

Abstract
There is significant interest in the potential for nebulised unfractionated heparin (UFH), as a novel therapy for patients with COVID‐19 induced acute hypoxaemic respiratory failure requiring invasive ventilation. The scientific and biological rationale for nebulised heparin stems from the evidence for extensive activation of coagulation resulting in pulmonary microvascular thrombosis in COVID‐19 pneumonia. Nebulised delivery of heparin to the lung may limit alveolar fibrin deposition and thereby limit progression of lung injury. Importantly, laboratory studies show that heparin can directly inactivate the SARS‐CoV‐2 virus, thereby prevent its entry into and infection of mammalian cells. UFH has additional anti‐inflammatory and mucolytic properties that may be useful in this context.
Methods and intervention
The Can nebulised HepArin Reduce morTality and time to Extubation in Patients with COVID‐19 Requiring invasive ventilation Meta‐Trial (CHARTER‐MT) is a collaborative prospective individual patient data analysis of on‐going randomised controlled clinical trials across several countries in five continents, examining the effects of inhaled heparin in patients with COVID‐19 requiring invasive ventilation on various endpoints.
Each constituent study will randomise patients with COVID‐19 induced respiratory failure requiring invasive ventilation. Patients are randomised to receive nebulised heparin or standard care (open label studies) or placebo (blinded placebo‐controlled studies) while under invasive ventilation. Each participating study collect a pre‐defined minimum dataset. The primary outcome for the meta‐trial is the number of ventilator‐free days up to day 28 day, defined as days alive and free from invasive ventilation.
Keywords: ARDS, COVID‐19, meta‐trial, nebulised heparin, pandemic, randomised study, RCT, respiratory failure, SARS, SARS‐CoV‐2, unfractionated heparin
What is already known about this subject
Unfractionated heparin (UFH) has antiviral, anti‐inflammatory and anticoagulant properties relevant to SARS‐CoV‐2 infection.
In pre‐pandemic clinical trials, nebulised unfractionated heparin limits lung injury progression and the development of ARDS and accelerates recovery in invasively ventilated patients with, or at risk of, acute respiratory distress syndrome.
There is a strong scientific rationale to investigate the therapeutic potential of inhaled nebulised UFH for patients with COVID‐19 who require invasive ventilation.
What this study adds
This meta‐trial is a prospective individual patient data analysis of on‐going randomised phase 1–‐3 clinical trials, testing whether a biologically plausible intervention improves outcomes in COVID‐19 patients requiring invasive ventilation.
The collective goal of this meta‐trial is to reach a conclusion about the efficacy of inhaled UFH in severe COVID‐19 as quickly as possible by pooling information from multiple clinical trials not originally configured as a network.
Individual studies contributing to the meta‐trial are conducted in multiple countries on different continents, which improves the external validity of the results.
1. INTRODUCTION
In December 2019, deaths from a novel coronavirus (severe acute respiratory syndrome coronavirus 2, SARS‐CoV‐2) infection were first reported from Wuhan, China. Coronavirus disease 2019 (COVID‐19) causes acute hypoxaemia in nearly 20% of patients infected, and this constitutes the primary reason for hospitalisation. 1 Of these hospitalised patients, a large number fulfil clinical criteria for acute respiratory distress syndrome (ARDS), with 12–24% requiring invasive mechanical ventilatory support. 2 , 3 , 4 , 5 , 6 , 7 ARDS is an acute condition characterised by a pro‐inflammatory response, leading to pulmonary oedema due to increased pulmonary vascular permeability, and loss of aerated lung tissue, and is seen in up to 23% of mechanically ventilated critically ill patients.
COVID‐19 causes diffuse severe lung injury via a pathophysiologic process involving hyperinflammation, coagulation dysfunction and DNA neutrophil extracellular traps (NETs), which lead to the development of a diffuse alveolar injury with hyaline membranes and microvascular thrombosis. 8 , 9 , 10 Between 35 and 46% of patients with ARDS die in hospital, depending on its severity. 11 , 12 Mortality rates in patients with COVID‐19 requiring invasive ventilation has been reported over a wide range (12–78%). 3 , 13 Systemic corticosteroid therapy has been reported to reduce 28‐day all‐cause mortality in severe COVID‐19 induced critical illness. 14
The scientific rationale and current pre‐clinical and clinical evidence for the use of nebulised unfractionated heparin (UFH) as a treatment for COVID‐19 has been outlined by our group in a comprehensive review article. 8 Nebulised UFH has anticoagulant, anti‐viral, anti‐inflammatory, and mucolytic effects that may be beneficial in reducing the severity of COVID‐19 lung injury. Nebulised delivery of UFH may limit intra‐alveolar fibrin deposition and reduce microvascular thrombosis, both key features of COVID‐19‐induced ARDS. Pre‐clinical studies suggest that UFH exerts anti‐viral effects, inhibiting SARS‐CoV‐2 Spike S1 protein receptor binding at tissue concentrations relevant for administration to humans. UFH binding to the Spike S1 protein induces a conformational change that prevents it from binding to the angiotensin converting enzyme 2 (ACE‐2) receptor. 15 , 16 When the spike protein binds to ACE‐2 on human epithelial cells, heparan sulphate (HS) is required as a co‐receptor, and this process is blocked by UFH, reducing the binding and infectivity of SARS‐CoV‐2 in human bronchial epithelial cells. 17 UFH directly inhibits SARS‐CoV‐2 infection in a dose‐dependent manner, with effects likely at clinically relevant concentrations. Importantly, UFH is more effective than low molecular weight heparins (LMWHs) in this regard. 18 The anti‐inflammatory effects of inhaled UFH may further attenuate pulmonary hyperinflammation and the generation of DNA NETs, each of which is important in the pathogenesis of COVID‐19 lung injury.
Studies in relevant pre‐clinical models of acute lung injury further support the therapeutic potential of inhaled heparin. Early phase clinical studies indicate that nebulised UFH limits alveolar deposition of fibrin, reduces the progression of acute lung injury and facilitates recovery. 19 , 20 , 21 , 22 Nebulised UFH decreased pulmonary dead space, reduced coagulation activation and microvascular thrombosis, attenuated the worsening of the Murray Lung Injury Score and reduced the need for invasive ventilatory support. 19 , 20 , 21 , 22 A recently published randomised double‐blind placebo‐controlled multi‐centre clinical trial of nebulised heparin in 256 patients with or at risk of developing ARDS, found that it reduced progression of lung injury, reduced development of ARDS and accelerated recovery with more survivors at home by Day 60. 23 We also recently reported the safety and efficacy potential of this treatment in 98 hospitalised patients with confirmed COVID‐19. 24
We hypothesise that inhaled nebulised UFH therapy will increase the number of days alive and free from invasive ventilation during the first 28 days in patients with COVID‐19 requiring invasive ventilation. The collective goal of the meta‐trial is to reach a conclusion about the efficacy of inhaled nebulised UFH in COVID‐19 as quickly as possible by pooling information from multiple clinical trials not originally configured as a network. 25 This meta‐trial is designed to closely parallel the approach and methodology of its sister study, INHALE‐HEP, in which the efficacy of nebulised heparin is being tested in patients with COVID‐19 that do not require invasive mechanical ventilation. 26 By design, there is considerable overlap in key methodologic details, with the only major difference being the study population. 26
This manuscript has been prepared in accordance with the Standard Protocol Items: Recommendations for Interventional Trials (SPIRIT) 2013 guideline (see Appendix I). 27
2. OBJECTIVES
The primary objective is to demonstrate that, in patients with severe COVID‐19 requiring invasive ventilation, nebulised UFH therapy increases the number of days alive and free from invasive ventilation during the first 28 days, compared to standard care alone or placebo.
3. STUDY CONCEPT AND DESIGN
The study concept has previously been described in our publication regarding the INHALE‐HEP trial. 26 Briefly, the term “meta‐trial” refers to a prospectively planned pooled analysis of data from multiple ongoing clinical trials, allowing for faster accumulation of data to enable rapid determination of efficacy for relevant clinical endpoints, a key advantage during the pandemic. The meta‐trial approach was recently used in a study investigating the effects of awake prone positioning for COVID‐19 acute hypoxaemic respiratory failure. 28 , 29 Our meta‐trial is designed as a collaborative individual participant prospective data analysis of investigator‐initiated randomised studies of nebulised UFH, either as blinded placebo‐controlled trials or as open‐label and early phase studies of nebulised UFH in addition to standard care compared to standard care alone in invasively ventilated ICU patients with confirmed COVID‐19 infection. 25 , 26 Each individual trial adheres to the highest methodological standards, thus ensuring data quality for the overall meta‐trial. Furthermore, by combining data from multiple trials carried out across different healthcare systems, the meta‐trial may provide evidence that has greater external validity and replicability compared to individual large‐scale trials. The statistical underpinnings of this design have been reported previously. 30
The primary outcome of the meta‐trial is ventilator‐free days during the first 28 days, defined as being alive and free from invasive mechanical ventilation. Individual studies may have other clinical, or biochemical, endpoints as primary outcomes and these are listed in the individual trial protocols.
4. PATIENT AND PUBLIC INVOLVEMENT
The emergent and rapidly evolving nature of the COVID pandemic necessitated that this research programme be developed without patient involvement. Patients were not invited to comment on the study design and were not consulted to develop patient‐relevant outcomes or interpret the results. We will invite patients to help us develop our dissemination strategy and to contribute to the writing or editing of the results manuscript for readability or accuracy. The review manuscript outlining the scientific rationale for this study has been shared with the World Health Organization to increase public access to this information. 8
5. STUDY SETTING
This meta‐trial includes studies of inhaled nebulised UFH in patients with severe COVID‐19 pneumonia who receive invasive ventilation in participating institutions. The list of trial sites for each constituent study will be available in each individual trial record on respective trial registries. Additional studies that meet the criteria for the meta‐trial (similar patient eligibility criteria, similar intervention, similar core set of outcome measures) may be added to this meta‐trial after publication of the meta‐trial protocol manuscript. All included studies agreed to contribute to the meta‐trial before designing and executing their study protocols.
6. STUDY ELIGIBILITY CRITERIA
Individual studies are eligible to be included in this prospective meta‐trial if they meet the following requirements:
Design: Prospective randomised clinical study in intensive care patients who need invasive ventilation for acute respiratory failure related to COVID‐19, confirmed by RT‐PCR, with an intervention group and a control group.
Patients: Inclusion and exclusion criteria as described in Table 1.
Intervention: Inhaled nebulised unfractionated heparin.
Data collection: Able to collect and provide data required for the meta‐trial outcomes.
Ethics: Approval of the protocols and related documents obtained from the relevant Human Research Ethics Committee (HREC) or Institutional Review Board (IRB) prior to the commencement of each individual study. Approval to contribute de‐identified individual patient data to the meta‐trial if required.
TABLE 1.
Patient eligibility criteria for enrolment in studies contributing to CHARTER‐MT
| Inclusion criteria | Age 18 years or older |
| Currently in an intensive care unit (ICU) or scheduled for transfer to the ICU. During the pandemic, critically ill inpatients might be cared for outside of the walls of the usual physical environment of ICU. For this reason, ICU is defined as an area designated for inpatient care of the critically ill where therapies including invasive mechanical ventilation can be provided. | |
| Endotracheal tube in place. | |
| Intubated less than 2 days prior to randomisation. | |
| PaO2 to FiO2 ratio less than or equal to 300 while intubated. | |
| There is a PCR‐positive sample for SARS‐CoV‐2 within the past 21 days. The sample can be a nasal or pharyngeal swab, sputum, tracheal aspirate, bronchoalveolar lavage, or another sample from the patient. | |
| Exclusion criteria | Heparin allergy or heparin‐induced thrombocytopaenia. |
| APTT > 120 seconds, not due to anticoagulant therapy and does not correct with administration of fresh frozen plasma. | |
| Platelet count < 20 × 109 per L. | |
| Pulmonary bleeding or uncontrolled bleeding. | |
| Pregnant or might be pregnant. | |
| Acute brain injury that may result in long‐term disability. | |
| Myopathy, spinal cord injury, or nerve injury or disease with a likely prolonged incapacity to breathe independently, e.g. Guillain‐Barre syndrome. | |
| Death is imminent or inevitable within 24 hours. | |
| Clinician objection. | |
| Refusal of participant (person responsible) consent. |
PCR, polymerase chain reaction; SARS‐CoV‐2, severe acute respiratory syndrome coronavirus 2; APTT, activated partial thromboplastin time; ICU, intensive care unit.
7. RECRUITMENT
The nature of the COVID‐19 pandemic brings significant uncertainty regarding the pace and pattern of patient enrolment, with considerable variation expected across geographic regions and over time. The meta‐trial approach should help address recruitment difficulties that arise in the specific trials. It also allows smaller feasibility/safety trials to be included in this analysis. Patients at study sites who met the inclusion criteria but are not enrolled will be documented, along with the reason for their exclusion.
8. INTERVENTIONS
Participants will be randomly allocated to receive either nebulised UFH in addition to the standard care required, or to receive standard care alone or placebo therapy, depending on the specific trial. Nebulised UFH (25 000 units in 5 mL) will be administered 6‐hourly via an Aerogen Solo vibrating mesh nebuliser (Aerogen, Ireland), while patients receive invasive ventilation in ICU. The duration of the administration of nebulised UFH is described in the individual study protocols. The set‐up of the nebuliser, ventilator circuit and expiratory filter is shown in Figure 1.
FIGURE 1.
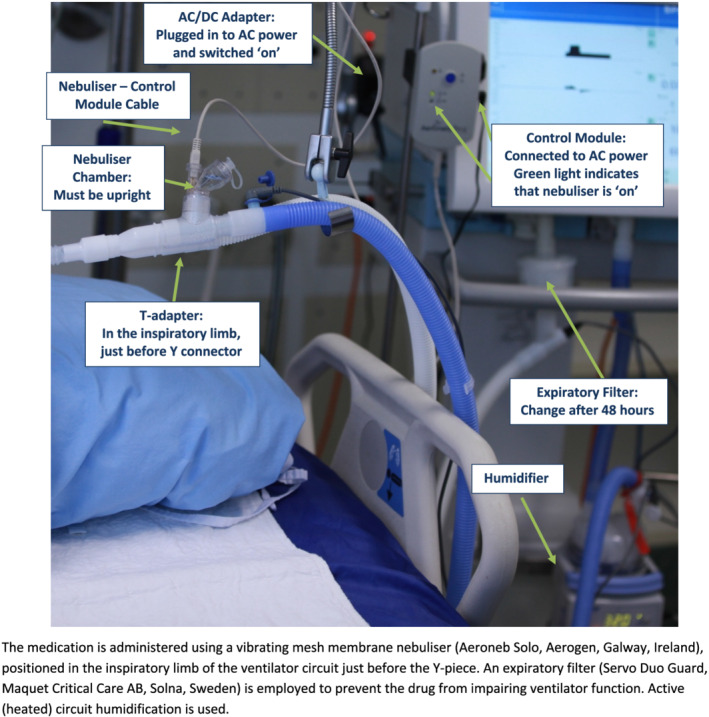
Ventilator circuit and nebuliser set‐up
Participants assigned to “standard care” will receive the standard care required as determined by the treating team while participants assigned to “placebo” will receive nebulised 0.9% sodium chloride (5 mL) administered 6‐hourly via an Aerogen Solo vibrating mesh nebuliser (Aerogen, Ireland), while invasively ventilated.
Reasons and mechanisms for unblinding in blinded prospective randomised controlled trials (PRCTs) will be described in the respective individual trial protocols.
Nebulised UFH (open label study) or any nebulised blinded treatment (blinded PRCT) will be withheld if any of the following occurs:
More than 10 days have elapsed since randomisation.
The patient is outside of ICU.
The patient is not receiving invasive ventilation.
The treating physician deems that there is a clinically unacceptable increase in activated partial thromboplastin time (APTT).
The treating physician deems that there is excessive bloodstaining of respiratory secretions.
There is pulmonary bleeding, major bleeding or suspected or confirmed heparin‐induced thrombocytopaenia (HIT).
Nebulised UFH (open label study) or any nebulised blinded treatment (blinded PRCT) should be recommenced if:
Having been withheld because the patient was outside the ICU, the patient returns to ICU.
Having been withheld because the patient was not invasively ventilated, invasive mechanical ventilation is reinstituted.
Having been withheld because the APTT was unacceptably prolonged, the APTT becomes acceptable.
Having been withheld because there was excessive bloodstaining of respiratory secretions, the bloodstaining of the respiratory secretions has resolved.
Having been withheld for pulmonary bleeding or major bleeding, the bleeding is definitively controlled.
Having been withheld for suspected HIT, the patient is found not to have this condition.
9. RELEVANT CONCOMITANT CARE PERMITTED OR PROHIBITED IN CONTRIBUTING STUDIES
The threshold dose at which inhaled UFH may produce systemic anticoagulation, increasing blood partial thromboplastin time and anti‐factor Xa activity, has been reported to be 150 000 units. 31 At the lower doses used in studies contributing to this meta‐trial, nebulised UFH only modestly increases peak APTT in patients who concomitantly receive intravenous (IV) or subcutaneous (SC) UFH and has no effect on the peak APTT in patients who receive IV or SC low molecular weight heparin (LMWH). 23
The following medications may be used in conjunction with nebulised heparin, subject to local regulatory approval, and do not constitute a reason to withhold study medication: deep vein thrombosis prophylaxis with UFH or LMWH; “full” therapeutic dose UFH or LMWH for a recognised clinical indication; non‐heparin anticoagulants; anti‐thrombotic medications; protamine; prone positioning; and inhaled nitric oxide.
10. PROVISIONS FOR POST‐TRIAL CARE
All enrolled patients will receive standard medical care post‐trial care, based on the care available in that relevant healthcare system and jurisdiction.
11. OUTCOMES
11.1. Primary outcome
The primary outcome is ventilator‐free days during the first 28 days, defined as being alive and free from mechanical ventilation. Non‐survivors to Day 28 are allocated a score of 0, and so counted as though not separated from invasive ventilation. If a patient achieves separation from invasive ventilation more than once, it is the final separation that is used to calculate the outcome.
In this study, ‘Day 0’ describes the period from randomisation to midnight on the day of enrolment, ‘Day 1’ the first calendar day after the day of enrolment, ‘Day 2’ the second calendar day after the day of enrolment, and so forth.
11.2. Secondary outcomes
Secondary efficacy outcomes are as follows:
The Alive and Ventilator Free score at Day 28. This hierarchical endpoint incorporates death and days after successful liberation from invasive ventilation at 28 days in such a manner that death constitutes a worse outcome than prolonged invasive ventilation and has been shown to preserve statistical power while improving face validity. Specifically, Alive and Ventilator Free is less prone to favour a treatment with discordant effects on survival and days free of ventilation. 32
Change in oxygenation index, driving pressure, mechanical power and ventilatory ratio to Day 4.
Change in white cell count, platelet count, C‐reactive protein, D‐dimer and INR to Day 10.
Number treated with neuromuscular blockers instituted after enrolment to Day 10.
Number treated with prone positioning instituted after enrolment to Day 10.
Number treated with extracorporeal membrane oxygenation (ECMO) instituted after enrolment to Day 10.
Number tracheotomised to Day 28.
Time to separation from invasive ventilation to Day 28, among survivors.
Time to separation from the ICU to Day 28, where non‐survivors to Day 28 are treated as though not separated from ICU.
Time to separation from the ICU to Day 28, among survivors.
During the pandemic critically ill inpatients might be cared for outside of the walls of the usual physical environment of ICU. For this reason, “ICU” is defined as an area designated for inpatient care of the critically ill where therapies including invasive mechanical ventilation can be provided.
Mechanical ventilation.
Survival to Day 28; survival to Day 60; and survival to hospital discharge, censored at Day 60.
Number residing at home or in a community setting at Day 60.
Number residing at home or in a community setting at Day 60, among survivors.
11.3. Safety outcomes
Safety outcomes are as follows:
Change in haemoglobin to Day 10.
Number transfused red blood cells (packed red cells and whole blood) to Day 10.
Volume of red blood cells (packed red cells and whole blood) transfused to Day 10.
Number who record major bleeding. Major bleeding is defined as: bleeding that results in death; and/or bleeding that is symptomatic and occurs in a critical area or organ (intra‐cranial, intra‐spinal, intra‐ocular, retroperitoneal, intra‐articular, or intramuscular with compartment syndrome); and/or bleeding that results in a fall in haemoglobin of 20 g/L or more, or results in transfusion of two or more units of whole blood or red cells.
Number who record pulmonary bleeding. Pulmonary bleeding is frank bleeding in the lungs, trachea or bronchi with repeated haemoptysis or requiring repeated suctioning and associated with acute deterioration in respiratory status.
Number who record HIT. HIT is defined as an unexplained fall in platelet count in combination with a positive heparin antibody test.
Number who record other adverse events and reactions. Adverse events and reactions are those that, in the site Principal Investigator's judgement, are not part of the expected clinical course and could be related (at least possibly) to the study and were medically significant or had serious sequelae for the patient.
11.4. Process of care assessments
Process of care assessments are as follows:
Time from intubation to randomisation.
Duration of illness to randomisation.
Total cumulative dose of nebulised heparin in ICU to Day 10.
Days of treatment with nebulised heparin in ICU to Day 10.
Mean APTT in ICU to Day 10 among all participants, and among those treated with IV or SC unfractionated heparin, and among those not treated with IV or SC unfractionated heparin.
Highest APTT in ICU to Day 10 among all participants, and among those treated with IV or SC unfractionated heparin, and among those not treated with IV or SC unfractionated heparin.
Days of treatment with each of the following therapies while in ICU to Day 10: unfractionated heparin, IV and SC; LMWH, IV and SC; warfarin; other anticoagulant therapy; lopinavir‐ritonavir; remdesivir; hydroxychloroquine; interferon‐β; interleukin antagonists; oseltamivir, laninamivir, zaninamivir or peramivir; macrolide; non‐macrolide antibacterial; antifungal; corticosteroid; inotrope or vasopressor infusion; neuromuscular blockade; and renal replacement.
11.5. Other outcomes
Individual studies may have additional and/or different primary and secondary outcomes, and these will be specified prospectively in the individual study protocols.
12. DATA COLLECTION
Trained staff will collect data at each site under the supervision of the site principal investigator using a case report form and data dictionary. Data will be collected at baseline, from Day 0–10 (blood tests, mechanical ventilation variables, therapies while in ICU); on Day 28 and Day 60 (ICU status, ventilation status, vital status, discharge status). The detailed list of collected data items and the schedule for data collection are provided in the individual study protocols.
Paper records, where used, will be stored in locked rooms accessible only to authorised study personnel. Electronic information will be kept on password protected computers accessible only to authorised personnel. Study‐related materials, including case report forms and the study database, will be retained after study conclusion for a minimum period of 15 years. Irrevocable disposal of paper study material will be performed by shredding using a commercial grade shredder or other similar means. Electronic data requiring disposal will be irrevocably erased from electronic media. While each participating centre will maintain a log of enrolled patients that includes patient identifiers, these identifiers are not transferred to the study coordinating centres, but will be retained locally where legally required.
13. DATA SAFETY MONITORING BOARD
The meta‐trial will not have a dedicated independent Data Safety Monitoring Board (DSMB), as this function will be carried out at individual study level where required.
14. DATA MANAGEMENT AND STATISTICAL ANALYSIS PLAN
14.1. Randomisation and allocation concealment
All contributing studies are randomised controlled studies. At randomisation, each participant is assigned to nebulised heparin or standard care in a one‐to‐one allocation ratio. Allocation concealment is performed at the level of each study as specified in the respective study protocols. Most contributing studies are open label by design. The meta‐trial data analysts are blinded. Unblinding is permissible when prespecified Bayesian stopping rules for efficacy or safety have been met.
14.2. Quality assurance monitoring
Conduct and progress of this meta‐trial is monitored on an ongoing basis by the meta‐trial Collaborative Research Group's executive committee.
14.3. Sample size
To demonstrate a clinically important 50% improvement in the hazard of ventilator separation with survival to Day 28 (the primary outcome), a sample of 270 is required. This assumes: 20% overall mortality, with these patients treated as though not separated from the ventilator; 5% overall who survive but fail to achieve ventilator separation; 5% overall withdrawal as might occur due to consent withdrawal; one‐to‐one allocation ratio to the intervention or standard care; power 80%; and two‐sided significance level of 0.05.
These assumptions were informed by an Australian registry that reported intensive care unit mortality of 22% for invasively ventilated patients with COVID‐19. 33 The assumptions are also supported by our pre‐pandemic trial of nebulised heparin in 256 mechanically ventilated intensive care patients. 23
Each individual contributing study may have a different sample size based on their primary outcome, which will have been reported on the individual trial registrations.
14.4. Statistical analysis
14.4.1. Principles
The analysis will mirror closely the analysis plan for the INHALE‐HEP study previously published. 26 This prospective meta‐analysis will be carried out on studies conducted in multiple countries, which increases effect size estimates across different conditions as well as the external validity of the results. We plan a prospective meta‐analysis of individual de‐identified patient‐level data. Common variables from all datasets will be combined to conduct the analysis.
If consent for participation is withdrawn or consent to continue is not given, the data will not be used unless consent to do so is obtained, including for all mortality time points. Analyses will be performed by intention‐to‐treat according to the participants' randomly allocated group, regardless of treatment compliance. These analyses will include participants for whom consent to continue is refused but the use of data already collected is allowed, including the primary outcome, and will exclude patients who do not fulfil the study entry criteria. 34
Missing data will not be imputed. The multilevel models described in the analysis are able to handle missing data due to loss to follow‐up. Where there are missing observations, the number of observations used will be reported. Two‐sided hypothesis testing at a significance level of 0.05 will be used. No adjustment for multiple tests will be made, with the interpretation of the significance of the tests being appropriate for the primary or secondary nature of the outcome. Analyses will be conducted using the Statistical Package for the Social Sciences (SPSS) Research Engine, Version 24.0 IBM SPSS Statistics or later, and “R” version 3.5.0 or later.
14.4.2. Monitoring and interim analyses
We plan to perform regular monitoring and analysis of the accumulating data, with use of Bayesian stopping rules that allow timely decisions without the penalties for multiple data looks and alpha spending associated with the classic randomised controlled trial monitoring approach. 25 , 35 , 36
At the first interim analysis, the prior distribution of the proportion of patients intubated will be multiplied by the likelihood of the observed data to give a posterior distribution of the proportion of patients intubated. At each subsequent interim analysis, the previous posterior distribution becomes the new prior, and a new posterior distribution of the proportion of patients who were intubated will be reported. The pooling of data into the prior distributions and the Bayesian updating of posterior distributions prevent the stopping rule from being overly influenced by potential bias from differential recruitment rates in different trials. Prespecified monitoring criteria will guide the recommendations of the meta‐trial's executive committee. If the probability of a difference in the primary outcome in the two groups rises above 0.90, then the executive committee can recommend that interim analyses be conducted, to support a decision to stop the meta‐trial for efficacy or to submit for publication the interim results. If the probability of a difference falls below 0.10, then the executive committee can recommend that interim analyses be conducted following the methods in the analyses section, to support a decision to stop the meta‐trial for futility.
14.4.3. Trial profile
Patient flow through the meta‐trial will be presented in a Consolidated Standards of Reporting Trials diagram (Figure 2). 37 We will report the number of patients who meet the trial eligibility criteria, the number of patients randomised, and the number of patients in the intention‐to‐treat dataset for whom data are available for evaluation of the primary outcome.
FIGURE 2.
Consolidated Standards of Reporting Trials (CONSORT) diagram for CHARTER‐MT
14.4.4. Participant characteristics and baseline comparisons
Patient characteristics at baseline will be tabulated by treatment group. The categorical variables will be presented as frequency counts (n) and as a proportion of the number of patients with available data (%). Continuous variables will be presented as summary statistics for location (mean or median) and variability (standard deviation or interquartile range). The total counts for variables with missing data will be indicated.
14.4.5. Analyses
Primary outcome
The primary outcome is ventilator‐free days during the first 28 days. Because of the meta‐trial design, we use regression modelling (patients nested in sites nested in trials), with site as a random effect and trial as a fixed effect, along with testing the effect of other covariates as collected in the common variable set. The fixed effect of dose and device will also be estimated across the sites which use different combinations. The fixed effect of study and/or country can also be assessed amongst the trials which use the same dose‐device combination.
We analyse binomial outcomes using multilevel logistic regression, reported as odds ratios and 95% confidence intervals. We analyse time to death using multilevel Cox proportional‐hazards regression, reported as hazard ratios and 95% confidence intervals. The analysis will compare the cause‐specific hazard in the treatment groups using the same multilevel Cox proportional hazards model. Continuous outcomes will be analysed using multilevel linear regression, reported as differences in means and 95% confidence intervals.
Secondary outcomes
Secondary outcomes will be analysed with the same analyses as described for the primary outcome.
14.4.6. Subgroup analyses
We plan to undertake subgroup analyses of the following variables: severity of COVID‐19 (according to the PaO2/FiO2 ratio and the modified ordinal scale), duration of intervention, time from admission to start of intervention, time from development of symptoms to start of intervention, administration of other therapies, age and sex of the patients.
14.4.7. Safety outcomes and adverse events
Adverse events are categorised as “not related”, “unlikely”, “possibly”, “probably” or “definitely related” to treatment, as determined by site investigators. Events will be tabulated by treatment group and reported as frequency counts (n) and proportions (%).
14.4.8. Future analyses
Individual studies contributing to the meta‐trial may be analysed and published separately as per the original protocols of these studies. We will consider conducting hypothesis‐generating exploratory analyses other than those prespecified above to further evaluate the impact of nebulised heparin on outcomes in this dataset. Any such analyses conducted after knowing the main results of the meta‐trial will be cautiously interpreted and clearly indicated in any subsequent publications.
15. ETHICS AND DISSEMINATION
All included studies will be performed in accordance with the ethical principles of the Declaration of Helsinki, with approval from the relevant Human Research Ethics Committee (HREC) or Institutional Review Board (IRB) prior to the commencement of each individual study. These authorisations include data inclusion in the meta‐trial and informing of participants of this data inclusion as per local HREC/IRB requirements. A list of HREC/IRB submissions and approvals of studies contributing to CHARTER MT, current at the time of submission of this manuscript, can be found in Appendix II. The investigators will ensure that all HREC/IRB conditions for the conduct of each study are met and that all requisite information is submitted to the responsible HREC/IRB. Any protocol modifications will be communicated in timely fashion to relevant parties, including investigators and HREC/IRBs.
The individual study protocols outline the process and requirements for obtaining patients' consent to participate in their study and as required by local laws and regulations.
The results of this meta‐trial will be published in peer‐reviewed medical journals and presented to the intensive care community and other stakeholders.
16. STUDY STATUS
At the time of submitting this meta‐trial protocol for publication, several contributing studies are recruiting patients and several studies are in preparation.
17. CONCLUSION
Nebulised UFH has a strong scientific and biological rationale, and warrants urgent investigation of its therapeutic potential, for patients with COVID‐19 requiring invasive mechanical ventilation. This investigator‐initiated international meta‐trial will investigate the efficacy and safety of nebulised UFH in this specific patient population.
17.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.
COMPETING INTERESTS
F.v.H., J.L., D.C., B.M., T.S., Q.N., E.G., A.G., S.S., R.P., R.S., J.S. and B.D. have nothing to declare. A.A. reports a research grant for preclinical research from Grifols and from Fisher&Paykel, and payment as Scientific Advisor for Grifols, outside the submitted work. C.P. reports receiving personal fees from Cipla, Immune Regulation, EpiEndo and Glycosynnovation, and has equity in Verona Pharma, outside of the submitted work.
SPONSOR META‐TRIAL
CHARTER‐MT Collaborative Research Group (CRG). Each individual investigator of every contributing trial is a member of the CHARTER‐MT CRG.
ROLE SPONSOR
The CHARTER‐MT CRG's executive committee, with representatives from each individual country/trial and chaired by the corresponding author of this manuscript, is responsible for the meta‐trial's study design; collection, management, analysis and interpretation of data; writing of the report; and the decision to submit the report for publication. Investigators from individual trials have ownership of their trial data. A data sharing agreement between investigators facilitates and governs the pooling and analysing of de‐identified individual patient data from individual trials, as well as outlines authorship.
ETHICS AND DISSEMINATION
The meta‐trial is registered at ClinicalTrials.gov ID NCT04545541. Each contributing study is individually registered and has received approval of the relevant ethics committee or institutional review board. Results of this study will be shared with the WHO, published in scientific journals, and presented at scientific meetings.
CONTRIBUTORS
F.v.H. drafted the manuscript. F.v.H., J.L., A.A., B.D., C.V. and M.S. are members of the CHARTER MT executive committee and co‐responsible for the meta‐trial protocol. A.R. and H.‐J.H. were responsible for the statistical analysis section, L.v.L. and R.S. were responsible for the data collection section. J.L., D.C. and B.M. contributed the Irish study to the manuscript and meta‐trial, T.S. is the PI for the US study, B.D., R.S., F.v.H., A.G., S.S., R.P. and J.S. contributed the Australian study to the manuscript and meta‐trial. All authors contributed to revision and finalisation of the manuscript. All authors read and approved the final draft for submission.
ACKNOWLEDGEMENT
No funding has been obtained for the meta‐trial. Individual contributing studies/countries are responsible for their own funding. Open access publishing facilitated by Australian National University, as part of the Wiley ‐ Australian National University agreement via the Council of Australian University Librarians.
Appendix I.
A.1. Reporting checklist for protocol of a clinical trial, based on the SPIRIT guidelines
Reporting checklist for protocol of a clinical trial.
Protocol: Can nebulised HepArin Reduce morTality and time to Extubation in Patients with COVID‐19 Requiring mechanical ventilation Meta‐Trial (CHARTER‐MT): Protocol and Statistical Analysis Plan for an investigator‐initiated international meta‐trial of prospective randomised clinical studies.
Based on the SPIRIT guidelines.
A.2. Instructions to authors
Complete this checklist by entering the page numbers from your manuscript where readers will find each of the items listed below.
Your article may not currently address all the items on the checklist. Please modify your text to include the missing information. If you are certain that an item does not apply, please write “n/a” and provide a short explanation.
Upload your completed checklist as an extra file when you submit to a journal.
In your methods section, say that you used the SPIRIT reporting guidelines, and cite them as:
Chan A‐W, Tetzlaff JM, Altman DG, Laupacis A, Gøtzsche PC, Krleža‐Jerić K, Hróbjartsson A, Mann H, Dickersin K, Berlin J, Doré C, Parulekar W, Summerskill W, Groves T, Schulz K, Sox H, Rockhold FW, Rennie D, Moher D. SPIRIT 2013 Statement: Defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200–207.
| Reporting item | Page number | ||
|---|---|---|---|
| Administrative information | |||
| Title | #1 | Descriptive title identifying the study design, population, interventions, and, if applicable, trial acronym | 1 |
| Trial registration | #2a | Trial identifier and registry name. If not yet registered, name of intended registry | 7 |
| Trial registration: Data set | #2b | All items from the World Health Organization trial registration data set | Yes |
| Protocol version | #3 | Date and version identifier | 2 |
| Funding | #4 | Sources and types of financial, material, and other support | 3 |
| Roles and responsibilities: Contributorship | #5a | Names, affiliations, and roles of protocol contributors | 1/2 |
| Roles and responsibilities: Sponsor contact information | #5b | Name and contact information for the trial sponsor | 3 |
| Roles and responsibilities: Sponsor and funder | #5c | Role of study sponsor and funders, if any, in study design; collection, management, analysis, and interpretation of data; writing of the report; and the decision to submit the report for publication, including whether they will have ultimate authority over any of these activities | 3 |
| Roles and responsibilities: Committees | #5d | Composition, roles, and responsibilities of the coordinating centre, steering committee, endpoint adjudication committee, data management team, and other individuals or groups overseeing the trial, if applicable (see item 21a for data monitoring committee) | 3 |
| Introduction | |||
| Background and rationale | #6a | Description of research question and justification for undertaking the trial, including summary of relevant studies (published and unpublished) examining benefits and harms for each intervention | 8–10 |
| Background and rationale: Choice of comparators | #6b | Explanation for choice of comparators | N/A |
| Objectives | #7 | Specific objectives or hypotheses | 10 |
| Trial design | #8 | Description of trial design including type of trial (e.g., parallel group, crossover, factorial, single group), allocation ratio, and framework (e.g., superiority, equivalence, non‐inferiority, exploratory) | 10–11 |
| Methods: Participants, interventions, and outcomes | |||
| Study setting | #9 | Description of study settings (eg, community clinic, academic hospital) and list of countries where data will be collected. Reference to where list of study sites can be obtained | 11 |
| Eligibility criteria | #10 | Inclusion and exclusion criteria for participants. If applicable, eligibility criteria for study centres and individuals who will perform the interventions (e.g., surgeons, psychotherapists) | 11–12 and Table 1 |
| Interventions: Description | #11a | Interventions for each group with sufficient detail to allow replication, including how and when they will be administered | 13 |
| Interventions: Modifications | #11b | Criteria for discontinuing or modifying allocated interventions for a given trial participant (e.g., drug dose change in response to harms, participant request, or improving/worsening disease) | 13–14 |
| Interventions: Adherence | #11c | Strategies to improve adherence to intervention protocols, and any procedures for monitoring adherence (e.g., drug tablet return; laboratory tests) | 18 |
| Interventions: Concomitant care | #11d | Relevant concomitant care and interventions that are permitted or prohibited during the trial | 13–14 |
| Outcomes | #12 | Primary, secondary, and other outcomes, including the specific measurement variable (e.g., systolic blood pressure), analysis metric (e.g., change from baseline, final value, time to event), method of aggregation (e.g., median, proportion), and time point for each outcome. Explanation of the clinical relevance of chosen efficacy and harm outcomes is strongly recommended | 15–18 |
| Participant timeline | #13 | Time schedule of enrolment, interventions (including any run‐ins and washouts), assessments, and visits for participants. A schematic diagram is highly recommended (see Figure 2) | 13 |
| Sample size | #14 | Estimated number of participants needed to achieve study objectives and how it was determined, including clinical and statistical assumptions supporting any sample size calculations | 20 |
| Recruitment | #15 | Strategies for achieving adequate participant enrolment to reach target sample size | 13 |
| Methods: Assignment of interventions (for controlled trials) | |||
| Allocation: Sequence generation | #16a | Method of generating the allocation sequence (e.g., computer‐generated random numbers), and list of any factors for stratification. To reduce predictability of a random sequence, details of any planned restriction (e.g., blocking) should be provided in a separate document that is unavailable to those who enrol participants or assign interventions | 19–20 |
| Allocation concealment mechanism | #16b | Mechanism of implementing the allocation sequence (e.g., central telephone; sequentially numbered, opaque, sealed envelopes), describing any steps to conceal the sequence until interventions are assigned | 20 |
| Allocation: Implementation | #16c | Who will generate the allocation sequence, who will enrol participants, and who will assign participants to interventions | 20 |
| Blinding (masking) | #17a | Who will be blinded after assignment to interventions (e.g., trial participants, care providers, outcome assessors, data analysts), and how | 10, 13 |
| Blinding (masking): Emergency unblinding | #17b | If blinded, circumstances under which unblinding is permissible, and procedure for revealing a participant's allocated intervention during the trial | 13 |
| Methods: Data collection, management, and analysis | |||
| Data collection plan | #18a | Plans for assessment and collection of outcome, baseline, and other trial data, including any related processes to promote data quality (e.g., duplicate measurements, training of assessors) and a description of study instruments (e.g., questionnaires, laboratory tests) along with their reliability and validity, if known. Reference to where data collection forms can be found, if not in the protocol | 19 |
| Data collection plan: Retention | #18b | Plans to promote participant retention and complete follow‐up, including list of any outcome data to be collected for participants who discontinue or deviate from intervention protocols | 19 |
| Data management | #19 | Plans for data entry, coding, security, and storage, including any related processes to promote data quality (e.g., double data entry; range checks for data values). Reference to where details of data management procedures can be found, if not in the protocol | 19 |
| Statistics: Outcomes | #20a | Statistical methods for analysing primary and secondary outcomes. Reference to where other details of the statistical analysis plan can be found, if not in the protocol | 20–21 |
| Statistics: Additional analyses | #20b | Methods for any additional analyses (e.g., subgroup and adjusted analyses) | 21 |
| Statistics: Analysis population and missing data | #20c | Definition of analysis population relating to protocol non‐adherence (e.g., as randomised analysis), and any statistical methods to handle missing data (e.g., multiple imputation) | 23 |
| Methods: Monitoring | |||
| Data monitoring: Formal committee | #21a | Composition of data monitoring committee (DMC); summary of its role and reporting structure; statement of whether it is independent from the sponsor and competing interests; and reference to where further details about its charter can be found, if not in the protocol. Alternatively, an explanation of why a DMC is not needed | 19 |
| Data monitoring: Interim analysis | #21b | Description of any interim analyses and stopping guidelines, including who will have access to these interim results and make the final decision to terminate the trial | 21 |
| Harms | #22 | Plans for collecting, assessing, reporting, and managing solicited and spontaneously reported adverse events and other unintended effects of trial interventions or trial conduct | 17 |
| Auditing | #23 | Frequency and procedures for auditing trial conduct, if any, and whether the process will be independent from investigators and the sponsor | 20 |
| Ethics and dissemination | |||
| Research ethics approval | #24 | Plans for seeking research ethics committee/institutional review board (REC/IRB) approval | 21–22 |
| Protocol amendments | #25 | Plans for communicating important protocol modifications (e.g., changes to eligibility criteria, outcomes, analyses) to relevant parties (e.g., investigators, REC/IRBs, trial participants, trial registries, journals, regulators) | 22 |
| Consent or assent | #26a | Who will obtain informed consent or assent from potential trial participants or authorised surrogates, and how (see item 32) | 22 |
| Consent or assent: Ancillary studies | #26b | Additional consent provisions for collection and use of participant data and biological specimens in ancillary studies, if applicable | N/A |
| Confidentiality | #27 | How personal information about potential and enrolled participants will be collected, shared, and maintained in order to protect confidentiality before, during, and after the trial | 3, 19 |
| Declaration of interests | #28 | Financial and other competing interests for principal investigators for the overall trial and each study site | 3–4 |
| Data access | #29 | Statement of who will have access to the final trial dataset, and disclosure of contractual agreements that limit such access for investigators | 3 |
| Ancillary and post‐trial care | #30 | Provisions, if any, for ancillary and post‐trial care, and for compensation to those who suffer harm from trial participation | 15 |
| Dissemination policy: Trial results | #31a | Plans for investigators and sponsor to communicate trial results to participants, healthcare professionals, the public, and other relevant groups (e.g., via publication, reporting in results databases, or other data sharing arrangements), including any publication restrictions | 22 |
| Dissemination policy: Authorship | #31b | Authorship eligibility guidelines and any intended use of professional writers | 3 |
| Dissemination policy: Reproducible research | #31c | Plans, if any, for granting public access to the full protocol, participant‐level dataset, and statistical code | 3 |
| Appendices | |||
| Informed consent materials | #32 | Model consent form and other related documentation given to participants and authorised surrogates | N/A |
| Biological specimens | #33 | Plans for collection, laboratory evaluation, and storage of biological specimens for genetic or molecular analysis in the current trial and for future use in ancillary studies, if applicable | N/A |
Note: The SPIRIT checklist is distributed under the terms of the Creative Commons Attribution License CC‐BY‐ND 3.0. This checklist can be completed online using https://www.goodreports.org/, a tool made by the EQUATOR Network in collaboration with Penelope.ai
Appendix II.
Trial registration and ethics approvals of studies contributing to CHARTER MT.
Trial registration, ethics submissions and approvals of studies contributing to CHARTER MT (Can nebulised HepArin Reduce morTality and time to Extubation in Patients with COVID‐19 Requiring invasive ventilation Meta‐Trial).
The following list is current at the time of manuscript submission on 6 January 2022.
The Australian study has been approved by the St Vincent's Hospital Melbourne Human Research Ethics Committee under the National Mutual Acceptance scheme in Australia, approval HREC 086/20. Trial registration: ICTRP https://anzctr.org.au/ACTRN12620000517976.aspx
The Frederick Health Hospital study was reviewed and approved by the Frederick Health Hospital Institutional Review Board, approval FHHep518. Trial registration: ClinicalTrials.gov NCT04397510.
The Irish study was reviewed and approved by the National Research Ethics Committee for COVID‐19‐related Health Research (NREC COVID‐19), approval 20‐NREC‐COV‐104. Trial registration: ClinicalTrials.gov NCT04511923.
The United Arab Emirates study is currently under review by the MOHAP Research Ethics Committee (Ministry of Health and Prevention, UAE).
The Coney Island Hospital study is currently under review by the New York City Department of Health and Mental Hygiene's Institutional Review Board.
The Indonesian study is currently under review by the local hospital and university ethics committees.
van Haren FMP, Laffey JG, Artigas A, et al. Can nebulised HepArin Reduce morTality and time to Extubation in patients with COVID‐19 Requiring invasive ventilation Meta‐Trial (CHARTER‐MT): Protocol and statistical analysis plan for an investigator‐initiated international meta‐trial of prospective randomised clinical studies. Br J Clin Pharmacol. 2022;88(7):3272-3287. doi: 10.1111/bcp.15253
The authors confirm that the Principal Investigator for this paper is Professor Frank M.P. van Haren and that he has direct responsibility for the described meta‐trial.
CHARTER Meta‐trial Protocol and Statistical Analysis Plan date and version: 4 January 2022, CHARTER meta‐trial version 3.0, ClinicalTrials.gov ID NCT04545541.
DATA AVAILABILITY STATEMENT
The datasets used for the current manuscript are available from the corresponding author on reasonable request.
REFERENCES
- 1. Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID‐19) outbreak in China: summary of a report of 72 314 cases from the Chinese Center for Disease Control and Prevention. JAMA. 2020;323(13):1239‐1242. doi: 10.1001/jama.2020.2648 [DOI] [PubMed] [Google Scholar]
- 2. Grasselli G, Zangrillo A, Zanella A, et al. Baseline characteristics and outcomes of 1591 patients infected with SARS‐CoV‐2 admitted to ICUs of the Lombardy Region, Italy. JAMA. 2020;323(16):1574‐1581. doi: 10.1001/jama.2020.5394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020;180(7):934–943. doi: 10.1001/jamainternmed.2020.0994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ranieri VM, Rubenfeld GD, Thompson BT, et al. Acute respiratory distress syndrome: the Berlin definition. JAMA. 2012;307(23):2526–2533. doi: 10.1001/jama.2012.5669 [DOI] [PubMed] [Google Scholar]
- 5. Wang D, Hu B, Hu C, et al. Clinical characteristics of 138 hospitalized patients with 2019 novel coronavirus‐infected pneumonia in Wuhan, China. JAMA. 2020;323(11):1061‐1069. doi: 10.1001/jama.2020.1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Richardson S, Hirsch JS, Narasimhan M, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID‐19 in the New York City area. JAMA. 2020;323(20):2052‐2059. doi: 10.1001/jama.2020.6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Petrilli CM, Jones SA, Yang J, et al. Factors associated with hospital admission and critical illness among 5279 people with coronavirus disease 2019 in New York City: prospective cohort study. BMJ. 2020;369:m1966. doi: 10.1136/bmj.m1966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Haren FMP, Page C, Laffey JG, et al. Nebulised heparin as a treatment for COVID‐19: scientific rationale and a call for randomised evidence. Crit Care. 2020;24(1):454. doi: 10.1186/s13054-020-03148-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Agnillo F, Walters KA, Xiao Y, et al. Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID‐19. Sci Transl Med. 2021;13(620):eabj7790. doi: 10.1126/scitranslmed.abj7790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ackermann M, Verleden SE, Kuehnel M, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid‐19. N Engl J Med. 2020;383(2):120‐128. doi: 10.1056/NEJMoa2015432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bellani G, Laffey JG, Pham T, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA. 2016;315(8):788‐800. doi: 10.1001/jama.2016.0291 [DOI] [PubMed] [Google Scholar]
- 12. Thompson BT, Chambers RC, Liu KD. Acute respiratory distress syndrome. N Engl J Med. 2017;377(6):562‐572. doi: 10.1056/NEJMra1608077 [DOI] [PubMed] [Google Scholar]
- 13. Tzotzos SJ, Fischer B, Fischer H, Zeitlinger M. Incidence of ARDS and outcomes in hospitalized patients with COVID‐19: a global literature survey. Crit Care. 2020;24(1):516. doi: 10.1186/s13054-020-03240-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. WHO Rapid Evidence Appraisal for COVID‐19 Therapies (REACT) Working Group , Sterne JAC, Murthy S, Diaz JV, et al. Association between administration of systemic corticosteroids and mortality among critically ill patients with COVID‐19: a meta‐analysis. JAMA. 2020;324(13):1330‐1341. doi: 10.1001/jama.2020.17023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mycroft‐West C, Su D, Elli S, et al. The 2019 coronavirus (SARS‐CoV‐2) surface protein (Spike) S1 Receptor Binding Domain undergoes conformational change upon heparin binding. bioRxiv. 2020.
- 16. Kwon PS, Oh H, Kwon SJ, et al. Sulfated polysaccharides effectively inhibit SARS‐CoV‐2 in vitro. Cell Discov. 2020;6(1):50. doi: 10.1038/s41421-020-00192-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clausen TM, Sandoval DR, Spliid CB, et al. SARS‐CoV‐2 infection depends on cellular heparan sulfate and ACE2. Cell. 2020;183(4):1043‐1057.e15. doi: 10.1016/j.cell.2020.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tree JA, Turnbull JE, Buttigieg KR, et al. Unfractionated heparin inhibits live wild‐type SARS‐CoV‐2 cell infectivity at therapeutically relevant concentrations. Br J Pharmacol. 2021;178(3):626‐635. doi: 10.1111/bph.15304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dixon B, Schultz MJ, Hofstra JJ, Campbell DJ, Santamaria JD. Nebulized heparin reduces levels of pulmonary coagulation activation in acute lung injury. Crit Care. 2010;14(5):445. doi: 10.1186/cc9269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dixon B, Campbell DJ, Santamaria JD. Elevated pulmonary dead space and coagulation abnormalities suggest lung microvascular thrombosis in patients undergoing cardiac surgery. Intensive Care Med. 2008;34(7):1216‐1223. doi: 10.1007/s00134-008-1042-7 [DOI] [PubMed] [Google Scholar]
- 21. Dixon B, Schultz MJ, Smith R, Fink JB, Santamaria JD, Campbell DJ. Nebulized heparin is associated with fewer days of mechanical ventilation in critically ill patients: a randomized controlled trial. Crit Care. 2010;14(5):R180. doi: 10.1186/cc9286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dixon B, Smith R, Santamaria JD, et al. A trial of nebulised heparin to limit lung injury following cardiac surgery. Anaesth Intensive Care. 2016;44(1):28‐33. doi: 10.1177/0310057X1604400106 [DOI] [PubMed] [Google Scholar]
- 23. Dixon B, Smith RJ, Campbell DJ, et al. Nebulised heparin for patients with or at risk of acute respiratory distress syndrome: a multicentre, randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet Respir Med. 2021;9(4):360‐372. doi: 10.1016/S2213-2600(20)30470-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Haren FM, van Loon LM, Steins A, et al. Inhaled nebulised unfractionated heparin for the treatment of hospitalised patients with COVID‐19: a multicentre case series of 98 patients. Br J Clin Pharmacol. 2022. doi: 10.1111/bcp.15212 [DOI] [PubMed] [Google Scholar]
- 25. Petkova E, Antman EM, Troxel AB. Pooling data from individual clinical trials in the COVID‐19 era. JAMA. 2020;324(6):543‐545. doi: 10.1001/jama.2020.13042 [DOI] [PubMed] [Google Scholar]
- 26. van Haren FMP, Richardson A, Yoon H‐J, et al. INHALEd nebulised unfractionated HEParin for the treatment of hospitalised patients with COVID‐19 (INHALE‐HEP): protocol and statistical analysis plan for an investigator‐initiated international metatrial of randomised studies. Br J Clin Pharmacol. 2021;87(8):3075‐3091. doi: 10.1111/bcp.14714 [DOI] [PubMed] [Google Scholar]
- 27. Chan AW, Tetzlaff JM, Altman DG, et al. SPIRIT 2013 statement: defining standard protocol items for clinical trials. Ann Intern Med. 2013;158(3):200‐207. doi: 10.7326/0003-4819-158-3-201302050-00583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li J, Pavlov I, Laffey JG, et al. Meta‐trial of awake prone positioning with nasal high flow therapy: invitation to join a pandemic collaborative research effort. J Crit Care. 2020;60:140‐142. doi: 10.1016/j.jcrc.2020.07.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ehrmann S, Li J, Ibarra‐Estrada M, et al. Lancet Respir Med. 2021;9(12):1387‐1395. doi: 10.1016/S2213-2600(21)00356-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tavernier E, Trinquart L, Giraudeau B. Finding alternatives to the dogma of power based sample size calculation: is a fixed sample size prospective meta‐experiment a potential alternative? PLoS ONE. 2016;11(6):e0158604. doi: 10.1371/journal.pone.0158604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Markart P, Nass R, Ruppert C, et al. Safety and tolerability of inhaled heparin in idiopathic pulmonary fibrosis. J Aerosol Med Pulm Drug Deliv. 2010;23(3):161‐172. doi: 10.1089/jamp.2009.0780 [DOI] [PubMed] [Google Scholar]
- 32. Novack V, Beitler JR, Yitshak‐Sade M, et al. Alive and Ventilator Free: a hierarchical, composite outcome for clinical trials in the acute respiratory distress syndrome. Crit Care Med. 2020;48(2):158‐166. doi: 10.1097/CCM.0000000000004104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burrell AJ, Pellegrini B, Salimi F, et al. Outcomes for patients with COVID‐19 admitted to Australian intensive care units during the first four months of the pandemic. Med J Aust. 2021;214(1):23‐30. doi: 10.5694/mja2.50883 [DOI] [PubMed] [Google Scholar]
- 34. Fergusson D, Aaron SD, Guyatt G, Hebert P. Post‐randomisation exclusions: the intention to treat principle and excluding patients from analysis. BMJ. 2002;325(7365):652‐654. doi: 10.1136/bmj.325.7365.652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lewis RJ, Angus DC. Time for clinicians to embrace their inner Bayesian?: Reanalysis of results of a clinical trial of extracorporeal membrane oxygenation. JAMA. 2018;320(21):2208‐2210. doi: 10.1001/jama.2018.16916 [DOI] [PubMed] [Google Scholar]
- 36. Saville BR, Connor JT, Ayers GD, Alvarez J. The utility of Bayesian predictive probabilities for interim monitoring of clinical trials. Clin Trials. 2014;11(4):485‐493. doi: 10.1177/1740774514531352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Schulz KF, Altman DG, Moher D, The CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomised trials. BMJ. 2010;340(1):c332. doi: 10.1136/bmj.c332 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used for the current manuscript are available from the corresponding author on reasonable request.