

Abstract
Astegolimab is a fully human immunoglobulin G2 monoclonal antibody that binds to the ST2 receptor and blocks the interleukin‐33 signaling. It was evaluated in patients with uncontrolled severe asthma in the phase 2b study (Zenyatta) at doses of 70, 210, and 490 mg subcutaneously every 4 weeks for 52 weeks. This work aimed to characterize astegolimab pharmacokinetics, identify influential covariates contributing to its interindividual variability, and make a descriptive assessment of the exposure‐response relationships. A population pharmacokinetic model was developed using data from 368 patients in the Zenyatta study. Predicted average steady‐state concentration was used in the subsequent exposure‐response analyses, which evaluated efficacy (asthma exacerbation rate) and biomarker end points including forced expiratory volume in 1 second, fraction exhaled nitric oxide, blood eosinophils, and soluble ST2. A 2‐compartment disposition model with first‐order elimination and first‐order absorption best described the astegolimab pharmacokinetics. The relative bioavailability for the 70‐mg dose was 15.3% lower. Baseline body weight, estimated glomerular filtration rate, and eosinophils were statistically correlated with pharmacokinetic parameters, but only body weight had a clinically meaningful influence on the steady‐state exposure (ratios exceeding 0.8‐1.25). The exposure‐response of efficacy and biomarkers were generally flat with a weak trend in favor of the highest dose/exposure. This study characterized astegolimab pharmacokinetics in patients with asthma and showed typical pharmacokinetic behavior as a monoclonal antibody–based drug. The exposure‐response analyses suggested the highest dose tested in the Zenyatta study (490 mg every 4 weeks) performed close to the maximum effect, and no additional response may be expected above it.
Keywords: astegolimab, asthma, exposure‐response, IL‐33, population pharmacokinetics, ST2
Asthma, a chronic respiratory disorder associated with allergic airway inflammation, affects over 300 million people worldwide. 1 , 2 Around 20% to 40% of them have persistent symptoms despite use of controller medications and are categorized as moderate to severe asthma patients. 3 , 4 , 5 , 6 These patients can experience recurrent acute episodes of asthma exacerbations, which can result in use of systemic corticosteroids, a hospital admission, or emergency room visit requiring systemic corticosteroids, 7 and can impose a substantial burden on their life. There remains a significant unmet medical need for new effective treatments to reduce the frequency and severity of asthma exacerbations.
Astegolimab is a fully human monoclonal antibody (mAb) of the immunoglobulin (Ig) G2 subclass that targets the ST2 receptor and blocks interleukin (IL)‐33 signaling. IL‐33, a member of the IL‐1 family of cytokines, 8 is considered an “alarmin” or a damage‐associated molecular pattern molecule, that is constitutively expressed on epithelial cells and released upon cell injury or stress from exposure to exogenous stimuli. IL‐33 activates various immune cells through its receptor ST2, also known as IL1RL1, 9 and typically promotes T‐helper type 2 cell responses mediated by innate and adaptive immune cells that reside in or infiltrate mucosal tissues in the lung. The ST2 receptor has a secreted soluble form (sST2) that arises from alternative splicing, which is elevated in settings of inflammation, and acts as a decoy to bind and inhibit released IL‐33. By blocking inflammation downstream of IL‐33, astegolimab is expected to benefit asthma patients.
The proof of concept for astegolimab was demonstrated in a previously reported randomized, placebo‐controlled, double‐blind, dose‐ranging phase 2b study in patients with severe asthma (Zenyatta study; NCT02918019). 10 Following a single‐blind administration of placebo, 502 patients were randomized to one of the study arms evaluating subcutaneous administrations of placebo (n = 127) or astegolimab as 70 (n = 127), 210 (n = 126), or 490 mg (n = 122) every 4 weeks. The treatment duration was 52 weeks, with an additional 16‐week duration of follow‐up. The selection of doses was determined by 3 factors: achieving an efficacious clinical response, providing a broad range of exposures to assess the dose/exposure‐response relationship, and ensuring patient safety. Based on data from various in vitro assays and inhibition of p38 activation, a downstream marker of IL‐33 signaling, in an ex vivo whole blood stimulation assay conducted in the phase 1 study, the minimum fully efficacious trough concentration was estimated at ≈8 μg/mL (unpublished data). From the pharmacokinetic (PK) results of the phase 1 study, the 70‐mg every‐4‐weeks regimen was predicted to achieve trough concentrations below this threshold and hence be a partially effective dose, whereas the 210‐ and 490‐mg every‐4‐weeks doses were expected to maintain trough concentrations above this threshold and be efficacious dose levels. The highest dose tested (490 mg every 4 weeks) significantly reduced the incidence of asthma exacerbations by 43% compared with placebo, meeting the primary end point of the study. As typically seen in clinical trials targeting patients with asthma, 11 , 12 , 13 , 14 the asthma exacerbation results showed large variability due to rarity of events, and further analyses were warranted to better understand the treatment effect of astegolimab in relation to its PK characteristics.
The objectives of this study were to (1) develop a population PK model to characterize the PK of astegolimab in patients enrolled in the Zenyatta study and to identify clinically relevant intrinsic and extrinsic factors that contribute to its interindividual variability (IIV); and (2) evaluate the exposure‐response relationship of astegolimab on efficacy and biomarker end points, including asthma exacerbation rate, forced expiratory volume in 1 second (FEV1), fraction exhaled nitric oxide (FeNO), blood eosinophils, and sST2, by a descriptive assessment.
Methods
Data and Study Design
The analysis data set included 502 patients with severe asthma who were randomized in the Zenyatta study. 10 To be enrolled in the study, the patients were required to have a history of ≥1 asthma exacerbation within 12 months before screening, and to be receiving medium‐ or high‐dose inhaled corticosteroids therapy and at least 1 additional controller medication. The study was approved by an ethics committee or institutional review board at each trial site and carried out in accordance with the International Conference on Harmonization Guideline for Good Clinical Practice. Informed consent was obtained from all subjects.
The data presented here included the data as of the primary readout. Incidence of asthma exacerbations was the primary efficacy end point; it was defined as new or increased asthma symptoms that resulted in either (1) hospitalization or an emergency department visit with administration of systemic corticosteroid treatment, or (2) treatment with systemic corticosteroids for ≥3 days or a long‐acting depot corticosteroid preparation with a therapeutic effectiveness of ≥3 days. Serum astegolimab concentrations, antidrug antibodies (ADAs), and clinical end points or biomarkers including FEV1, FeNO, blood eosinophils, and sST2 were measured at protocol‐defined time points. One PK sample was taken just before the very first dose, and trough PK samples were to be taken 4, 8, 12, 24, 36, 48, 52, and 68 weeks after the first dosing of the study drug. A subgroup of the patients had additional visits for intense PK sampling to better characterize the PK of astegolimab (48, 46, and 36 patients from the 70, 210, and 490 mg every‐4‐weeks arms, respectively). In this group, additional PK samples were to be taken 3, 7, and 14 days after the very first dose, as well as 3, 7, and 10 days after the seventh dose. Samples to assess ADA were to be taken at baseline and 12, 24, 36, 52, and 68 weeks after the subject's very first dose. Prebronchodilator FEV1 and blood eosinophils were to be measured at baseline and 2, 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, 44, 48, 52, and 68 weeks after the subject's very first dose. Measurements of FeNO were to be made at screening baseline and 2, 4, 12, 24, 36, and 52 weeks after the subject's very first dose, and those of sST2 were scheduled at baseline and 2, 8, 12, 36, and 68 weeks after the subject's first dose. These measurements were also performed at an early‐termination visit in case of early discontinuation.
Total serum astegolimab concentrations were determined using a validated sandwich enzyme‐linked immunosorbent assay, with a lower limit of quantification (LLOQ) of 0.15 μg/mL. The presence of ADAs was determined using a validated homogenous bridging enzyme‐linked immunosorbent assay. Patients were considered to be ADA positive if they were ADA negative at baseline but developed an ADA following study drug administration (treatment‐induced ADA), or if they were ADA positive at baseline and the titer of ≥1 postbaseline samples was at least 4‐fold greater than the titer of the baseline sample (treatment‐enhanced ADA). Otherwise, patients were considered to be ADA negative. Total sST2 in serum were measured as indicator of target binding.
Graphical Exploration of PK Data
Serum astegolimab concentration–time profiles were graphically explored to see the overall tendency of the PK profile in each arm. Plots of serum concentrations (either as absolute or dose‐normalized values) vs time, stratified by the type of observation and dose group, were investigated to inform the subsequent population PK modeling strategy.
Population PK Modeling
Population PK analyses were conducted with nonlinear mixed‐effect modeling using the PK data from all patients with at least 1 PK observation that (1) occurred after the start of treatment (ie, excluding all patients randomly assigned to placebo), and (2) was not below the limit of quantification (BLQ). First, a structural model was developed to describe the overall astegolimab serum concentration‐time profile. First‐order absorption models and 1‐ and 2‐compartmental disposition models with first‐order elimination from the central compartment were evaluated.
IIVs were evaluated on all relevant PK parameters and were, generally, added in an exponential form, assuming the PK parameters were log‐normally distributed:
| (1) |
where TVP is the typical value of the parameter P, Pi is the individual value of the parameter, and ηi is a normally distributed random variable with a mean of 0 and standard deviation of ω. For IIV on relative bioavailability (Frel), a Box‐Cox distribution 15 was considered since deviation from the log‐normal distribution was anticipated. The Box‐Cox distribution was implemented as:
| (2) |
where θBox‐Cox is a shape parameter for the Box‐Cox distribution. Note that this ηBox‐Cox was introduced in the exponent by replacing ηi in Equation (1).
Additive, proportional, and a combination of additive and proportional residual error models were explored to incorporate unexplained residual variability. The model selection was based on inspection of graphical diagnostics and changes in the objective function value (ΔOFV). For nested models, a difference of –3.84 in OFV corresponds to approximately P < .05 for 1 degree of freedom. The model that provided the best description of the data without showing any unacceptable trends in the goodness‐of‐fit plots was referred to as the base model.
Subsequently, covariate model building was conducted using a stepwise covariate modeling (SCM) approach. 16 The SCM was executed in 2 stepwise phases, a forward inclusion phase and a backward elimination phase, where the forward selection P value was set to .01 and the backward elimination P value to .001. The P values were derived from ΔOFVs. In an initial step of SCM, baseline body weight (BWT) was added as a structural covariate to clearance and volume of distribution parameters, separately from the other covariates, with 1 shared coefficient acting on both central volume and peripheral volume, and another shared coefficient on both drug clearance (CL) and intercompartmental clearance. These structural covariate coefficients were included without regard to statistical significance but subsequently were subject to testing of significance in the backward elimination. The 70‐mg dose (vs the other 2 doses combined) was tested as a categorical covariate on Frel and first‐order absorption rate constant based on the observed tendency of slightly lower trough concentrations when compared on the dose‐normalized scale (Figure 1). Other categorical covariates (sex, race, and ADA status) and continuous covariates (baseline age, albumin, alanine aminotransferase, bilirubin, sST2, estimated glomerular filtration rate [eGFR], and eosinophils) were tested on clearance and distribution‐volume parameters.
Figure 1.
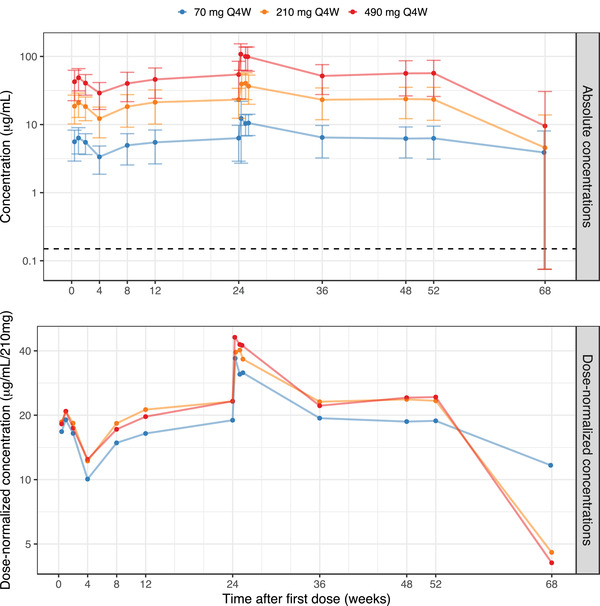
Observed mean astegolimab serum concentrations versus time, based on the PK analysis data set, on a semilogarithmic scale. The upper panel shows absolute concentrations and the lower panel concentrations normalized to the 210‐mg dose. Filled circles represent the mean per dose and visit, with error bars as ± SD (upper panel). All sampling time points within a treatment arm have been connected by a line. Sampling time points are at trough except for the PK profiles after the first dose and after the week 24 dose, as well as the sample at week 68 (20 weeks after the most recent dose). For the week 68 visit the SD was larger than the mean, in all 3 arms, but the lower limits were depicted at LLOQ/2. The horizontal broken line marks the LLOQ (upper panel). LLOQ, lower limit of quantification; Q4W, every 4 weeks; SD, standard deviation.
Relationships for continuous covariates were coded as power models, and for categorical covariates were coded as a fractional difference vs the most common category, as shown in Equations (3) and (4) below, respectively:
| (3) |
| (4) |
where Covref is a reference covariate value for covariate m, to which the covariate model is normalized (median or mode). The total effect of covariates on parameter P was then the product of the n covariate terms:
| (5) |
where for subject i, TVPi is the typical value of the parameter P that will replace TVP in Equation (1), and Ppop is the typical parameter value for a subject with reference covariate values. The covariate model building resulted in the final model.
First‐order conditional estimation with interaction method was used for maximum likelihood estimation. Model evaluation was performed by inspection of graphical diagnostics, including goodness‐of‐fit plots and visual predictive checks (VPCs). In the VPC, data were simulated 2000 times using the doses, PK sampling times, and covariate data from the subjects that were used in the analysis data set. Observations below the LLOQ were included and used in the calculation of the relevant statistics (the median and the 90% prediction intervals). The observed and simulated dependent variable vs time profiles of these statistics were compared graphically. A 95% confidence interval (CI) was included for each of the percentiles in the VPCs.
Exposure‐Response Analysis
Relationships between astegolimab exposure and various efficacy and biomarker end points including annualized asthma exacerbation rate during the 52‐week treatment period, FEV1, FeNO, blood eosinophils, and sST2 were analyzed. Individual predictions of average astegolimab concentration at steady state (Css,av) based on assigned dose and individual PK parameters from the population PK analysis (eg, apparent CL) were used as exposure metrics. All end points were graphically explored by dividing exposure into tertiles: subjects in placebo arm were compared with subjects from the active treatment arms who were grouped in tertiles. The impact of covariates on the exposure‐response relationship for each end point was also evaluated by stratifying graphical output. The covariates of interest were baseline eosinophil level (above or below 150 cells/μL or 300 cells/μL) and IL1RL1 genotype status (positive or negative).
Asthma exacerbation rate was summarized for placebo and each exposure tertile as a mean weighted exacerbation rate based on each individual patient data, which was calculated by taking the total number of exacerbations observed during the treatment period divided by the total patient years at risk on treatment. For the biomarker end points (FEV1, FeNO, blood eosinophil, and sST2), the changes from baseline in biomarkers over time since the first astegolimab dose were first investigated. In addition, to reduce random noise, subjects’ average change from baseline during weeks 8 to 52 was investigated. From week 8 onward, the responses seem to reach steady state. For each subject, the mean change from baseline in this time period was calculated, and this was displayed both as a continuous smooth curve vs astegolimab exposure, and also as means (across the individual means) and 95%CI of the mean, for placebo and each exposure tertile. Since FeNO was not measured at week 8, samples during weeks 12 to 52 have been used for this end point.
The 95%CI of the mean weighted exacerbation rate was calculated using equations for the exact 95%CI of the mean of a Poisson distribution. For the other end points, the mean response for each exposure quartile was calculated using the observed data and the 95%CI of the mean was derived using the mean ± 1.96 × standard error.
Software
Population PK analyses were performed using NONMEM version 7.3.0 (ICON Development Solutions, Hanover, Maryland) assisted by Perl‐speaks NONMEM version 4.8.1 (http://psn.sourceforge.net/). All the graphical analyses were performed using R version 3.3.3 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Graphical Exploration of PK Data
Mean serum astegolimab concentration time‐profiles are shown in Figure 1. Notice that at the follow‐up visit that occurs 20 weeks after the last dose, the concentration values for the majority of patients were below the LLOQ. In Figure 1, the BLQ values have been excluded, which has a profound impact on the calculated mean and standard deviations at this visit.
The exposure of astegolimab increased dose dependently, although in the dose‐normalized scale, the 70‐mg dose showed slightly lower concentrations compared with the other dose groups, in particular for trough samples.
Population PK Model Development
The population PK model of astegolimab was developed on the basis of data from 368 patients with 3113 quantifiable postdose PK observations. Postdose BLQ observations were about 7% of the total postdose PK observations and were mostly from the follow‐up period. Table 1 summarizes the baseline demographic and laboratory covariates that were used in the modeling. A 2‐compartment model with first‐order absorption was selected as the base model: the model included log‐normally distributed IIV on first‐order absorption rate constant and drug CL from the central compartment, whereas IIV in Frel was estimated as a Box‐Cox distribution. The residual error model was parameterized as a combined additive and proportional error model.
Table 1.
Summary of Baseline Demographics and Other Characteristics of the PK Analysis Data Set (n = 368)
| Continuous Covariates | Units | Median (Range) |
|---|---|---|
| Body weight | kg | 79.0 (43.0‐130) |
| BMI | kg/m2 | 28.1 (18.4‐37.0) |
| Age | y | 53.0 (18.0‐75.0) |
| Albumin | g/L | 45.0 (37.0‐54.0) |
| ALT | U/L | 19.0 (7.00‐117) |
| Bilirubin | μmol/L | 7.00 (1.50‐35.0) |
| sST2 | ng/mL | 11.7 (1.56‐222) |
| eGFR | mL/min/1.73 m2 | 87.9 (29.3‐161) |
| Eosinophils | cells/μL | 180 (5.00‐1930) |
| Categorical covariates | N (%) | |
| Dose | ||
| 70 mg every 4 wks | 124 (33.7) | |
| 210 mg every 4 wks | 124 (33.7) | |
| 490 mg every 4 wks | 120 (32.6) | |
| Sex | ||
| Male | 122 (33.2) | |
| Female | 246 (66.8) | |
| Race | ||
| Asian | 17 (4.6) | |
| Black | 20 (5.4) | |
| Native American | 17 (4.6) | |
| Multiple | 5 (1.4) | |
| White | 309 (84.0) | |
| Subject‐level ADA status a | ||
| Negative | 341 (92.7) | |
| Positive | 27 (7.3) |
ADA, antidrug antibody; ALT, alanine aminotransferase; BMI, body mass index; eGFR, estimated glomerular filtration rate; PK, pharmacokinetic; sST2, soluble ST2; wks, weeks.
Patients are considered to be ADA positive if they had either treatment‐induced ADA or treatment‐enhanced ADA; otherwise, they were considered to be ADA negative.
As a result of the SCM, baseline eGFR and blood eosinophil levels on CL and 70‐mg dose on Frel were identified as statistically significant covariates influential for astegolimab PK. Effect of BWT on clearance and distribution‐volume parameters were also retained in the model after the backward elimination step.
The population PK parameter estimates of the final astegolimab PK model are presented in Table 2. The relative bioavailability was found to be 15.3% lower (95%CI, 9.3%‐21.2%) for the 70‐mg dose. Terminal elimination half‐life for the reference subject with the reference covariates (79 kg, baseline eGFR of 87.9 mL/min/1.73 m2, and baseline blood eosinophil level of 180 cell count/μL) corresponds to 19.6 days. Overall, the final model well described the observed serum astegolimab concentration‐time profiles during the treatment period (Figure 2). The impact of the covariates on the steady‐state exposure for 490‐mg every‐4‐weeks dosing of astegolimab is displayed in Figure 3. The results indicate that BWT was the most influential covariate affecting the steady‐state exposure of astegolimab, with relative ratios of 1.510 (95%CI, 1.385‐1.648) and 0.721 (95%CI, 0.674‐0.773) in average concentration, and 1.509 (95%CI, 1.378‐1.652) and 0.722 (95%CI, 0.672‐0.776) in trough concentration, for BWT 52 kg and 110 kg (2.5th and 97.5th percentiles of the BWT in the data set), respectively, when compared to the reference subject (79 kg). Other covariates retained in the final model (baseline eGFR and eosinophils) had mild impact, and relative ratios of the steady‐state exposure were mostly kept within the 0.8 to 1.25 range.
Table 2.
Final Population PK Model Parameter Estimates
| PK Parameter | Unit | Estimate | RSE (%) | SHR (%) |
|---|---|---|---|---|
| ka | day−1 | 0.0437 | 6.03 | |
| CL/F | L/day | 0.244 | 2.57 | |
| Vc/F | L | 0.614 | 9.58 | |
| Vp/F | L | 2.74 | 7.92 | |
| Q/F | L/day | 0.171 | 12.7 | |
| BoxCoxIIV,Frel a | –2.81 | 24.3 | ||
| BWT on CL and Q b | Exponent | 0.986 | 10.5 | |
| BWT on Vtot b | Exponent | 1.02 | 14.9 | |
| BEGFR on CL | Exponent | 0.431 | 19.9 | |
| Dose70mg on F | Rel. change | –0.153 | 19.8 | |
| BEOS on CL | Exponent | 0.0905 | 25.1 | |
| IIVKa | CV | 0.477 | 12.0 | 20.3 |
| IIVCL | CV | 0.224 | 11.4 | 37.4 |
| IIVFrel | SD | 0.243 | 12.3 | 26.1 |
| RUVpropotional | CV | 0.198 | 4.90 | 9.06 |
| RUVadditive | μg/mL | 0.603 | 13.3 | 9.06 |
BEGFR, estimated glomerular filtration rate at baseline; BEOS, blood eosinophil level at baseline; BWT, body weight at baseline; CL, clearance; CV, coefficient of variation; F, bioavailability; Frel, relative bioavailability; IIV, interindividual variability; ka, first‐order absorption rate constant; PK, pharmacokinetic; Q, intercompartmental clearance; RSE, relative standard error; RUV, residual unexplained variability; SHR, shrinkage; Vc, central volume of distribution; Vp, peripheral volume of distribution; Vtot, Vc and Vp.
The RSE for IIV and RUV parameters are reported on the approximate CV scale.
BoxCoxIIV,Frel is the shape parameter in Box‐Cox transformation (Equation (2) in the Methods section) of the eta distribution for Frel. A negative parameter value indicates the distribution is left‐skewed in comparison to a log‐normal IIV distribution.
BWT had a shared coefficient for volumes of distribution (acting on both Vc and Vp), and another shared coefficient for clearances (acting on both CL and Q).
Figure 2.
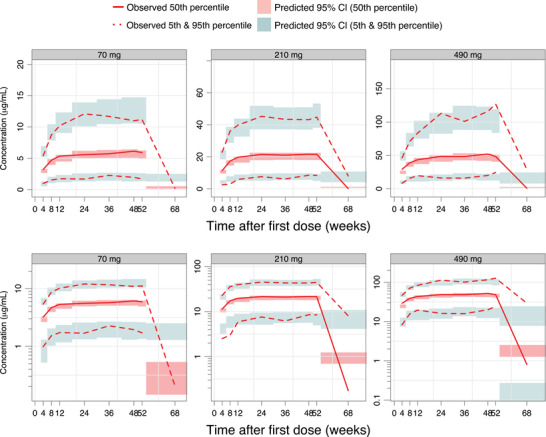
Visual predictive check of astegolimab trough and washout concentrations, for the final PK model, stratified by dose arm. Astegolimab concentrations are displayed versus time after first dose, on a linear scale (upper panels) and on a semilogarithmic scale (bottom panels). The solid and dashed red lines represent the median, 5th and 95th percentiles of the observations; the shaded red and blue areas represent the 95%CI of the median, 5th and 95th percentiles predicted by the model. Observed concentrations below the LLOQ have been included in the calculation of percentiles, but for statistics where the observed percentile is below the LLOQ, the percentile for observed concentration has been excluded from the graph (occurs at follow‐up). LLOQ, lower limit of quantification; PK, pharmacokinetic.
Figure 3.
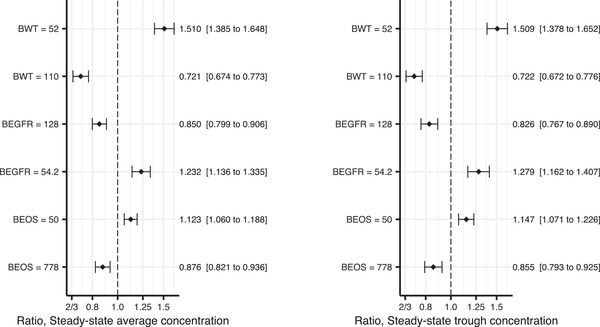
Forest plot showing the final population PK model estimates of covariate effects on the steady‐state exposure for 490‐mg every‐4‐weeks dosing: average concentration in left panel and trough concentration in the right panel. The impact has been shown for the 2.5th and 97.5th percentiles of the covariate (across the subjects in the PK analysis data set), and in relation to the median covariate value. The vertical, dashed line is marking no change (a ratio of 1), compared to the reference patient. The x‐axis is on log scale, since ratios 2/3 and 1.5 are of corresponding impact, just as ratios 0.8 and 1.25 are. The closed symbols represent the point estimates and the whiskers represent the 95%CI, based on the NONMEM covariance matrix (10 000 bootstrap samples). The numbers on the right hand of the plot represent point estimates (95%CIs) of the change in the parameter compared to the reference patient (with baseline values: 79 kg body weight, 87.9 mL/min/1.73 m2 eGFR and 180 eosinophil cell count/μL). BEGFR, estimated glomerular filtration rate at baseline; BEOS, blood eosinophil level at baseline; BWT, body weight at baseline; CL, clearance; Q, intercompartmental clearance; Vc, central volume of distribution; Vp, peripheral volume of distribution. *The effect of body weight was implemented as a shared effect (coefficient) between CL and Q, and between Vc and Vp, respectively.
Exposure‐Response Analysis on Asthma Exacerbation Rate
The mean weighted exacerbation rates were 0.74 (95%CI, 0.60‐0.91), 0.49 (95%CI, 0.38‐0.63), 0.53 (95%CI, 0.41‐0.68), and 0.43 (95%CI, 0.32‐0.56) events/year in the placebo group, exposure low‐, middle‐, and high‐tertile groups, respectively. Slightly lower mean‐weighted exacerbation rates were observed for all the exposure tertile groups with patients on active treatment vs placebo patients as indicated by 95%CIs being completely below the mean rate for placebo (Figure 4). However, among the Css,av tertiles, the CIs overlap, and no clear separation was observed for the weighted mean exacerbation rate among the 3 tertiles in patients on active treatment. Subgroup analyses of the exacerbation rate with the covariates of interest did not identify any trends (data not shown). The results of exacerbation rate vs exposure indicated 28% to 42% reduced exacerbation rate for active groups compared to placebo, while no exposure‐response trend was apparent between the active groups. Modest (8%‐13%) higher treatment effect was observed for the highest‐tertile group in the relative reduction in exacerbation rate compared with those in the exposure low‐ or middle‐tertile groups.
Figure 4.
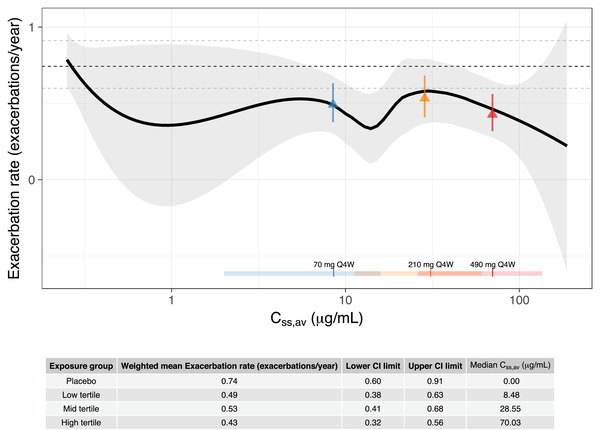
Exacerbation rate versus Css,av on a semilogarithmic scale. The solid line is a loess smooth where the shaded area represents the 95%CI of the smooth. The vertical lines and horizontal bars, at the bottom of the graph, are the median and range (2.5th and 97.5th percentiles) of the distribution of individual predicted Css,av in the 70‐mg every‐4‐weeks (blue), 210‐mg every‐4‐weeks (yellow), and 490‐mg every‐4‐weeks (red) dose groups, respectively. The dashed, black horizontal line shows weighted mean exacerbation rate for the placebo group and the dashed gray lines show the corresponding 95%CI (exact Poisson CI). The triangles show weighted mean exacerbation rate in Css,av low (blue), mid (yellow), and high (red) tertile groups with error bars showing the corresponding 95%CI (exact Poisson CI). Css,av, average concentration at steady state; Q4W, every 4 weeks.
Exposure‐Response Analysis on Biomarkers
The changes from baseline in biomarkers (FEV1, FeNO, blood eosinophil, and sST2) over time since the first astegolimab dose are shown in Figure 5. Separation among placebo and exposure tertiles were observed for FEV1, eosinophil, and sST2, and seems to reach steady state around 8 weeks after the start of treatment. To reduce between‐visit variability, Figure 6 illustrates the overall exposure‐response for (planned) samples during weeks 8 or 12 to 52. The observed changes in FEV1 over time indicates an exposure‐response trend with a slight increase at high exposures. Mean absolute changes in FEV1 during weeks 8 to 52 were 0.11 (95%CI, 0.06‐0.16), 0.12 (95%CI, 0.07‐0.16), 0.16 (95%CI, 0.10‐0.22), and 0.18 (95%CI, 0.11‐0.24) L in the placebo group, exposure low‐, middle‐, and high‐tertile groups, respectively, which showed separation for placebo and the lowest tertile compared with the 2 higher tertiles. A decrease in eosinophil levels over time was indicated with a slight trend of separation between placebo and exposure tertiles. Mean absolute changes in eosinophil levels during weeks 8 to 52 were –8.9 (95%CI, –39 to 21), –59 (95%CI, –89 to –29), –41 (95%CI, –63 to –20), and –57 (95%CI, –81 to –34) cells/μL in the placebo group, exposure low‐, middle‐, and high‐tertile groups, respectively. Treatment effect in the active groups compared with placebo was observed, but without any apparent trend between the exposure tertiles suggesting absence of exposure‐response in the tested exposure range. There was no indication of response to treatment for FeNO. There was an increase in total sST2 over time for all tertiles of Css,av compared with placebo. Mean absolute changes in sST2 levels during weeks 8 to 52 were 0.29 (95%CI, –1.1 to 1.7), 280 (95%CI, 260‐300), 310 (95%CI, 280‐330), and 320 (95%CI, 300‐350) ng/mL in the placebo group, exposure low‐, middle‐, and high‐tertile groups, respectively. Clear accumulation of sST2 was observed only in the active groups as expected, due to formation of antibody‐target complex, and within the tertiles of Css,av there was a trend of higher increase in sST2 for higher Css,av tertiles.
Figure 5.
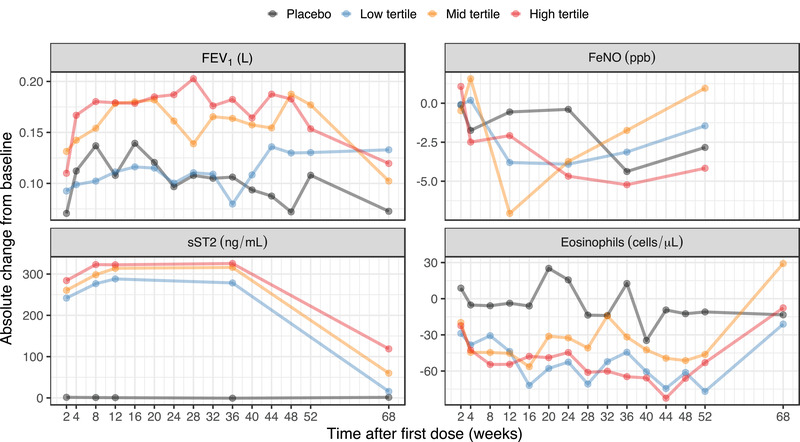
The time course of the biomarkers presented as observed mean absolute change from baseline since first dose, grouped by placebo and tertile of Css,av. Measurements from unscheduled visits are not included in the plot. Css,av, average concentration at steady state; FeNO, fraction exhaled nitric oxide; FEV1, forced expiratory volume in 1 second; sST2, soluble ST2.
Figure 6.
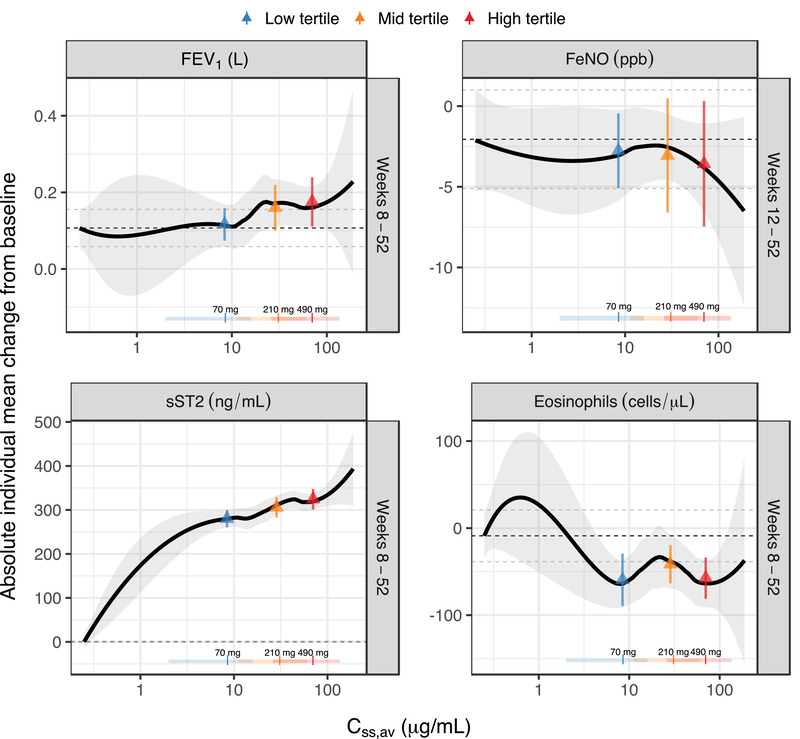
Individual mean absolute FEV1, FeNO, sST2, and eosinophils change from baseline for weeks 8 to 52 (weeks 12‐52 for FeNO, as there was no measurement for week 8), versus Css,av on a semilogarithmic scale. The solid line is a loess smooth where the shaded area represents the 95%CI of the smooth. The vertical lines and horizontal bars, at the bottom of the graph, are the median and range (2.5th and 97.5th percentiles), respectively, of the distribution of individual predicted Css,av for the 70‐ (blue), 210‐ (orange), and 490‐mg (red) every‐4‐weeks regimen. The dashed black horizontal line shows mean change from baseline for the placebo group and the dashed gray lines show the corresponding 95%CI. The triangles show mean change from baseline with error bars showing the corresponding 95%CI. Css,av, average concentration at steady state; FeNO, fraction exhaled nitric oxide; FEV1, forced expiratory volume in 1 second; Q4W, every 4 weeks; sST2, soluble ST2.
Discussion
This is the first study to characterize the PK and evaluate the exposure‐response relationship of a novel mAb therapy targeting ST2 receptor, astegolimab. Astegolimab PK following subcutaneous dosing of 70 to 490 mg every 4 weeks in patients with asthma was described by a 2‐compartment disposition model with first‐order elimination and first‐order absorption; terminal elimination half‐life for the reference subject was estimated to be 19.6 days and was comparable to other humanized antibodies. 17
The individual characteristics identified as having a statistically significant impact on disposition parameters were BWT, eGFR, and eosinophils at baseline. Volumes (central and peripheral) and clearances (drug clearance and intercompartmental clearance) all increased close to proportional with subject's BWT, which is commonly seen in mAb‐based drugs. 18 In addition, clearance decreased with lower eGFR and increased with higher eosinophils at baseline, while the impact was considered not to be clinically meaningful given the mild influence of these covariates on the steady‐state exposure of astegolimab (Figure 3). In general, noncatabolic routes of elimination, such as renal and biliary excretion, are considered to be negligible for the elimination of a mAb. 18 However, eGFR at baseline was identified as a significant covariate for astegolimab in this study. Currently, there is no known physiological mechanism to explain this finding, but it should be noted that there were only a limited number of patients with eGFR <60 mL/min/1.73 m2 (N = 19) or >120 mL/min/1.73 m2 (N = 17) in the Zenyatta study. Since astegolimab is a mAb, the relation to blood eosinophil level at baseline may reflect increased protein turnover with increased inflammatory state. 18 , 19
The population PK model adequately described profiles of astegolimab serum concentrations over time during the treatment period. The concentrations at follow‐up (20 weeks after the subject's last dose) were overpredicted (Figure 2); this may be a consequence of the model not accounting for a potential target mediated drug‐disposition, which would result in a nonlinear elimination of antibody‐based drugs. 17 , 20 , 21 This would also be in line with the lower‐than‐dose‐proportional exposure for the 70‐mg dose compared with the 2 higher doses (Figure 1), which was found to be 15.3% lower (95%CI, 9.3%‐21.2%) in the relative bioavailability in the population PK model. On the other hand, a different extent and/or rate of absorption of the lowest dose may also be related to a smaller injection volume of active drug ingredient at the 70‐mg dose. In the Zenyatta study, each dose of the study drug was administered as four subcutaneous injections, one into each quadrant of the abdomen, one 1‐mL injection and three 2‐mL injections for a total of 7 mL, of either astegolimab solution (70 mg/mL) or placebo. Thus, there were differences in actual injection volumes containing the active drug ingredient. Model diagnostics for the final population PK model otherwise indicated a satisfactory predictive performance and model fit. The parameters were estimated in a good precision (relative standard error <30%) and with acceptable shrinkage (<40%), supporting its suitability to derive individual predictions of exposure during the treatment period for the subsequent exposure‐response analysis.
The graphical exposure‐response analysis of asthma exacerbation rate confirmed the clinically meaningful treatment effect of astegolimab for patients with severe uncontrolled asthma as described by the primary analysis results. 10 However, the analysis was generally not indicative of any apparent exposure‐response trends within the active treatment groups (Figure 4). There was no indication of any significant exposure‐response relationships between exposure tertiles among patients receiving active treatment.
For the biomarkers, close to full treatment effect appeared to be established within 8 to 12 weeks and stayed stable thereafter (Figure 5). Exposure‐response was evident for sST2, which is considered to be the target engagement biomarker of astegolimab. Clear separation between placebo and active groups indicated the binding of astegolimab to its target in blood, and higher exposures led to higher increases in sST2. However, the increments in sST2 response for each exposure tertile (among patients receiving active treatment) was small, which indicates that the target engagements of astegolimab are almost saturated at the observed exposure ranges, at least in the systemic blood circulation. The caveat is that it is unclear if the target engagement is also enough in lung tissue, which is the site of action of the drug. An exposure‐response trend existed also for FEV1, where the lowest exposure tertile had similar response as placebo, but the higher exposure tertiles had a higher response. This is in line with the observation for asthma exacerbation rate and supported the treatment effect of astegolimab to improve lung function. For blood eosinophils, the exposure‐response analysis indicated a slight degree of separation between patients receiving placebo and patients receiving active treatment. However, there was no indication of any significant exposure‐response relationships between exposure tertiles among patients receiving active treatment. For FeNO there was neither separation between patients receiving placebo and patients receiving active treatment nor exposure‐response relationship between exposure tertiles among patients receiving active treatment. This lack of treatment effect and exposure‐response trend in FeNO, a downstream biomarker of type 2 cytokines such as IL‐4 and IL‐13, suggests potential redundancies in this pathway in asthma.
Although the exposure‐safety relationship is another important consideration when informing the dose recommendation of a novel drug, it was not investigated in the current study given the overall favorable safety profile of astegolimab. The clinical safety results of the Zenyatta study 10 suggested there was even no treatment effect of astegolimab on safety; proportion of patients who experienced ≥1 adverse event(s) were similar across all cohorts (77.2%, 70.9%, 72.2%, and 72.1% in placebo; and 70‐mg, 210‐mg, and 490‐mg every‐4‐weeks arms, respectively). Injection site reaction, which was the most common drug‐related adverse event reported more frequently in the astegolimab groups than in the placebo group, was also reported with similar (or even numerically slightly lower with increasing dose level) rates across the three dose levels (7.9%, 6.3%, and 4.9% in 70‐mg, 210‐mg, and 490‐mg every‐4‐weeks arms, respectively). From these results, astegolimab is expected to have no significant safety concern up to 490 mg every 4 weeks.
As described above, separation between the placebo and active treatment groups were present for several end points, but the difference within the exposure tertile‐groups was generally small, and there was no clear tendency of exposure‐response relationships from an efficacy point of view. Considering the available clinical results altogether, the highest dose of 490 mg every 4 weeks may be the recommended dose for further clinical evaluation for the following reasons: (1) astegolimab appears to be safe and was generally well tolerated in the tested dose/exposure range 10 ; (2) exacerbation rates showed large variability across the dose/exposure tested and were numerically better in the highest dose tested; (3) the observed exposure‐response trend in FEV1 is supportive for better lung function improvement in the higher exposure range; and (4) close to maximum target engagement was suggested by the response of sST2 (acknowledging the significant caveat about unknown relationship between sST2 in blood circulation and membrane bound ST2 at the site of action in tissue). Based on the observation that sST2 response almost reached its plateau, no additional response may be expected even if the dose is increased above 490 mg.
Conclusions
In conclusion, this study characterized the PK of astegolimab and showed that it is as expected for a typical mAb in the linear exposure range. The identified covariates were BWT, eGFR, and eosinophils, of which BWT is the most influential on astegolimab PK. None of the covariates are deemed clinically relevant or warrant a dose adjustment given the overall PK variability and the preferential safety profile of astegolimab. Hence, a fixed subcutaneous dose of astegolimab is supported.
The exposure‐response relationships for asthma exacerbation, FEV1, and sST2, albeit a weak trend, was numerically favorable for the highest dose. The highest dose of 490 mg every 4 weeks performed close to the maximum effect, and no additional response may be expected even if the every‐4‐weeks dose of astegolimab were to be increased above this dose level.
Data Sharing Statement
Qualified researchers may request access to individual patient level data through the clinical study data request platform (https://vivli.org/). Further details on Roche's criteria for eligible studies are available here (https://vivli.org/members/ourmembers/). For further details on Roche's Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see here (https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm).
Conflicts of Interest
N.K. is a current employee of Chugai Pharmaceutical Co., Ltd. and was working at Genentech Inc. at the time of this study. M.D. was an employee of Genentech at the time of the study and owned stock in Roche Holding Ltd. (current affiliation: Roche Products Australia, Millers Point, NSW Australia). R.J.S. was an employee of Pharmetheus at the time of the study (current affiliation: the Swedish Medical Products Agency). J.R. is a current employee of Pharmetheus. L.E.F. is a professor at Uppsala University and a paid consultant to Pharmetheus. J.R. and L.E.F. own stocks in Pharmetheus. S.V., D.C., T.S., G.S., and J.J. are current Genentech employees and Roche shareholders. W.S.P. was an employee of Genentech at the time of the study and owned stock in Roche Holding Ltd. (current affiliation: Clinical Pharmacology and DMPK, Ultragenyx Pharmaceutical, Novato, USA). A.Q. was an employee of Genentech at the time of the study and owned stock in Roche Holding Ltd. (current affiliation: Clinical Pharmacology and Quantitative Pharmacology, AstraZeneca, Gothenburg, Sweden).
Funding
This research was sponsored by Genentech Inc. (South San Francisco, California), a member of the Roche group.
Acknowledgments
The authors thank all the patients and their families who participated in the clinical studies, investigators, and staff at the study institutions, and colleagues at Genentech Inc., especially Wiebke Theess and Xiaoying Yang for valuable comments.
*A.Q. was an employee at the time of this study (current affiliation: Clinical Pharmacology and Quantitative Pharmacology, AstraZeneca, Gothenburg, Sweden).
**W.S.P. was an employee at the time of this study (current affiliation: Clinical Pharmacology and DMPK, Ultragenyx Pharmaceutical, Novato, California, USA).
***M.D. was an employee at the time of this study (current affiliation: Roche Products Australia, Millers Point, NSW Australia).
References
- 1. Global strategy for asthma management and prevention, 2020 . Global Initiative for Asthma. www.ginasthma.org. Accessed November 19, 2021.
- 2. To T, Stanojevic S, Moores G, et al. Global asthma prevalence in adults: findings from the cross‐sectional world health survey. BMC Public Health. 2012;12:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Peters SP, Ferguson G, Deniz Y, Reisner C. Uncontrolled asthma: a review of the prevalence, disease burden and options for treatment. Respir Med. 2006;100:1139‐1151. [DOI] [PubMed] [Google Scholar]
- 4. Zeiger RS, Schatz M, Li Q, et al. High blood eosinophil count is a risk factor for future asthma exacerbations in adult persistent asthma. J Allergy Clin Immunol Pract. 2014;2:741‐50. [DOI] [PubMed] [Google Scholar]
- 5. Hermosa JL, Sánchez CB, Rubio MC, Mínguez MM, Walther JL. Factors associated with the control of severe asthma. J Asthma. 2010;47:124‐130. [DOI] [PubMed] [Google Scholar]
- 6. Cazzoletti L, Marcon A, Corsico A, et al. Asthma severity according to Global Initiative for Asthma and its determinants: an international study. Int Arch Allergy Immunol. 2010;151:70‐79. [DOI] [PubMed] [Google Scholar]
- 7. Reddel HK, Taylor DR, Bateman ED, et al. An official American Thoracic Society/European Respiratory Society statement: asthma control and exacerbations: standardizing endpoints for clinical asthma trials and clinical practice. Am J Respir Crit Care Med. 2009;180:59‐99. [DOI] [PubMed] [Google Scholar]
- 8. Sims JE, Smith DE. The IL‐1 family: regulators of immunity. Nat Rev Immunol. 2010;10:89‐102. [DOI] [PubMed] [Google Scholar]
- 9. Nabe T. Interleukin (IL)‐33: new therapeutic target for atopic diseases. J Pharmacol Sci. 2014;126:85‐91. [DOI] [PubMed] [Google Scholar]
- 10. Kelsen SG, Agache IO, Soong W et al. Astegolimab (anti‐ST2) efficacy and safety in adults with severe asthma: a randomized clinical trial. J Allergy Clin Immunol. 2021;148(3):790–798. [DOI] [PubMed] [Google Scholar]
- 11. Hanania NA, Korenblat P, Chapman KR, et al. Efficacy and safety of lebrikizumab in patients with uncontrolled asthma (LAVOLTA I and LAVOLTA II): replicate, phase 3, randomised, double‐blind, placebo‐controlled trials. Lancet Respir Med. 2016;4:781‐796. [DOI] [PubMed] [Google Scholar]
- 12. Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate‐to‐severe uncontrolled asthma. N Engl J Med. 2018;378:2486‐2496. [DOI] [PubMed] [Google Scholar]
- 13. Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377:936‐946. [DOI] [PubMed] [Google Scholar]
- 14. Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198‐1207. [DOI] [PubMed] [Google Scholar]
- 15. Petersson KJ, Hanze E, Savic RM, Karlsson MO. Semiparametric distributions with estimated shape parameters. Pharm Res. 2009;26:2174‐2185. [DOI] [PubMed] [Google Scholar]
- 16. Jonsson EN, Karlsson MO. Automated covariate model building within NONMEM. Pharm Res. 1998;15:1463‐1468. [DOI] [PubMed] [Google Scholar]
- 17. Dostalek M, Gardner I, Gurbaxani BM, Rose RH, Chetty M. Pharmacokinetics, pharmacodynamics and physiologically‐based pharmacokinetic modelling of monoclonal antibodies. Clin Pharmacokinet. 2013;52:83‐124. [DOI] [PubMed] [Google Scholar]
- 18. Dirks NL, Meibohm B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin Pharmacokinet. 2010;49:633‐659. [DOI] [PubMed] [Google Scholar]
- 19. Ordás I, Mould DR, Feagan BG, Sandborn WJ. Anti‐TNF monoclonal antibodies in inflammatory bowel disease: pharmacokinetics‐based dosing paradigms. Clin Pharmacol Ther. 2012;91:635‐646. [DOI] [PubMed] [Google Scholar]
- 20. Peletier LA, Gabrielsson J. Dynamics of target‐mediated drug disposition: characteristic profiles and parameter identification. J Pharmacokinet Pharmacodyn. 2012;39:429‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84:548‐558. [DOI] [PubMed] [Google Scholar]