

Abstract
Background and Aims
Hepatic ischemia‐reperfusion (HIR) injury, a common clinical complication of liver transplantation and resection, affects patient prognosis. Ring finger protein 5 (RNF5) is an E3 ubiquitin ligase that plays important roles in endoplasmic reticulum stress, unfolded protein reactions, and inflammatory responses; however, its role in HIR is unclear.
Approach and Results
RNF5 expression was significantly down‐regulated during HIR in mice and hepatocytes. Subsequently, RNF5 knockdown and overexpression of cell lines were subjected to hypoxia‐reoxygenation challenge. Results showed that RNF5 knockdown significantly increased hepatocyte inflammation and apoptosis, whereas RNF5 overexpression had the opposite effect. Furthermore, hepatocyte‐specific RNF5 knockout and transgenic mice were established and subjected to HIR, and RNF5 deficiency markedly aggravated liver damage and cell apoptosis and activated hepatic inflammatory responses, whereas hepatic RNF5 transgenic mice had the opposite effect compared with RNF5 knockout mice. Mechanistically, RNF5 interacted with phosphoglycerate mutase family member 5 (PGAM5) and mediated the degradation of PGAM5 through K48‐linked ubiquitination, thereby inhibiting the activation of apoptosis‐regulating kinase 1 (ASK1) and its downstream c‐Jun N‐terminal kinase (JNK)/p38. This eventually suppresses the inflammatory response and cell apoptosis in HIR.
Conclusions
We revealed that RNF5 protected against HIR through its interaction with PGAM5 to inhibit the activation of ASK1 and the downstream JNK/p38 signaling cascade. Our findings indicate that the RNF5‐PGAM5 axis may be a promising therapeutic target for HIR.
Abbreviations
- AddnASK1
ASK1 dominant‐negative mutant adenovirus
- ALT
alanine aminotransferase
- ASK1
apoptosis‐regulating kinase 1
- AST
aspartate aminotransferase
- Bad
Bcl2‐associated agonist of cell death
- Bax
Bcl2‐associated x protein
- Bcl2
B‐cell leukemia/lymphoma
- Bcl‐XL
B‐cell lymphoma/leukemia XL gene
- BSA
bovine serum albumin
- C‐Caspase‐3
cleaved‐caspase‐3
- Ccl2
chemokine (C‐C motif) ligand 2
- cDNA
complementary DNA
- Co‐IP
coimmunoprecipitation
- Cxcl10
chemokine (C‐X‐C motif) ligand 10
- ER
endoplasmic reticulum
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GSEA
gene set enrichment analysis
- H&E
hematoxylin and eosin
- HIR
hepatic ischemia‐reperfusion
- HR
hypoxia‐reoxygenation
- IF
immunofluorescence
- IHC
immunohistochemistry
- IKKβ
inhibitory kappa B kinase
- IP
immunoprecipitation
- IR
ischemia‐reperfusion
- IκBα
inhibitor of kappa B alpha
- JNK
c‐Jun N‐terminal kinase
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
knockout
- Ly6G
lymphocyte antigen 6 complex locus G
- MAPK
mitogen‐activated protein kinase
- MS
mass spectrometry
- NTG
nontransgenic
- PGAM5
phosphoglycerate mutase family member 5
- RNA‐seq
RNA sequencing
- RNF5
ring finger protein 5
- RNF5‐HKO
hepatocyte‐specific RNF5‐knockout
- RNF5‐HTG
hepatocyte‐specific RNF5 transgenic
- TG
transgenic
- TUNEL
terminal deoxynucleotidyl transferase–mediated deoxyguanosine triphosphate (dUTP) nick‐end labeling
- WT
wild type
INTRODUCTION
Ischemia‐reperfusion (IR) injury refers to the tissue and organ damage caused when blood supply is restored after a period of ischemia.[ 1 ] Hepatic ischemia‐reperfusion (HIR) injury involves a biphasic process of ischemia‐induced cell damage and reperfusion‐induced inflammatory response, which is mainly observed in partial hepatectomy, liver transplantation, trauma, and hypovolemic shock.[ 2 , 3 ] Liver damage caused by IR may cause abnormal liver function, acute liver failure, multiple organ failure, and even death.[ 4 ] Unfortunately, there is no effective prevention or treatment of HIR injury in clinical practice. At present, the mechanism of HIR injury is not well understood; thus, it is imperative to elucidate its mechanism to develop targets for treatment.
The mechanism of HIR injury is complex and involves various cells and diverse processes.[ 5 ] HIR is a dynamic process, and the accompanying inflammatory response plays a key role. In the early stage of liver ischemia, insufficient glucose and oxygen supply, reduced ATP production, and cell metabolic disorders can directly cause hepatocyte damage and activate Kupffer cells, dendritic cells, and natural killer cells, resulting in inflammatory damage.[ 6 ] During reperfusion, these activated immune cells can drive neutrophils and lymphocytes to infiltrate liver tissues by releasing cytokines, chemokines, and cell adhesion molecules, thus aggravating liver cell damage.[ 6 , 7 ] HIR injury is characterized by progressive hepatocyte cell damage, apoptosis/necrosis, and acute inflammation.
Ring finger protein 5 (RNF5), an E3 ubiquitin ligase localized in the endoplasmic reticulum (ER) and mitochondrial membranes, has been implicated in the regulation of substrate stability and localization through K63‐ or K48‐linked ubiquitination.[ 8 ] RNF5 plays a critical role in various cellular processes, including cell motility regulation,[ 9 ] ER‐associated protein degradation,[ 8 ] innate immunity through MITA ubiquitination,[ 10 ] ER quality control through ubiquitination of misfolded proteins,[ 11 ] and ubiquitination‐dependent relocalization of the c‐Jun N‐terminal kinase (JNK)‐associated membrane protein, a proteasome adaptor protein.[ 12 ] Furthermore, RNF5 is involved in the regulation of viral and bacterial infections.[ 13 , 14 ] In a model of inflammatory bowel disease, RNF5‐knockout (KO) mice exhibited more‐severe colitis symptoms, and the lesion‐site changes associated with enhanced inflammation in colonic tissue included altered proliferation and cell‐death programs.[ 15 ] However, the function of RNF5 in HIR injury remains open for study.
In this study, we found that RNF5 expression was remarkably decreased during HIR. Using hepatic RNF5‐KO and transgenic (TG) mice, we demonstrated that RNF5 reduced inflammatory response, cell apoptosis, and liver damage in HIR. Mechanistically, RNF5 interacts with and ubiquitinates phosphoglycerate mutase family member 5 (PGAM5) during HIR. Ubiquitination of PGAM5 by RNF5 led to its degradation and inhibition of apoptosis‐regulating kinase 1 (ASK1) and the downstream signaling pathways of JNK/p38 activation, reducing apoptosis and inflammation. Our findings suggest that RNF5 may serve as a potential therapeutic target for reducing HIR injury.
MATERIALS AND METHODS
Animals
Male mice (8–10 weeks old) were obtained from Vital River (Beijing, China). All mice had free access to food and water and were housed in a specific pathogen‐free facility with 12/12‐hour light/dark cycles. Mice had a 1‐week period of acclimatization before sham or ischemia reperfusion treatment; all animal experiments were approved by the Animal Care Committee of the First Affiliated Hospital of Zhengzhou University and were conducted in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85‐23, revised 1996). The construction of hepatocyte‐specific RNF5 knockout and transgenic mice has been described in detail in the Supporting Material.
HIR surgery
We established a partial (70%) warm HIR model as described.[ 16 ] Mice were anesthetized with 1% pentobarbital sodium (50 mg/kg), and the abdominal cavity was exposed at the midline of the abdomen with the blood vessels separated around the livers. A microvascular clamp was used to clamp the blood vessels supplying the left and middle of the liver in the IR group, whereas vascular clamping was not performed in the sham‐operated group. After ischemia for 1 hour and reperfusion for different times, blood and liver tissues were collected for subsequent analysis.
Measurements of liver injury
Serum concentrations of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured using the ADVIA 2400 Chemistry System (Siemens, Tarrytown, NY), according to the manufacturer's instructions.
H&E staining
H&E staining was used to evaluate liver necrosis as described.[ 17 ] Liver tissue samples were fixed in 10% neutral‐buffered formalin, dehydrated, and embedded in paraffin. Paraffin‐embedded liver tissue was cut into 5‐μm‐thick continuous sections and stained with H&E (Hematoxylin, G1004, Servicebio, Wuhan, China; Eosin, BA‐4024, Baso, Zhuhai, China). Images were captured by using a light microscope (ECLIPSE 80i; Nikon, Tokyo, Japan).
Immunofluorescence staining and immunohistochemistry
For CD11b and Ly6G immunofluorescence (IF) staining, paraffinized sections were subjected to deparaffinization, rehydration, and EDTA antigen retrieval. Sections were blocked with 10% bovine serum albumin (BSA) at 37°C for 1 hour, then rinsed three times with PBS, and incubated with primary antibodies against mouse CD11b (BM3925, 1:12000 dilution; Boster, Pleasanton, CA) or lymphocyte antigen 6 complex locus G (Ly6G; GB11229; 1:200 dilution; Servicebio, Wuhan, China) overnight at 4°C. After washing with PBS, they were incubated with corresponding secondary antibodies (Alexa Flour 568 goat anti‐rabbit IgG [H+L], A11036; Invitrogen, Carlsbad, CA) for 1 hour at 37°C and washed with PBS. Finally, cells were labeled with DAPI, and images were captured under a fluorescent microscope (BX51; Olympus, Tokyo, Japan).
Paraffin liver sections were stained with cleaved‐caspase‐3 (C‐Caspase‐3) after deparaffinization and rehydration. Sections were repaired in EDTA at high temperature for 20 minutes, then blocked in BSA. Next, primary antibodies against mice C‐Caspase‐3 (9664, 1:150 dilution; CST, Danvers, MA) were used and incubated overnight at 4°C. After rewarming sections for 30 minutes at 37℃ and washing with PBS, they were incubated with corresponding secondary antibodies (PV‐9001; ZSGB‐bio, Beijing, China). Finally, 3′‐diaminobenzidine were used to visualize the section. Hematoxylin was used to label nuclei.
Terminal deoxynucleotidyl transferase–mediated deoxyguanosine triphosphate (dUTP) nick‐end labeling staining
According to the manufacturer’s instructions, paraffin‐embedded sections were subjected to deparaffinization, rehydration, and rinsed with PBS and then incubated with proteinase‐K working solution (20 μg/mL) at room temperature for 10 minutes. After rinsing twice with PBS, terminal deoxynucleotidyl transferase–mediated deoxyguanosine triphosphate (dUTP) nick‐end labeling (TUNEL) was added (11684817910; Roche Diagnostics, Indianapolis, IN) to the reaction mixture and incubated at room temperature for 1 hour. Finally, sections were rinsed three times with PBS before DAPI staining. Numbers of cell apoptosis were counted under a fluorescent microscope.
Construction of plasmids and stable cell lines
Full‐length or truncated RNF5 and PGAM5 were amplified from homo complementary DNA (cDNA), and they were connected to different vectors by the in‐fusion method to acquire overexpression plasmids. HEK293T cells were cotransfected with packaging plasmids (pMD2.G and psPAX2) and indicated plasmids for 48 hours. Supernatant of HEK 293T cells were collected and used to infect L02 hepatocytes with polybrene (H9268; Sigma‐Aldrich, St. Louis, MO) at a concentration of 10 μg/mL. Infected L02 hepatocytes were treated with puromycin (A1113803; Gibco, Grand Island, NY) at a concentration of 2 μg/mL to acquire stable cell lines. Finally, RT‐PCR and western blotting were used to identify stable cell lines. The RNF5 adenovirus was purchased from Hanbio Biotechnology Co., Ltd. (Shanghai, China) and used to infect primary hepatocytes. The primers used are listed in Table S1.
Cell culture and hypoxia‐reoxygenation experiments
Hepatocyte L02 and HEK293T cell lines were purchased from the ATCC (American Type Culture Collection, Manassas, VA) without mycoplasma contamination; primary hepatocytes were obtained from mice by using a modified two‐step collagenase perfusion method and were cultured in DMEM supplemented with 10% fetal bovine serum (FBS) and 1% penicillin‐streptomycin at 37°C with an environment of 5% CO2. For hypoxia‐reoxygenation (HR) experiments, rapid‐growth‐phase cells were subjected to hypoxia (94% N2, 1% O2, and 5% CO2) in sugar‐free, serum‐free DMEM for 6 hours. Then, cells were cultured with 10% FBS medium under normal air conditions (95% air, 5% CO2) at 37°C for 6 hours. The HR experiment of primary hepatocytes was performed as described before.[ 16 ] For the inhibitory experiment of ASK1, L02 cells were cultured with an 80‐μM ASK1 inhibitor (GS4997) to inhibit the activation of ASK1 before the HR experiments.
Immunoprecipitation and mass spectrometry assays
The immunoprecipitation (IP) assay was performed as described.[ 18 ] HEK293T or L02 cells were cotransfected with the indicated plasmids for 24 hours and lysed with IP buffer. After an ultrasonic bath and centrifugation (12,000g for 5 minutes), supernatants were incubated with protein A/G agarose beads (catalog no.: AA104307; Bestchrom, Shanghai, China) and anti‐tag antibody overnight at 4°C. Beads were washed with NaCl buffer and boiled with SDS loading buffer for 15 minutes at 95°C before western blotting. For the mass spectrometry (MS) assay, L02 cells were transfected with indicated plasmid and subjected to HR experiments. Samples were prepared according to the description above, and 12% of the gel was used for electrophoresis. Liquid chromatography with tandem MS (LC‐MS/MS) analysis was performed on the gel after silver staining according to the Pierce Silver Stain Kit (24600; Thermo Fisher, Rockford, IL), according to the manufacturer’s instructions.
Ubiquitination assays
L02 cells were cotransfected with the indicated plasmids for 24 hours and lysed with 80 μl of IP buffer and 10 μl of 10% SDS, then heated at 95°C for 15 minutes. After 10× dilution with cold buffer, the sample preparation was completed according to the above IP methods and then performed western blotting analysis.
Real‐time quantitative PCR
Total RNA was extracted from animal tissues and cells using the TRIzol reagent (Invitrogen). RNA concentration and purity were determined using NanoDrop 2000 (Thermo Fisher Scientific, Madison, WI, USA). RNA (2 µg) was used to synthesize cDNA, and RT‐qPCR assays were performed with specific primers in a Real‐Time PCR System (LightCycler 480 Instrument II; Roche, Basel, Switzerland), according to the manufacturer’s instructions. Primer sequences used are listed in Table S2.
Western blotting analysis
Liver tissues or cell samples were lysed in radioimmunoprecipitation assay lysis buffer (P0013E; Beyotime Biotechnology, Shanghai, China). Next, the supernatant was collected and quantified with a bicinchoninic acid protein assay kit (catalog no.: 23225; Thermo Fisher Scientific, Waltham, MA), and then boiled with SDS loading buffer for 15 minutes at 95°C. Protein samples were separated by SDS‐PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (IPVH00010; Millipore, Billerica, MA). PVDF membranes were incubated at 4°C overnight with primary antibodies, followed by incubation with the corresponding secondary antibodies. A ChemiDoc MP imaging system (Bio‐Rad Laboratories, Hercules, CA) was used to detect protein signals. The antibodies used are listed in Table S3.
RNA sequencing and analysis
For the RNA‐sequencing (RNA‐seq) analysis, total RNA was extracted from RNF5‐Flox and RNF5‐HKO mice live tissue samples. Then, cDNA libraries were constructed; the single‐ended libraries were sequenced using BGISEQ 500. Reads were matched to reference genome sequences (mm10/GRCm38) using HISAT2 software (version 2.1.0), using SamTools (version 1.4) to convert the obtained file into a binary BAM format and using StringTie software (version 1.3.3b) and default parameters to calculate the mapping value per million reads per kilobase fragment of the exon model. DESeq2 software (version 1.2.10) was used to calculate differential gene expression according to the following two criteria: (1) folding change >1.5 and (2) adjusted p value < 0.05.
Hierarchical clustering analysis
The hierarchical clustering analysis was performed to analyze global sample distribution profiles by constructing a clustering tree based on data from RNA‐seq by the hclust function of R‐packet.
Gene set enrichment analysis
Each known Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway or biological process term involving genes from the Gene Ontology (GO) database were defined as a gene set. The Java gene set enrichment analysis (GSEA; version 3.0) platform, using the “Signal2Noise” metric, was performed to analyze gene set enrichment based on the degree of differential expression genes. Gene sets with p values < 0.05 and false discovery rate values < 0.25 were considered statistically significant.
KEGG pathway enrichment analysis
A KEGG pathway enrichment analysis was performed to analyze the differential expression genes through Fisher’s exact test. KEGG pathway annotations were downloaded from the KEGG database, and p values < 0.05 were considered statistically significant enrichment pathways.
Statistical analysis
Data are expressed as the mean ± SD. Normally distributed data between two groups were compared using the Student t test or Mann‐Whitney U test for nonparametric tests. Multiple group comparisons were analyzed using one‐way ANOVA. When data met the conditions of normal distribution, a Bonferroni post hoc test (data with homogeneity of variance) or Tamhane T2 post hoc test (data without homogeneity of variance) was used. A Kruskal‐Wallis nonparametric statistical test was used when the data showed non‐normal distribution. Statistical significance was set at p < 0.05.
RESULTS
RNF5 suppresses hepatocyte apoptosis and inflammation under HR challenge
To investigate the role of RNF5 in HIR, we initially determined whether RNF5 levels changed in mouse liver tissues after HIR. mRNA levels of RNF5 were decreased in a time‐dependent manner after HIR (Figure 1A). Consistent with these findings, protein level of RNF5 was significantly decreased in the group subjected to an HIR operation compared with the sham group (Figure 1B). Moreover, RNF5 mRNA and protein levels were also significantly reduced in hepatocytes in response to HR stimulation compared with normal culture hepatocytes (Figure 1C,D). Subsequently, we constructed the RNF5 knockdown cell line to evaluate the effect of RNF5 knockdown on hepatocyte apoptosis and inflammation after HR stimulation. RT‐PCR confirmed that shRNF5#2 cells had a higher knockdown efficiency of RNF5 and were used for the following experiments (Figure 1E). Results showed that RNF5 knockdown increased the expression of Bax and decreased the expression of Bcl2 at both mRNA and protein levels and increased the level of C‐Caspase‐3 after HR challenge (Figure 1F,G). Furthermore, RNF5 knockdown also aggravated the inflammatory process, as manifested by the activation of NF‐κB signaling pathways and the increase of proinflammatory factors (Il6, Tnf, Il1β, chemokine [C‐X‐C motif] ligand 10 [Cxcl10], Il8, and chemokine [C‐C motif] ligand 2 [Ccl2]) expression after HR challenge (Figure 1H,I). Meanwhile, we constructed an RNF5 overexpression cell line by lentivirus infection and found that RNF5 overexpression had the opposite results compared to RNF5 deficiency after HR challenge (Figure 1J‐N). Additionally, knockdown or overexpression of RNF5 by adenovirus infection in primary hepatocytes also had the opposite effect on inflammatory response and apoptosis (Figure S1A,B). Collectively, these results indicate that RNF5 suppresses hepatocyte apoptosis and inflammation under HR challenge.
FIGURE 1.

Changes and roles of RNF5 in HIR. (A) RT‐PCR analysis of the mRNA levels of RNF5 in WT mice subjected to sham treatment or 1 hour of ischemia followed by reperfusion for 1, 3, 6, or 24 hours (n = 4 per group). (B) Western blotting analysis and quantification of RNF5 proteins in mouse liver after 1 hour of ischemia followed by reperfusion for 6 hours (n = 3 per group). (C) RT‐PCR analysis of the mRNA levels of RNF5 in L02 cells subjected to HR challenge (n = 4 independent experiments). (D) Western blotting analysis and quantification of RNF5 in hepatocytes after HR challenge (n = 3 independent experiments). (E) RT‐PCR analysis of RNF5 expression in shPLKO.1 and shRNF5 cells (n = 3 independent experiments). (F) RT‐PCR analysis and quantification of mRNA expression of Bax, and Bcl2 in shPLKO.1 and shRNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). (G) Western blotting analysis and quantification of the levels of Bax, Bcl2, and C‐Caspase‐3 in the indicated groups after HR challenge (n = 3 independent experiments). (H) Western blotting analysis and quantification of the levels of NF‐κB signaling pathway‐related proteins (p‐IKKβ, p‐p65, and IkBα) in hepatocytes from shPLKO.1 and shRNF5 cells subjected to HR challenge (n = 3 independent experiments). (I) RT‐PCR analysis of the mRNA levels of Il6, Tnf, Il1β, Cxcl10, Il8, and Ccl2 in the indicated groups after HR challenge (n = 3 independent experiments). (J) Western blotting analysis of RNF5 protein levels in hepatocytes infected with Flag‐Ctrl or Flag‐RNF5 lentivirus (n = 3 independent experiments). (K) RT‐PCR analysis and quantification of mRNA expression of Bax and Bcl2 in Flag‐Ctrl and Flag‐RNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). (L) Western blotting analysis and quantification of the levels of Bax, Bcl2, and C‐Caspase‐3 in Flag‐Ctrl and Flag‐RNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). (M) Western blotting analysis and quantification of the levels of NF‐κB signaling pathway‐related proteins (p‐IKKβ, p‐p65, and IkBα) in hepatocytes from Flag‐Ctrl and Flag‐RNF5 cells subjected to HR challenge (n = 3 independent experiments). (N) RT‐PCR analysis of the mRNA levels of Il6, Tnf, Il1β, Cxcl10, Il8, and Ccl2 in Flag‐Ctrl and Flag‐RNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). GAPDH was used as the loading control. Data are shown as the mean ± SD. For statistical analysis, the Student t test, ANOVA, or Kruskal‐Wallis nonparametric statistical test was used (panels A–D, *p < 0.05; **p < 0.01 sham vs. IR/HR; panel E, **p < 0.01 shPLKO.1 vs. shRNF5; panels F–I, *p < 0.05, **p < 0.01 shPLKO.1(H0R0) vs. shPLKO.1(H6R6); # p < 0.05, ## p < 0.01 shPLKO.1 vs. shRNF5; panels K–N, *p < 0.05, **p < 0.01 Flag‐Ctrl (H0R0) vs. Flag‐Ctrl (H6R6); # p < 0.05, ## p < 0.01 Flag‐Ctrl vs. Flag‐RNF5). Ctrl, control; p‐IKKβ, phosphorylated IKKβ; p‐p65, phosphorylated p65
RNF5 deficiency aggravates HIR‐induced liver damage and apoptosis
To investigate the function of RNF5 in HIR, hepatocyte‐specific RNF5‐knockout (herein, RNF5‐HKO) mice and RNF5 Flox/Flox (herein, Flox) control mice were generated and subjected to IR treatment (Figure 2A). Serum AST and ALT levels were comparable between RNF5‐HKO mice and Flox mice under sham conditions, indicating that RNF5 deficiency has no effect on liver injury. However, after HIR, serum ALT and AST levels of RNF5‐HKO mice were significantly higher than those of Flox mice (Figure 2B). Histological analysis showed more‐severe necrosis areas in livers of RNF5‐HKO mice than those in Flox mice (Figure 2C). To examine the function of RNF5 on cell death in HIR, we performed TUNEL staining analysis and found that RNF5 deficiency significantly promoted hepatocyte apoptosis compared to Flox mice (Figure 2D). Furthermore, the results of C‐Caspase‐3 immunohistochemistry (IHC) staining showed that C‐Caspase‐3‐positive cells in the HKO 6‐hour group were significantly greater than in the Flox group (Figure 2E). Compared with Flox mice, RNF5‐deficient mice had down‐regulated antiapoptotic factor B‐cell leukemia/lymphoma (Bcl2) and significantly up‐regulated proapoptotic factors Bcl2‐associated x protein (Bax) and Bcl2‐associated agonist of cell death (Bad) at mRNA levels (Figure 2F). Consistently, RNF5‐deficient mice showed increased protein levels of Bax and C‐Caspase‐3, with decreased levels of Bcl2 and B‐cell lymphoma/leukemia XL gene (Bcl‐XL; Figure 2G). Collectively, these results demonstrate that RNF5 deficiency aggravates HIR‐induced liver damage and apoptosis.
FIGURE 2.

RNF5 deficiency aggravates liver damage and apoptosis in HIR. (A) Western blotting analysis of the levels of RNF5 in RNF5‐HKO mice and RNF5 Flox/Flox mice liver tissue samples (n = 3 per group). (B) Levels of serum ALT and AST in RNF5‐Flox and RNF5‐HKO mice under sham conditions and after 1 hour of ischemia, followed by reperfusion for 3 and 6 hours (n = 8 per group). (C) Representative H&E staining and quantification of necrotic areas of liver tissues from RNF5‐Flox and RNF5‐HKO mice at 3 and 6 hours after reperfusion or sham treatment (n = 6 per group). Scale bar, 200 μm. (D) Representative TUNEL staining of liver tissues from RNF5‐Flox and RNF5‐HKO mice at 3 and 6 hours after reperfusion (n = 4 per group). Scale bar, 100 μm. (E) Representative C‐Caspase‐3 IHC staining of liver tissues from RNF5‐Flox and RNF5‐HKO mice (n = 5–6 per group). Scale bar, 200 μm. (F) RT‐PCR analysis of the mRNA levels of Bad, Bax, and Bcl2 in RNF5‐Flox and RNF5‐HKO mice after reperfusion for 3 and 6 hours (n = 4 per group). (G) Western blotting analysis and quantification of Bax, Bcl2, Bcl‐XL, and C‐Caspase‐3 proteins in RNF5‐Flox and RNF5‐HKO mice after reperfusion for 6 hours (n = 3 per group). GAPDH was used as the loading control. Data are shown as the mean ± SD. For statistical analysis, the Student t test was used (*p < 0.05; **p < 0.01). HPF, high‐power field [Color figure can be viewed at wileyonlinelibrary.com]
RNF5 deficiency aggravates HIR‐induced inflammation
Excessive sterile inflammation plays a significant role in HIR injury.[ 19 ] To examine the function of RNF5 in the inflammation of HIR injury, we analyzed the infiltration of inflammatory cells in liver tissues. Under HIR, the level of infiltrated inflammatory cells was significantly increased in livers of the RNF5‐HKO group compared with Flox group (Figure 3A,B). Additionally, increased expression of proinflammatory genes, including Tnf, Il6, Il1β, and Ccl2, were observed in livers of RNF5‐HKO mice subjected to HIR (Figure 3C). Moreover, activation of NF‐κB signaling was enhanced in RNF5‐HKO mice compared with Flox mice after HIR, as shown by the increased phosphorylation of inhibitory kappa B kinase (IKKβ) and p65 and decreased levels of total inhibitor of kappa B alpha (IkBα; Figure 3D). These findings suggested that RNF5 deficiency aggravates HIR‐induced inflammation.
FIGURE 3.

RNF5 deficiency aggravates inflammation response in HIR. (A,B) IF staining of CD11b‐ and Ly6G‐positive cells (red) of ischemic liver sections of RNF5‐Flox and RNF5‐HKO mice at 3 and 6 hours after reperfusion (n = 4 per group). Scale bar, 100 μm. (C) RT‐PCR analysis of the mRNA levels of Tnf, Il6, Il1β, and Ccl2 in RNF5‐Flox and RNF5‐HKO mice after reperfusion for 3 and 6 hours (n = 4 per group). (D) Western blotting analysis and quantification of NF‐κB signaling‐pathway proteins in RNF5‐Flox and RNF5‐HKO mice after reperfusion for 6 hours (n = 3 per group). GAPDH was used as the loading control. Data are shown as the mean ± SD. For statistical analysis, the Student t test was used (*p < 0.05; **p < 0.01). HPF, high‐power field; p‐IKKβ, phosphorylated IKKβ; p‐p65, phosphorylated p65 [Color figure can be viewed at wileyonlinelibrary.com]
RNF5 overexpression ameliorates liver damage, apoptosis, and inflammation induced by HIR
Considering that RNF5 deficiency can aggravate HIR injury, we generated hepatocyte‐specific RNF5 transgenic (RNF5‐HTG) mice to further confirm the function of RNF5 in IR injury (Figure 4A). RNF5 overexpression did not affect basal liver injury under sham conditions (Figure 4B,C). However, compared with nontransgenic (NTG) controls, RNF5‐HTG mice had reduced serum levels of ALT and AST and necrotic areas after HIR (Figure 4B,C). Additionally, the number of TUNEL‐ and C‐Caspase‐3‐positive cells in the RNF5‐HTG group was significantly less than that in the NTG group (Figure 4D‐E). Consistently, after HIR, RNF5 overexpression significantly down‐regulated proapoptotic factors (Bax and Bad) and up‐regulated the antiapoptotic factor, Bcl2, at mRNA levels (Figure 4F). Furthermore, RNF5 overexpression also decreased the protein levels of C‐Caspase‐3 and Bax and increased the protein levels of Bcl2 and Bcl‐XL (Figure 4G). We next examined the effect of RNF5 overexpression on inflammation during HIR. RNF5‐HTG mice showed less infiltration of CD11b+ and Ly6G+ inflammatory cells, decreased expression levels of Il6, Tnf, Il1β, and Ccl2, and a suppressed NF‐κB signaling compared with NTG mice after IR treatment (Figure 5A‐D). Collectively, these results demonstrated that RNF5 overexpression suppressed liver injury, apoptosis, and inflammation after HIR.
FIGURE 4.

RNF5 overexpression alleviates liver damage and apoptosis in HIR. (A) Western blotting analysis of the level of RNF5 in RNF5‐HTG mice and RNF5‐NTG mice liver tissue samples (n = 3 per group). (B) Levels of serum ALT and AST in RNF5‐NTG and RNF5‐HTG mice under sham treatment and after 1 hour of ischemia followed by reperfusion for 3 and 6 hours (n = 8 per group). (C) Representative H&E staining and quantification of necrotic areas of liver tissue from RNF5‐NTG and RNF5‐HTG mice at 3 and 6 hours after reperfusion or sham treatment (n = 6 per group). Scale bar, 200 μm. (D) Representative TUNEL staining of liver tissues from RNF5‐NTG and RNF5‐HTG mice at 3 and 6 hours after reperfusion (n = 4 per group). Scale bar, 100 μm. (E) Representative C‐Caspase‐3 IHC staining of liver tissues from RNF5‐NTG and RNF5‐HTG mice (n = 5–6 per group). Scale bar, 200 μm. (F) RT‐PCR analysis of mRNA levels of Bad, Bax, and Bcl2 in RNF5‐NTG and RNF5‐HTG mice after reperfusion for 3 and 6 hours (n = 4 per group). (G) Western blotting analysis and quantification of Bax, Bcl2, Bcl‐XL, and C‐Caspase‐3 proteins in RNF5‐NTG and RNF5‐HTG mice after reperfusion for 3 and 6 h (n = 3 per group). GAPDH was used as the loading control. Data are shown as the mean ± SD. For statistical analysis, the Student t test or Mann‐Whitney U test was used (*p < 0.05; **p < 0.01) [Color figure can be viewed at wileyonlinelibrary.com]
FIGURE 5.

RNF5 overexpression inhibits inflammation response in HIR. (A,B) IF staining of CD11b‐ and Ly6G‐positive cells (red) of ischemic liver sections of RNF5‐NTG and RNF5‐HTG mice at 3 and 6 hours after reperfusion (n = 4 per group). Scale bar, 100 μm. (C) RT‐PCR analysis of the mRNA levels of Tnf, Il6, Il1β, and Ccl2 in RNF5‐NTG and RNF5‐HTG mice after reperfusion for 3 and 6 hours (n = 4 per group). (D) Western blotting analysis and quantification of NF‐κB signaling‐pathway proteins in RNF5‐NTG and RNF5‐HTG mice after reperfusion for 6 hours (n = 3 per group). GAPDH was used as the loading control. Data are shown as the mean ± SD. For statistical analysis, the Student t test was used (*p < 0.05; **p < 0.01). HPF, high‐power field; p‐IKKβ, phosphorylated IKKβ; p‐p65, phosphorylated p65 [Color figure can be viewed at wileyonlinelibrary.com]
RNF5 inhibits the ASK1‐JNK‐p38 signaling pathways during HIR
Next, we elucidated the mechanism underlying the protective role of RNF5 during HIR. We performed RNA‐Seq on livers of RNF5‐HKO and Flox mice at 6 hours after HIR. Cluster analysis confirmed that the gene expression pattern was significantly different between RNF5‐HKO and RNF5‐Flox mice (Figure 6A). GSEA of GO analysis showed that RNF5 deficiency systemically activated apoptosis‐ and inflammation‐related pathways and genes (Figure 6B‐D). KEGG analysis revealed that mitogen‐activated protein kinase (MAPK) signaling was the most up‐regulated signaling pathway by RNF5 deficiency, indicating that RNF5 may protect against HIR injury by suppressing MAPK signaling (Figure 6E). Next, we determined the effect of RNF5 on MAPK signaling. We found that the JNK/p38 signaling pathway was significantly activated in hepatocytes of shRNF5 cells compared with PLKO.1 cells, but was significantly inhibited in hepatocytes of Flag‐RNF5 cells compared with Flag‐Ctrl cells after HR challenge (Figure 6F,G). ASK1 is a member of the MAPK family, which can activate downstream JNK/p38 signaling and is the main upstream inducer of HIR.[ 20 ] We found that the phosphorylation levels of ASK1 and JNK/p38 in RNF5‐HKO mice were significantly increased after HIR, but the total levels of ASK1 and JNK/p38 did not change. Opposite results were observed in RNF5‐HTG mice subjected to HIR (Figure 6H,I). However, there was no significant change in total and phosphorylation levels of TGFβ activated kinase 1 and TANK‐binding kinase 1 (Figure S2A,B). These results demonstrate that RNF5 suppresses the ASK1‐JNK‐p38 signaling pathway in response to HIR.
FIGURE 6.

RNF5 inhibits ASK1‐JNK/p38 signaling pathways in HIR. (A) Hierarchical clustering tree analysis of the global sample distribution profiles from RNA‐seq (n = 3 per group). (B) GSEA showed that RNF5 deficiency caused changes in the biological process of cell apoptosis and inflammation (n = 3 per group). (C) Heatmap shown the expression of apoptosis‐related genes in livers of RNF5‐Flox and RNF5‐HKO mice subjected to IR treatment and detected by RNA‐seq analyses (n = 3 per group). (D) Heatmap shown the expression of inflammation‐related genes in livers of RNF5‐Flox and RNF5‐HKO mice subjected to IR treatment and detected by RNA‐seq analyses (n = 3 per group). (E) KEGG enrichment analysis of RNA‐seq data showing the significantly enriched pathways contributing to RNF5 function from RNF5‐Flox and RNF5‐HKO mice subjected to IR treatment (n = 3 per group). (F) Western blotting analysis and quantification of the levels of MAPK signaling pathway‐related proteins (p‐JNK, p‐p38) in PLKO.1 and shRNF5 L02 hepatocytes subjected to HR challenge (n = 3 independent experiments). (G) Western blotting analysis and quantification of the levels of MAPK signaling pathway‐related proteins (p‐JNK, p‐p38) in Flag‐Ctrl and Flag‐RNF5 L02 hepatocytes subjected to HR challenge (n = 3 independent experiments). (H) Western blotting analysis and quantification of ASK1, p‐ASK1, JNK, p‐JNK, p38, and p‐p38 levels from RNF5‐Flox and RNF5‐HKO mice liver after 1 hour of ischemia and reperfusion for 6 hours (n = 3 per group). (I) Western blotting analysis and quantification of ASK1, p‐ASK1, JNK, p‐JNK, p38, and p‐p38 levels from RNF5‐NTG and RNF5‐HTG mice liver after 1 hour of IR for 6 hours (n = 3 per group). GAPDH as the loading control. Data are shown as mean ± SD. For statistical analysis, The Student t test was used (*p < 0.05; **p < 0.01). Abr, ABR activator of RhoGEF and GTPase; Ano6, anoctamin 6; Bak1, BCL2 antagonist/killer 1; Ccr2, C‐C motif chemokine receptor 2; Cxcl2, C‐X‐C motif chemokine ligand 2; Cxcr3, C‐X‐C motif chemokine receptor 3; Cyba, cytochrome B‐245 alpha chain; DEGs, differentially expressed genes; Dock2, dedicator of cytokinesis 2 Fcer1g, Fc epsilon receptor Ig; Fcr, Fc receptor; Hand2, heart and neural crest derivatives expressed 2; Hck, HCK proto‐oncogene, Src family tyrosine kinase; HIF‐1, hypoxia‐inducible factor 1; Itga2, integrin subunit alpha 2; Jak3, Janus kinase 3; Map2k3, mitogen‐activated protein kinase kinase 3; Map3k6, mitogen‐activated protein kinase kinase kinase 6; Map3k14, mitogen‐activated protein kinase kinase kinase 14; Map4k3, mitogen‐activated protein kinase kinase kinase kinase 3; Nckap1l, Nck‐associated protein 1‐like; p‐p38, phosphorylated p38; p‐ASK1, phosphorylated ASK1; p‐JNK, phosphorylated JNK; Pldl, phospholipase D1; Ppara, peroxisome proliferator activated receptor alpha; Ppp2r1b, protein phosphatase 2 scaffold subunit Abeta; Pycard, PYD and CARD domain containing; Relt, RELT TNF receptor; Robo2, roundabout guidance receptor 2; Slc11a1, solute carrier family 11 member 1; Slit2, Slit guidance ligand 2; Spi1, Spi‐1 proto‐oncogene; Tlr, Toll‐like receptor; Tnfai6, TNF‐alpha‐induced protein 6; Tnfrsf1a, TNF receptor superfamily member 1a; Tnfrsf8, TNF receptor superfamily member 8; Tnfrsf12a, TNF receptor superfamily member 12a; Tnfrsf19, TNF receptor superfamily member 19; Tub, tubulin [Color figure can be viewed at wileyonlinelibrary.com]
RNF5 binds to and ubiquitinates PGAM5
To explore the mechanism by which RNF5 regulates ASK1 activity, we performed mass spectrometry assay to search for RNF5‐binding proteins (Figure 7A). Results identified PGAM5 as the candidate that interacted with RNF5, which has been reported to activate ASK1 and its downstream JNK/p38 kinases by dephosphorylation[ 21 ]. Co‐IP analysis confirmed the interaction of RNF5 and PGAM5 (Figure 7B). GST pull‐down assay results confirmed the direct interaction between RNF5 and PGAM5 (Figure 7C). According to mapping results, we found that the N‐terminal domain of RNF5 binds to PGAM5, and the deletion of the transmembrane region (Δ118‐138) of RNF5 seriously affects the binding of RNF5 to PGAM5 (Figure 7D). Then, we found that RNF5 overexpression decreased the level of PGAM5 and inhibited the phosphorylation of ASK1(T838), whereas RNF5 deficiency had the opposite results (Figure 7E,F). Next, considering that RNF5 is an E3 ligase, we investigated whether RNF5 regulated the degradation of PGAM5. Results showed that RNF5 overexpression decreased the PGAM5 protein, and this effect was blocked by treatment with proteasome inhibitor MG132, indicating that RNF5 promoted the proteasome‐dependent PGAM5 degradation (Figure 7G). Furthermore, we examined whether RNF5 ubiquitinates PGAM5. We found that RNF5 can ubiquitinate and degrade PGAM5 through proteasome degradation by inducing K48‐linked ubiquitination (Figure 7H,I). Therefore, our findings showed that RNF5 interacts with PGAM5 and promotes its ubiquitination and degradation.
FIGURE 7.
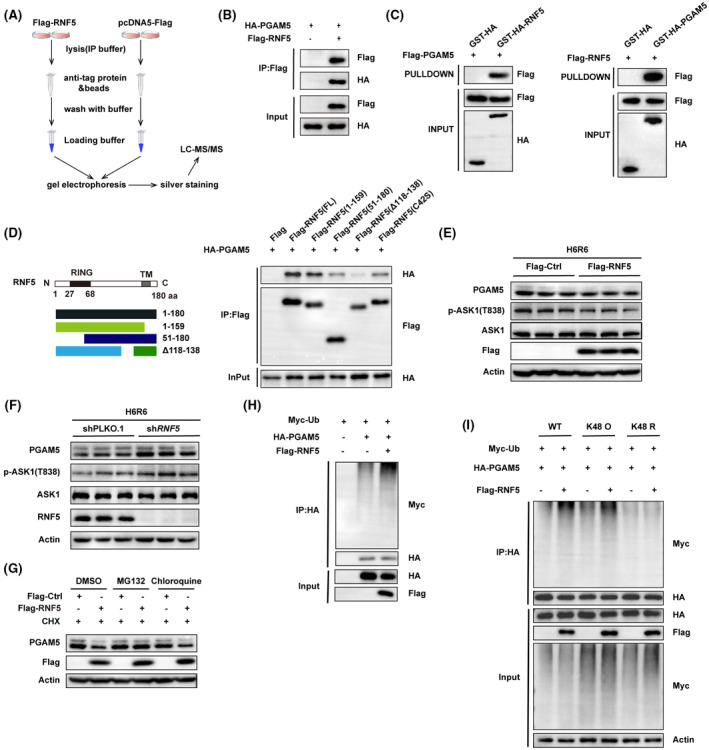
RNF5 directly interacts with and ubiquitinates PGAM5. (A) LC‐MS/MS analysis of the RNF5‐binding proteins from Flag‐Ctrl and Flag‐RNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). (B) Cotransfection of Flag‐RNF5‐expressing and HA‐PGAM5‐expressing plasmid into 293T cells. Co‐IP analysis of the interaction between RNF5 and PGAM5 (n = 3 independent experiments). (C) Interaction between RNF5 and PGAM5 was assayed by GST precipitation, and purified GST‐HA was used as the control (n = 3 independent experiments). (D) HEK293 cells were cotransfected with PGAM5 (HA‐tagged) plasmid and various truncated forms of RNF5 (Flag‐tagged) plasmids. Western blotting of the mapping analysis showing the binding domains of RNF5 to PGAM5 (n = 3 independent experiments). (E) Western blotting analysis of the levels of ASK1, p‐ASK1(T838), PGAM5, and RNF5 proteins in Flag‐Ctrl and Flag‐RNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). (F) Western blotting analysis of the levels of ASK1, p‐ASK1 (T838), PGAM5, and RNF5 proteins in shPLKO.1 and shRNF5 L02 hepatocytes after HR challenge (n = 3 independent experiments). (G) L02 hepatocytes were cotransfected with pcDNA5‐Flag or Flag‐RNF5 and treated with CHX , DMSO, MG132 (25 μmol, 12 hours), or chloroquine (50 μmol, 12 hours). Western blotting analysis of the levels of PGAM5 and RNF5 expression (n = 3 independent experiments). (H) Western blotting analysis of the effect of RNF5 on the ubiquitination level of PGAM5 (n = 3 independent experiments). (I) L02 hepatocytes were cotransfected with Myc‐Ub (WT, K48 O, or K48 R) and HA‐PGAM5 with or without Flag‐RNF5. Western blotting analysis on the ubiquitination level of PGAM5 with an anti‐Myc antibody (n = 3 independent experiments). β‐actin was used as the loading control. CHX, cycloheximide; Ctrl, control; GST, glutathione S‐transferase; HA, hemagglutinin; p‐ASK1, phosphorylated ASK1; TM, tunicamycin; Ub, ubiquitin [Color figure can be viewed at wileyonlinelibrary.com]
Inhibiting ASK1 rescues proinflammatory and ‐apoptosis phenotype in RNF5‐knockdown hepatocytes
To further clarify whether ASK1 mediates the role of RNF5 in HIR, we inhibited ASK1 activation using GS4997, an ASK1 inhibitor, in hepatocytes subjected to HR challenge. Compared with the control group, the phosphorylation of ASK1 and its downstream JNK/p38 significantly decreased in RNF5‐knockdown hepatocytes treated with GS4997 after HR challenge (Figure 8A). Furthermore, results showed that inflammation and apoptosis levels of the GS4997 group were significantly decreased compared to those in the DMSO group after HR challenge (Figure 8B‐E). In addition, we infected primary hepatocytes from wild‐type (WT) mice and RNF5‐KO mice with an ASK1 dominant‐negative mutant adenovirus (AddnASK1) to inhibit ASK1 function. ASK1 and its downstream JNK and p38 phosphorylation results showed that this AddnASK1 performed its function efficiently (Figure S3A). Furthermore, RT‐PCR and western blotting were used to detect the inflammatory response and apoptosis of primary hepatocytes induced by HR stimulation. Consistent with the findings of using ASK1 inhibitor GS4997, AddnASK1 alleviated the inflammatory response and apoptosis caused by RNF5 deficiency (Figure S3B‐E). These results indicate that ASK1 mediates the protective role of RNF5 in HIR.
FIGURE 8.

Inhibition of ASK1 rescues RNF5 deficiency‐induced adverse effects in HIR. (A) Western blotting analysis and quantification of ASK1, p‐ASK1, JNK, p‐JNK, p38, and p‐p38 levels in shPLKO.1 and shRNF5 hepatocytes treated with DMSO or GS4997 before HR challenge. (B) RT‐PCR analysis of the mRNA expression of proinflammatory factors (Il6, Tnf, Il1β, Cxcl10, and Il8) in shPLKO.1 and shRNF5 hepatocytes treated with DMSO or GS4997 before HR challenge. (C) Western blotting analysis of NF‐κB signaling‐related proteins in shPLKO.1 and shRNF5 hepatocytes treated with DMSO or GS4997 before HR challenge. (D) RT‐PCR analysis of the mRNA levels of Bax and Bcl2 in shPLKO.1 and shRNF5 hepatocytes treated with DMSO or GS4997 before HR challenge. (E) Western blotting analysis of apoptosis‐related proteins Bax, Bcl2, and Bcl‐XL and C‐Caspase‐3 in shPLKO.1 and shRNF5 hepatocytes treated with DMSO or GS4997 before HR challenge. GAPDH was used as the loading control. Data are shown as the mean ± SD. For statistical analysis, ANOVA or the Kruskal‐Wallis nonparametric statistical test was used (*p < 0.05; **p < 0.01 shPLKO.1 vs. shRNF5; ## p < 0.01 shRNF5+ GS4997 vs. shRNF5+ DMSO). GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; p‐p38, phosphorylated p38; p‐p65, phosphorylated p65; p‐ASK1, phosphorylated ASK1; p‐JNK, phosphorylated JNK; p‐IKKβ, phosphorylated IKKβ [Color figure can be viewed at wileyonlinelibrary.com]
DISCUSSION
Liver transplantation is the most effective method for the treatment of end‐stage liver disease.[ 22 ] However, almost all liver grafts experience IR injury, which may lead to liver cell death, bile duct stenosis, and graft dysfunction, thus increasing the incidence of acute and chronic rejection after liver transplantation and can result in primary liver graft failure. In this study, we found that RNF5, which was remarkably down‐regulated during HIR, plays a crucial role in the regulation of liver damage in HIR. Importantly, we revealed that RNF5 interacts with PGAM5 and regulates the activation of ASK1 and the downstream JNK/p38 signaling cascade induced by HIR, thereby exerting an overall protective effect against inflammatory response and apoptosis in HIR.
HIR injury occurs in two stages: cell damage during ischemia and inflammation during reperfusion.[ 23 , 24 ] Normally, ischemia damage is easily tolerated by the liver, whereas reperfusion‐induced inflammation is the main cause of liver damage.[ 25 ] Therefore, recent therapeutic studies have focused on the direct inhibition of inflammation and cell death during the reperfusion stage.[ 26 , 27 ] Increasing evidence shows that liver injury could be effectively alleviated by regulating the inflammatory response and cell death during IR. RNF5 is involved in the regulation of viral and bacterial infection processes. RNF5‐KO mice showed more‐severe colitis symptoms, and the lesion‐site changes associated with enhanced inflammation in the colonic tissue included altered proliferation and cell‐death programs.[ 15 ] Consistently, we observed that RNF5‐deficient mice exhibit more‐severe infiltration of inflammatory lymphocytes in liver, up‐regulated inflammatory factor transcription, and enhanced activation of the classical inflammatory signaling pathway, NF‐κB, compared with Flox control mice. However, we also found that RNF5 overexpression can inhibit inflammatory activation by reducing the expression of proinflammatory factors and chemokines and suppressing inflammatory infiltration. These results indicate that RNF5 has a protective effect against HIR injury.
HIR injury is related to apoptosis; during HIR, the main cause of hepatocyte death is proinflammatory cytokine‐mediated apoptosis and reactive oxygen species–induced cell necrosis.[ 28 ] Apoptosis is a major contributor to reperfusion injuries. Because of the shortage of oxygen and energy supply and the depletion of ATP during ischemia, the intracellular environment is changed, and cell apoptosis is triggered. RNF5 regulates the stability and clearance of proteins that function in various cellular processes. Reduced RNF5 expression has been reported to limit the degree of apoptosis in advanced breast cancer cells; RNF5 deficiency enhanced breast cancer apoptosis induced by chemotherapeutic drugs.[ 29 ] Previous studies have shown that apoptosis is a key factor in HIR injury, and reducing apoptosis has been shown to have a hepatoprotective effect.[ 30 ] Consistently, we found that RNF5 can ameliorate HIR injury by inhibiting apoptosis.
PGAM5 is a serine/threonine phosphatase belonging to the PGAM family and is localized in the mitochondrial membrane or ER‐mitochondria interface.[ 31 ] PGAM5 plays an important role in mitochondrial fission,[ 32 ] cell death,[ 33 , 34 , 35 ] cellular senescence,[ 36 ] immune response,[ 37 ] inflammation,[ 38 ] lipid metabolism,[ 39 ] and redox regulation.[ 40 , 41 ] PGAM5 functions by binding to the outer membrane proteins of the mitochondria or cytosolic proteins.[ 41 , 42 , 43 ] In addition, PGAM5 could activate ASK1 by dephosphorylation, resulting in up‐regulated JNK/p38 MAPK signaling.[ 21 ]
MAPK signaling pathways play a role in HIR injury, and their inhibition can alleviate HIR injury.[ 44 , 45 , 46 ] We found that RNF5 overexpression results in decreased levels of PGAM5, decreased ASK1 phosphorylation, and the down‐regulation of JNK/p38 expression. As an E3 ubiquitin ligase, RNF5 can ubiquitinate its substrate. Through ubiquitination experiments, we found that RNF5 can degrade PGAM5 through proteasomal degradation by inducing K48‐linked ubiquitination. Collectively, we revealed that RNF5 plays a protective role in HIR injury by mediating the ubiquitination of PGAM5 to affect the activation of ASK1, thereby inhibiting the activation of its downstream MAPK signaling pathway.
In conclusion, we demonstrated that RNF5 ameliorates hepatic inflammation, suppresses cell apoptosis, and protects against liver damage induced by HIR. The protective role of RNF5 is largely mediated by its interaction with PGAM5 that specifically regulates the ASK1‐JNK/p38 signaling pathway. Our findings provide a theoretical basis for future studies exploring potential therapeutic targets for reducing HIR injury.
CONFLICT OF INTEREST
None of the authors have any potential conflict of interests to disclose.
AUTHOR CONTRIBUTIONS
Ming‐Jie Ding, Hao‐Ran Fang, Jia‐Kai Zhang, Xiao‐Jing Zhang, and Wen‐Zhi Guo. participated in research design. Ming‐Jie Ding, Hao‐Ran Fang, Jia‐Kai Zhang., Ji‐Hua Shi, Pei‐Hao Wen, Zhi‐Hui Wang, Sheng‐Li Cao, Yi Zhang, Xiao‐Yi Shi, Hua‐Peng Zhang, Yu‐Ting He, Bing Yan, Hong‐Wei Tang, Dan‐Feng Guo, Zhen Liu, Li Zhang, and Jie Gao. conducted experiments. All authors performed data analysis and interpretation. Ming‐Jie Ding, Hao‐Ran Fang, and Jia‐Kai Zhang. drafted the manuscript. Xiao‐Jing Zhang and Wen‐Zhi Guo supervised the study, and all authors read and approved the final manuscript.
Supporting information
Supplementary Material
Ding M‐J, Fang H‐R, Zhang J‐K, Shi J‐H, Yu X, Wen P‐H, et al. E3 ubiquitin ligase ring finger protein 5 protects against hepatic ischemia reperfusion injury by mediating phosphoglycerate mutase family member 5 ubiquitination. Hepatology. 2022;76:94–111. 10.1002/hep.32226
Ming‐Jie Ding, Hao‐Ran Fang, and Jia‐Kai Zhang contributed equally to this work.
Funding information
Supported by grants from the National Natural Science Foundation of China (81971881, U2004122); the Foreign Intelligence Introduction Project of Henan Province (GZS2020004); and the Leading talents of scientific and technological innovation in the Central Plains (214200510027)
Contributor Information
Xiao‐Jing Zhang, Email: zhangxjing@whu.edu.cn.
Wen‐Zhi Guo, Email: fccguowz@zzu.edu.cn.
REFERENCES
- 1. Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, et al. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia‐reperfusion. J Exp Med. 2005;201:1135–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bamboat ZM, Balachandran VP, Ocuin LM, Obaid H, Plitas G, DeMatteo RP. Toll‐like receptor 9 inhibition confers protection from liver ischemia‐reperfusion injury. Hepatology. 2010;51:621–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang XJ, Cheng XU, Yan ZZ, Fang J, Wang X, Wang W, et al. An ALOX12‐12‐HETE‐GPR31 signaling axis is a key mediator of hepatic ischemia‐reperfusion injury. Nat Med. 2018;24:73–83. [DOI] [PubMed] [Google Scholar]
- 4. de Rougemont O, Lehmann K, Clavien PA. Preconditioning, organ preservation, and postconditioning to prevent ischemia‐reperfusion injury to the liver. Liver Transpl. 2009;15:1172–82. [DOI] [PubMed] [Google Scholar]
- 5. Kan C, Ungelenk L, Lupp A, Dirsch O, Dahmen U. Ischemia‐reperfusion injury in aged livers‐the energy metabolism, inflammatory response, and autophagy. Transplantation. 2018;102:368–77. [DOI] [PubMed] [Google Scholar]
- 6. Shen X, Reng F, Gao F, Uchida Y, Busuttil RW, Kupiec‐Weglinski JW, et al. Alloimmune activation enhances innate tissue inflammation/injury in a mouse model of liver ischemia/reperfusion injury. Am J Transplant. 2010;10:1729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Abu‐Amara M, Yang SY, Tapuria N, Fuller B, Davidson B, Seifalian A. Liver ischemia/reperfusion injury: processes in inflammatory networks—a review. Liver Transpl. 2010;16:1016–32. [DOI] [PubMed] [Google Scholar]
- 8. Kuang E, Qi J, Ronai Z. Emerging roles of E3 ubiquitin ligases in autophagy. Trends Biochem Sci. 2013;38:453–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Didier C, Broday L, Bhoumik A, Israeli S, Takahashi S, Nakayama K, et al. RNF5, a RING finger protein that regulates cell motility by targeting paxillin ubiquitination and altered localization. Mol Cell Biol. 2003;23:5331–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhong B, Zhang Y, Tan B, Liu TT, Wang YY, Shu HB. The E3 ubiquitin ligase RNF5 targets virus‐induced signaling adaptor for ubiquitination and degradation. J Immunol. 2010;184:6249–55. [DOI] [PubMed] [Google Scholar]
- 11. Younger JM, Chen L, Ren HY, Rosser MFN, Turnbull EL, Fan CY, et al. Sequential quality‐control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell. 2006;126:571–82. [DOI] [PubMed] [Google Scholar]
- 12. Tcherpakov M, Delaunay A, Toth J, Kadoya T, Petroski MD, Ronai ZA. Regulation of endoplasmic reticulum‐associated degradation by RNF5‐dependent ubiquitination of JNK‐associated membrane protein (JAMP). J Biol Chem. 2009;284:12099–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuang E, Okumura CYM, Sheffy‐Levin S, Varsano T, Shu V‐W, Qi J, et al. Regulation of ATG4B stability by RNF5 limits basal levels of autophagy and influences susceptibility to bacterial infection. PLoS Genet. 2012;8:e1003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhong BO, Zhang LU, Lei C, Li Y, Mao AP, Yang Y, et al. The ubiquitin ligase RNF5 regulates antiviral responses by mediating degradation of the adaptor protein MITA. Immunity. 2009;30:397–407. [DOI] [PubMed] [Google Scholar]
- 15. Fujita YU, Khateb A, Li Y, Tinoco R, Zhang T, Bar‐Yoseph H, et al. Regulation of S100A8 stability by RNF5 in intestinal epithelial cells determines intestinal inflammation and severity of colitis. Cell Rep. 2018;24:3296–311.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yan ZZ, Huang YP, Wang X, Wang HP, Ren F, Tian RF, et al. Integrated omics reveals Tollip as an regulator and therapeutic target for hepatic ischemia‐reperfusion injury in mice. Hepatology. 2019;70:1750–69. [DOI] [PubMed] [Google Scholar]
- 17. Wang PX, Zhang R, Huang L, Zhu LH, Jiang DS, Chen HZ, et al. Interferon regulatory factor 9 is a key mediator of hepatic ischemia/reperfusion injury. J Hepatol. 2015;62:111–20. [DOI] [PubMed] [Google Scholar]
- 18. Wang J, Ma J, Nie H, Zhang XJ, Zhang P, She ZG, et al. Hepatic regulator of G protein signaling 5 ameliorates nonalcoholic fatty liver disease by suppressing transforming growth factor beta‐activated kinase 1‐c‐Jun‐N‐terminal kinase/p38 signaling. Hepatology. 2021;73:104–25. [DOI] [PubMed] [Google Scholar]
- 19. Ahmed O, Robinson MW, O’Farrelly C. Inflammatory processes in the liver: divergent roles in homeostasis and pathology. Cell Mol Immunol. 2021;18:1375–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boldorini R, Clemente N, Alchera E, Carini R. DUSP12 acts as a novel endogenous protective signal against hepatic ischemia‐reperfusion damage by inhibiting ASK1 pathway. Clin Sci (Lond). 2021;135:161–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Takeda K, Komuro Y, Hayakawa T, Oguchi H, Ishida Y, Murakami S, et al. Mitochondrial phosphoglycerate mutase 5 uses alternate catalytic activity as a protein serine/threonine phosphatase to activate ASK1. Proc Natl Acad Sci U S A. 2009;106:12301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crismale JF, Ahmad J. Expanding the donor pool: hepatitis C, hepatitis B and human immunodeficiency virus‐positive donors in liver transplantation. World J Gastroenterol. 2019;25:6799–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brenner C, Galluzzi L, Kepp O, Kroemer G. Decoding cell death signals in liver inflammation. J Hepatol. 2013;59:583–94. [DOI] [PubMed] [Google Scholar]
- 24. Ju C, Colgan SP, Eltzschig HK. Hypoxia‐inducible factors as molecular targets for liver diseases. J Mol Med (Berl). 2016;94:613–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van Golen RF, van Gulik TM, Heger M. The sterile immune response during hepatic ischemia/reperfusion. Cytokine Growth Factor Rev. 2012;23:69–84. [DOI] [PubMed] [Google Scholar]
- 26. Datta G, Fuller BJ, Davidson BR. Molecular mechanisms of liver ischemia reperfusion injury: insights from transgenic knockout models. World J Gastroenterol. 2013;19:1683–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125:917–36. [DOI] [PubMed] [Google Scholar]
- 28. Chen K, Li JJ, Li SN, Feng J, Liu T, Wang F, et al. 15‐Deoxy‐Δ(12,14)‐prostaglandin J(2) alleviates hepatic ischemia‐reperfusion injury in mice via inducing antioxidant response and inhibiting apoptosis and autophagy. Acta Pharmacol Sin. 2017;38:672–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jeon Y, Khelifa S, Ratnikov B, Scott D, Feng Y, Parisi F, et al. Regulation of glutamine carrier proteins by RNF5 determines breast cancer response to ER stress‐inducing chemotherapies. Cancer Cell. 2015;27:354–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin FS, Shen SQ, Chen ZB, Yan RC. 17β‐estradiol attenuates reduced‐size hepatic ischemia/reperfusion injury by inhibition apoptosis via mitochondrial pathway in rats. Shock. 2012;37:183–90. [DOI] [PubMed] [Google Scholar]
- 31. Bernkopf DB, Jalal K, Brückner M, Knaup KX, Gentzel M, Schambony A, et al. Pgam5 released from damaged mitochondria induces mitochondrial biogenesis via Wnt signaling. J Cell Biol. 2018;217:1383–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen G, Han Z, Feng DU, Chen Y, Chen L, Wu H, et al. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor‐mediated mitophagy. Mol Cell. 2014;54:362–77. [DOI] [PubMed] [Google Scholar]
- 33. Wang Z, Jiang H, Chen S, Du F, Wang X. The mitochondrial phosphatase PGAM5 functions at the convergence point of multiple necrotic death pathways. Cell. 2012;148:228–43. [DOI] [PubMed] [Google Scholar]
- 34. Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, et al. Oxeiptosis, a ROS‐induced caspase‐independent apoptosis‐like cell‐death pathway. Nat Immunol. 2018;19:130–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He GW, Günther C, Kremer AE, Thonn V, Amann K, Poremba C, et al. PGAM5‐mediated programmed necrosis of hepatocytes drives acute liver injury. Gut. 2017;66:716–23. [DOI] [PubMed] [Google Scholar]
- 36. Yu B, Ma J, Li J, Wang D, Wang Z, Wang S. Mitochondrial phosphatase PGAM5 modulates cellular senescence by regulating mitochondrial dynamics. Nat Commun. 2020;11:2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang YJ, Bang BR, Han KH, Hong L, Shim EJ, Ma J, et al. Regulation of NKT cell‐mediated immune responses to tumours and liver inflammation by mitochondrial PGAM5‐Drp1 signalling. Nat Commun. 2015;6:8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moriwaki K, Farias Luz N, Balaji S, De Rosa MJ, O’Donnell CL, Gough PJ, et al. The mitochondrial phosphatase PGAM5 is dispensable for necroptosis but promotes inflammasome activation in macrophages. J Immunol. 2016;196:407–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sekine S, Yao A, Hattori K, Sugawara S, Naguro I, Koike M, et al. The ablation of mitochondrial protein phosphatase Pgam5 confers resistance against metabolic stress. EBioMedicine. 2016;5:82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu Y, Fang F, Miriyala S, Crooks PA, Oberley TD, Chaiswing L, et al. KEAP1 is a redox sensitive target that arbitrates the opposing radiosensitive effects of parthenolide in normal and cancer cells. Cancer Res. 2013;73:4406–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lo SC, Hannink M. PGAM5 tethers a ternary complex containing Keap1 and Nrf2 to mitochondria. Exp Cell Res. 2008;314:1789–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Panda S, Srivastava S, Li Z, Vaeth M, Fuhs S, Hunter T, et al. Identification of PGAM5 as a mammalian protein histidine phosphatase that plays a central role to negatively regulate CD4(+) T cells. Mol Cell. 2016;63:457–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, et al. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson’s‐like movement disorder. Nat Commun. 2014;5:4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Qin JJ, Mao W, Wang X, Sun P, Cheng D, Tian S, et al. Caspase recruitment domain 6 protects against hepatic ischemia/reperfusion injury by suppressing ASK1. J Hepatol. 2018;69:1110–22. [DOI] [PubMed] [Google Scholar]
- 45. Guo WZ, Fang HB, Cao SL, Chen SY, Li J, Shi JH, et al. Six‐transmembrane epithelial antigen of the prostate 3 deficiency in hepatocytes protects the liver against ischemia‐reperfusion injury by suppressing transforming growth factor‐β‐activated kinase 1. Hepatology. 2020;71:1037–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chen SY, Zhang HP, Li J, Shi JH, Tang HW, Zhang YI, et al. Tripartite motif‐containing 27 attenuates liver ischemia/reperfusion injury by suppressing transforming growth factor β‐activated kinase 1 (TAK1) by TAK1 binding protein 2/3 degradation. Hepatology. 2021;73:738–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material