] [Google Scholar]
- 38b. Homaei A. A., Sariri R., Vianello F., Stevanato R., J. Chem. Biol. 2013, 6, 185–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dippe M., Bauer A.-K-, Porzel A., Funke E., Müller A. O., Schmidt J., Beier M., Wessjohann L., Adv. Synth. Catal. 2019, 361, 5346–5350. [Google Scholar]
- 40. Bradford M. M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 41. Studier F. W., Protein Expression Purif. 2005, 41, 207–234. [DOI] [PubMed] [Google Scholar]
- 42. Kelley L. A., Sternberg M. J. E., Nat. Protoc. 2009, 4, 363–371. [DOI] [PubMed] [Google Scholar]
- 43. Dippe M., Ulbrich-Hofmann R., J. Am. Oil Chem. Soc. 2010, 87, 1005–1011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information