

Abstract
An expansion of the solvent‐free synthetic toolbox is essential for advances in the sustainable chemical industry. Mechanochemical reactions offer a superior safety profile and reduced amount of waste compared to conventional solvent‐based synthesis. Herein a new mechanochemical method was developed for nucleophilic substitution of alcohols using fluoro‐N,N,N′,N′‐tetramethylformamidinium hexafluorophosphate (TFFH) and K2HPO4 as an alcohol‐activating reagent and a base, respectively. Alcohol activation and reaction with a nucleophile were performed in one milling jar via reactive isouronium intermediates. Nucleophilic substitution with amines afforded alkylated amines in 31–91 % yields. The complete stereoinversion occurred for the SN2 reaction of (R)‐ and (S)‐ethyl lactates. Substitution with halide anions (F−, Br−, I−) and oxygen‐centered (CH3OH, PhO−) nucleophiles was also tested. Application of the method to the synthesis of active pharmaceutical ingredients has been demonstrated.
Keywords: Alcohols, amines, isouronium, mechanochemistry, nucleophilic substitution
Greener amine synthesis: Nucleophilic substitution of alcohols via reactive isouronium salts under mechanochemical one‐jar reaction conditions is presented for the first time. The method opens access towards biologically active amines, including active pharmaceutical ingredients.

Introduction
The synthesis of active pharmaceutical ingredients (APIs) driven by green chemistry concepts plays a pivotal role en route to a cleaner and safer pharmaceutical industry.[ 1 , 2 ] Notably, a great deal of waste and safety concerns commonly originate from the use of solvents, typically responsible for 80–90 % of total mass consumption in a given industrial process. [3] Besides environmental and safety issues, solvents can also generate dangerous contaminants in the produced APIs, such as cancerogenic nitrosamines derived from trace secondary amine impurities in N,N‐dimethylformamide.[ 4 , 5 ] In this regard, mechanochemistry[ 6 , 7 , 8 ] as an essentially solvent‐free technique can beneficially contribute to the ongoing green renovation of the pharma industry.[ 9 , 10 , 11 ] Despite the rapid development of mechanochemical organic synthesis over the past decades,[ 12 , 13 , 14 , 15 ] its synthetic portfolio still does not entirely cover the diverse range of transformations required for APIs production and needs to be expanded.
Nitrogen‐containing functional groups prevail in the existing immense number of bioactive molecules, including APIs. [16] As a consequence, almost 80 % of heteroatom alkylation and arylation reactions used in the synthesis of drug candidates involve the formation of C−N bonds. [17] In addition to the mainstream developments like amide synthesis,[ 18 , 19 , 20 , 21 , 22 , 23 ] the state‐of‐the‐art mechanochemical synthetic toolbox offers several opportunities to construct C−N bonds [24] in amines via alkylation[ 25 , 26 , 27 ] and arylation [28] with organic halides, including transition metal‐catalyzed couplings.[ 29 , 30 , 31 , 32 , 33 , 34 ] However, the use of alcohols as ubiquitous starting materials remains nearly untapped. [35] Nucleophilic substitution of the hydroxy group in alcohols is one of the most fundamental and widespread chemical transformations. Applied to amine synthesis, alcohols can serve as safer replacements to lacrimatic and potentially genotoxic organic halides. However, the direct nucleophilic substitution of alcohols remains a challenge,[ 2 , 36 , 37 , 38 , 39 , 40 ] and prior transformation of hydroxy group into a better leaving group (e. g., sulfonate ester) is commonly required.
Recently, our group reported a new mechanochemical amidation approach based on the use of reactive uronium‐type amide coupling reagents such as (1‐cyano‐2‐ethoxy‐2‐oxoethylidenaminooxy)dimethylaminomorpholinocarbenium hexafluorophosphate (COMU) or chloro‐N,N,N′,N′‐tetramethylformamidinium hexafluorophosphate (TCFH) and K2HPO4 as a base (Scheme 1A). [41] The reaction proceeded via the generation of activated derivatives of carboxylic acids, such as acyl uroniums and acyl phosphates.
Scheme 1.

Mechanochemical synthesis of amides via in‐situ activation of carboxylic acids [41] and preparation of amines from alcohols.
Inspired by these findings, we planned to extend the same activation methodology towards nucleophilic substitution of alcohols (Scheme 1B). Indeed, O‐alkyl isouronium salts can be obtained by the reactions of alcohols with halouronium salts [e. g., TCFH or its analogue fluoro‐N,N,N′,N′‐tetramethylformamidinium hexafluorophosphate (TFFH)], a process commonly utilized for the preparation of amide coupling reagents.[ 42 , 43 ] Similarly to O‐alkyl isoureas generated from alcohols and carbodiimides,[ 44 , 45 , 46 , 47 , 48 ] isouronium salts can act as reactive intermediates in nucleophilic displacement reactions.[ 49 , 50 ] However, none of these alcohol‐activation techniques have been previously attempted under solvent‐free conditions or utilized for the synthesis of amines. Here we report for the first time the application of an air‐stable and non‐hygroscopic solid TFFH reagent[ 50 , 51 , 52 ] for activation of alcohols towards nucleophilic substitution with amines, halogens, and some oxygen nucleophiles under essentially solvent‐free conditions provided by mechanochemistry.
Results and Discussion
Optimization of coupling of alcohols with amines
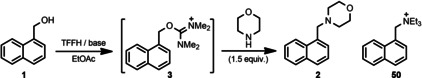
The reaction between 1‐naphthalenemethanol (1) and morpholine as a nitrogen nucleophile was selected as a model transformation to find the optimal reaction conditions leading to tertiary amine product 2 (Scheme 2). Ball milling experiments were performed in a Form‐Tech Scientific FTS1000 shaker mill operating at 30 Hz using a 14 mL zirconia‐coated milling jar and a single 10 mm milling ball. In contrast to amide synthesis, [41] attempted in‐situ activation of alcohol 1 with TCFH and TFFH reagents in the presence of morpholine resulted in a very low yield of 2 (less than 8 %; see Table S1 in the Supporting Information). This result is an outcome of a faster competitive reaction of halouronium reagents with morpholine itself.[ 52 , 53 ] To overcome the problem, a stepwise reaction in a single milling jar was considered, which involved the generation of isouronium salt 3 by the reaction of 1 with a halouronuim reagent before adding morpholine. To our delight, this technique afforded high 88–91 % conversions to 3 by milling alcohol 1 with TFFH reagent in the presence of K2HPO4 as a base for 1 h (Scheme 2). The addition of morpholine to the same reaction jar resulted in fast (less than 20 min) and quantitative transformation of 3 into amine 2, which was isolated in 82 % yield. Milling of solid reactants and addition of liquid morpholine resulted in formation of viscous paste‐like reaction mixture (see photos on Scheme 2).
Scheme 2.

Optimized conditions for nucleophilic substitution of hydroxy group in 1 with morpholine and appearance of the reaction mixture after milling (2.8 mmol experiment). Selected optimization experiments (performed with 0.16 mmol of alcohol 1 as a limiting substrate) and kinetic studies. TEA = trimethylamine; NMI = N‐methylimidazole; DBU=1,8‐diazabicyclo[5.4.0]undec‐7‐ene; DIC = N,N′‐diisopropylcarbodiimide; CPME = cyclopentyl methyl ether; DMIS = dimethyl isosorbide. The complete dataset of optimization experiments is provided in the Supporting Information (Table S2).
Selection of base, liquid additive for liquid‐assisted grinding (LAG),[ 6 , 7 ] and coupling reagent were identified as a trio of most crucial parameters affecting the reaction efficiency. No reaction of 1 and TFFH occurred without a basic additive. Concerning the choice of base (Scheme 2A), replacement of organic bases like triethylamine (TEA), N‐methylimidazole (NMI), and diazabicyclo(5.4.0)undec‐7‐ene (DBU) with inexpensive and less hazardous inorganic salts was superior (e. g., K2HPO4 or K3PO4) or at least as efficient (K2CO3 vs. TEA). Potassium fluoride (KF) and dipotassium phosphate (K2HPO4) were identified as the most efficient bases during the initial screening, affording 73 and 77 % conversions to 3, respectively. In the case of K2HPO4, a slight increase of TFFH amount (1.5 equiv.) to compensate for pyrophosphate formation (see the Supporting Information, Figure S2) delivered the highest conversion values (88–91 %).
A minor amount of liquid additive usually accelerates mechanochemical reactions, a technique known as LAG. It is empirically characterized by the parameter η, expressing the ratio of the volume of liquid [μL] added to the total amount of solid [mg]. [54] Although LAG requires a subtle amount of liquid (fixed at 0.2 μL mg−1 in our experiments), we preferred to screen several green solvents as LAG additives,[ 55 , 56 ] in addition to several conventional polar solvents (Scheme 2B; see Table S2 in the Supporting Information for additional data). In our hands, green solvents like ethyl acetate (EtOAc), cyclopentyl methyl ether (CPME), and dimethyl isosorbide (DMIS) served as excellent LAG additives offering the best conversions in the 72–77 % range and outperforming other polar solvents.
Among the activating reagents, TFFH was identified as the best activator, producing isouronium salt 3 in the highest yield. In comparison, COMU delivered lower 65 % yield of the corresponding isouronium intermediate (Scheme 2C). In contrast to TFFH, the corresponding chlorine reagent TCFH was found almost unreactive towards alcohol 1, with the same order of reactivity also observed in acetonitrile solution (see Table S6 in the Supporting Information). The use of TFFH is also preferable to TCFH or COMU in view of the lowest molecular weight and, therefore, better atom economy for the former. The reaction of alcohol 1 with N,N′‐diisopropylcarbodiimide (DIC) led to the formation of the corresponding O‐alkyl isourea [46] but did not result in any further nucleophilic substitution reaction (see Table S2 in the Supporting Information for the details). Note that at least 60 min milling time was required to achieve the highest conversion (88–91 %) of starting alcohol 1 into intermediate 3, as a kinetic study revealed (Scheme 2D).
Scope and limitations
With the optimal conditions established, the substrate scope of various alcohols was examined (Scheme 3). First, the above‐mentioned amine 2 was successfully prepared in 80 % yield (1.03 g) in an upscaled preparative run performed in two simultaneously shaken milling jars. Other morpholine derivatives 4–6 were flawlessly prepared in 77–91 % yields from the corresponding benzylic‐type primary alcohols (the reaction scope was further examined at 0.25–0.60 mmol scale, see section 2 in the Supporting Information). Substitution of the aromatic ring with electron‐donating (OMe) or electron‐withdrawing (CF3) groups did not substantially affect the reaction efficacy (5 and 6, Scheme 3).
Scheme 3.

Scope of alcohols and limitations of amine synthesis (performed with 0.25–0.60 mmol of alcohols as limiting substrates, unless stated otherwise). [a] A gram‐scale reaction was run in two simultaneously shaken jars with 0.45 g (2.8 mmol) of alcohol 1 in each jar. [b] 85 % ee after 15 min milling with TFFH/K2HPO4. [c] Indoline was used instead of morpholine. [d] Reaction time with morpholine: 3 h. (S)‐23 was prepared from (R)‐lactate with the same yield and optical purity.
Coming to secondary benzylic substrates, (R)‐phenylglycine derivative 7 was obtained from optically pure (>99 % ee) methyl (S)‐mandelate in high 84 % yield, but with reduced optical purity (76 % ee). Although phenylglycine is known as one of the most epimerization‐prone amino acids, [57] we found that optical purity of 7 remained almost unchanged over variation of milling time with morpholine (30–120 min; see Table S5 in the Supporting Information). However, shortening the duration of the alcohol activation step to 15 min resulted in a noticeably better 85 % ee. These results indicate that partial epimerization of the corresponding isouronium intermediate could occur (presumably via enolization [58] ) while its SN2 reaction with the amine is faster and dominant. At the same time, secondary benzylic alcohols like (S)‐α‐methyl‐2‐naphthalenemethanol (8) and diphenylmethanol (9) produced ethers 10 and 11 during the milling with TFFH/K2HPO4. Ether 10 was obtained as an almost equimolecular mixture of (S,S)‐ and (R,S)‐diastereomers according to 1H NMR spectroscopy and HPLC analysis on a chiral stationary phase, indicating that the reaction followed the SN1 mechanism and occurred via the corresponding benzylic cation. Tertiary benzylic alcohol 12 was unreactive and did not form the corresponding isouronium salt. Notably, highly chemoselective amination of primary alcohol 1 was achieved in the presence of tertiary substrate 12 due to such a high difference in reactivity. This finding was applied in the preparation of amine 13, which had an intact tertiary alcoholic moiety. Amines 14 and 15 were successfully prepared from the corresponding propargylic and allylic alcohols in 86 and 56 % yields, respectively. However, other tested allylic substrates like alcohols 16 and 17 failed to render the corresponding amine products due to several dominant side processes, such as ether formation (i. e., the transformation of 17 into 18).
Isouronium salts were also quickly (within 1 h) generated from most of the tested primary and secondary aliphatic alcohols, with the only exception of totally unreactive menthol 19. However, in contrast to activated benzylic and allylic derivatives, aliphatic substrates showed insufficient reactivity in the subsequent nucleophilic displacement reaction with morpholine. The reaction required at least 3 h milling time to reach acceptable 40–50 % conversions. Although longer milling is technically possible to increase the yields, we consider it less practical especially in view of the observed slow decomposition of isouronium intermediates (see below). Thus, 3‐phenylpropyl amine 20 was produced from the corresponding activated alcoholic precursor in 40 % yield after 3 h milling with morpholine. Likewise, 6‐chlorohexan‐1‐ol afforded amine 21 in a similar low yield, while the rest of the reaction mixture was represented by unreacted isouronium derivative (see Figure S7 in the Supporting Information). No nucleophilic substitution of chlorine was observed showing that isouronium moiety is a better leaving group. Secondary aliphatic alcohol like cyclohexanol produced completely unreactive isouronium salts 22. Intriguingly, ethyl (S)‐ and (R)‐lactates were quite reactive secondary alcoholic substrates, producing alanine derivative 23 in 50 % yield and excellent optical purity (>99 % ee). The stereochemical outcome indicates complete stereoinversion via the SN2 mechanism. Surprisingly, a derivative of aspartic acid 24 was obtained from dimethyl (S)‐malate with a total loss of optical purity. This result, along with a substantial amount of dimethyl fumarate detected in the reaction mixture (see Figure S8 in the Supporting Information), indicates that substitution of the hydroxy group probably occurred via an elimination‐addition mechanism in this case and elimination can compete with nucleophilic substitution process.
The scope of amines was explored in the substitution reactions of 2‐naphthylmethanol 25 (Scheme 4). Secondary aliphatic amines like thiomorpholine and piperidine delivered the corresponding amines 26 and 27 in 79 and 52 % yields, respectively. Methyl ester of d‐proline was used as solid hydrochloric salt and rendered amine 28 in a modest 42 % yield. Since solid and liquid substrates might have different reactivity under mechanochemical conditions, [59] an additional reaction with a solid nucleophilic amine (N‐Boc piperazine) was tested and produced the corresponding product 29 in a respectable 71 % yield. Notably, less nucleophilic aromatic amine (indoline) delivered a high 86 % yield of the corresponding amine product 30, while carbazole 31 was already too poor nucleophile to show any reactivity. Primary amines, for example, 3,5‐bis(trifluoromethyl)benzylamine (32), were found reacting stepwise under the standard reaction conditions, producing a mixture of mono‐ and dialkylated products 33 and 34, respectively. Full conversion into the tertiary amine product can be achieved using stoichiometric amounts (0.5 equiv.) of a primary amine; for example, amine 35 was prepared from benzylamine in 77 % yield under these conditions.
Scheme 4.

Scope of amines prepared from 2‐naphthalenemethanol (25). (Performed with 0.25 mmol of 25 as a limiting substrate). [a] Amine hydrochloride was used as starting material. [b] With 0.5 equiv. of benzylamine.
Application to the synthesis of APIs and bioactive amines
The knowledge acquired during the optimization studies and examination of substrate scope prompted us to implement the synthesis of several bioactive amines (including APIs) using mechanochemical C−N bond‐forming reactions as the key steps. Thus, a stimulant drug 1‐methyl‐4‐benzylpiperazine (36, MBZP) was prepared by alkylation of N‐methylpiperazine (37) with benzylic alcohol and isolated in 54 % yield by crystallization of its hydrochloric salt from methanol (Scheme 5). Antidepressant piberaline (38) was prepared from picolinic acid (39) via stepwise mechanochemical amide coupling, [41] solvent‐free Boc‐deprotection, [60] and mechanochemical amination steps. Derivatives of 4‐(hydroxymethyl)benzoic acid (42) have multiple pharmaceutical uses, including anticancer agents (e. g., Imanitib, [61] Masitinib [62] ), histone deacetylase inhibitors, [63] histamine receptor antagonists (e. g., Bavisant). [64] The observed low reactivity of TCFH reagent towards alcohols compared to TFFH (see Scheme 2C) and, conversely, the high amide coupling potency of the TCFH/K2HPO4 reagent system [41] was utilized to perform protecting group‐free mechanochemical amidation of carboxylic group in 42 (Scheme 6) while leaving the benzylic hydroxy group intact. Switching to the TFFH reagent at the next step resulted in activation of benzylic alcohol 43 and yielded the morpholine derivative [64] 44 in 80 % yield.
Scheme 5.

Mechanochemical C−N bond‐forming reactions in the synthesis of antidepressants 1‐methyl‐4‐benzylpiperazine (36, MBZP) and piberaline (38).
Scheme 6.

Examples of pharmacologically active derivatives of 4‐(hydroxymethyl)benzoic acid (42) and preparation of amine 44 via chemoselective activation of hydroxy groups in 42.
Nucleophilic substitution with halogens and oxygen nucleophiles
In addition to amines and their derivatives, oxygen‐ and halogen‐containing moieties occupy a spacious fraction of pharmaceutically relevant functional groups. [16] This fact inspired us to examine the efficacy of the developed OH‐activation protocol towards incorporating halogens and oxygen‐containing functionalities. The corresponding nucleophilic substitution reactions were performed with 1‐ and 2‐naphthalenemethanols (1 and 25, Scheme 7). Activation of the hydroxy group in 25 with the TFFH reagent followed by milling the produced isouronium intermediate with solid MgBr2 or KI afforded the corresponding bromo and iodo derivatives in high yields [45 and 46; Eqs. (1) and (2)]. Notably, no fluoro derivative was produced during the milling with TFFH/KF, both serving as a source of a fluoride anion. Only replacing KF with CsF resulted in the generation of fluorine derivative 47 [Eq. (3)], although in a moderate 40 % yield. This result stays in line with previous observations,[ 65 , 66 ] showing that nucleophilic reactivity of anions under solvent‐free conditions is strongly controlled by their counter‐ion pairing. The effect was interpreted in the framework of hard‐soft acid‐base (HSAB) theory, [14] explaining the enhanced nucleophilicity of F− in CsF as a result of a mismatched soft‐hard ion pair. Higher nucleophilic reactivity of F− in CsF compared to KF also correlates with lower lattice energy for the former (CsF, 740 kJ mol−1; KF, 821 kJ mol−1). [67] Use of potassium phenolate as the nucleophile resulted in a modest 46 % yield of the corresponding substitution product 48 [Eq. (4)]. At the same time, the isouronium salt generated from 2‐naphthalenemethanol was highly sensitive towards solvolysis in a nucleophilic solvent such as methanol and produced methyl ether 49 in high 91 % yield [Eq. (5)] after 3 h.
Scheme 7.

Substitution reactions of naphthalenemethanols with various nucleophiles. [a] 1H NMR yield.
Comparison of mechanochemical and solution‐based amine synthesis
For the synthesis of amine 2, the efficacy of the developed mechanochemical protocol was compared with the same reaction carried out in ethyl acetate solution (Table 1). It worth noting that the performance of the solution‐based reaction was not dedicatedly optimized, so we kept its conditions close to those of mechanochemical transformation for comparative reasons only (additional experimental data for the solution reactions is provided in the Supporting Information, section 5). First, a homogeneous solution reaction with trimethylamine as a base quickly (>1 h) produced isouronium compound 3. The latter delivered amine 2 in 72 % yield after addition of morpholine (entry 1). However, the reaction was accompanied by the formation of quaternary ammonium salt 50 from Et3N. Use of solid inorganic base (K2HPO4) gave cleaner conversion to 3 but in much slower rate (entry 2). The highest 74 % yield of isouronium compound 3 (and, consequently, amine 2) was attained after 6 h reaction with TFFH and further increase of the reaction time did not improve yield due to slow decomposition of 3. The reaction became even more sluggish when performed with larger amount of reactants (starting from 5.7 mmol of alcohol 1) and produced amine 2 in only 65 % yield (46 % yield of isolated product, entry 3). On the contrary, as it was shown above, the mechanochemical protocol was upscaled without any difficulties and delivered 2 in faster reaction rate and higher 80 % yield in comparison with the solution reaction (entry 4). Ball milling was essential for attaining high yields and fast reaction rates since the reaction performed by slurry stirring was much less efficient (entry 5).
Table 1.
Comparison of mechanochemical and solution‐based reactions.
|
| ||||||
|---|---|---|---|---|---|---|
|
Entry |
Reaction conditions[a] |
Scale [mmol of 1] |
Time[b] [h] |
Yield of 2[c] [%] |
PMI[d] |
|
|
1 |
solution |
TFFH (1 equiv.)/Et3N (1 equiv.) |
0.16 |
1 |
(72)[e] |
– |
|
2 |
TFFH (1.5 equiv.)/K2HPO4 (2 equiv.) |
0.16 |
6 |
(74) |
– |
|
|
3 |
TFFH (1.5 equiv.)/K2HPO4 (2 equiv.) |
5.7 |
24 |
46 (65) |
117 |
|
|
4 |
ball milling |
TFFH (1.5 equiv.)/K2HPO4 (2 equiv.) |
2×2.8[f] |
1 |
80 (86) |
41.4 |
|
5 |
slurry stirring |
TFFH (1.5 equiv.)/K2HPO4 (2 equiv.) |
0.16 |
6 |
(76) |
– |
[a] Approximately 0.15 m concentration of 1 was used in the solution reactions, η=0.2 μL mg−1 for ball milling and slurry stirring. [b] For the reaction of 1 with TFFH. The subsequent reaction of isouronium intermediate 3 with morpholine was fast (>1 h) and quantitative. [c] Yield of isolated amine product or its 1H NMR yield (given in parenthesis). [d] PMI=process mass intensity, excluding column chromatography. See the Supporting Information (section 6) for calculation details and additional green metrics. [e] Around 15 % of quaternary salt 50 was formed as main by‐product. [f] The reaction was run in two jars simultaneously and then combined for work‐up.
Green chemistry metrics calculated [68] for the upscaled solution and mechanochemical reactions (entries 3 and 4) manifest additional advantages of the latter as producing less waste due to better yield and lower solvent consumption. Thus, process mass intensity (PMI) of the mechanochemical reaction was almost 3 times lower than that of solution‐based synthesis (see section 6 in the Supporting Information for additional green metrics).
The presented comparison of solution‐based protocol and mechanochemical synthesis highlights the benefits of the latter approach in performing fast reactions with insoluble reactants and unstable reaction intermediates.
Conclusion
We have developed a new mechanochemical protocol for nucleophilic substitution of alcohols by applying fluoro‐N,N,N′,N′‐tetramethylformamidinium hexafluorophosphate (TFFH) as an activator and K2HPO4 as a non‐toxic inorganic base. This combination of reagents enables fast (within 1 h) and effective conversion of alcohols into reactive O‐alkyl isouronium salts under ball milling conditions with sustainable solvents (ethyl acetate, cyclopentyl methyl ether) as liquid‐assisted grinding (LAG) additives. We showed for the first time that O‐alkyl isouronium salts could act as highly reactive activated derivatives of alcohols in nucleophilic displacement reactions with amines, under mechanochemical conditions or in solution. Thus, mechanochemically generated isouronium intermediates afforded a range of N‐alkylated amine products in 31–91 % yield after ball milling with amines in the same reaction vessel. Complete stereoinversion was attained in the substitution reactions of ethyl (S)‐ and (R)‐lactates with morpholine, while enolizable α‐hydroxy esters could result in reduced or even fully demolished optical purity of the product. The developed substitution protocol is particularly efficient for reactive alcoholic substrates like primary benzylic alcohols. However, steric hindrances (tertiary/secondary substrates) or reduced reactivity in nucleophilic displacement reactions (aliphatic substrates) could substantially reduce the yield of amine products or even prevent the reaction from occurring. These limitations were advantageous when performing chemoselective transformations, such as substituting a primary benzylic alcohol in the presence of a tertiary alcoholic function. A striking difference in the chemoselectivity of fluoro‐ and chlorouronium coupling reagents [TFFH vs. chloro‐N,N,N′,N′‐tetramethylformamidinium hexafluorophosphate (TCFH)] allowed performing amide coupling of 4‐(hydroxymethyl)benzoic acid with an unprotected benzylic alcohol moiety. The developed process expands the synthetic portfolio of mechanochemical reactions. It can serve as a beneficial supplement to the existing C−N bond‐forming protocols, which are suitable for multistep mechanosynthesis of active pharmaceutical ingredients with a decreased environmental impact and a better safety profile.
Experimental Section
General procedure for mechanochemical synthesis of amines from alcohols
Alcohol (1 equiv.), TFFH (1.5 equiv.), and K2HPO4 (2 equiv.) were placed into a 14 mL ZrO2‐coated milling jar charged with a single 10 mm ZrO2 milling ball. Ethyl acetate (η=0.2 μL mg−1) was added and the jar was set to mill at 30 Hz for 60 min. Amine (1.5–3 equiv.) was added to the formed reaction mixture, and the jar was set to mill at 30 Hz for additional 60 min. The resulting crude reaction mixture was diluted with ethyl acetate (10–15 mL), filtered, and concentrated under reduced pressure before purification by silica gel chromatography to afford the amine product.
Detailed information about optimization studies, experimental methods, and compounds characterization data are given in the Supporting Information.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The research was supported by the European Union's H2020‐FETOPEN grant 828779 (INITIO). Funding from the Estonian Research Council grant PRG399 and support from COST Action CA18112 “Mechanochemistry for Sustainable Industry”, the ERDF CoE in Molecular Cell Engineering 2014‐2020.4.01.15‐0013, EU Regional Development Fund and Dora Plus Program are gratefully acknowledged. We are also grateful to Dr. Alexander‐Mati Müürisepp (TalTech) for measuring IR spectra.
T. Dalidovich, J. V. Nallaparaju, T. Shalima, R. Aav, D. G. Kananovich, ChemSusChem 2022, 15, e202102286.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.33774/chemrxiv‐2021‐kgcx5).
Contributor Information
Prof. Dr. Riina Aav, Email: riina.aav@taltech.ee.
Dr. Dzmitry G. Kananovich, Email: dzmitry.kananovich@taltech.ee.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Constable D. J. C., Dunn P. J., Hayler J. D., Humphrey G. R., J. L. Leazer, Jr. , Linderman R. J., Lorenz K., Manley J., Pearlman B. A., Wells A., Zaks A., Zhang T. Y., Green Chem. 2007, 9, 411–420. [Google Scholar]
- 2. Bryan M. C., Dunn P. J., Entwistle D., Gallou F., Koenig S. G., Hayler J. D., Hickey M. R., Hughes S., Kopach M. E., Moine G., Richardson P., Roschangar F., Steven A., Weiberth F. J., Green Chem. 2018, 20, 5082–5103. [Google Scholar]
- 3. Constable D. J. C., Jimenez-Gonzalez C., Henderson R. K., Org. Process Res. Dev. 2007, 11, 133–137. [Google Scholar]
- 4. López-Rodríguez R., McManus J. A., Murphy N. S., Ott M. A., Burns M. J., Org. Process Res. Dev. 2020, 24, 1558–1585. [Google Scholar]
- 5. Sörgel F., Kinzig M., Abdel-Tawab M., Bidmon C., Schreiber A., Ermel S., Wohlfart J., Besa A., Scherf-Clavel O., Holzgrabe U., J. Pharm. Biomed. Anal. 2019, 172, 395–405. [DOI] [PubMed] [Google Scholar]
- 6. Do J.-L., Friščić T., ACS Cent. Sci. 2017, 3, 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Do J. L., Friščić T., Synlett 2017, 28, 2066–2092. [Google Scholar]
- 8. James S. L., Adams C. J., Bolm C., Braga D., Collier P., Friščić T., Grepioni F., Harris K. D. M., Hyett G., Jones W., Krebs A., Mack J., Maini L., Orpen A. G., Parkin I. P., Shearouse W. C., Steed J. W., Waddell D. C., Chem. Soc. Rev. 2012, 41, 413–447. [DOI] [PubMed] [Google Scholar]
- 9. Tan D., Loots L., Friščić T., Chem. Commun. 2016, 52, 7760–7781. [DOI] [PubMed] [Google Scholar]
- 10. Colacino E., Porcheddu A., Charnay C., Delogu F., React. Chem. Eng. 2019, 4, 1179–1188. [Google Scholar]
- 11. Ying P., Yu J., Su W., Adv. Synth. Catal. 2021, 363, 1246–1271. [Google Scholar]
- 12. Howard J. L., Cao Q., Browne D. L., Chem. Sci. 2018, 9, 3080–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tan D., Friščić T., Eur. J. Org. Chem. 2018, 1, 18–33. [Google Scholar]
- 14. Andersen J., Mack J., Green Chem. 2018, 20, 1435–1443. [Google Scholar]
- 15. Porcheddu A., Colacino E., De Luca L., Delogu F., ACS Catal. 2020, 10, 8344–8394. [Google Scholar]
- 16. Ertl P., Altmann E., McKenna J. M., J. Med. Chem. 2020, 63, 8408–8418. [DOI] [PubMed] [Google Scholar]
- 17. Roughley S. D., Jordan A. M., J. Med. Chem. 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- 18. Declerck V., Nun P., Martinez J., Lamaty F., Angew. Chem. Int. Ed. 2009, 48, 9318–9321; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 9482–9485. [Google Scholar]
- 19. Métro T.-X., Bonnamour J., Reidon T., Sarpoulet J., Martinez J., Lamaty F., Chem. Commun. 2012, 48, 11781–11783. [DOI] [PubMed] [Google Scholar]
- 20. Štrukil V., Bartolec B., Portada T., Đilović I., Halasz I., Margetić D., Chem. Commun. 2012, 48, 12100–12102. [DOI] [PubMed] [Google Scholar]
- 21. Gonnet L., Tintillier T., Venturini N., Konnert L., Hernandez J.-F., Lamaty F., Laconde G., Martinez J., Colacino E., ACS Sustainable Chem. Eng. 2017, 5, 2936–2941. [Google Scholar]
- 22. Ardila-Fierro K. J., Crawford D. E., Körner A., James S. L., Bolm C., Hernández J. G., Green Chem. 2018, 20, 1262–1269. [Google Scholar]
- 23. Nicholson W. I., Barreteau F., Leitch J. A., Payne R., Priestley I., Godineau E., Battilocchio C., Browne D. L., Angew. Chem. Int. Ed. 2021, 60, 21868–21874. [DOI] [PubMed] [Google Scholar]
- 24. Margetić D., Štrukil V., Mechanochemical Organic Synthesis, Elsevier, Boston, 2016, pp. 141–233. [Google Scholar]
- 25. Nun P., Martin C., Martinez J., Lamaty F., Tetrahedron 2011, 67, 8187–8194. [Google Scholar]
- 26. Métro T.-X., Salom-Roig X. J., Reverte M., Martinez J., Lamaty F., Green Chem. 2015, 17, 204–208. [Google Scholar]
- 27. Briš A., Đud M., Margetić D., Beilstein J. Org. Chem. 2017, 13, 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andersen J. M., Starbuck H. F., J. Org. Chem. 2021, 86, 13983–13989. [DOI] [PubMed] [Google Scholar]
- 29. Martina K., Rinaldi L., Baricco F., Boffa L., Cravotto G., Synlett 2015, 26, 2789–2794. [Google Scholar]
- 30. Kubota K., Seo T., Koide K., Hasegawa Y., Ito H., Nat. Commun. 2019, 10, 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kubota K., Takahashi R., Uesugi M., Ito H., ACS Sustainable Chem. Eng. 2020, 8, 16577–16582. [Google Scholar]
- 32. Kubota K., Endo T., Uesugi M., Hayashi Y., Ito H., ChemSusChem 2021, DOI: 10.1002/cssc.202102132. [DOI] [PubMed] [Google Scholar]
- 33. Cao Q., Nicholson W. I., Jones A. C., Browne D. L., Org. Biomol. Chem. 2019, 17, 1722–1726. [DOI] [PubMed] [Google Scholar]
- 34. Shao Q.-L., Jiang Z.-J., Su W.-K., Tetrahedron Lett. 2018, 59, 2277–2280. [Google Scholar]
- 35. Lingome C. E., Pourceau G., Gobert-Deveaux V., Wadouachi A., RSC Adv. 2014, 4, 36350–36356. [Google Scholar]
- 36. Wetzel A., Wöckel S., Schelwies M., Brinks M. K., Rominger F., Hofmann P., Limbach M., Org. Lett. 2013, 15, 266–269. [DOI] [PubMed] [Google Scholar]
- 37. Watson A. J. A., Maxwell A. C., Williams J. M. J., J. Org. Chem. 2011, 76, 2328–2331. [DOI] [PubMed] [Google Scholar]
- 38. Charville H., Jackson D. A., Hodges G., Whiting A., Wilson M. R., Eur. J. Org. Chem. 2011, 2011, 5981–5990. [Google Scholar]
- 39. Zhang G., Yin Z., Zheng S., Org. Lett. 2016, 18, 300–303. [DOI] [PubMed] [Google Scholar]
- 40. Mastalir M., Tomsu G., Pittenauer E., Allmaier G., Kirchner K., Org. Lett. 2016, 18, 3462–3465. [DOI] [PubMed] [Google Scholar]
- 41. Dalidovich T., Mishra K. A., Shalima T., Kudrjašova M., Kananovich D. G., Aav R., ACS Sustainable Chem. Eng. 2020, 8, 15703–15715. [Google Scholar]
- 42. El-Faham A., Albericio F., J. Org. Chem. 2008, 73, 2731–2737. [DOI] [PubMed] [Google Scholar]
- 43. El-Faham A., Albericio F., J. Pept. Sci. 2010, 16, 6–9. [DOI] [PubMed] [Google Scholar]
- 44. Poelert M. A., Kellogg R. M., Hulshof L. A., Recl. Trav. Chim. Pays-Bas 1994, 113, 365–368. [Google Scholar]
- 45. Chighine A., Crosignani S., Arnal M. C., Bradley M., Linclau B., J. Org. Chem. 2009, 74, 4753–4762. [DOI] [PubMed] [Google Scholar]
- 46. Mathias L. J., Synthesis 1979, 8, 561–576. [Google Scholar]
- 47. Sood D. E., Champion S., Dawson D. M., Chabbra S., Bode B. E., Sutherland A., Watson A. J. B., Angew. Chem. Int. Ed. 2020, 59, 8460–8463; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 8538–8541. [Google Scholar]
- 48. Li Z., Crosignani S., Linclau B., Tetrahedron Lett. 2003, 44, 8143–8147. [Google Scholar]
- 49. Merad J., Matyašovský J., Stopka T., Brutiu B. R., Pinto A., Drescher M., Maulide N., Chem. Sci. 2021, 12, 7770–7774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bellavance G., Dubé P., Nguyen B., Synlett 2012, 2012, 569–574. [Google Scholar]
- 51. Pittelkow M., Kamounah F. S., Boas U., Pedersen B., Christensen J. B., Synthesis 2004, 15, 2485–2492. [Google Scholar]
- 52. El-Faham A., Khattab S. N., Synlett 2009, 2009, 886–904. [Google Scholar]
- 53. Barton D. H. R., Elliott J. D., Géro S. D., J. Chem. Soc. Chem. Commun. 1981, 21, 1136–1137. [Google Scholar]
- 54. Friščić T., Childs S. L., Rizvi S. A. A., Jones W., CrystEngComm 2009, 11, 418–426. [Google Scholar]
- 55. Prat D., Hayler J., Wells A., Green Chem. 2014, 16, 4546–4551. [Google Scholar]
- 56. Prat D., Wells A., Hayler J., Sneddon H., McElroy C. R., Abou-Shehada S., Dunn P. J., Green Chem. 2016, 18, 288–296. [Google Scholar]
- 57. Liang C., Behnam M. A. M., Sundermann T. R., Klein C. D., Tetrahedron Lett. 2017, 58, 2325–2329. [Google Scholar]
- 58. Silverman R. B., Organic Chemistry of Enzyme-Catalyzed Reactions (Second Edition), Academic Press, San Diego, 2002, pp. 367–368. [Google Scholar]
- 59. Seo T., Kubota K., Ito H., J. Am. Chem. Soc. 2020, 142, 9884–9889. [DOI] [PubMed] [Google Scholar]
- 60. Bonnamour J., Métro T.-X., Martinez J., Lamaty F., Green Chem. 2013, 15, 1116–1120. [Google Scholar]
- 61. Druker B. J., Tamura S., Buchdunger E., Ohno S., Segal G. M., Fanning S., Zimmermann J., Lydon N. B., Nat. Med. 1996, 2, 561–566. [DOI] [PubMed] [Google Scholar]
- 62. Hahn K. A., Oglivie G., Rusk T., Devauchelle P., Leblanc A., Legendre A., Powers B., Leventhal P. S., Kinet J.-P., Palmerini F., Dubreuil P., Moussy A., Hermine O., J. Vet. Intern. Med. 2008, 22, 1301–1309. [DOI] [PubMed] [Google Scholar]
- 63. Suzuki T., Ando T., Tsuchiya K., Fukazawa N., Saito A., Mariko Y., Yamashita T., Nakanishi O., J. Med. Chem. 1999, 42, 3001–3003. [DOI] [PubMed] [Google Scholar]
- 64. Letavic M. A., Aluisio L., Apodaca R., Bajpai M., Barbier A. J., Bonneville A., Bonaventure P., Carruthers N. I., Dugovic C., Fraser I. C., Kramer M. L., Lord B., Lovenberg T. W., Li L. Y., Ly K. S., Mcallister H., Mani N. S., Morton K. L., Ndifor A., Nepomuceno S. D., Pandit C. R., Sands S. B., Shah C. R., Shelton J. E., Snook S. S., Swanson D. M., Xiao W., ACS Med. Chem. Lett. 2015, 6, 450–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Vogel P., Figueira S., Muthukrishnan S., Mack J., Tetrahedron Lett. 2009, 50, 55–56. [Google Scholar]
- 66. Ortiz-Trankina L. N., Crain J., Williams C., Mack J., Green Chem. 2020, 22, 3638–3642. [Google Scholar]
- 67.W.-P. Leung, Y.-C. Chan, in Encyclopedia of Inorganic and Bioinorganic Chemistry, (Ed.: R. A. Scott), 2014, DOI: 10.1002/9781119951438.eibc0003.pub2. (Article in online Encyclopedia).
- 68. McElroy C. R., Constantinou A., Jones L. C., Summerton L., Clark J. H., Green Chem. 2015, 17, 3111–3121. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.