

Abstract
Aims
Cancer drugs are increasingly approved through expedited regulatory pathways including the European conditional marketing authorization (CMA). Whether, when taking CMA post‐approval confirmatory trials into account, the level of evidence and clinical benefit between CMA and standard approved (SMA) drugs differs remains unknown.
Methods
We identified all CMA cancer indications converted to SMA in 2006–2020 and compared these to similar SMA indications with regard to pivotal trial and CMA post‐approval confirmatory trial design, outcomes and demonstrated clinical benefit (per the European Society for Medical Oncology Magnitude of Clinical Benefit Scale). We tested for differences in clinical benefit and whether substantial clinical benefit was demonstrated. To account for the clinical benefit of unconverted CMA indications, we performed sensitivity analyses.
Results
We included 15 SMA and 15 converted CMA cancer indications (17 remained unconverted). Approval of 11 SMA (73%) and four CMA indications (27%) was supported by a controlled trial. Improved overall survival (OS) was demonstrated for four SMA indications (27%). Improved quality of life (QoL) was demonstrated for three SMA (20%) and one CMA indication(s) (7%). Of subsequent CMA post‐approval confirmatory trials, 11 were controlled (79%), one demonstrated improved OS (7%) and five improved QoL (36%). After conversion, CMA indications were associated with similar clinical benefit (P = .31) and substantial clinical benefit as SMA indications (risk ratio 1.4, 95% confidence interval 0.57–3.4).
Conclusion
While CMA cancer indications are initially associated with less comprehensive evidence than SMA indications, levels of evidence and clinical benefit are similar after conversion from CMA to SMA.
Keywords: benefit–risk assessment, clinical benefit, clinical trials, conditional marketing authorization, drug regulation, European Medicines Agency
What is already known about this subject
In Europe, cancer drugs are often approved without evidence on overall survival (OS) and quality of life (QoL), and increasingly granted conditional marketing authorization (CMA).
Whether, when taking CMA post‐approval confirmatory trials into account, levels of evidence and clinical benefit differ between CMA and standard approved (SMA) drugs remains unknown.
What this study adds
At approval, OS and QoL benefits were observed for few SMA and even fewer CMA cancer indications.
After conversion of CMA indications to SMA, levels of evidence and clinical benefit were comparable thanks to CMA post‐approval confirmatory trials.
These levels may be substantially lower than expected by physicians and patients.
1. INTRODUCTION
Cancer contributes substantially to the global disease burden, ranking third among the major causes of disability‐adjusted life years. 1 Moreover, it is the primary cause of premature death in highly developed countries. 2 Although new cancer drug treatments continuously become available, a high unmet medical need for additional and more effective treatments remains.
To address unmet medical needs, drug regulatory authorities commonly use expedited regulatory pathways for approval of promising cancer drugs. These include the conditional marketing authorization (CMA) pathway of the European Medicines Agency (EMA) and the accelerated approval (AA) pathway of the United States Food and Drug Administration (US FDA). 3 , 4 Expedited pathways allow approval based on less comprehensive evidence than normally required, leaving important uncertainties about efficacy and safety to be addressed by post‐approval confirmatory trials. Thereafter, a standard marketing authorization (SMA) may be granted. 5
While physicians may expect that newly approved cancer drugs provide substantial clinical value such as improvements in overall survival (OS) and quality of life (QoL) as compared to current standards of care, these expectations are often too high. 6 Moreover, although the EMA prefers OS as efficacy endpoint for SMA cancer drugs, 7 conclusive evidence on OS (and QoL) is often lacking when new cancer drugs and their indication(s) are approved. For example, for a cohort of 68 EMA‐approved cancer indications, Davis et al. reported that OS and QoL benefits had initially been demonstrated for 24 (35%) and seven (10%) of them. A median 5.4 years after approval, OS and QoL benefits were demonstrated for another three (4%) and five (7%) indications, respectively. For 33 (49%) of the indications, no OS or QoL benefits had been demonstrated, including for all ten that had been approved through the CMA expedited pathway. 8 Similar figures have been reported for the US. 9 , 10
The combination of (i) infrequent demonstration of OS and QoL benefits upon approval and (ii) regulatory acceptance of higher levels of uncertainty raises the question whether cancer drugs approved via expedited pathways live up to their initial promise of providing substantial value in clinical practice. An important validated instrument that may help to address these questions is the European Society for Medical Oncology (ESMO) Magnitude of Clinical Benefit Scale (MCBS). 11 Such an instrument enables the assessment of clinical benefit taking into account clinical endpoints of efficacy, but also surrogate endpoints such as progression‐free survival (PFS) and overall response rate (ORR), as well as toxicity profiles. Davis et al. applied the ESMO‐MCBS v1.0 to 23 EMA‐approved solid cancer indications for which OS benefits had been demonstrated and showed that these were of substantial clinical benefit in only 11 (48%) cases due to too small effects on OS in the other cases. 8 However, the authors did not assess the clinical benefit of indications without OS benefits, did not specifically focus on expedited pathways, and were not able to evaluate single arm trials, which has only recently become possible with the updated ESMO‐MCBS v1.1. 11
We aimed to compare the availability of evidence and demonstrated clinical benefit of CMA versus SMA cancer indications in Europe, taking into account the contribution of post‐approval confirmatory trials.
2. METHODS
2.1. Study design and cohort selection
We performed a retrospective cohort study consisting of three groups of cancer indications. First, we identified all cancer drugs initially approved through the EMA's CMA pathway and converted to SMA up to 31 December 2020, indicating that sufficient confirmatory evidence had been provided to fulfil so‐called “specific obligations”. For these drugs, we included all initial indications (“converted CMA indications”). Second, to benchmark their evidence and clinical benefit characteristics against SMA cancer indications, we identified an equal number of SMA cancer drugs. To allow a fair comparison that takes into account that the ability to conduct controlled clinical trials may differ between specific cancer types and their rarity, that toxicity may depend on whether drugs are targeted or not, and that evidence requirements and the availability of alternative drugs may change over time, we identified SMA drugs that were as similar as possible to the converted CMA drugs with respect to: (i) pharmacotherapeutic group, based on the first five characters of the ATC code (Index 2020, www.whocc.no); (ii) cancer type they were initially approved for; (iii) initial approval date, and (iv) whether the EMA had granted orphan status at their initial approval. We were kindly supported therein by a clinical assessor from the Dutch Medicines Evaluation Board (see acknowledgements). This approach is similar to previous research. 12 For these drugs, we also included all initial indications (“SMA indications”). Third, we identified all CMA cancer drugs that remained unconverted on 31 December 2020 and included their indications (“unconverted CMA indications”) as a separate group to perform sensitivity analyses (see below). We excluded generics and biosimilars. As we did not include patients or volunteers, ethics approval was not required.
2.2. Extraction of trial data
For all SMA and CMA indications, we identified the trials that formed the main evidence base for their initial approval (“pivotal trials”), as per the European Public Assessment Reports (EPARs) at www.ema.europa.eu. We extracted trial design characteristics and evidence concerning efficacy endpoints, QoL and toxicity. For converted CMA indications, we also identified post‐approval confirmatory trials imposed by the EMA as specific obligations. Since their characteristics and provided evidence are generally not available in EPARs, we extracted these from the EMA's confidential assessment reports that were accessed through a Memorandum of Understanding with the Dutch Medicines Evaluation Board. To ensure assessment of evidence availability and demonstrated clinical benefit for the initially approved indication, we only included post‐approval confirmatory trials that delivered evidence on clinical and surrogate endpoints of survival, ORR, QoL and/or toxicity and that had been performed in the approved or a highly related treatment setting, e.g. combination rather than monotherapy.
2.3. Scoring of trial data using the ESMO‐MCBS
For each trial, we applied the ESMO‐MCBS v1.1 to assess the demonstrated clinical benefit. The ESMO‐MCBS offers multiple forms to differentiate between trial designs, endpoints and magnitudes of effects. It allows a higher score for randomized controlled trials (RCTs) and clinical endpoints as compared to single arm trials and surrogate endpoints, ranging from 5 to 1 (non‐curative settings) or A to C (curative settings). Scores 4, 5, A and B indicate substantial clinical benefit. We validated our assessments of clinical benefit against those published by the ESMO at www.esmo.org/guidelines/esmo-mcbs for solid cancer indications, and against a recent publication by the European Hematology Association (EHA) for haematological cancer indications. 13
2.4. Data analysis
We used descriptive statistics to compare the availability of evidence at the initial approval of SMA and CMA indications with respect to, for example, pivotal trial design, endpoints and number of patients studied. Furthermore, for converted CMA indications, we compared the availability of evidence at conversion to SMA, i.e., the evidence derived from post‐approval confirmatory trials, to that at initial approval. Finally, we compared the availability of evidence at conversion for CMA indications to that at initial approval for SMA indications, i.e., the moment that the EMA considers that comprehensive evidence is available.
We considered one clinical benefit score at initial approval for each SMA and CMA indication, taking the highest in case of multiple pivotal trials. Similarly, for converted CMA indications, we considered the highest score available after conversion to SMA. To allow numerical comparisons between groups, we used clinical benefit score 5 for one indication that was categorized as “A”, as both reflect the highest clinical benefit score. We then compared the scores available after conversion of CMA indications to (i) those at initial approval of CMA indications and (ii) those at initial approval of SMA indications. First, we tested for differences in overall scores using the Mann–Whitney U‐test. Second, we compared the chance that substantial clinical benefit (score ≥4) was demonstrated by calculating risk ratios (RR) and 95% confidence intervals (CI). Additionally, we visualized the contribution of post‐approval confirmatory trials to the clinical benefit of converted CMA indications through a Sankey diagram.
The analyses described above only consider clinical benefit scores for the converted CMA cancer indications, which may introduce bias by skewing the findings towards the successful CMA indications. To address this, we performed sensitivity analyses that consider the potential impact of unconverted CMA cancer indications, using two scenarios. In scenario 1, we added clinical benefit scores for three types of unconverted CMA indications for which CMA conversion could have been reasonably expected in a counterfactual situation: (i) indications that had been unconverted longer than the median time to conversion of the converted CMA indications; (ii) indications that were ultimately found to lack efficacy, leading to revocation of the CMA; and (iii) indications for which no specific obligations had been required. We added their last known clinical benefit score, i.e., a zero for those that lacked efficacy and the clinical benefit score at initial approval for the other indications. In scenario 2, we added clinical benefit scores for all unconverted CMA indications, i.e., also for those that had been unconverted shorter than the median time to conversion of the converted CMA indications.
3. RESULTS
3.1. Description of the cohort
In 2006–2020, 30 CMA cancer drugs had been approved. Of these, 15 (50%) had been converted to SMA. Of the 15 converted CMA drugs, one had been approved with two indications: sunitinib (Sutent) for renal cell cancer and gastrointestinal stromal cancer. Subsequently, we identified similar SMA drugs for 14 converted CMA drugs; all except pixantrone (Pixuvri) which was therefore excluded. Of the SMA drugs, also one drug had been approved for two indications: idelalisib (Zydelig) for chronic lymphocytic leukaemia (CLL) and follicular lymphoma. Only neratinib (Nerlynx) was approved for use in a curative setting: adjuvant treatment of early‐stage hormone receptor positive HER2‐overexpressed breast cancer. An overview of the included SMA and converted CMA drugs and indications is provided in Tables S1 and S2. In addition, an overview of the 15 unconverted CMA drugs and their 17 indications (including two for brentuximab vedotin [Adcetris]: Hodgkin lymphoma and systemic anaplastic large cell lymphoma; and two for entrectinib [Rozlytrek]: non‐small cell lung cancer [NSCLC] and a tumour agnostic indication) is provided in Table S3. The main characteristics of all 15 SMA and 32 CMA indications are presented in Table 1.
TABLE 1.
Characteristics of included SMA and CMA cancer indications
| SMA indications (N = 15) | Converted a CMA indications (N = 15) | Unconverted a CMA indications (N = 17) | ||||
|---|---|---|---|---|---|---|
| Pharmacotherapeutic group | ||||||
| Monoclonal antibodies (ATC code L01XC) | 5 | 33% | 5 | 33% | 6 | 35% |
| Protein kinase inhibitors (ATC code L01XE) | 8 | 53% | 7 | 47% | 8 | 47% |
| Other antineoplastic agents (ATC code L01XX) | 2 | 13% | 3 | 20% | 3 | 18% |
| Year of approval | ||||||
| 2004–2008 | 2 | 13% | 4 | 27% | 0 | 0% |
| 2009–2013 | 3 | 20% | 4 | 27% | 4 | 24% |
| 2014–2018 | 10 | 67% | 7 | 47% | 4 | 24% |
| 2019–2020 | 0 | 0% | 0 | 0% | 9 | 53% |
| Indication at initial approval | ||||||
| Solid tumours | 10 | 67% | 11 | 73% | 10 | 59% |
| ‐ Breast cancer | 1 | 7% | 1 | 7% | 0 | 0% |
| ‐ Basal cell cancer | 1 | 7% | 1 | 7% | 0 | 0% |
| ‐ Colorectal cancer | 1 | 7% | 1 | 7% | 0 | 0% |
| ‐ Cutaneous squamous cell cancer | 0 | 0% | 0 | 0% | 1 | 6% |
| ‐ Epithelial ovarian/fallopian tube/peritoneal cancer | 0 | 0% | 0 | 0% | 1 | 6% |
| ‐ Gastrointestinal stromal cancer | 0 | 0% | 1 | 7% | 1 | 6% |
| ‐ Melanoma | 1 | 7% | 0 | 0% | 0 | 0% |
| ‐ Merkel cell cancer | 0 | 0% | 1 | 7% | 0 | 0% |
| ‐ Non‐small cell lung cancer | 4 | 27% | 4 | 27% | 2 | 12% |
| ‐ Renal cell cancer | 2 | 13% | 2 | 13% | 0 | 0% |
| ‐ Soft tissue sarcoma | 0 | 0% | 0 | 0% | 1 | 6% |
| ‐ Thyroid cancer | 0 | 0% | 0 | 0% | 2 | 12% |
| ‐ Tissue agnostic | 0 | 0% | 0 | 0% | 2 | 12% |
| Hematological tumours | 5 | 33% | 4 | 27% | 7 | 41% |
| ‐ Leukaemia | 0 | 0% | 0 | 0% | 1 | 6% |
| ‐ Lymphoma | 4 | 27% | 3 | 20% | 4 | 24% |
| ‐ Multiple myeloma | 1 | 7% | 1 | 7% | 2 | 12% |
| Curative setting | 1 | 7% | 0 | 0% | 0 | 0% |
| Monotherapy | 10 | 67% | 14 | 93% | 14 | 82% |
| First‐line/line‐agnostic treatment | 3 | 20% | 4 | 27% | 6 | 35% |
| Orphan designation at initial approval | 4 | 27% | 6 | 40% | 11 | 65% |
| US FDA approval | ||||||
| Accelerated approval | 4 | 27% | 11 | 73% | 11 | 65% |
| Standard approval | 10 | 67% | 4 | 27% | 6 | 35% |
| Not approved | 1 | 7% | 0 | 0% | 0 | 0% |
| Time (median months, IQR) | ||||||
| To conversion (n = 15) | NA | 32 | 17–48 | NA | ||
| Unconverted (n = 16) | NA | NA | 19 | 7–73 | ||
| To revocation (n = 1) | NA | NA | 32 | NA | ||
| Amended indication after conversion to SMA | NA | 3 | 20% | NA | ||
ATC code, Anatomical Therapeutic Chemical code; CMA, conditional marketing authorization; IQR, interquartile range; NA, not applicable; SMA, standard marketing authorization; US FDA, United States Food and Drug Administration.
(Un)converted as at 31 December 2020.
3.2. Availability of evidence and clinical benefit at initial approval (SMA and converted CMA indications)
For SMA and converted CMA indications, we identified 19 and 18 pivotal trials, respectively, i.e., one pivotal trial for 12 indications in each group (80%) and two or more for the other indications. Their characteristics are presented in Table 2. A detailed overview of trial level is provided in Table S4.
TABLE 2.
Characteristics of pivotal trials at initial approval and post‐approval confirmatory trials at conversion, per indication
| SMA indications (initial approval, N = 15) | Converted a CMA indications (initial approval, N = 15) | Converted a CMA indications (conversion, N = 14) | Unconverted a CMA indications (initial approval, N = 17) | |||||
|---|---|---|---|---|---|---|---|---|
| Number of trials | ||||||||
| Median, range | 1 | 1–3 | 1 | 1–2 | 1 | 1–3 | 1 | 1–1 |
| Availability of at least a: | ||||||||
| Phase III trial | 11 | 73% | 4 | 27% | 11 | 79% | 3 | 18% |
| Controlled trial | 11 | 73% | 4 | 27% | 11 | 79% | 5 | 29% |
| Trial with OS as primary endpoint | 4 | 27% | 0 | 0% | 2 | 14% | 0 | 0% |
| Trial with PFS/TTP or DFS as primary endpoint | 7 | 47% | 4 | 27% | 9 | 64% | 4 | 24% |
| Trial with ORR as primary endpoint | 4 | 27% | 11 | 73% | 3 b | 21% | 13 c | 76% |
| Trial in which OS data were collected | 15 | 100% | 15 | 100% | 14 | 100% | 17 | 100% |
| Trial in which QoL data were collected | 14 | 93% | 7 | 47% | 13 | 93% | 10 | 59% |
| Total number of patients included in trials | ||||||||
| Median, IQR | 356 | 278–1109 | 196 | 116–356 | 414 | 320–1053 | 97 | 74–139 |
CMA, conditional marketing authorization; DFS, disease‐free survival; IQR, interquartile range; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; QoL, quality of life; SMA, standard marketing authorization; TTP, time to progression.
(Un)converted as at 31 December 2020.
One formally defined as ORR with a duration of at least 6 months, i.e., durable response rate.
Two formally defined as major cytogenetic response rate and complete response rate, respectively.
While OS data were collected in all pivotal trials, four SMA indications (27%) and no converted CMA indications were supported by a pivotal trial with OS as (co‐)primary endpoint. For two of these SMA indications (13%), acute lymphatic leukaemia (ALL) of inotuzumab ozogamicin (Besponsa) and renal cell cancer of sorafenib (Nexavar), a statistically significant increase in OS was demonstrated. For the other two SMA indications, NSCLC of gefitinib (Iressa) and melanoma of pembrolizumab (Keytruda), two pivotal trials had OS as primary endpoint. For gefitinib, these failed to show a difference in OS and demonstrated non‐inferiority to treatment with docetaxel, respectively. For pembrolizumab, these had planned interim analyses of the co‐primary endpoint PFS that mainly supported approval while OS data were not yet mature. When also considering OS as secondary endpoint, for two further SMA indications (13%) a statistically significant increase in OS was demonstrated. For the remaining nine SMA and all 15 converted CMA indications, the main evidence of efficacy was based on the primary endpoints DFS (one SMA, 7%), PFS or TTP (four SMA, 27%; four CMA, 27%), and ORR (four SMA, 27%; 11 CMA, 73%). Additionally, for 14 SMA indications (93%) and seven converted CMA indications (47%), QoL data were collected, which demonstrated a statistically significant increase for three (20%) and one (7%), respectively. Lastly, for one SMA indication (7%), NSCLC of gefitinib, there was evidence of significantly reduced grade 3 or 4 toxicities. These evidential aspects and the resulting demonstrated clinical benefit are presented in Table 3.
TABLE 3.
Clinical benefit qualifications and underlying evidence aspects
| Clinical benefit (score) | Trial design | Endpoint scored | Preliminary score a | Increased toxicity | Reduced toxicity a | Improved QoL | SMA indications (initial approval, N = 15) | Converted b CMA indications (initial approval, N = 15) | Converted b CMA indications (conversion c , N = 15) | Unconverted b CMA indications (initial approval, N = 17) |
|---|---|---|---|---|---|---|---|---|---|---|
| Substantial | 5 (33%) | 1 (7%) | 7 (47%) | 1 (6%) | ||||||
| 5/A | Controlled | OS | 4/4 | +1 | +1 | 1 | ||||
| Controlled | DFS | A | 1 | |||||||
| 4 | Controlled | OS | 4/4 | 1 | 1 | |||||
| Controlled | PFS/TTP | 3/3 | +1 | 2 | 4 | |||||
| Controlled | PFS/TTP | 3/3 | +1 | 1 | ||||||
| Controlled, non‐inferiority | OS | 4/4 | x d | x d | 1 | |||||
| Controlled, non‐inferiority | PFS/TTP | 4/4 | x d | 1 | ||||||
| Single arm | ORR | 3/3 | +1 | 1 | ||||||
| Moderate | 6 (40%) | 8 (53%) | 7 (47%) | 12 (71%) | ||||||
| 3 | Controlled | OS | 3/4 | 1 | ||||||
| Controlled | PFS/TTP | 3/3 | 3 | 3 | 4 | 2 | ||||
| Single arm | ORR | 3/3 | 2 | 5 | 3 | 10 | ||||
| Low | 4 (27%) | 6 (40%) | 1 (7%) | 4 (24%) | ||||||
| 2 | Controlled | OS | 2/4 | 1 | ||||||
| Controlled | PFS | 3/3 | −1 | 1 | ||||||
| Controlled | CRR | 2/2 | 1 | |||||||
| Single arm | ORR | 3/3 | −1 | 2 | ||||||
| Single arm | ORR | 2/3 | 1 | 5 | ||||||
| 1 | Controlled | PFS/TTP | 1/4 | 1 | 1 | |||||
| Controlled | OS | 1/4 | 1 | |||||||
| Single arm | ORR | 1/3 | 1 | |||||||
| Distribution of clinical benefit scores (median [IQR]) | 3.0 (2.5–4.0) | 3.0 (2.0–3.0) | 3.0 (3.0–4.0) | 3.0 (3.0–3.0) | ||||||
CMA, conditional marketing authorization; CRR, complete response rate; DFS, disease‐free survival; IQR, interquartile range; ORR, overall response rate; OS, overall survival; PFS, progression‐free survival; QoL, quality of life; SMA, standard marketing authorization; TTP, time to progression.
Grade 3/4, impacting daily well‐being.
(Un)converted as at 31 December 2020.
Including the gastrointestinal stromal cancer indication of sunitinib.
Preliminary score already includes improved QoL and/or reduced toxicity: this indicates the clinical benefit in the context of the non‐inferior efficacy outcome.
The ESMO‐MCBS preliminary clinical benefit score that is reflected by the available evidence on the scored endpoint. This score may be adjusted in case of increased toxicity, reduced toxicity and/or improved QoL.
3.3. Availability of evidence and clinical benefit after conversion to SMA (converted CMA indications)
No specific obligations had been required by the EMA for the gastrointestinal stromal cancer indication of sunitinib since sufficient evidence was considered already available. 14 For the remaining 14 converted CMA indications, we identified 36 specific obligations. Of these, we excluded 18 specific obligations—mostly trials in non‐approved treatment settings, of which 15 had been required for the metastatic colorectal carcinoma (CRC) indication of panitumumab (Vectibix). The remaining 18 specific obligations (Table S1) comprised 19 post‐approval confirmatory trials (at least one for each of the 14 converted CMA indications with specific obligations) that we included in our analyses.
For these 14 converted CMA indications, the characteristics of post‐approval confirmatory trials are presented in Table 2 and Table S4. In 11 cases (79%), the evidence initially provided at their approval was supplemented post‐approval by a controlled phase III confirmatory trial, of which nine had been ongoing at time of initial approval. For two indications (14%), OS was the primary endpoint, i.e., CRC of panitumumab and ALL of blinatumomab (Blincyto). These trials demonstrated non‐inferiority to treatment with cetuximab and superiority to standard of care chemotherapy, respectively. For the remaining 12 converted CMA indications (86%), OS was a secondary endpoint, but no differences were demonstrated. Rather, the main evidence of efficacy was based on the primary endpoints PFS or TTP (nine indications, 64%), or ORR (three indications, 21%). For these latter three indications, Merkel cell cancer of avelumab (Bavencio), CLL of venetoclax (Venclyxto) and basal cell cancer of vismodegib (Erivedge), all pivotal and post‐approval confirmatory trials were single arm phase II trials. Additionally, for 13 converted CMA indications (93%), QoL data were collected in their post‐approval confirmatory trial, which demonstrated a statistically significant increase for five (36%). Also, for three converted CMA indications (21%), there was evidence of significantly reduced grade 3 or 4 toxicities. In total, the post‐approval confirmatory trials included more than twice as many patients as included in the pivotal trials for converted CMA indications: median 414 versus 196 patients.
For all but one converted CMA indication, the clinical benefit demonstrated by post‐approval confirmatory trials was equal to or higher than that demonstrated by the pivotal trials (Figure 1). The only exception was the CRC indication of panitumumab for which the ASPECCT trial demonstrated non‐inferiority in the absence of QoL or toxicity benefits and therefore a lack of clinical benefit as compared to treatment with cetuximab. 15 We thus retained the score for panitumumab at initial approval since this was the highest. At conversion to SMA, the demonstrated clinical benefit (Table 3) was higher than at initial CMA approval (P = .0074). Moreover, the chance to be associated with substantial clinical benefit increased from 7% to 47%; RR 7.0 (95% CI 1.0–50). After completion of the post‐approval confirmatory trials for CMA indications and their subsequent conversion to SMA, their median clinical benefit scores were similar to those of SMA indications at initial approval (P = .31). Similarly, we identified no difference in their chance to be associated with substantial clinical benefit, although it was numerically higher for converted CMA indications than for SMA indications: 47% versus 33%; RR 1.4 (95% CI 0.57–3.4; Table 4).
FIGURE 1.
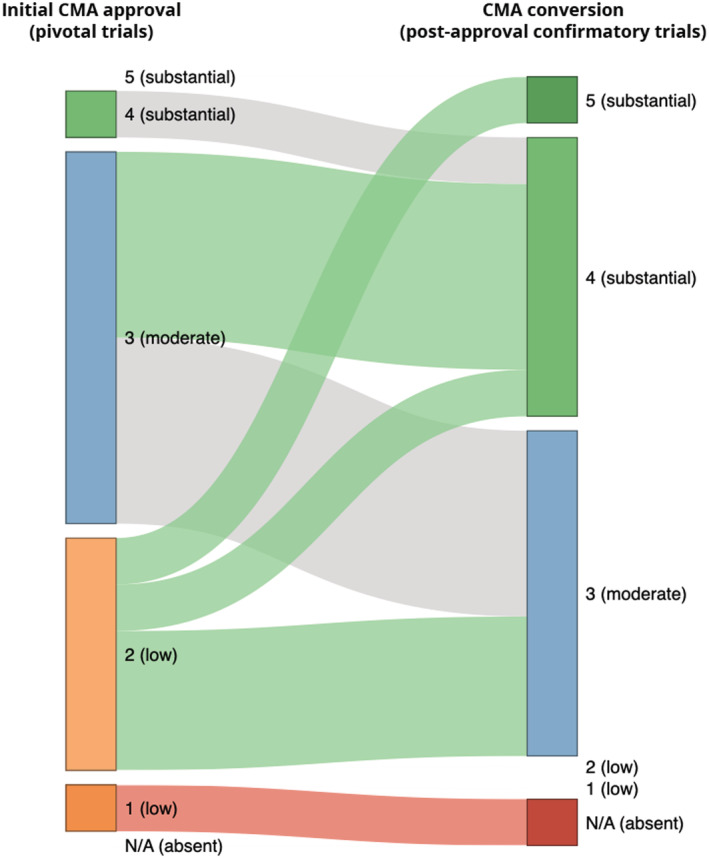
Contribution of post‐approval confirmatory trials to the demonstrated clinical benefit of converted CMA cancer indications, reflected by change in ESMO‐MCBS score Colours of the connections between the time points indicate whether clinical benefit increased (green), remained the same (grey), or decreased (red). ESMO‐MCBS, European Society for Medical Oncology Magnitude of Clinical Benefit Scale
TABLE 4.
Sensitivity analyses including unconverted CMA cancer indications
| Median clinical benefit score (IQR) | P‐value | Substantial clinical benefit (N) | Risk ratio (95% CI) | |
|---|---|---|---|---|
| SMA indications | 3.0 (2.5–4.0) | Ref. | 5/15 (33%) | Ref. |
|
Original analysis Converted CMA indications |
3.0 (3.0–4.0) | 0.31 | 7/15 (47%) | 1.4 (0.57–3.4) |
| Scenario 1 (+ 8 unconverted CMA indications) | 3.0 (3.0–4.0) | 0.79 | 7/23 (30%) | 0.91 (0.35–2.3) |
| Scenario 2 (+ 17 unconverted CMA indications) | 3.0 (3.0–3.0) | 0.87 | 7/32 (22%) | 0.66 (0.25–1.7) |
CI, confidence interval; IQR, interquartile range.
3.4. Sensitivity analyses: Inclusion of unconverted CMA indications
The characteristics of the pivotal trials that supported the initial approval of unconverted and converted CMA indications were similar, with the exception that fewer patients were included in the pivotal trials of unconverted CMA indications (Table 2 and Table S4). This was also reflected by differences in orphan designations (Table 1). In addition, the clinical benefit scores of the two groups of CMA indications were also similar at initial approval: 3.0 (IQR 3.0–3.0) versus 3.0 (IQR 2.0–3.0) for unconverted and converted CMA indications, respectively. Six versus seven percent of indications were associated with substantial clinical benefit (Table 3).
After initial approval, one unconverted CMA indication was found to lack efficacy and subsequently revoked by the European Commission: the soft tissue sarcoma indication of olaratumab (Lartruvo). 16 , 17 In addition, for one indication, no specific obligations had been required—the NSCLC indication of entrectinib—and six indications remained unconverted longer than the median time to conversion of the converted CMA indications (32 months)—those of bosutinib (Bosulif; 93 months), brentuximab vedotin (n = 2; 98 months), cabozantinib (Cometriq; 81 months), ixazomib (Ninlaro; 49 months) and vandetanib (Caprelsa; 106 months). When including these eight indications in the analyses, the median clinical benefit scores of CMA indications remained similar to those of SMA indications, but the point estimate of the association with substantial clinical benefit decreased (RR 0.91, 95% CI 0.35–2.3; Table 4, scenario 1). When including all 17 unconverted CMA indications in the analyses, the point estimate decreased further (RR 0.66, 95% CI 0.25–1.7; Table 4, scenario 2).
4. DISCUSSION
We aimed to compare the availability of evidence and clinical benefit for CMA versus SMA cancer indications in Europe, taking into account the contribution of post‐approval confirmatory trials. Our results indicate that after conversion of CMA cancer indications to SMA, which took place for almost half of all CMA cancer indications, the availability of evidence and the demonstrated clinical benefit is similar to that at initial approval of SMA cancer indications. This was mainly due to the CMA post‐approval confirmatory trials that increased the available evidence and improved the demonstrated clinical benefit as compared to the CMA pivotal trials. If confirmatory evidence for unconverted indications never materializes, the clinical benefit of indications approved through the CMA pathway would at best remain comparable to SMA indications, with one third or less showing substantial benefit.
CMA approval was often supported by single arm trials with ORR as primary endpoint. Consistent with the EMA's CMA guideline, 5 this is indeed less “comprehensive” evidence than the controlled trials with OS or a surrogate survival endpoint that mostly supported SMA approval. Similarly, around 80% of cancer indications granted AA by the FDA are supported by single arm trials and ORR data. 18 However, it should be noted that for our cohort of indications, the types of EMA and FDA approval did not perfectly match.
For most converted CMA indications, a post‐approval confirmatory trial with similar characteristics was conducted. This suggests that “comprehensive evidence” is similarly defined for approval of SMA indications and conversion of CMA indications. Nonetheless, still only few cancer indications were supported by statistically significant increases in OS and QoL, as reported before for both Europe 8 , 19 and the US. 9 , 10 , 19 These are important findings that highlight that regulators' definitions of comprehensive evidence are not necessarily in line with those of physicians, 6 nor patients' 20 perceptions of clinically relevant evidence.
After conversion of CMA indications, there was no difference in demonstrated clinical benefit as compared to SMA indications, although the chance to provide substantial clinical benefit was numerically higher (47% vs 33%). In the sensitivity analyses that included unconverted CMA indications, this increase disappeared (30% vs 33% in scenario 1 that also included all CMA indications for which conversion could be expected and 22% vs 33% in scenario 2 that also included all unconverted CMA indications). Scenario 2 was the most conservative in assuming that none of the 17 unconverted CMA indications would ultimately demonstrate a clinical benefit higher than at their initial approval. This seems unlikely given that nine of the 15 converted CMA indications did demonstrate a higher clinical benefit. Scenario 1, for which an unchanged clinical benefit compared to approval was only assumed for seven indications, including the six indications that remained unconverted longer than 32 months, provides a more likely estimate. Both scenarios considered that the soft tissue sarcoma indication of olaratumab was not associated with any clinical benefit (see also below).
Our findings demonstrate that the context of unmet medical need that is addressed by CMA indications appeared not necessarily associated with a high clinical benefit. Reasons may be that the concept “unmet medical need” is not necessarily defined taking clinical benefit into account, 21 or treatment effects proved smaller than expected. The latter occurred very recently when avelumab was converted to SMA, although durable response rate, ORR and PFS decreased substantially as compared to initial approval. 22 This was deemed acceptable as no other approved drug treatments existed. Therefore, in contexts of unmet medical need, it seems that the existence of a positive treatment effect is ultimately considered more important than its actual size.
Our observation that substantial clinical benefit is demonstrated for one‐third to half of the CMA cancer indications raises the question whether expedited pathways are of added value: is the glass half full or half empty? On the one hand, expedited pathways likely shortened pre‐approval clinical development and thereby time to approval, 18 , 23 probably due to reliance on uncontrolled pivotal trials with ORR as primary endpoint. 18 , 24 Estimates of the degree of shortening range from around 2 years in Europe 23 to 3.5 years in the US, 18 often leading to approvals in the US first. 25 On the other hand, post‐approval evidence generation has been considered insufficient, in both Europe and the US, because of confirmatory trials that are uncontrolled and/or include surrogate endpoints, 4 , 9 , 10 , 26 leaving patients exposed to uncertainties and risks. In addition, cancer indications approved through the FDA's AA pathway have been suggested to be at increased risk for post‐approval safety‐related label changes. 27 However, studies investigating the EMA's CMA pathway did not report similar findings, 23 potentially because oncology dossiers are submitted later to the EMA than the FDA to include additional or more mature evidence. 28
To adequately address uncertainties and identify risks while allowing timely approval of new cancer indications, comprehensive evidence should come available shortly after approval, preferably from RCTs. 24 , 29 To prevent feasibility issues from leading to significantly delayed, downgraded or terminated RCTs, a suggested best practice is that they should be initiated pre‐approval and recruitment should be well underway. 3 , 4 , 9 , 30 Notably, EMA‐ and FDA‐expedited approvals for olaratumab for treatment of soft tissue sarcoma were recently withdrawn because the post‐approval ANNOUNCE trial could not confirm benefits suggested by the pivotal phase II trial. 16 , 17 As recruitment for this RCT was almost completed at initial approval, it could unambiguously inform further regulatory decision‐making relatively shortly after. The olaratumab case concerned the first ever withdrawal of a CMA for such reason and few other withdrawals of AAs for cancer indications are known. 24 However, we recognize that with increasing approvals of drugs based on early phase evidence, unambiguous results from RCTs might not always be available. In these cases, it is imperative that when addressing uncertainties, regulators explicitly draw on available knowledge concerning benefit–risk of, for example, comparable drugs—in the same drug class or with a comparable mechanism—or in comparable patient populations. Moreover, performing an RCT may not always be feasible, especially in the context of (ultra)orphan disease. 31 Perhaps here, the principles underlying the ESMO‐MCBS for single arm trials should be followed, restricting approval to situations where effect estimates are large enough to suggest at least a moderate clinical benefit.
Our study has several strengths. It was the first to comprehensively assess the availability of evidence for a cohort of cancer indications that had received expedited versus non‐expedited approval and that were broadly similar, allowing control for disease characteristics that may affect evidence generation. Also, it was the first to apply ESMO‐MCBS v1.1 to dynamically assess the clinical benefit of a cohort of cancer indications, including those approved on the basis of single‐arm trials, and both at initial approval and after completion of post‐approval confirmatory trials for expedited approvals. This approach may be beneficial for future studies that address other questions regarding evidence generation for cancer drugs. Finally, since we based our assessments of clinical benefit on trial results available in regulatory documents, including the EMA's confidential assessment reports, we were able to determine the impact of regulatory decision‐making on clinical benefit. Notably, although different data cut‐offs and reporting standards between regulatory and scientific data sources may have influenced the availability of trial results that we based our analyses of clinical benefit on, the validation of our assessments of clinical benefit against the scores published by ESMO and EHA showed that 33 of the 35 available scores corresponded (94%). The reasons for divergent scores were our use of: (i) data from predefined analyses 32 rather than retrospective biomarker subgroup analyses published years after initial approval 33 (trial 20 020 408 for the CRC indication of panitumumab), and (ii) EPAR data with an early data cut‐off 34 rather than main trial results that included long‐term follow‐up data 35 (ELOQUENT‐2 trial for the multiple myeloma indication of elotuzumab [Empliciti]).
Our study also has limitations. First, we had to make assumptions about the ultimately demonstrated clinical benefit of yet unconverted CMA cancer indications. However, the results of the sensitivity analysis based on these assumptions supported our main findings. Second, we applied the ESMO‐MCBS to solid and haematological cancer indications. However, although the ESMO‐MCBS has not yet been validated for haematological cancer indications, a recent feasibility study indicated that it was widely applicable to the vast majority of evaluated haematological trials and corresponded with the opinion of clinical experts. 13 Third, we selected the SMA indications based on their similarity to the converted CMA indications. We recognize that it is, by definition, impossible to obtain two perfectly alike groups. For example, we included a Merkel cell cancer CMA indication and a melanoma SMA indication. However, these were the most similar indications when also taking into account characteristics such as orphan designation, pharmacotherapeutic group and moment of approval in the ever‐evolving regulatory and medical landscapes. Fourth, we studied a small cohort of cancer indications of which the majority concerned NSCLC and haematological cancers. While we included all CMA cancer indications that have been approved to date and formal statistical significance testing is thus not necessary when studying their clinical benefit, our findings may not be generalizable to future CMA cancer indications, especially when these comprise different cancer types.
In conclusion, we found that after conversion of CMA cancer indications to SMA, both the availability of evidence and the demonstrated clinical benefit are similar to that at initial approval of SMA cancer indications. This suggests that the definition of the regulatory concept “comprehensive evidence” is similar for cancer indications that received standard and expedited approval. To ensure swift availability of comprehensive evidence, we stress that expedited approvals should preferably be granted only if well‐designed confirmatory RCTs are ongoing.
COMPETING INTERESTS
Dr Sonke reports institutional research support from AstraZeneca, Merck, Novartis and Roche outside the scope of this manuscript. Dr Leufkens reports that he is a member of the Lygature Leadership Team. All remaining authors have declared no conflicts of interest.
CONTRIBUTORS
L.T.B., R.E.B., A.K.M.T. and J.H. designed the study; L.T.B. and R.E.B. collected and analysed the data; L.T.B., R.E.B. and J.H. verified the data; L.T.B., R.E.B., A.K.M.T., M.E.E., G.S.S., O.H.K., H.G.M.L. and J.H. interpreted the data and wrote the manuscript.
OPEN RESEARCH BADGES
This article has earned an Open Data badge for making publicly available the digitally‐shareable data necessary to reproduce the reported results. The data is available at www.ema.europa.eu.
Supporting information
TABLE S1 CMA drugs that were converted as at 31 December 2020, their initially approved indications and specific obligations for post‐approval confirmatory trials
TABLE S2 Included SMA drugs and their initially approved indication
TABLE S3 CMA drugs that were unconverted as at 31 December 2020, their initially approved indications and specific obligations for post‐approval confirmatory trials
TABLE S4 Characteristics of pivotal trials at initial approval and post‐approval confirmatory trials at conversion
ACKNOWLEDGEMENTS
The authors wish to thank dr. B.O. Fabriek (Dutch Medicines Evaluation Board, Utrecht, the Netherlands) for her contribution to the construction of the study cohort, and dr. N.I. Cherny (Shaare Zedek Medical Center, Jerusalem, Israel) and Ms. N.J. Latino (European Society for Medical Oncology) for their insightful comments on how to apply the ESMO‐MCBS.
Bloem LT, Bot RE, Mantel‐Teeuwisse AK, et al. Pre‐approval and post‐approval availability of evidence and clinical benefit of conditionally approved cancer drugs in Europe: A comparison with standard approved cancer drugs. Br J Clin Pharmacol. 2022;88(5):2169-2179. doi: 10.1111/bcp.15141
The views expressed in this article are the personal views of the authors and must not be understood or quoted as being made on behalf of or reflecting the position of the Dutch Medicines Evaluation Board or the European Medicines Agency or any of their committees.
DATA AVAILABILITY STATEMENT
The data are publicly available at www.ema.europa.eu, except for data from post‐approval confirmatory trials for CMA indications. These data were obtained from the EMA's unpublished CMA annual renewal and conversion reports through a Memorandum of Understanding with the Dutch Medicines Evaluation Board.
REFERENCES
- 1. World Health Organization . Global Health Estimates 2016: Disease burden by Cause, Age, Sex, by Country and by Region, 2000–2016. Geneva, Switzerland: World Health Organization; 2018. [Google Scholar]
- 2. Wild CP, Weiderpass E, Stewart BW (Eds). World Cancer Report: Cancer Research for Cancer Prevention. Lyon, France: International Agency for Research on Cancer; 2020. [Google Scholar]
- 3. Bloem LT, Mantel‐Teeuwisse AK, Leufkens HGM, De Bruin ML, Klungel OH, Hoekman J. Postauthorization changes to specific obligations of conditionally authorized medicines in the European Union: a retrospective cohort study. Clin Pharmacol Ther. 2019;105(2):426‐435. [DOI] [PubMed] [Google Scholar]
- 4. Naci H, Smalley KR, Kesselheim AS. Characteristics of preapproval and postapproval studies for drugs granted accelerated approval by the US Food and Drug Administration. JAMA. 2017;318(7):626‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. European Medicines Agency . CHMP Guideline on the scientific application and the practical arrangements necessary to implement Commission Regulation (EC) No 507/2006 on the conditional marketing authorisation for medicinal products for human use falling within the scope of Regulation (EC) No 726/2004 (EMA/CHMP/509951/2006, Rev. 1). 25 February 2016.
- 6. Kesselheim AS, Woloshin S, Eddings W, Franklin JM, Ross KM, Schwartz LM. Physicians' knowledge about FDA approval standards and perceptions of the "breakthrough therapy" designation. JAMA. 2016;315(14):1516‐1518. [DOI] [PubMed] [Google Scholar]
- 7. European Medicines Agency . CHMP Guideline on the evaluation of anticancer medicinal products in man (EMA/CHMP/205/95, Rev. 5). 22 September 2017.
- 8. Davis C, Naci H, Gurpinar E, Poplavska E, Pinto A, Aggarwal A. Availability of evidence of benefits on overall survival and quality of life of cancer drugs approved by European Medicines Agency: retrospective cohort study of drug approvals 2009–13. BMJ. 2017;359:j4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim C, Prasad V. Cancer drugs approved on the basis of a surrogate end point and subsequent overall survival: an analysis of 5 years of US Food and Drug Administration approvals. JAMA Intern Med. 2015;175(12):1992‐1994. [DOI] [PubMed] [Google Scholar]
- 10. Gyawali B, Hey SP, Kesselheim AS. Assessment of the clinical benefit of cancer drugs receiving accelerated approval. JAMA Intern Med. 2019;179(7):906‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cherny NI, Dafni U, Bogaerts J, et al. ESMO‐Magnitude of Clinical Benefit Scale version 1.1. Ann Oncol. 2017;28(10):2340‐2366. [DOI] [PubMed] [Google Scholar]
- 12. Elsallab M, Bravery CA, Kurtz A, Abou‐El‐Enein M. Mitigating deficiencies in evidence during regulatory assessments of advanced therapies: a comparative study with other biologicals. Mol Ther Methods Clin Dev. 2020;18:269‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kiesewetter B, Cherny NI, Boissel N, et al. EHA evaluation of the ESMO‐Magnitude of Clinical Benefit Scale version 1.1 (ESMO‐MCBS v1.1) for haematological malignancies. ESMO Open. 2020;5:e000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. European Medicines Agency . CHMP Assessment Report for Sutent 2006.
- 15. Price TJ, Peeters M, Kim TW, et al. Panitumumab versus cetuximab in patients with chemotherapy‐refractory wild‐type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open‐label, non‐inferiority phase 3 study. Lancet Oncol. 2014;15(6):569‐579. [DOI] [PubMed] [Google Scholar]
- 16. Tap WD, Wagner AJ, Schöffski P, et al. Effect of doxorubicin plus olaratumab vs doxorubicin plus placebo on survival in patients with advanced soft tissue sarcomas: The ANNOUNCE randomized clinical trial. JAMA. 2020;323(13):1266‐1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Herold R, Camarero J, Melchiorri D, Sebris Z, Enzmann H, Pignatti F. Revocation of the conditional marketing authorisation of a cancer medicine: the olaratumab experience. Eur J Cancer. 2019;123:25‐27. [DOI] [PubMed] [Google Scholar]
- 18. Yamashita K, Kaneko M, Narukawa M. Regulatory characteristics and pivotal study design of US Food and Drug Administration approval of drugs for major vs. minor cancer. Eur J Clin Pharmacol. 2019;75(9):1193‐1200. [DOI] [PubMed] [Google Scholar]
- 19. Salcher‐Konrad M, Naci H, Davis C. Approval of cancer drugs with uncertain therapeutic value: a comparison of regulatory decisions in Europe and the United States. Milbank Q. 2020;98(4):1219‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schwartz LM, Woloshin S. Communicating uncertainties about prescription drugs to the public: a national randomized trial. Arch Intern Med. 2011;171(16):1463‐1468. [DOI] [PubMed] [Google Scholar]
- 21. Vreman RA, Heikkinen I, Schuurman A, et al. Unmet medical need: an introduction to definitions and stakeholder perceptions. Value Health. 2019;22(11):1275‐1282. [DOI] [PubMed] [Google Scholar]
- 22. European Medicines Agency . CHMP Assessment Report for Bavencio (EMA/496529/2017). 20 July 2017.
- 23. Boon WP, Moors EH, Meijer A, Schellekens H. Conditional approval and approval under exceptional circumstances as regulatory instruments for stimulating responsible drug innovation in Europe. Clin Pharmacol Ther. 2010;88(6):848‐853. [DOI] [PubMed] [Google Scholar]
- 24. Beaver JA, Howie LJ, Pelosof L, et al. A 25‐year experience of US Food and Drug Administration accelerated approval of malignant hematology and oncology drugs and biologics: a review. JAMA Oncol. 2018;4(6):849‐856. [DOI] [PubMed] [Google Scholar]
- 25. Uyl‐de Groot CA, Heine R, Krol M, Verweij J. Unequal access to newly registered cancer drugs leads to potential loss of life‐years in Europe. Cancers (Basel). 2020;12(8):2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Banzi R, Gerardi C, Bertele V, Garattini S. Conditional approval of medicines by the EMA. BMJ. 2017;357:j2062. [DOI] [PubMed] [Google Scholar]
- 27. Mostaghim SR, Gagne JJ, Kesselheim AS. Safety related label changes for new drugs after approval in the US through expedited regulatory pathways: retrospective cohort study. BMJ. 2017;358:j3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kashoki M, Hanaizi Z, Yordanova S, et al. A comparison of EMA and FDA decisions for new drug marketing applications 2014–2016: concordance, discordance, and why. Clin Pharmacol Ther. 2020;107(1):195‐202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gyawali B, Kesselheim AS. Reinforcing the social compromise of accelerated approval. Nat Rev Clin Oncol. 2018;15(10):596‐597. [DOI] [PubMed] [Google Scholar]
- 30. Woloshin S, Schwartz LM, White B, Moore TJ. The fate of FDA postapproval studies. N Engl J Med. 2017;377(12):1114‐1117. [DOI] [PubMed] [Google Scholar]
- 31. Kesselheim AS, Myers JA, Avorn J. Characteristics of clinical trials to support approval of orphan vs nonorphan drugs for cancer. JAMA. 2011;305(22):2320‐2326. [DOI] [PubMed] [Google Scholar]
- 32. European Medicines Agency . CHMP Assessment Report for Vectibix (EMEA/CHMP/597531/2007). 19 December 2007.
- 33. Peeters M, Oliner KS, Parker A, et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res. 2013;19(7):1902‐1912. [DOI] [PubMed] [Google Scholar]
- 34. European Medicines Agency . CHMP Assessment Report for Empliciti (EMA/129497/2015). 28 January 2016.
- 35. Dimopoulos MA, Lonial S, Betts KA, et al. Elotuzumab plus lenalidomide and dexamethasone in relapsed/refractory multiple myeloma: extended 4‐year follow‐up and analysis of relative progression‐free survival from the randomized ELOQUENT‐2 trial. Cancer. 2018;124(20):4032‐4043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 CMA drugs that were converted as at 31 December 2020, their initially approved indications and specific obligations for post‐approval confirmatory trials
TABLE S2 Included SMA drugs and their initially approved indication
TABLE S3 CMA drugs that were unconverted as at 31 December 2020, their initially approved indications and specific obligations for post‐approval confirmatory trials
TABLE S4 Characteristics of pivotal trials at initial approval and post‐approval confirmatory trials at conversion
Data Availability Statement
The data are publicly available at www.ema.europa.eu, except for data from post‐approval confirmatory trials for CMA indications. These data were obtained from the EMA's unpublished CMA annual renewal and conversion reports through a Memorandum of Understanding with the Dutch Medicines Evaluation Board.