

Abstract
Abdominal aortic aneurysms (AAAs) elicit massive inflammatory leukocyte recruitment to the aorta. CD4+ T cells, which include regulatory T cells (Tregs) and conventional T cells (Tconvs), are involved in the progression of AAA. Tregs have been reported to limit AAA formation. However, the function and phenotype of the Tconvs found in AAAs remain poorly understood. We characterized aortic Tconvs by bulk RNA sequencing and discovered that Tconvs in aortic aneurysm highly expressed Cxcr6 and Csf2. Herein, we determined that the CXCR6/CXCL16 signaling axis controlled the recruitment of Tconvs to aortic aneurysms. Deficiency of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), encoded by Csf2, markedly inhibited AAA formation and led to a decrease of inflammatory monocytes, due to a reduction of CCL2 expression. Conversely, the exogenous administration of GM‐CSF exacerbated inflammatory monocyte infiltration by upregulating CCL2 expression, resulting in worsened AAA formation. Mechanistically, GM‐CSF upregulated the expression of interferon regulatory factor 5 to promote M1‐like macrophage differentiation in aortic aneurysms. Importantly, we also demonstrated that the GM‐CSF produced by Tconvs enhanced the polarization of M1‐like macrophages and exacerbated AAA formation. Our findings revealed that GM‐CSF, which was predominantly derived from Tconvs in aortic aneurysms, played a pathogenic role in the progression of AAAs and may represent a potential target for AAA treatment.
Keywords: abdominal aortic aneurysm, CXCR6, GM‐CSF, Tconvs
Abbreviations
- AAAs
abdominal aortic aneurysms
- GM‐CSF
granulocyte‐macrophage colony‐stimulating factor
- IRF5
interferon regulatory factor 5
- LNs
lymph nodes
- Tconvs
conventional T cells
- Th
T helper cells
- Tregs
regulatory T cells
1. INTRODUCTION
Abdominal aortic aneurysms (AAAs) are progressive enlargements of the infrarenal aortas to diameters 1.5‐times larger than normal or an enlargement of the abdominal aortas to a diameter ≥3 cm. 1 The main complication of AAAs is aortic rupture, which is estimated to cause 150,000–200,000 deaths each year worldwide. 2 , 3 At present, open or endovascular surgery is the only widely used treatment for this condition, and efficient medical treatments for AAAs are still lacking. Hence, it is important to understand the cellular and molecular mechanisms underlying the pathogenesis of AAAs.
The histological features of AAAs include smooth muscle cell apoptosis, elastin fragmentation, ae well as chronic adventitial and medial inflammatory cell infiltration. 4 Inflammation is considered to play a crucial pathogenic role in AAA formation. Chemokines are important for the directed recruitment of immune cells to the sites of inflammation. 5 , 6 Currently, little is known about how conventional CD4+ T cells (Tconvs) are attracted to the site of the aorta in AAAs. CXCR6, also known as Bonzo, STRL33 or TYMSTR, was originally described as a coreceptor of human immunodeficiency virus‐1 and simian immunodeficiency virus 7 ; however, it was subsequently found to promote the homing of lymphocytes to nonlymphoid tissues. 8 Deficiency of CXCR6 in apoE−/− mice resulted in reduced T cell numbers and macrophage infiltration within the lesion, decreasing atherosclerosis. 9 A requirement for CXCR6 in the progression of AAA, and in particular, its influence on Tconv recruitment to the aortas, has not been established.
Previous animal studies have reported that in the absence of CD4+ T cells, mice were resistant to the induction of aneurysm. 10 Regarding to the roles of CD4+ T cell subsets in AAA formation, the most extensively investigated and consistent finding has been that T regulatory cells (Tregs) protect against AAA development by decreasing inflammation. 11 However, the role of Tconvs in AAA development remains controversial. 10 , 12 , 13 , 14 Activated CD4+ T cells secrete a set of cytokines depending on the site of inflammation. 15 Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF), encoded by Csf2, was originally described to play a role in hematopoiesis, and it can stimulate stem cells to differentiate into granulocytes (neutrophils, eosinophils, and basophils) and monocytes. 16 Recent work has indicated that GM‐CSF is largely produced by T cells and then sensed exclusively by myeloid cells, positioning GM‐CSF as a critical molecule that links the adaptive and innate arms of the immune system. 17 , 18 , 19 Several findings had shown that specific transcription factor defects lead to upregulation of GM‐CSF expression in cells, exacerbating vascular diseases. 20 , 21 Eiketsu et al. suggested that GM‐CSF expression in experimental aneurysms was correlated with aneurysm diameters in humans. 22 However, the precise mechanism by which GM‐CSF, especially which produced by Tconvs, functions in AAA development remains elusive.
In the present study, we demonstrated that Tconvs were recruited to the aortas through the CXCR6/CXCL16 axis and expressed high levels of GM‐CSF in aortic aneurysms. Furthermore, our results showed that GM‐CSF exacerbated inflammatory monocyte infiltration into the aorta and induced the polarization of macrophages toward the M1 phenotype, worsening AAA severity.
2. METHODS
2.1. Mice
Male C57BL/6J (wild‐type, WT) and CD45.1 mice aged 8–12 weeks were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). Foxp3‐GFP‐expressing mice on a C57BL/6 background (stock# 006772) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mice deficient in GM‐CSF (Csf2−/−) were kindly provided by Dr Benkang Shi from the Department of Urology, Qilu Hospital of Shandong University (Shandong, China). CXCR6GFP/GFP mice were kindly provided by Professor Hong Zhou (The First Affiliated Hospital of USTC, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, China). As male mice are more susceptible to AAA formation than female mice, 23 male mice were used for AAA induction or cell isolation. All the male mice were housed, and the experiments were conducted following procedures approved by the guidelines of the Institutional Animal Care and Use Committee of Huazhong University of Science and Technology and performed in accordance with the guidelines from Directive 2010/63/EU of the European Parliament on the protection of animals used for scientific purposes.
2.2. Abdominal aortic aneurysms models
2.2.1. Elastase model
The periadventitial elastase model was used to establish experimental AAA in mice at the age of 8 to 12 weeks as previously described. 24 Briefly, the mice were anesthetized by the injection of pentobarbital sodium (50 mg/kg) intraperitoneally, and the infrarenal aortas were exposed and isolated from the renal vein to the iliac bifurcation. After anatomical identification, the aortas were bathed in 10 μl of 100% porcine pancreatic elastase (PPE; Sigma‐Aldrich Co., St. Louis, MO) or heat‐inactivated elastase (control) for 10 min. After exposure to elastase, the abdominal contents were replaced, and the incision was closed with a 4–0 suture. The mice were placed in a recovery area with thermal support until full recovery. After 7 or 14 days of surgery, mouse was euthanized with intraperitoneal sodium pentobarbital (200 mg/kg), and the abdominal aortas were collected.
2.2.2. Calcium phosphate (CaPO4) model
AAA was induced by CaPO4 in mice at the age of 8 to 12 weeks as previously described. 25 The mice were anesthetized by the injection of pentobarbital sodium (50 mg/kg) intraperitoneally, and the infrarenal aortas were exposed and isolated from the renal vein to the iliac bifurcation. A small piece of gauze soaked in 0.5 M CaCl2 was applied around the aorta. After 10 min, the gauze was replaced with another piece of PBS‐soaked gauze, which was allowed to remain in place for 5 min. The control mice were similarly treated with 0.5 M NaCl for 15 min. Sham‐operated mice were used as experimental controls. The mice were placed in a recovery area with thermal support until full recovery. After 7 days of surgery, mouse was euthanized with intraperitoneal sodium pentobarbital (200 mg/kg), and the abdominal aortas were collected.
2.3. Flow cytometry assay
To analyze the mouse leukocytes, the mice were euthanized by CO2 overdose with a displacement rate of 50% chamber volume per minute; and then, peripheral blood cells were collected; spleen were mechanically crushed and filtered to obtain single‐cell suspensions; bone marrow (BM) cells were harvested by flushing the femurs; and infrarenal aortas were used to obtain enzyme‐digested aortic single‐cell suspension; these cell populations were subjected to flow cytometric analysis. For intracellular cytokine staining, the cells were cultured in media containing the indicated cytokines in the presence of the Cell Stimulation Cocktail for 4 hr at 37°C in a 5% CO2 incubator. The cells were first stained with antibodies against surface markers for 30 min on ice, and then, the cells were fixed, permeabilized, and stained with antibodies against intracellular antigens before analysis by flow cytometry (FACS Aria III, BD).
2.4. RNA‐sequencing analysis
Single‐cell suspensions were produced from the spleen and infrarenal aortas of AAA mice for Tconv analysis, 26 which was validated in advs.202104338. RNA was isolated with an miRNeasy Micro kit (Qiagen) according to the manufacturer’s protocol. Amplified cDNA was prepared using the SMART‐Seq V4 ultralow input RNA kit (Takara) according to the manufacturer’s instructions. Sequencing libraries were generated using the NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, USA) following the manufacturer’s recommendations, and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly‐T oligo‐attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEBNext First‐Strand Synthesis Reaction Buffer (5×). First‐strand cDNA was synthesized using random hexamer primers and M‐MuLV reverse transcriptase (RNase H−). Second‐strand cDNA synthesis was subsequently performed using DNA polymerase I and RNase H. The remaining overhangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of the 3′ ends of DNA fragments, NEBNext adaptors with hairpin loop structures were ligated to prepare for hybridization. To preferentially select cDNA fragments that were preferentially 250~300 bp in length, the library fragments were purified with the AMPure XP system (Beckman Coulter, Beverly, USA). Then, size‐selected, adaptor‐ligated cDNA was incubated with 3 µl of USER Enzyme (NEB, USA) at 37°C for 15 min followed by 95°C for 5 min before PCR. Then, PCR was performed with Phusion high‐fidelity DNA polymerase, universal PCR primers and Index (X) Primer. Finally, the PCR products were purified (AMPure XP system), and the library quality was assessed with the Agilent Bioanalyzer 2100 system.
Clustering of the index‐coded samples was performed on a cBot Cluster Generation System using the TruSeq PE Cluster Kit v3‐cBot‐HS (Illumina) according to the manufacturer’s instructions. After cluster generation, the libraries were sequenced on an Illumina platform, and 125‐bp to 150‐bp paired‐end reads were generated.
Differential expression analysis of two conditions/groups (two biological replicates per condition) was performed using the DESeq2 R package (1.1 6.1). DESeq2 provides statistical routines for identifying differential expression in digital gene expression data using a model based on the negative binomial distribution. The resulting p‐values were adjusted using Benjamini and Hochberg’s approach for controlling the false discovery rate. Genes with a p‐value <.05 as determined by DESeq2 were considered to be differentially expressed.
2.5. Parabiosis experiments
Mice were joined by parabiosis as previously described. 27 The mice were anesthetized with 2% isoflurane and injected twice daily with buprenorphine (0.1 mg/kg i.p.) for 3 days, starting on the day of the surgery. Experiments began 14 days after the parabiosis surgery, as this was the time required to establish a shared circulation.
2.6. Treatments
To block CXCL16, mice were treated with intraperitoneal injections of 100 μg of monoclonal rat anti‐mouse CXCL16 neutralizing antibody every 3 days (R&D Systems, Clone #142417, Germany), and the control group was treated with an isotype control Ab. Recombinant murine GM‐CSF (1.5 μg/mouse) or vehicle solution (PBS) was administered daily by intraperitoneal injection for 2 weeks after the induction of AAA.
2.7. Adoptive transfer
For Tconvs transfers, CD4+CD25− Tconvs were isolated from the spleens of Csf2−/− mice or WT mice by flow cytometry. A total of 5 × 106 WT or Csf2−/− Tconvs were intravenously (i.v.) injected via the tail vein into each Csf2−/− recipient mouse, and at the same time, a PPE‐induced AAA model was established in the recipient mice.
2.8. Elisa
CCL2 and GM‐CSF levels were measured with ELISA kits according to the manufacturer’s instructions (NeoBioscience Technology).
2.9. Histological and immunohistochemicalanalyses
After perfusion and fixation with 4% paraformaldehyde, the infrarenal abdominal aortas from the experimental mice were processed for paraffin embedding. Serial sections (4 μm) were prepared by cutting the abdominal aorta into two equal halves and sectioning throughout the tissue. Abdominal aorta sections were subjected to hematoxylin and eosin and Verhoeff‐Van Gieson staining at regular intervals (200 μm) to detect elastic fibers for the histoarchitectural evaluation of aneurysms as previously described. The serial tissue sections obtained from these mice were further subjected to immunohistochemistry. For immunohistochemistry, the abdominal aortas were stained with antibodies against CD68, CD3, Mmp2 and Mmp9 as described. All the staining was imaged under an OLYMPUS BX51 microscope. The intensity of the immunostaining was evaluated as previously described by obtaining at least four images from random areas of interest from each tissue. The immunostaining was quantified using ImageJ. All the examinations were conducted by two independent investigators, and images were blinded to reduce bias during quantification.
2.10. Murine bone marrow–derived macrophage isolation and differentiation
Six‐ to eight‐week‐old mice (C57BL/6) were euthanized by CO2 overdose with a displacement rate of 50% chamber volume per minute, and femurs were isolated. BM cells were obtained by flushing the femurs. The cells were differentiated (7 days) in Dulbecco’s modified Eagle’s medium (Sigma‐Aldrich, St. Louis, MO, USA) supplemented with 25 ng ml−1 M‐CSF (Peprotech Inc, 315‐02), 10% heat‐inactivated fetal bovine serum, and antibiotics and cultured at 37°C in a 5% CO2 incubator. To obtain differentiated macrophages, 5 ng/ml macrophage colony‐stimulating factor (Peprotech Inc, 315‐02) was added every 2 days for 6 days.
2.11. IRF5 siRNA transfection in bone marrow–derived macrophages
The siRNA sequence targeting t mouse IRF5 (siRNA sequence, 5′‐GAAGAATGGCCTGATGTCA‐3′) and the negative control sequence (NC) were purchased from Guangzhou RiboBio. IRF5‐siRNA and NC were transfected into the BMDMs via Lipofectamine 3000 (Invitrogen). Western blotting analysis and qPCR were used to evaluate the siRNA efficiency.
2.12. Western blot
Tissue was harvested for analysis from the suprarenal aorta, and the tissues included aneurysms if present. Briefly, protein lysates were obtained from the corresponding regions of the aneurysmal tissue. Tissue lysates were separated by electrophoresis on SDS‐polyacrylamide gels and transferred to PVDF (polyvinylidene fluoride) membranes. The membranes were immunoblotted with antibodies against CXCL16 (1:1000; Proteintech; 60123‐1‐Ig), IRF5 (1:1000; Abcam; ab181553) and CCL2 (1:1000; Proteintech; 66272‐1‐Ig) at 4°C overnight. The blots were washed three times and incubated with HRP‐conjugated species‐appropriate secondary antibodies (1:3000) for 2 h at room temperature. An enhanced chemiluminescence kit (Pierce, Rockford, IL) was used to detect all the protein bands. The expression of glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) was detected as a loading control. The expression levels of the proteins were quantified by scanning densitometry and normalized to the expression level of GAPDH.
2.13. Quantitative PCR
Briefly, tissue and cellular mRNA was extracted using TRIzol reagent (No. 15596018; Invitrogen, Carlsbad, CA). mRNA was reverse transcribed into cDNA with the PrimeScript RT Reagent Kit (No. RR064B; Takara Biotechnology, Dalian, China). Real‐time RT‐PCR was performed using SYBR Green Master Mix (No. RR066A; Takara, Japan) on an ABI PRISM 7900 Sequence Detector System (Applied Biosystems, Foster City, CA). The primers used were as follows: MMP2: 5′‐CAAGTTCCCCGGCGATGTC‐3′ 5′‐TTCTGGTCAAGGTCACCTGTC‐3′; MMP9: 5′‐CTGGACAGCCAGACACTAAAG‐3′ 5′‐CTCGCGGCAAGTCTTCAGAG‐3′; CCL2: 5′‐CTTCTGGGCCTGCTGTTCA‐3′ 5′‐CCAGCCTACTCATTGGGATCA‐3′; iNOS: 5′‐GTTCTCAGCCCAACAATACAAGA‐3′ 5′‐GTGGACGGGTCGATGTCAC‐3′; TNF‐α: 5′‐CCCTCACACTCAGATCATCTTCT‐3′ 5′‐GCTACGACGTGGGCTACAG‐3′; IL‐6: 5′‐TAGTCCTTCCTACCCCAATTTCC‐3′ 5′‐TTGGTCCTTAGCCACTCCTTC‐3′; CD206: 5′‐CTCTGTTCAGCTATTGGACGC‐3′5′‐CGGAATTTCTGGGATTCAGCTTC‐3′; Fizz1: 5′‐CCAATCCAGCTAACTATCCCTCC‐3′5′‐ACCCAGTAGCAGTCATCCCA‐3′; Arg1: 5′‐CAGAAGAATGGAAGAGTCAG‐3′5′‐CAGATATGCAGGGAGTCACC‐3′; IRF5: 5′‐ AGAGACAGGGAAGTACACTGAAG‐3′5′‐TGGAAGTCACGGCTTTTGTTAAG‐3′ and GAPDH: 5′‐GCAGTGGCAAAGTGGAGATT‐3′5′‐CGCTCCTGGAAGATGGTGAT‐3′. In all cases, the PCR conditions were 95°C for 5 min, 95°C for 30 s, 60°C for 30 s, 72°C for 30 s, and 72°C for 5 min with 40 cycles of amplification.
2.14. Statistical analysis
The data are expressed as the mean ± SEM. For two‐group comparisons, an unpaired t test was applied to normally distributed variables. The statistical significance of differences between more than two groups was calculated using one‐ or two‐way ANOVA followed by the Bonferroni posttest when the data were normal distribution. The nonparametric tests, such as Kruskal–Wallis test or Mann–Whitney U test, were used when the data were abnormal distribution. The data were analyzed, and the graphs were prepared using Prism 8.0 (GraphPad software, San Diego, CA, USA). Differences were considered statistically significant at p < .05.
3. RESULTS
3.1. Aorta infiltrating Tconvs exhibit distinct transcriptomes during the development of abdominal aortic aneurysm
A large number of Tconvs infiltrated the aortas, and the numbers of infiltrating Tconvs peaked 7 days after PPE‐mediated induction of AAA (Figure 1A). Ki67 staining assays revealed that the proliferation of vascular Tconvs was higher than Tconvs under basal conditions and in peripheral circulation (Figure 1B). To comprehensively elucidate the characteristics of Tconvs found in aortic aneurysms and how they affect the progression of AAA, RNA sequencing analysis was performed on Tconvs from the aneurysmal aortic wall and from the spleen counterparts. We identified 1061 differentially expressed genes (DEGs), including 469 upregulated DEGs and 592 downregulated DEGs (Figure 1C). Gene ontology analysis revealed that the DEGs in aortic Tconvs were primarily enriched in chemokine‐mediated signaling pathways, cytokine‐mediated signaling pathways, inflammatory response, and positive regulation of leukocyte migration (Figure 1D). KEGG pathway enrichment analysis indicated that these DEGs were involved in the cytokine–cytokine receptor interaction pathway (Figure 1E). After extracting genes from the KEGG term “cytokine–cytokine receptor interaction pathway (mmu04060),” inflammatory cytokines such as Csf2, Il22, Il5, Il15, and Il18, were found to be highly expressed in the aorta Tconvs (Figure 1F), and the high expression of these cytokines might be associated with Tconv‐dependent inflammatory responses in AAAs. Moreover, we also observed that a series of chemotaxis‐related genes were expressed by aortic Tconvs (Figure 1F), which possibly revealed the mechanism by which Tconvs migrated to the aortic aneurysms. Altogether, these results revealed that Tconvs proliferated in aortic aneurysms and had different transcriptional profiles compared with those of their splenic counterparts.
FIGURE 1.
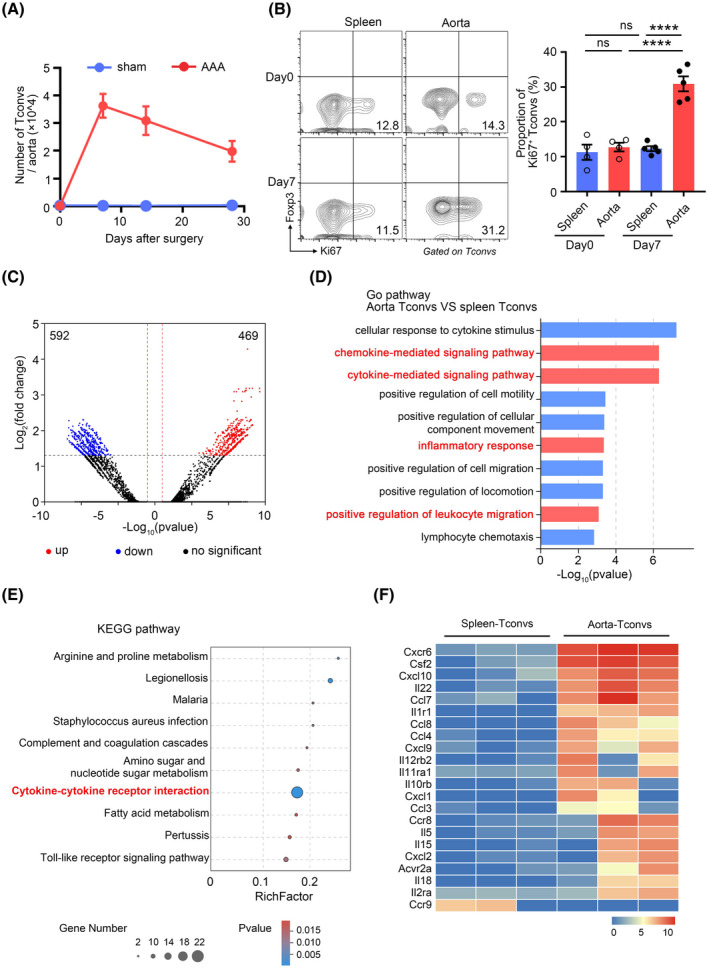
Tconvs infiltrate aortic aneurysms and exhibit a different transcriptome than the splenic Tconvs. (A) The numbers of Tconvs in the aortic aneurysms at day 0, day 7, day 14, and day 28 after abdominal aortic aneurysms (AAA) or sham operation. n = 4–5 per group. (B) Representative flow cytometry plots (left) and quantification (right) of Ki67 expression by Tconvs in aortic aneurysms and spleens 7 days after AAA. n = 4–5 per group. ****p < .0001 according to one‐way ANOVA. (C) RNA‐seq volcano plot of differential gene expression between Tconvs in aortic aneurysms and spleens. (D) The bar plot presents the enrichment scores (−Log10(p value)) of the top 10 significantly enriched Gene Ontology terms. (E) Top 10 enrichment scores of KEGG pathway enrichment analysis. The rich factor represents the degree of enrichment of the differentially expressed genes. The p‐value is represented by a color scale. (F) Heat map of the indicated gene (KEGG Term “Cytokine–cytokine receptor interaction” [KEGG: mmu04060]) expression of Tconvs in aortic aneurysms and spleens
3.2. The CXCR6/CXCL16 axis is necessary for the recruitment of Tconvs to aortic aneurysms
CD4+ T cells can be activated and induced to express chemokine receptors in lymph node (LN), then, these cells exit the LN via efferent vessels and are guided by chemokines to sites of infection and inflammation to perform their immune functions. 28 Additionally, it has also been proposed that naïve CD4+ T cells are constitutively present in the aorta and can be directly primed in the vessel wall. 29 To determine the source of the Tconvs in AAA, we administered the drug FTY720 to block the migration of Tconvs from the LN into circulation (Figure S1A). The results indicated that FTY720 treatment significantly reduced Tconvs in aortic aneurysms, which suggested that recruitment from circulation contributed to the accumulation of Tconvs in aortic aneurysms (Figure S1B). Furthermore, we conducted a parabiosis experiment to determine the extent of Tconvs in the aortic aneurysms that originated from the circulation (Figure S1C). We confirmed that approximately 76.5% of the Tconvs in aneurysms were recruited from peripheral circulation (Figure S1D). Therefore, the results described above showed that Tconvs in aortic aneurysms were mainly recruited from peripheral circulation.
To elucidate the mechanism underlying the infiltration of Tconvs into the aortic aneurysms, we analyzed the changes in the expression of chemokine receptors by Tconvs and found that Cxcr6 was the most significantly upregulated DEG in aortic Tconvs (Figure 2A). Flow cytometry assays confirmed that Tconvs in aortic aneurysms expressed higher levels of CXCR6 than those in the spleen (Figure 2B). During the progression of AAAs, expression of CXCL16, the only ligand of CXCR6, gradually increased in the aneurysm aorta at the transcript (Figure 2C) and protein levels (Figure 2D). To explore the cellular source of CXCL16 in aortic aneurysms, we analyzed single‐cell gene expression profiles (GSE152583) from aortic aneurysms in mice with PPE‐induced AAA. 30 It revealed that CXCL16 might mainly come from macrophages in AAA (Figure S2A). To confirm it, first, CD45+ cells and CD45− cells were isolated from aortic aneurysms and the expression of CXCL16 was further confirmed by RT‐PCR. Our results revealed that the expression of CXCL16 in CD45+ cells was higher than that in CD45− cells (Figure S2B). Among the CD45+ cells, we further sorted macrophages and non‐macrophages and found that the macrophages were the main source of CXCL16 in aneurysm tissue (Figure S2C). Thus, our results revealed that macrophages were the main cellular source of CXCL16 in aortic aneurysm tissue at day 7 post‐AAA. Then, we speculated that the CXCR6/CXCL16 axis might be responsible for the recruitment of Tconvs into the aneurysm aorta from circulation. To verify this hypothesis, we treated mice with PEE‐induced AAA with an anti‐CXCL16 antibody to block CXCL16. Anti‐CXCL16 antibody‐treated mice showed a marked reduction in the numbers of Tconvs in aortic aneurysms (Figure 2E). Furthermore, blocking CXCL16 mainly affected CXCR6+ Tconvs, the proportion of which was reduced in aortic aneurysms (Figure 2F). Similar to anti‐CXCL16 antibody treatment, CXCR6‐deficient mice (CXCR6GFP/GFP) exhibited decreased Tconvs in aortic aneurysms (Figure 2G). The aortic diameter was markedly smaller in CXCR6GFP/GFP mice than in CXCR6GFP/+ mice (Figure 2H) which was probably due to the effect of Tconvs. Taken together, these results suggested that the CXCR6/CXCL16 axis was necessary for the recruitment of Tconvs into aortic aneurysms.
FIGURE 2.
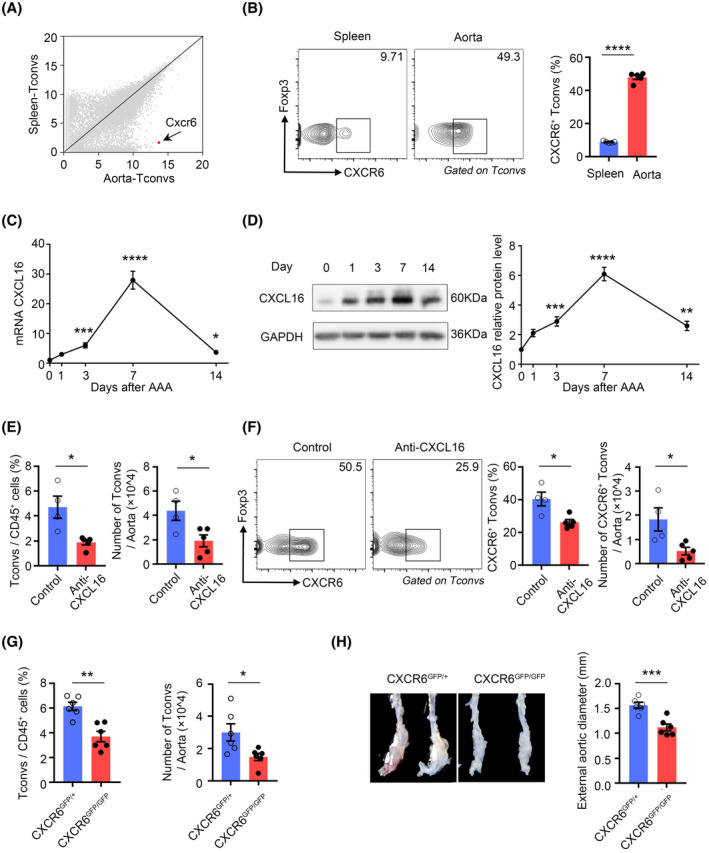
The CXCL16/ CXCR6 axis mediates the recruitment of Tconvs in abdominal aortic aneurysm (AAA). (A) Scatter plots comparing gene expression quantified by RNA‐sequencing of aortic aneurysm Tconvs versus splenic Tconvs. The Cxcr6 gene is highlighted in red. (B) Representative flow cytometry plots (left) and quantification (right) of CXCR6 expression of Tconvs in aortic aneurysms and spleens 7 days after AAA induction. n = 5 per group. ****p < .0001 according to unpaired two‐tailed t test. (C) CXCL16 transcripts in aortic aneurysms were quantified by RT‐PCR on day 0, day 1, day 3, day 7, and day 14 after AAA induction. n = 8–12 per group. *p < .05, ***p < .001, ****p < .0001 versus day 0 according to Kruskal–Wallis test. (D) CXCL16 protein levels in aortic aneurysms were quantified by western blotting at day 0, day 1, day 3, day 7, and day 14 after AAA induction. Left: representative immunoblot. Right: summary data. n = 6 per group. **p < .01, ***p < .001, ****p < .0001 versus day 0 according to one‐way ANOVA. (E) Proportion (left) and number (right) of Tconvs in aortic aneurysms after treatment with IgG (control) or CXCL16 antibody. n = 4–5 per group. *p < .05 according to Mann–Whitney test on proportion and unpaired two‐tailed t test on number. (F) Representative flow cytometry plots (left) and quantification (right) of CXCR6+ Tconvs in aortic aneurysms. n = 4–5 per group. *p < .05 according to unpaired two‐tailed t test. (G) Proportion (left) and number (right) of Tconvs in aortic aneurysms of CXCR6GFP/+ and CXCR6GFP/GFP mice at day 7 after AAA induction. n = 6 per group. *p < .05, **p < .01 according to unpaired two‐tailed t test. (H) Representative pictures (left) of abdominal aortic fragments and quantification of maximal diameters (right) from CXCR6GFP/+ and CXCR6GFP/GFP mice 2 weeks after porcine pancreatic elastase –induced AAA. n = 6 per group. ***p < .001 according to unpaired two‐tailed t test
3.3. Tconvs are the major source ofgranulocyte‐macrophage colony‐stimulating factor (GM‐CSF) within aortic aneurysms
After elucidating the mechanism of Tconvs infiltration, we further explored the function of these cells in AAAs. When comparing the transcriptomes of splenic Tconvs and aortic Tconvs, it was revealed that aortic Tconvs exhibited significantly increased expression of Csf2 (Figure 3A). The cytokine GM‐CSF, which is encoded by Csf2, was initially classified as a hematopoietic growth factor. However, the majority of myeloid cells do not require GM‐CSF for steady‐state myelopoiesis. Instead, under inflammatory conditions, GM‐CSF serves as a conduit of communication between tissue‐infiltrating lymphocytes and myeloid cells. 31 To verify the expression of GM‐CSF by aortic Tconvs, we performed RT‐PCR assays, and the results revealed that aortic Tconvs expressed significantly higher levels of Csf2 than splenic Tconvs (Figure 3B). Flow cytometry analysis further confirmed the enhanced expression of GM‐CSF by aortic aneurysm Tconvs compared with splenic and control aortic Tconvs (Figures S3A and 3C). Next, we sorted Tconvs from aneurysm aortic wall and spleen by FACS 7 days after PPE‐induced AAA. The secretion of GM‐CSF in the medium supernatant was detected by ELISA. In consistent with the flow cytometry results, Tconvs from aortic aneurysm secreted more GM‐CSF than the spleen (Figure S3B). Although T cells appear to be the most prominent producers of GM‐CSF in inflammation tissue, numerous other sources, such as parenchyma cells, B cells and myeloid cells et al., have also been described. 32 To examine whether other cells were potential sources of GM‐CSF in AAAs, we performed flow cytometry to detect the source of GM‐CSF in aortic aneurysms. The result showed that GM‐CSF was mainly produced by CD45+ cells in AAAs and that CD4+ cells were the major producers of GM‐CSF among CD45+ cells at day 7 (Figure 3D). Our results revealed that Tregs in aneurysm aorta barely expressed GM‐CSF (Figure 3E). Thus, these data indicated that Tconvs were the predominant source of GM‐CSF in AAAs.
FIGURE 3.
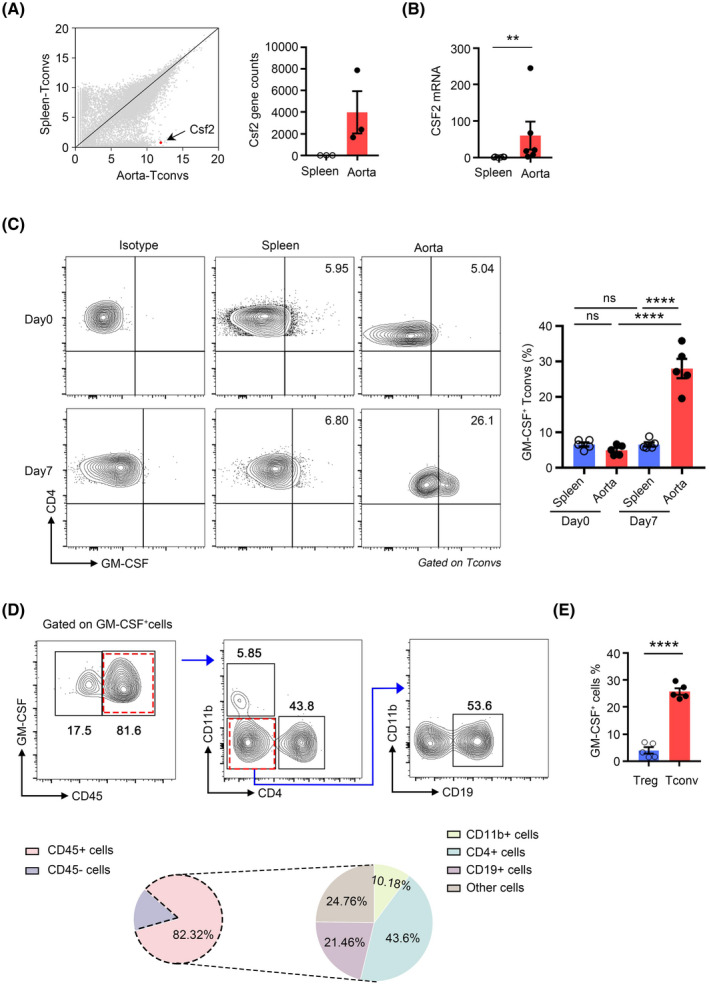
Tconvs are potent producers of granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) during abdominal aortic aneurysm (AAA) formation. (A) Right: Scatter plots comparing the quantification of gene expression by RNA‐sequencing of aortic aneurysm Tconvs versus splenic Tconvs. The Csf2 gene is highlighted in red. Left: Raw gene counts of the Csf2 transcript were quantified by RNA sequencing. (B) CSF2 transcripts of Tconvs in aortic aneurysms and spleen were quantified by RT‐PCR. n = 6 per group. **p < .01 according to Mann–Whitney test. (C) Representative flow cytometry plots (left) and quantification (right) of GM‐CSF expression by Tconvs in aortic aneurysms and spleens 7 days after AAA induction. n = 5 per group. ****p < .0001 according to one‐way ANOVA. (D) Representative flow cytometry plots depicting strategies for gating different GM‐CSF‐positive cells populations (top); relative frequency of different cell populations out of all GM‐CSF‐positive cells in the aorta on day 7 after AAA induction (bottom). n = 5 per group. (E) Quantification of GM‐CSF expression on Tregs and Tconvs in the aortic aneurysms. n = 5 per group. ****p < .0001 according to unpaired two‐tailed t test
3.4. GM‐CSF deficiency limits the porcine pancreatic elastase –induced development of AAA
To address the role of GM‐CSF in the development of AAA, we established PPE‐induced AAA in GM‐CSF−/− mice and WT littermate controls. The abdominal aortic diameter was significantly smaller in the GM‐CSF−/− mice than in the control WT mice (Figure 4A). Along with AAA formation, a substantial decrease in the elastin degradation score was observed in the GM‐CSF−/− mice, as shown by Verhoeff‐Van Gieson staining (Figure 4B). The reduction in elastin degradation in GM‐CSF−/− mice was associated with a decrease in MMP2 and MMP9 expression in the aneurysmal lesions (Figures 4C and S3C). Moreover, T cell (CD3+) and macrophage (CD68+) infiltration into the aortic wall were markedly decreased in the GM‐CSF−/− mice compared with the WT mice (Figure 4D).
FIGURE 4.
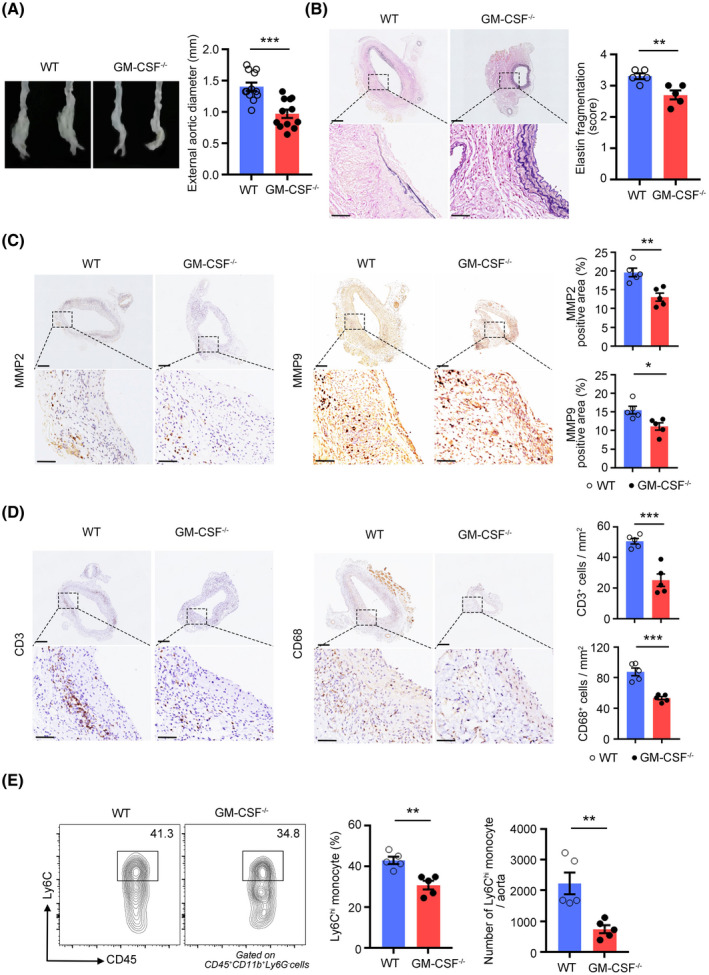
GM‐CSF deficiency reduces the porcine pancreatic elastase (PPE)–induced development of abdominal aortic aneurysm (AAAs). (A) Representative images of abdominal aortic fragments (left) and quantification of maximal diameters (right) of wild‐type and GM‐CSF−/− mice 2 weeks after PPE‐mediated AAA induction. n = 12 per group. ***p < .001 according to unpaired two‐tailed t test. (B) Representative photomicrographs (left) and quantification (right) of elastin layers in the aortic wall by elastic Verhoeff‐Van Gieson staining at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. **p < .01 according to unpaired two‐tailed t test. (C) Representative photomicrographs (left) and quantification (right) of MMP2 and MMP9‐positive area in the aorta at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. *p < .05, **p < .01 according to unpaired two‐tailed t test. (D) Representative photomicrographs (left) and quantification (right) of CD3‐positive T cells and CD68‐positive macrophages in the aorta at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. ***p < .001 according to unpaired two‐tailed t test. (E) Representative flow cytometry images (left) and quantification (right) of Ly6Chi monocytes in aortic aneurysms at day 7. n = 5 per group. **p < .01 according to unpaired two‐tailed t test for proportion and Mann–Whitney test for number
Because nearly all myeloid cells express the GM‐CSF receptor and are likely candidates for the critical cell targets of GM‐CSF, 33 we, therefore, analyzed myeloid cells in AAAs. Although we did not observe a significant difference in the proportions of neutrophils between the WT and GM‐CSF−/− mice (Figure S3D,E), we found that GM‐CSF deficiency limited the infiltration of inflammatory Ly6Chi monocytes into the aortic aneurysms (Figure 4E) and peripheral blood (Figure S4A). The numbers of inflammatory Ly6Chi monocytes in the BM and spleen were also decreased in the GM‐CSF−/− mice (Figure S4B,C). Recent studies revealed that GM‐CSF can stimulate the expression of CCL2, which mediates monocyte recruitment into inflamed tissues through its interaction with the CCR2 chemokine receptor. Thus, we detected the expression of CCL2 in GM‐CSF−/− mice and found that it was reduced both in the serum (Figure S4D) and aortic aneurysms (Figure S4E,F); this result might explain the decrease in the numbers of Ly6Chi monocytes in the aorta and blood.
The effect of GM‐CSF on AAA development was also confirmed in another model of AAA, namely, that induced by CaPO4, and we observed similar findings, that is, aortic dilation was also reduced in the GM‐CSF−/− mice (Figure S4G).
3.5. Treatment with GM‐CSF exacerbates abdominal aortic aneurysm formation
To confirm the pathological role of GM‐CSF in AAA development, mice were treated daily with recombinant GM‐CSF (rmGM‐CSF) or PBS after PPE‐mediated AAA induction. Exogenous rmGM‐CSF administration in vivo worsened AAA formation, as shown by increased aneurysm growth (Figure 5A), elastic fiber disruption (Figure 5B), MMP2 and MMP9 (Figures 5C and S5A) and leukocyte and macrophage accumulation in the aortic wall (Figure 5D). There was no significant difference in neutrophil infiltration between the two groups (Figure S5B,C). Ly6Chi monocyte infiltration within the aortic wall and blood was increased in the rmGM‐CSF treated group compared with the PBS‐treated group (Figures 5E and S6A). The exogenous administration of GM‐CSF correspondingly increased the expression of CCL2 in the serum (Figure S6B) and aortic aneurysms (Figure S6C,D). It has been proposed that GM‐CSF controls monocyte production by acting on a specific BM progenitor. 34 Consistently, we found that mice treated with rmGM‐CSF had higher levels of inflammatory monocytes in their spleen and BM than control mice (Figure S6E,F).
FIGURE 5.
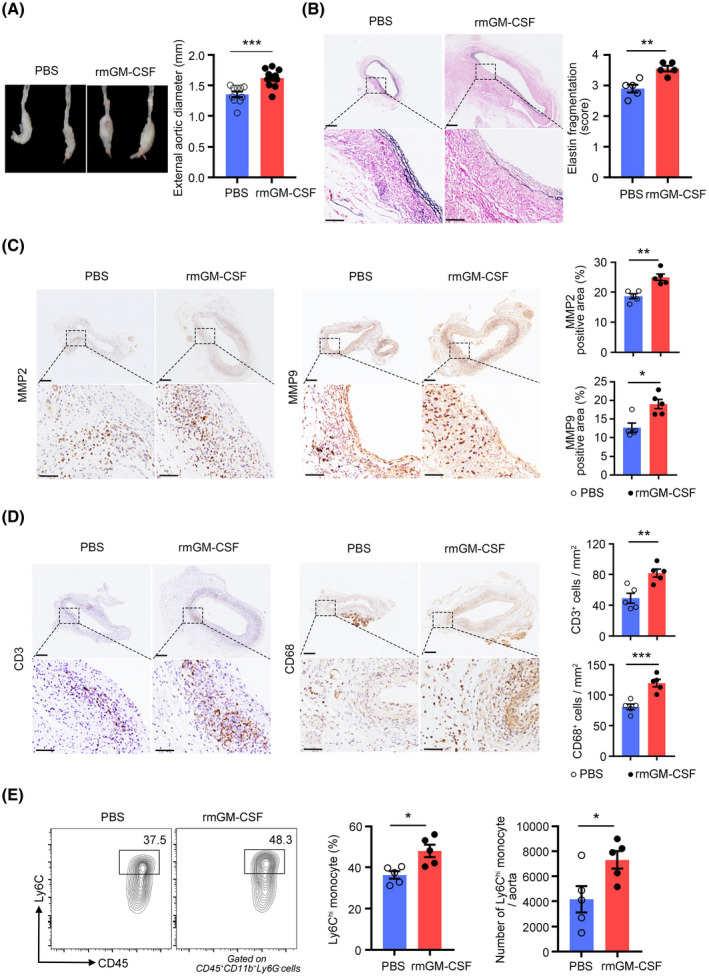
Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) treatment exacerbates porcine pancreatic elastase (PPE)–induced abdominal aortic aneurysm (AAA) development. (A) AAA was induced in mice by PPE, and the mice were intraperitoneally injected with rmGM‐CSF or PBS. Representative images of abdominal aortic fragments (left) and quantification of maximal diameters (right). n = 10–11 per group. ***p < .001 according to unpaired two‐tailed t test. (B) Representative photomicrographs (left) and quantification (right) of elastin layers in the aortic wall by elastic Verhoeff‐Van Gieson staining at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. **p < .01 according to unpaired two‐tailed t test. (C) Representative photomicrographs (left) and quantification (right) of MMP2 and MMP9‐positive areas in the aorta at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. **p < .01 according to unpaired two‐tailed t test for MMP2 and Mann–Whitney test for MMP9. (D) Representative photomicrographs (left) and quantification (right) of CD3‐positive T cells and CD68‐positive macrophages in the aorta at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. **p < .01, ***p < .001 according to unpaired two‐tailed t test. (E) Representative flow cytometry images (left) and quantification (right) of Ly6Chi monocytes in aortic aneurysms at day 7. n = 5 per group. *p < .05 according to unpaired two‐tailed t test
3.6. GM‐CSF promotes M1‐like macrophage polarization in abdominal aortic aneurysm formation
The effects of GM‐CSF priming on the proinflammatory phenotype of macrophage have already been investigated by several groups. 35 , 36 Inducible NO synthase (iNOS), interleukin‐6 (IL‐6), and tumor necrosis factor α (TNF‐α) mRNA expression was downregulated in macrophages sorted from the aortic lesions of GM‐CSF−/− mice compared with those sorted from the aortic lesions of WT littermate mice (Figure 6A). Moreover, there was no change in the mRNA expression of the M2‐associated molecules CD206, Arg1, and Fizz1 (Figure S7A). Flow cytometry results showed a decrease in the numbers of CD11c+ M1 macrophages (Figure 6B), while the numbers of CD206+ M2 macrophages did not change (Figure S7B). In contrast, the expression of M1‐related genes was upregulated (Figure S7C) in macrophages isolated from aortic aneurysms after rmGM‐CSF treatment, and the expression of M2‐related genes did not change (Figure S7D). The proportion of CD11c+ macrophages increased 7 days after GM‐CSF administration (Figure 6C), and the proportion of CD206+ macrophages did not change (Figure S7E). Conclusively, rmGM‐CSF leads to M1‐like macrophage polarization, exacerbating the expansion of aortic aneurysms.
FIGURE 6.

Granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) modulates the polarization of macrophage in the aortic wall after porcine pancreatic elastase (PPE)‐mediated abdominal aortic aneurysm (AAA) induction by upregulating the expression of IRF5. (A) mRNA expression of the M1‐associated genes iNOS, IL‐6 and TNF‐α in CD45+CD11b+F4/80+ macrophages sorted from aortic aneurysms 7 days post AAA. n = 5 per group. **p < .01, ****p < .0001 according to unpaired two‐tailed t test or Mann–Whitney test. (B) Representative flow cytometry images (left) and quantification (right) of the proportion and number of CD11c+ macrophage in wild‐type and GM‐CSF−/− mice after 7 days of PPE‐induced AAA. n = 5 per group. *p < .05 according to Mann–Whitney test. (C) AAA was induced in mice by PPE, and the mice were intraperitoneally injected with rmGM‐CSF or PBS. Representative flow cytometry images (left) and quantification (right) of the proportion and number of CD11c+ macrophages in aortic aneurysms 7 days after PPE‐mediated AAA formation. n = 5 per group. *p < .05, **p < .01 according to unpaired two‐tailed t test. (D) scRNA‐Seq datasets from murine 30 aneurysm lesions were analyzed and tSNE plots showed the expression of Irf5 and Cd68. (E) The protein expression of IRF5 in macrophages in AAAs. Left: histogram plots displaying rmGM‐CSF (red), PBS (blue), and isotype control (gray). Right: quantification of the mean fluorescence intensity of IRF5. n = 5 per group. *p < .05 according to unpaired two‐tailed t test. (F) Bone marrow–derived macrophages were transfected with IRF5 siRNA or control siRNA. Expression of the M1‐associated genes iNOS, IL‐6 and TNF‐α was measured with real‐time PCR. n = 6 per group. **p < .01, ***p < .001, ****p < .0001 versus Control; #p < .05, ###p < .001 versus rmGM‐CSF+siIRF5 according to two‐way ANOVA
We next explored the molecular pathway(s) used by GM‐CSF to promote M1‐like macrophage polarization in AAA development. IRF5 is a transcription factor previously shown to play a key role in promoting the differentiation of Ly6Chi monocytes into the inflammatory CD11c+ macrophage phenotype. 37 However, the expression of IRF5 has been reported in monocytes, macrophages, B‐ and T‐cells, fibroblasts and endothelial cells, which can be found in aortic aneurysms and have been implicated in promoting inflammatory macrophage polarization. To assess IRF5 expression in AAA‐associated cells, we analyzed single‐cell gene expression profiles from aortic aneurysms in mice with PPE‐induced AAA. 30 Single‐cell analysis showed that IRF5 expression was significantly enriched in CD68+ cells, that is, the subsets of macrophages found in AAA (Figure 6D). Furthermore, we quantified the expression of IRF5 by flow cytometry staining and found that IRF5 expression was significantly increased in macrophages after rmGM‐CSF treatment (Figure 6E). We speculated that GM‐CSF might promote the polarization of macrophages toward a proinflammatory phenotype by upregulating the expression of IRF5. To confirm this hypothesis, the expression of IRF5 in macrophages was knocked down by siRNA. The efficacy of siIRF5 transfection was assessed both at the mRNA and protein levels; compared with siCON‐transfected cells, siIRF5‐transfected cells exhibited a significant decrease in IRF5 expression (Figure S7F,G). Silencing the IRF5 gene in macrophages significantly decreased the GM‐CSF‐induced production of M1‐associated proinflammatory cytokines (Figure 6F). Thus, in addition to affecting the recruitment of inflammatory monocytes, we elucidated that GM‐CSF can also promote M1‐like macrophage polarization through upregulation of IRF5 expression to exacerbate AAA development.
3.7. Tconvs‐derived GM‐CSF promotesthe progression of abdominal aortic aneurysm
To confirm the direct role of Tconv‐derived GM‐CSF in AAA progression, we performed adoptive transfer of Tconvs from WT or GM‐CSF−/− mice into GM‐CSF−/− mice while recipient mice were conducted PPE‐induced AAAs (Figure 7A). Compared with those from WT mice, the adoptive transfer of Tconvs from GM‐CSF−/− mice resulted in significantly smaller aortic diameters (Figure 7B) and fewer elastic fragmentations (Figure 7C). Moreover, attenuated aneurysm formation was accompanied by decreased CD3+ T cell and macrophage infiltration (Figure 7D), as well as decreased MMP2 and MMP9 expression (Figure S8A). These results indicated that GM‐CSF derived from Tconvs played a critically detrimental role in the progression of AAA.
FIGURE 7.
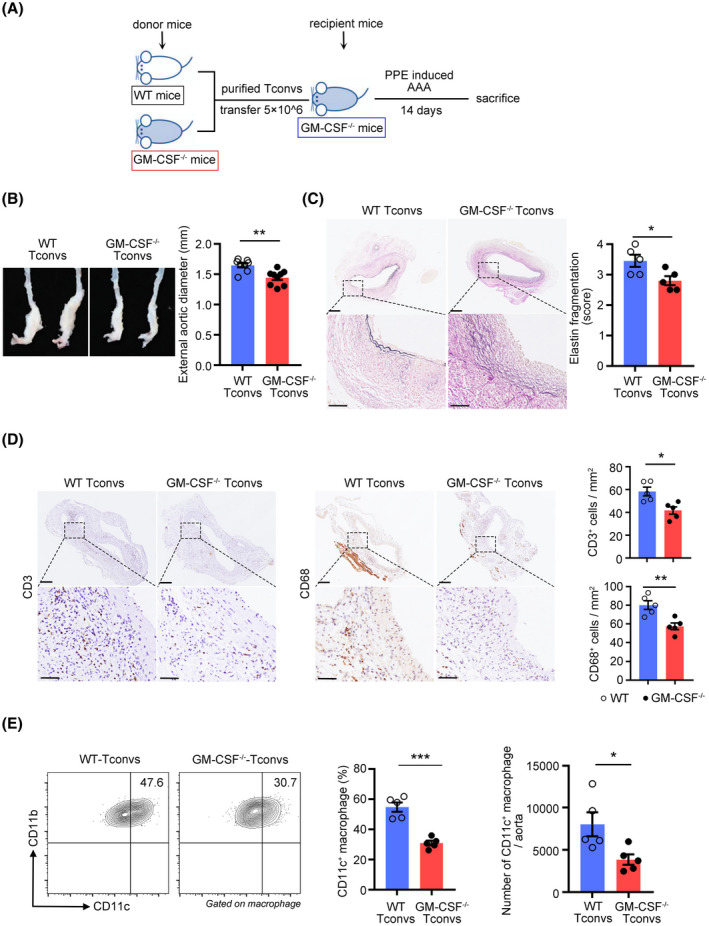
Tconvs‐derived granulocyte‐macrophage colony‐stimulating factor (GM‐CSF) controls the progression of abdominal aortic aneurysm (AAA). (A) Experimental strategy for the transfer of donor wild‐type or GM‐CSF−/− Tconvs into GM‐CSF−/− recipient mice, followed by porcine pancreatic elastase –mediated AAA induction. (B) Representative pictures of abdominal aortic fragments (left) and quantification of maximal diameters (right). n = 8–9 per group. **p < .01 according to unpaired two‐tailed t test. (C) Representative photomicrographs (left) and quantification (right) of elastin layers in the aortic wall according to elastic Verhoeff‐Van Gieson staining at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. *p < .05 according to unpaired two‐tailed t test. (D) Representative photomicrographs (left) and quantification (right) of CD3‐positive T cells and CD68‐positive macrophages in the aorta at day 14, scale bar = 200 µm for whole aortic sections; scale bar = 50 µm for magnified areas. n = 5 per group. *p < .05, **p < .01 according to unpaired two‐tailed t test. (E) Representative flow cytometry images (left) and quantification (right) of CD11c+ macrophage in aortic aneurysms at day 7. n = 5 per group. *p < .05, ***p < .001 according to unpaired two‐tailed t test
The adoptive transfer of Tconvs from GM‐CSF−/− mice also caused a decrease in the proportion of CD11c+ M1 macrophages (Figure 7E), whereas there was no change in the proportion of CD206+ M2 macrophages in aortic aneurysms (Figure S8B). Therefore, all these results indicated that GM‐CSF derived from Tconvs could promote the progression of AAA by inducing macrophage polarization.
4. DISCUSSION
In this study, we demonstrated for the first time that the CXCR6/CXCL16 axis controlled the migration of Tconvs into aortic aneurysms and identified GM‐CSF as a Tconv product that promoted macrophage polarization to exacerbate AAA.
It is well understood that T cells, particularly CD4+ T cells, are required for the formation of AAA; the specific effector functions of CD4+ T cells that lead to the formation of AAA remain controversial 38 and there is currently no literature on the cellular and molecular cues that enable CD4+ T cells to migrate to aortic aneurysms. Consistently, our results verified the accumulation of Tconvs during the progression of AAA. However, the specific mechanism by which Tconvs accumulate during the progression of AAA is still unclear. Previous studies have shown that the unique characteristics and cellular distribution of CXCR6/CXCL16 suggest that these molecules play important role in T‐cell trafficking and cell‐cell contact during inflammation. 39 Some reports have verified that CXCR6 supports the migration of T helper cells (Th), which are involved in atherosclerosis, hepatocarcinogenesis, and colitis. 40 , 41 , 42 In addition, A. Collado et al. 43 demonstrated that deficiency in CXCR6 markedly attenuated AAA formation, which was similar to our results. They revealed that decreased lesion formation was associated with a reduction in macrophage, CD3+, and CXCR6+ cell infiltration and neovascularization. Consistent with these studies, we found that CXCR6 was highly expressed by aortic Tconvs, and mice lacking CXCR6 or blocking its ligand, CXCL16, decreased the infiltration of Tconvs in AAAs, demonstrating a unique role of the CXCR6/CXCL16 axis in regulating the migration of Tconvs to aortic aneurysms. Given the multiple cell types expressing CXCR6, 44 the reduction of AAA in CXCR6GFP/GFP mice may not be able to exclude the contribution of other immune cells in AAA. Aida Collado et al. revealed that in CXCR6GFP/GFP mice, CD8+CXCR6+ lymphocyte infiltration was reduced in AAA lesions, which might also contribute to the reduction AAA formation. 43 In addition, multiple studies have shown that CXCR6‐dependent infiltration of NK cells and NK T cells (NKTs) leads to an accentuated inflammatory response in mice. 45 , 46 , 47 Thus, the specific mechanism of AAA reduction in CXCR6‐deficient mice remains to be further explored. Besides, CXCR6 knockout only partially decreased the Tconv numbers, which suggested that other chemokine receptors might also contribute to the recruitment of Tconvs in AAA. Ocaña et al. have revealed that the CXCR4/CXCL12 interaction participated in the recruitment and retention of inflammatory lymphocytes that infiltrate the aorta in AAA. 48 Whether other chemotactic axes have an impact on Tconv recruitment needs further investigate.
In addition to Treg‐derived IL‐10 playing a protective role in the development of experimental AAA, 49 the function of cytokines produced by other CD4+ T cells in the development of AAA remains controversial. GM‐CSF‐producing CD4+ T cells have been shown to drive chronic inflammatory diseases. 50 IL‐1 and IL‐23 drive the differentiation of Th1 and Th17 effector cells into highly pathological GM‐CSF‐producing CD4+ T cells, exacerbating experimental autoimmune encephalomyelitis (EAE). 51 , 52 Moreover, Sheng et al. 18 put forward a novel subset of Th cells (TH‐GM) and verified that STAT5 programmed TH‐GM differentiation in the context of EAE. In addition to STAT5, recent reports identified a new transcriptional regulator, Bhlhe40, that governs GM‐CSF production by CD4+ T cells. 17 , 53 We confirmed that Tconvs in aortic aneurysms expressed high levels of GM‐CSF, which indicated a critical role of the GM‐CSF produced by Tconvs in AAA formation. However, in AAA formation, whether these GM‐CSF+ CD4+ T cells represent a unique Th cell lineage and which signaling pathways might regulate the differentiation of these cells remain unclear and require further study.
Although virtually all myeloid cells are capable of sensing GM‐CSF, their individual contribution to inflammation depends on the tissue microenvironment of the affected organs. 50 GM‐CSF initiated cardiac disease by modulating local macrophage responses, 54 and tissue damage in GM‐CSF‐driven chronic colitis was proposed to be mediated by activated eosinophils. 55 Among all myeloid cells, however, only Ly6Chi monocytes require GM‐CSF to acquire the pathological signature required for the coordination of tissue inflammation in EAE. 56 Previous studies have suggested a role of GM‐CSF in the pathogenesis of aortic disease. Smad3‐deficient mice have a vascular phenotype similar to aneurysm‐osteoarthritis syndrome, which was related to Smad3−/− CD4+ T cells secreting more GM‐CSF than Smad3+/+ CD4+ T cells. 21 KLF6 represses expression of GM‐CSF, mice with macrophages that were deficient in KLF6 manifested aortic dissection/intramural hematoma. 20 However, the precise mechanism underlying the function of GM‐CSF in AAA development is unclear. In our research, we found that GM‐CSF exacerbated AAA formation by promoting inflammatory monocyte recruitment and inducing macrophage polarization toward the M1 phenotype, and these effects could be further promoted by exogenous GM‐CSF administration and inhibited in GM‐CSF−/− mice. Therefore, targeting GM‐CSF might be an effective treatment for patients with AAA.
Thomas et al. suggested a critical role for IRF5 in M1 macrophage polarization. 57 In fact, our data showed that GM‐CSF could upregulate the expression of IRF5 in macrophages found in aortic aneurysms, and the effect of GM‐CSF on inducing macrophage polarization disappeared after silencing the expression of IRF5 by macrophages was silenced, indicating that GM‐CSF might activate the transcription factor IRF5 to promote macrophage polarization toward the M1 type and exacerbate AAA formation. However, the specific mechanism by which GM‐CSF regulates the IRF5 signaling pathway is still unclear, and further study should be conducted.
In conclusion, our study confirmed that in a PPE‐induced model of AAAs, the CXCR6/CXCL16 axis mediated the infiltration of Tconvs into the aortic aneurysms. GM‐CSF derived from Tconvs promoted the recruitment of inflammatory monocytes and activated the transcription factor IRF5 to polarize macrophages toward the M1 phenotype, exacerbating the progression of AAA. Our research provides a theoretical basis for GM‐CSF targeted therapy in patients with AAA.
DISCLOSURES
The authors have no relevant conflicts of interest.
AUTHOR CONTRIBUTIONS
Dan Li and Jingyong Li designed of the study and wrote the paper. Henan Liu and Luna Zhai performed experiments and collected data. Wangling Hu, Ni Xia, Tingting Tang, Jiao Jiao, Shaofang Nie, Yuhua Liao, Xiangping Yang, and Guo‐Ping Shi analyzed, interpreted, and discussed data. Xiang Cheng developed the conception, organized, designed, and wrote the paper. All authors contributed to the article and approved the submitted version.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Supporting information
Fig S1‐S8
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81720108005, 82030016 to X.C.; Nos. 81400364, 81770503 to N.X.; No. 81974037 to T.T.T.; and No. 82000443 to J.Y.L.) and grants from the National Heart, Lung, and Blood Institute (Nos. HL151627, HL157073 to G.‐P.S.), and the National Institute of Neurological Disorders and Stroke (No. AG063839 to G.‐P.S.).
Li D, Li J, Liu H, et al. Pathogenic Tconvs promote inflammatory macrophage polarization through GM‐CSF and exacerbate abdominal aortic aneurysm formation. FASEB J. 2022;36:e22172. doi: 10.1096/fj.202101576R
Dan Li, Jingyong Li and Henan Liu are contributed equally to this work.
REFERENCES
- 1. Shimizu K, Mitchell RN, Libby P. Inflammation and cellular immune responses in abdominal aortic aneurysms. Arterioscler Thromb Vasc Biol. 2006;26:987‐994. [DOI] [PubMed] [Google Scholar]
- 2. Sampson UK, Norman PE, Fowkes FG, et al. Estimation of global and regional incidence and prevalence of abdominal aortic aneurysms 1990 to 2010. Glob Heart. 2014;9:159‐170. [DOI] [PubMed] [Google Scholar]
- 3. GBD 2013 Mortality and Causes of Death Collaborators . Global, regional, and national age‐sex specific all‐cause and cause‐specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2015;385:117‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sakalihasan N, Michel JB, Katsargyris A, et al. Abdominal aortic aneurysms. Nat Rev Dis Primers. 2018;4:34. [DOI] [PubMed] [Google Scholar]
- 5. Thelen M, Stein JV. How chemokines invite leukocytes to dance. Nat Immunol. 2008;9:953‐959. [DOI] [PubMed] [Google Scholar]
- 6. Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. 2013;50:7‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alkhatib G, Liao F, Berger EA, Farber JM, Peden KW. A new SIV co‐receptor, STRL33. Nature. 1997;388:238. [DOI] [PubMed] [Google Scholar]
- 8. Kim CH, Kunkel EJ, Boisvert J, et al. Bonzo/CXCR6 expression defines type 1‐polarized T‐cell subsets with extralymphoid tissue homing potential. J Clin Invest. 2001;107:595‐601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Galkina E, Harry BL, Ludwig A, et al. CXCR6 promotes atherosclerosis by supporting T‐cell homing, interferon‐gamma production, and macrophage accumulation in the aortic wall. Circulation. 2007;116:1801‐1811. [DOI] [PubMed] [Google Scholar]
- 10. Xiong W, Zhao Y, Prall A, Greiner TC, Baxter BT. Key roles of CD4+ T cells and IFN‐gamma in the development of abdominal aortic aneurysms in a murine model. J Immunol. 2004;172:2607‐2612. [DOI] [PubMed] [Google Scholar]
- 11. Yodoi K, Yamashita T, Sasaki N, et al. Foxp3+ regulatory T cells play a protective role in angiotensin II‐induced aortic aneurysm formation in mice. Hypertension. 2015;65:889‐895. [DOI] [PubMed] [Google Scholar]
- 12. Shimizu K, Shichiri M, Libby P, Lee RT, Mitchell RN. Th2‐predominant inflammation and blockade of IFN‐gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114:300‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sharma AK, Lu G, Jester A, et al. Experimental abdominal aortic aneurysm formation is mediated by IL‐17 and attenuated by mesenchymal stem cell treatment. Circulation. 2012;126:S38‐S45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Romain M, Taleb S, Dalloz M, et al. Overexpression of SOCS3 in T lymphocytes leads to impaired interleukin‐17 production and severe aortic aneurysm formation in mice—brief report. Arterioscler Thromb Vasc Biol. 2013;33:581‐584. [DOI] [PubMed] [Google Scholar]
- 15. Eberl G, Pradeu T. Towards a general theory of immunity? Trends Immunol. 2018;39:261‐263. [DOI] [PubMed] [Google Scholar]
- 16. Burgess AW, Metcalf D. The nature and action of granulocyte‐macrophage colony stimulating factors. Blood. 1980;56:947‐958. [PubMed] [Google Scholar]
- 17. Piper C, Zhou V, Komorowski R, et al. Pathogenic Bhlhe40+ GM‐CSF+ CD4+ T cells promote indirect alloantigen presentation in the GI tract during GVHD. Blood. 2020;135:568‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sheng W, Yang F, Zhou Y, et al. STAT5 programs a distinct subset of GM‐CSF‐producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014;24:1387‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Komuczki J, Tuzlak S, Friebel E, et al. Fate‐mapping of GM‐CSF expression identifies a discrete subset of inflammation‐driving T helper cells regulated by cytokines IL‐23 and IL‐1beta. Immunity. 2019;50(1289‐1304):e1286. [DOI] [PubMed] [Google Scholar]
- 20. Son BK, Sawaki D, Tomida S, et al. Granulocyte macrophage colony‐stimulating factor is required for aortic dissection/intramural haematoma. Nat Commun. 2015;6:6994. [DOI] [PubMed] [Google Scholar]
- 21. Ye P, Chen W, Wu J, et al. GM‐CSF contributes to aortic aneurysms resulting from SMAD3 deficiency. J Clin Invest. 2013;123:2317‐2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sho E, Sho M, Hoshina K, Kimura H, Nakahashi TK, Dalman RL. Hemodynamic forces regulate mural macrophage infiltration in experimental aortic aneurysms. Exp Mol Pathol. 2004;76:108‐116. [DOI] [PubMed] [Google Scholar]
- 23. Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS, Smith JD. Consideration of sex differences in design and reporting of experimental arterial pathology studies‐statement from ATVB council. Arterioscler Thromb Vasc Biol. 2018;38:292‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li J, Xia N, Wen S, et al. IL (Interleukin)‐33 suppresses abdominal aortic aneurysm by enhancing regulatory T‐cell expansion and activity. Arterioscler Thromb Vasc Biol. 2019;39:446‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yamanouchi D, Morgan S, Stair C, et al. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J Vasc Surg. 2012;56:455‐461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J, Xia N, Li D, et al. Aorta regulatory T cells with a tissue‐specific phenotype and function promote tissue repair through Tff1 in abdominal aortic aneurysms. Adv Sci. 2022:2104338. doi: 10.1002/advs.202104338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robbins CS, Hilgendorf I, Weber GF, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Garcia Nores GD, Ly CL, Cuzzone DA, et al. CD4(+) T cells are activated in regional lymph nodes and migrate to skin to initiate lymphedema. Nat Commun. 2018;9:1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. MacRitchie N, Grassia G, Noonan J, et al. The aorta can act as a site of naive CD4+ T‐cell priming. Cardiovasc Res. 2020;116:306‐316. [DOI] [PubMed] [Google Scholar]
- 30. Zhao G, Lu H, Chang Z, et al. Single‐cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc Res. 2021;117:1402‐1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhan Y, Lew AM, Chopin M. The pleiotropic effects of the GM‐CSF rheostat on myeloid cell differentiation and function: more than a numbers game. Front Immunol. 2019;10:2679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hamilton JA. GM‐CSF in inflammation. J Exp Med. 2020;217:e20190945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Greter M, Helft J, Chow A, et al. GM‐CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36:1031‐1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Anzai A, Choi JL, He S, et al. The infarcted myocardium solicits GM‐CSF for the detrimental oversupply of inflammatory leukocytes. J Exp Med. 2017;214:3293‐3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lacey DC, Achuthan A, Fleetwood AJ, et al. Defining GM‐CSF‐ and macrophage‐CSF‐dependent macrophage responses by in vitro models. J Immunol. 2012;188:5752‐5765. [DOI] [PubMed] [Google Scholar]
- 36. Fleetwood AJ, Lawrence T, Hamilton JA, Cook AD. Granulocyte‐macrophage colony‐stimulating factor (CSF) and macrophage CSF‐dependent macrophage phenotypes display differences in cytokine profiles and transcription factor activities: implications for CSF blockade in inflammation. J Immunol. 2007;178:5245‐5252. [DOI] [PubMed] [Google Scholar]
- 37. Corbin AL, Gomez‐Vazquez M, Berthold DL, et al. IRF5 guides monocytes toward an inflammatory CD11c(+) macrophage phenotype and promotes intestinal inflammation. Sci Immunol. 2020;5:eaax6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dale MA, Ruhlman MK, Baxter BT. Inflammatory cell phenotypes in AAAs: their role and potential as targets for therapy. Arterioscler Thromb Vasc Biol. 2015;35:1746‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Latta M, Mohan K, Issekutz TB. CXCR6 is expressed on T cells in both T helper type 1 (Th1) inflammation and allergen‐induced Th2 lung inflammation but is only a weak mediator of chemotaxis. Immunology. 2007;121:555‐564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Butcher MJ, Wu CI, Waseem T, Galkina EV. CXCR6 regulates the recruitment of pro‐inflammatory IL‐17A‐producing T cells into atherosclerotic aortas. Int Immunol. 2016;28:255‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tao H, Chen X. Role of CXCR6‐deficient natural killer T cells and CD4 T cells in hepatocarcinogenesis. Gastroenterology. 2019;157:1169‐1170. [DOI] [PubMed] [Google Scholar]
- 42. Mandai Y, Takahashi D, Hase K, et al. Distinct roles for CXCR6(+) and CXCR6(‐) CD4(+) T cells in the pathogenesis of chronic colitis. PLoS One. 2013;8:e65488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Collado A, Marques P, Escudero P, et al. Functional role of endothelial CXCL16/CXCR6‐platelet‐leucocyte axis in angiotensin II‐associated metabolic disorders. Cardiovasc Res. 2018;114:1764‐1775. [DOI] [PubMed] [Google Scholar]
- 44. Izquierdo MC, Martin‐Cleary C, Fernandez‐Fernandez B, et al. CXCL16 in kidney and cardiovascular injury. Cytokine Growth Factor Rev. 2014;25:317‐325. [DOI] [PubMed] [Google Scholar]
- 45. Germanov E, Veinotte L, Cullen R, Chamberlain E, Butcher EC, Johnston B. Critical role for the chemokine receptor CXCR6 in homeostasis and activation of CD1d‐restricted NKT cells. J Immunol. 2008;181:81‐91. [DOI] [PubMed] [Google Scholar]
- 46. Mossanen JC, Kohlhepp M, Wehr A, et al. CXCR6 Inhibits hepatocarcinogenesis by promoting natural killer T‐ and CD4(+) T‐cell‐dependent control of senescence. Gastroenterology. 2019;156(1877‐1889):e1874. [DOI] [PubMed] [Google Scholar]
- 47. Paust S, Gill HS, Wang BZ, et al. Critical role for the chemokine receptor CXCR6 in NK cell‐mediated antigen‐specific memory of haptens and viruses. Nat Immunol. 2010;11:1127‐1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ocana E, Perez‐Requena J, Bohorquez JC, Brieva JA, Rodriguez C. Chemokine receptor expression on infiltrating lymphocytes from abdominal aortic aneurysms: role of CXCR4‐CXCL12 in lymphoid recruitment. Atherosclerosis. 2008;200:264‐270. [DOI] [PubMed] [Google Scholar]
- 49. Zhou Y, Wu W, Lindholt JS, et al. Regulatory T cells in human and angiotensin II‐induced mouse abdominal aortic aneurysms. Cardiovasc Res. 2015;107:98‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Becher B, Tugues S, Greter M. GM‐CSF: from growth factor to central mediator of tissue inflammation. Immunity. 2016;45:963‐973. [DOI] [PubMed] [Google Scholar]
- 51. Codarri L, Gyulveszi G, Tosevski V, et al. RORγt drives production of the cytokine GM‐CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12:560‐567. [DOI] [PubMed] [Google Scholar]
- 52. El‐Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL‐1‐ and IL‐23‐induced production of the cytokine GM‐CSF. Nat Immunol. 2011;12:568‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lin CC, Bradstreet TR, Schwarzkopf EA, et al. Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat Commun. 2014;5:3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stock AT, Hansen JA, Sleeman MA, McKenzie BS, Wicks IP. GM‐CSF primes cardiac inflammation in a mouse model of Kawasaki disease. J Exp Med. 2016;213:1983‐1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Griseri T, Arnold IC, Pearson C, et al. Granulocyte macrophage colony‐stimulating factor‐activated eosinophils promote interleukin‐23 driven chronic colitis. Immunity. 2015;43:187‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Croxford AL, Lanzinger M, Hartmann FJ, et al. The cytokine GM‐CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. 2015;43:502‐514. [DOI] [PubMed] [Google Scholar]
- 57. Krausgruber T, Blazek K, Smallie T, et al. IRF5 promotes inflammatory macrophage polarization and TH1‐TH17 responses. Nat Immunol. 2011;12:231‐238. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S8
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.