

Abstract
Fabry disease (FD) is an X‐linked genetic disease due to pathogenic variants in GLA. The phenotype varies depending on the GLA variant, alpha‐galactosidase residual activity, patient's age and gender and, for females, X chromosome inactivation. Over 1000 variants have been identified, many through screening protocols more susceptible to disclose non‐pathogenic variants or variants of unknown significance (VUS). This, together with the non‐specificity of some FD symptoms, challenges physicians attempting to interpret GLA variants. The traditional way to interpreting pathogenicity is based on a combined approach using allele frequencies, genomic databases, global and disease‐specific clinical databases, and in silico tools proposed by the American College of Medical Genetics and Genomics. Here, a panel of FD specialists convened to study how expertise may compare with the traditional approach. Several GLA VUS, highly controversial in the literature (p.Ser126Gly, p.Ala143Thr, p.Asp313Tyr), were re‐analyzed through reviews of patients' charts. The same was done for pathogenic GLA variants with some specificities. Our data suggest that input of geneticists and physicians with wide expertise in disease phenotypes, prevalence, inheritance, biomarkers, alleles frequencies, disease‐specific databases, and literature greatly contribute to a more accurate interpretation of the pathogenicity of variants, bringing a significant additional value over the traditional approach.
Keywords: ACMG criteria, experts, Fabry disease, genetic variants, pathogenicity interpretation, variants of unknown significance
1. INTRODUCTION
Fabry disease (FD; OMIM #301500) is a rare X‐linked inborn error of glycosphingolipid metabolism, caused by pathogenic variants in the GLA gene (Gene Entrez: 2717; NCBI reference sequence: NM_000169.3; OMIM #300644; Locus Reference Genomic record LRG_672), which encodes the lysosomal enzyme α‐galactosidase A (α‐Gal A, EC 3.2.1.22; Uniprot P06280). Quantitative/functional α‐Gal A deficiency leads to progressive accumulation of its undegraded substrates globotriaosylceramide (Gb3) and its deacylated derivative globotriaosylsphingosine (lyso‐Gb3), in tissues and body fluids. 1
FD has two main forms, “classic” and “later‐onset.” 1 , 2 “Classic” FD typically manifests during childhood, progressing to life‐threatening renal, cardiac, and/or neurological complications, with hampered quality of life and reduced life expectancy. 1 , 2 “Later‐onset” FD typically has cardiac manifestations. 3 , 4 FD phenotypes primarily depend upon the disease form, nature of the GLA variant, age, gender and residual α‐Gal A activity. 1
High‐throughput next generation sequencing (NGS)‐based screening programs in high‐risk populations and newborns have identified several novel GLA variants. Understanding variant pathogenicity is the key to accurate prevalence estimation, diagnosis, and management of FD. Revised interpretation of GLA variants, previously inaptly classified “pathogenic,” has decreased the estimates of FD prevalence (Table 1). 5 , 6 , 7 , 8
TABLE 1.
Revised prevalence of Fabry disease in various screening populations after re‐interpretation of pathogenicity
| Patient population | Previous prevalence 5 , 6 | Revised prevalence 7 , 8 | Absolute prevalence 7 , 8 |
|---|---|---|---|
| Newborn males 5 , 7 | 0.03% 5 | 0.014% 7 | 1 in 6883 7 |
| Adult males 6 , 8 | |||
| Hemodialysis | 0.33% | 0.21% | 1 in 476 |
| Renal transplant | 0.38% | 0.25% | 1 in 400 |
| Hypertrophic cardiomyopathy | 2.67% | 0.94% | 1 in 106 |
| Cryptogenic strokes | 4.23% | 0.13% | 1 in 769 |
| Adult females 6 , 8 | |||
| Hemodialysis | 0.10% | 0.15% | 1 in 667 |
| Renal transplant | 0% | 0% | |
| Hypertrophic cardiomyopathy | 2.8% | 0.90% | 1 in 111 |
| Cryptogenic strokes | 2.13% | 0.14% | 1 in 714 |
Over 1000 GLA variants have been reported, 9 , 10 , 11 , 12 most of them private, including some variants of unknown significance (VUS). 13 Furthermore, the non‐specificity of FD symptoms presents a challenge for physicians attempting to interpret the clinical relevance of a VUS.
A panel of French physicians involved in the diagnosis and management of FD (clinical and molecular geneticists, nephrologists, and internal medicine specialists), convened to discuss the diagnostic challenges posed by GLA VUS. This paper illustrates the challenges in interpretating pathogenicity of GLA VUS through real‐life cases, highlights the input of highly specialized experts and provide practical recommendations for accurate diagnosis of FD.
2. TERMINOLOGY
In FD, the term “variant” had been historically used to describe the two main forms/phenotypes, creating confusion between genetic and clinical levels when the Human Genome Variation Society (HGVS) advised to replace “mutation” by the term “variant.” 14 We therefore recommend keeping the term “variant” to describe GLA alleles and using “form” or “phenotype” for clinical description of the disease (i.e., classic or later‐onset form/phenotype of FD). The terms “mutation” and “polymorphism” should be avoided, due to implied pathogenicity and benignity, respectively. Variants are categorized as benign (class 1), likely benign (class 2), of uncertain significance (VUS, class 3), likely pathogenic (class 4), or pathogenic (class 5). 13
3. PHENOTYPES AND UNDERLYING BIOCHEMICAL MECHANISMS
3.1. Clinical phenotypes
The “classic” phenotype typically manifests during childhood, 15 progressing in adulthood to life‐threatening complications, hampered quality of life and reduced life expectancy (Figure 1). 1 , 2 The later‐onset FD phenotype mostly exhibits cardiac manifestations, which appear later in life. 1 , 3 , 4 , 16
FIGURE 1.
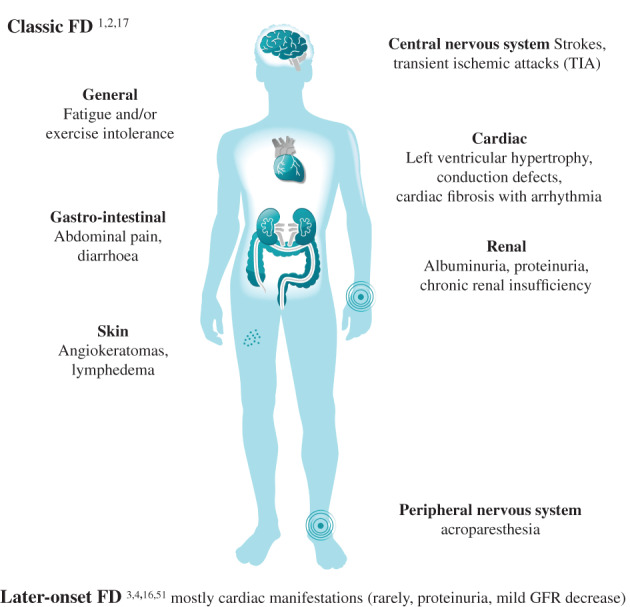
Main clinical manifestations of Fabry disease (FD)
Absent/very low residual α‐Gal A activity is associated with the more severe classic phenotype, earlier onset of symptoms, and multi‐organ involvement. 1 , 17 , 18 However, as many genetic diseases, FD has a highly variable phenotypic expressivity.
3.2. Biochemical phenotypes
Alpha‐Gal A activity can be measured in leukocytes, plasma, or dried blood spot (DBS) cards. Reference values vary widely depending on pre‐analytical and technical conditions. The activity of another lysosomal enzyme, e.g., β‐galactosidase or hexosaminidase, should be simultaneously determined for quality control. Alpha‐Gal A deficiency is expressed in micromole/L/h. 19 , 20 In males, substrate accumulation starts once α‐Gal A activity drops below 20%–25% of normal levels. 1 In severe deficiency, α‐Gal A levels are <1%–3% of mean control values. Although low α‐Gal A activity in females is a good indicator of pathogenicity, 21 its diagnostic value in females is diminished due to normal α‐Gal A activity in several heterozygous females with confirmed FD, depending on X‐chromosome inactivation (XCI). 21
Globotriaosylsphingosine or lyso‐Gb 3 (α‐d‐galactopyranosyl‐(1,4)‐β‐d‐galactopyranosyl‐(1,4)‐β‐d‐glucopyranosyl‐(1,1)‐(2S,3R,4E)‐2‐amino‐octadec‐4‐ene‐1,3‐diol) is the deacylated form of Gb3. Increased plasma lyso‐Gb3 levels in male FD patients were first reported in 2008. 22 This amphipathic lipid, found at high concentration in plasma of male FD patients, has been attributed to the action of acid ceramidase on Gb3. Lyso‐Gb3 has progressively superseded Gb3 as the main biomarker for diagnosing and monitoring FD.
3.2.1. Is lyso‐Gb3 causally involved in the pathogenesis of FD?
Increasing evidence implicates lyso‐Gb3 in the pathogenesis of FD, possibly through inflammation. 23 , 24 The addition of lyso‐Gb3 to cultured smooth muscle cells induces cell proliferation, suggesting its role in the vascular remodeling characteristic of FD. 20 The inhibitory effect of lyso‐Gb3 on endothelial nitric oxide synthase may also contribute to vascular dysfunction in FD patients. 25 Lyso‐Gb3 may induce glomerular damage through activation of Notch1 signaling and TGF‐β1‐mediated production of extracellular matrix by podocytes. 26 Recent studies suggest an activation of innate immunity and possibly of adaptive immunity by lyso‐Gb3 in target tissues including myocardium and kidney. In a recently proposed model of cardiac FD staging, myocardial inflammation can also precede the effects of Gb3 storage on cardiac wall thickness and function. 27
3.2.2. Can lyso‐Gb3 be used as a biomarker in FD?
Plasma lyso‐Gb3 levels are dramatically increased in male patients with classic FD (up to 100‐fold control values). 22 , 28 , 29 , 30 , 31 , 32 Concentrations in males with a mild phenotype and in heterozygous classic females are lower, but nonetheless abnormal. 28 , 29 , 30 , 31 , 32 In male FD patients, lyso‐Gb3 levels are higher in those with frameshift and nonsense variants than in those with missense variants. 33 Plasma lyso‐Gb3 can reliably distinguish classically affected male and female FD patients from individuals without FD. 30 Normal lyso‐Gb3 levels are insufficient to exclude FD in women, but are highly unlikely in male FD patients. Interestingly, several studies have demonstrated normal plasma lyso‐Gb3 levels in individuals carrying benign GLA variants. 32 , 34 , 35 A moderate increase in plasma lyso‐Gb3 levels was reported to indicate FD in patients with an uncertain diagnosis of FD carrying a VUS, while normal plasma lyso‐Gb3 levels were observed in individuals with negative tissue biopsies. 30
Multiple studies have investigated the association between plasma lyso‐Gb3 levels and clinical FD manifestations. In both male and female FD patients, a correlation between plasma lyso‐Gb3 exposure and disease severity was reported. 29 Plasma lyso‐Gb3 concentration was identified as an independent risk factor for cerebrovascular white matter lesions in male patients and left ventricular hypertrophy in female patients. 29 A study of patients with genetic variants associated with classic FD found a correlation between lyso‐Gb3 concentrations and left ventricular mass index, but not kidney function. 36 In later‐onset FD patients carrying the c.644A>G; p.(Asn215Ser)/p.N215S variant, lyso‐Gb3 levels correlated with left ventricular mass, glomerular filtration rate, and overall disease severity. 16 Finally, a correlation between highly elevated plasma lyso‐Gb3 levels and severe clinical events has been recently reported. 37 Urinary lyso‐Gb3 levels are also increased in FD patients, and correlate with Gb3 levels. 38 Like urinary Gb3 concentration, total urinary concentration of lyso‐Gb3 and its structural analogs represent a specific diagnostic biomarker, and is elevated in both classical and non‐classical FD patients. 39
3.2.3. Does lyso‐Gb3 have a prognostic value in therapeutic drugs monitoring in FD patients?
In hemizygous FD patients, enzyme replacement therapy (ERT) markedly reduces (though does not always normalize) plasma lyso‐Gb3 levels within 3 months, thereafter, remaining relatively stable. 40 , 41 Similar reduction or stabilization of lyso‐Gb3 levels was reported for female FD patients. 41 Later treatment initiation results in less effective lyso‐Gb3 clearance. In male patients with classic FD, lyso‐Gb3 levels a year after ERT initiation were lower in patients who began ERT before age 25, versus those who started later in life. 42 An early initiation of ERT in two yet clinically asymptomatic children with classic FD completely normalized lyso‐Gb3. 43 In FD patients, treatment with oral chaperone migalastat for 1–2 years led to reduced, but not normalized, plasma lyso‐Gb3 levels. 44 , 45 Regular monitoring of biochemical response to chaperone therapy (e.g., plasma lyso‐Gb3 and α‐Gal A activity) is crucial to adjust treatment approach, and patient clinical control at least every 6–12 months. 46 In summary, state‐of‐the‐art knowledge indicates that lyso‐Gb3 is a useful biomarker both for diagnosing and monitoring FD patients.
3.3. Additional factors influencing phenotype in Fabry disease
Multiple factors may contribute to the phenotypic heterogeneity associated with a given GLA variant. 47 Theoretically, modifier genes may alter the expression of GLA or another disease‐causing gene influencing FD phenotype. 48 As in other genetic diseases, environmental factors (e.g., hypertension, smoking) may also contribute to phenotypic variability.
The clinical presentation is more variable in heterozygous females than hemizygous males, likely due to epigenetics. 1 , 21 , 49 The inactivation of one of the two X‐chromosomes is generally random in females, though some females have preferential inactivation of the same chromosome in >80% of cells (skewed XCI), affecting the clinical phenotype and prognosis of FD. 21 Epigenetic regulation, therefore, largely explains the variable expressivity observed for a same GLA variant in female FD patients.
4. GLA ALLELIC HETEROGENEITY: DEALING WITH UNCERTAINTY
Diagnosing FD is often challenging due to GLA high allelic heterogeneity (Figure 2), possible absence of male cases in the family and consequent enzymatic values and non‐specificity of (early‐onset) symptoms.
FIGURE 2.
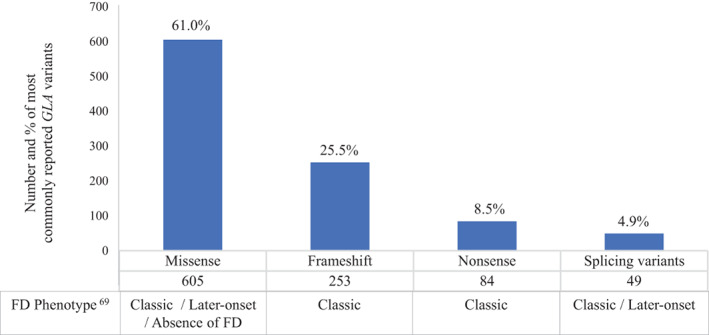
Most commonly reported types of GLA variants in the Human Gene Mutation Database (HGMD) and related Fabry disease (FD) phenotypes in hemizygous patients
Classic FD with absent or negligible α‐Gal A activity is associated with several nonsense GLA pathogenic variants including but not limited to CpG hotspots [e.g. c.658C>T; p.(Arg220*), c.679C>T; p.(Arg227*), c.901C>T; p.(Arg301*)] and missense variants impacting the active site or key residues of the enzyme [e.g. c.509A>G; p.(Asp170Gly), c.508G>C; p.(Asp170His), c.508G>A; p.(Asp170Asn), c.509A>T; p.(Asp170Val)]. 9 , 11 , 12 Other missense or splice‐site GLA variants may, in contrast, be associated with later‐onset FD and a residual enzyme activity between 3% and 25% of normal in male patients, including c.337T>C; p.(Phe113Leu)/p.(F113L) 50 and c.644A>G; p.(Asn215Ser)/p.(N215S), 51 both prevalent in Caucasian populations, and c.639+919G>A (also known as c.640‐801G>A or IVS4+919G>A), the most prevalent variant in Chinese populations. 17 , 52
Some GLA variants have been classified as VUS, partly because many of them are private to one or few families and because ACMG‐AMP classification is difficult to implement in routine practice. Interpreting variants is particularly complex in the later‐onset phenotype, which may mimic isolated HCM and lack other FD features. 47 The pathogenicity of several missense GLA variants has been extensively discussed in the literature. However, for some of them it remains unclear, requiring insights from the experts for an accurate interpretation.
Hereafter, eight cases illustrating the challenges in interpretation of GLA variants' pathogenicity, and the insights from field experts, are presented. Written informed consent was obtained for all cases.
4.1. GLA variants not resulting in FD diagnosis
4.1.1. c.427G>A; p.(Ala143Thr)/p.(A143T)
A 41‐year‐old man was referred for transient ischemic attack (TIA) with dysarthria and central paralysis of the left VII cranial nerve. Brain magnetic resonance imaging (MRI) revealed sequelae of a recent minor stroke and multifocal punctate white matter lesions, mainly in the periventricular area. Medical history included hypertension and tobacco use. Classic FD signs (angiokeratoma or cornea verticillata), renal or ear involvement were all absent. Holter‐ECG (24‐h) was normal. Cardiac MRI revealed a normal left ventricular mass (81 g/m2) and gadolinium late‐enhancement showed subendocardial (not intramural) fibrosis of inferolateral, anterolateral, and septal walls, indicative of ischemia rather than FD. Significant obstructive coronary disease was absent. Plasma α‐Gal A levels were slightly reduced (6.7 μkat/kg) with normal plasma lyso‐Gb3 (0.3 ng/ml; normal values <1.8 ng/ml) and urinary Gb3. Molecular genetic analyses identified a hemizygous missense GLA variant c.427G>A; p.(Ala143Thr).
The patient was initially diagnosed with FD and disease‐specific treatment was initiated. He remained stable for >7 years. Family screening and segregation analysis revealed his 68‐year‐old mother and 31‐year‐old brother as asymptomatic p.(Ala143Thr) carriers, suggesting the non‐pathogenicity of this GLA variant. The patient's brother had only slightly decreased α‐Gal A activity, and neither cardiovascular nor FD signs or symptoms.
Considering the low specificity of a single disease symptom, high residual enzyme activity and recent literature strongly questioning the pathogenicity of the p.(Ala143Thr) variant, diagnosis of FD in this patient was reconsidered and finally ruled out/excluded. Other etiologies for cardiac fibrosis are being investigated.
A 44‐year‐old female patient with a history of pain due to polyarthralgia and rachialgia, and ischemic stroke of unknown etiology consulted a rheumatology department. Molecular GLA analysis identified the p.(Ala143Thr) variant. Investigation revealed arthritis of the second and third metacarpophalangeal joints, but no signs of FD. Alpha‐Gal A activity and plasma lyso‐Gb3 (0.3 ng/ml; normal values <1.8 ng/ml) were normal. Family screening identified p.(Ala143Thr) in the patient's 15‐ and 21‐year‐old sons and 35‐year‐old sister. Neither of her sons had FD signs/symptoms and therefore refused further investigations including enzymatic assay, which would have been diagnostic. Extensive investigations in the patient's sister, who had multiple sclerosis, did not reveal any sign of FD. The index case was subsequently diagnosed with spondylarthritis, while FD was excluded.
A 25‐year‐old male patient with moderate HCM was referred for further analyses. Septal wall thickness was 17 mm. Ventricular obstruction, dilation and rhythm anomalies were absent. Left ventricular ejection fraction was 60%. There were no features of FD. Family history revealed that patient's maternal uncle, with the p.(Ala143Thr) variant, had been considered 12 years earlier with a possible diagnosis of FD, which was subsequently reconsidered and excluded on the basis of α‐Gal A activity only reduced to 35% (in a male) and normal urinary Gb3 (1.8 nmol/mmol creatinine; normal values <8 nmol/mmol creatinine). Extensive analysis of a panel of sarcomeric genes involved in hereditary cardiomyopathies revealed c.6327_6332del; p.(2109_2111del), a pathogenic variant in the FLNC gene (known to cause sarcomeric autosomal dominant cardiomyopathies). This FLNC variant was inherited from his 57‐year‐old mother, who will be further investigated.
The missense p.(Ala143Thr) variant was first described in 1997 53 in an asymptomatic 2‐month‐old infant with low enzymatic activity but no family history of FD. Software tools predict p.(Ala143Thr) to impact the biological function of α‐Gal A. Databases associate p.(Ala143Thr) to FD, except dbFGP (International Fabry Disease Genotype–Phenotype Database) and LOVD (Leiden Open Variation Database) (Table 3). The variant p.(Ala143Thr) has long been considered pathogenic and disease‐modifying treatments were unnecessarily indicated. 8 As in the case of the 41‐year‐old male patient described above, various studies of patients hemizygotes for p.(Ala143Thr) with stroke/TIA demonstrated only slightly decreased α‐Gal A activity when compared to other GLA missense variants. Those patients had normal lyso‐Gb3 levels, no typical FD symptoms, Gb3 negative biopsies and generally an absence of stroke/TIA in other family members carrying p.(Ala143Thr). 35 , 54 Alpha‐Gal A residual activity in males with p.(Ala143Thr) ranges between 25% and 72% of normal, 30 in line with 46% reported for the 41‐year‐old male and 35% for the 72‐year‐old male described above. This expert panel considers p.(Ala143Thr) to be likely benign. An important evidence is that the prevalence of p.(Ala143Thr) in the non‐Finish European population (0.095%, Table 3) is higher than the prevalence of FD itself. Interestingly, in individuals with p.(Ala143Thr) identification in high‐risk population screenings, the only association with FD is the event that motivated the screening, in favor of a result driven by inclusion bias. For example, TIA/stroke was found in p.(Ala143Thr) patients, when screening population with TIA/stroke of unknown cause, but no higher frequency of end‐stage renal disease was found in that population; the same observation applies for other entry points that motivated screening. 35 , 54 Likewise, due to its not so rare prevalence in the general population (0.051% and 0.13% in the Europeans and Americans, respectively) (https://evs.gs.washington.edu/), p.(Ala143Thr) is the paradigm of a coincidental finding while exploring a variety of genetic diseases, such as MYH‐associated polyposis, achromatopsia, hereditary cancer‐predisposing syndrome, and renal dysplasia, where it should not be considered causal (similarly to the aforementioned strokes/TIA). 55
TABLE 3.
Characteristics of common controversial GLA variants according to genetic databases and in silico prediction softwares
| c.427G>A; p.(Ala143Thr) / p.(A143T) / Thr143 | c.937G>T; p.(Asp313Tyr) / p.(D313Y) / Tyr313 | c.196G>C; p.(Glu66Gln) / p.(E66Q) / Gln66 | c.352C>T; p.(Arg118Cys) / p.(R118C) / Cys118 | c.376A>G; p.(Ser126Gly) / p.(S126G) / Gly126 | |
|---|---|---|---|---|---|
| GnomAD v2.1.1 | |||||
| AF (%) in exomes, genomes (total) | 0.055, 0.018 (0.051) | 0.30, 0.31 (0.30) | 0.012, 0.0045 (0.011) | 0.022, 0.032 (0.023) | 0.033, 0.063 (0.036) |
| Highest AF (%) by population | 0.095 in European (non‐Finnish) |
0.69 in Ashkenazi Jewish 0.45 in European (non‐Finnish) |
0.15 in East Asian | 0.044 in European (non‐Finnish) | 0.074 in European (non‐Finnish) |
| Pathogenicity according to FD‐specific databases | |||||
| dbFGP | Benign | Benign | Benign | Benign | Likely benign |
| The Japanese Fabry Database | LO [5]; classic [4]; B [3]; VUS [1]; np [8] | Classic [5]; B [2]; LO [1]; np [9] | B [5]; classic [5]; LO [3]; np [3] | LO [2]; np [5] | LO [1]; np [6] |
| Pathogenicity according to general databases | |||||
| ClinVar | VUS [10]; LP [4]; P [2] | LB [13]; VUS [3]; B [2] | VUS [4]; LB [2] | VUS [12]; LP [2]; LB [1] | LB [6]; VUS [4]; B [1] |
| LOVD | LB [2]; VUS [1] | LB [3]; B [2]; VUS [1] | np | VUS [3]; P [1] | 2 LB [2]; VUS [1] |
| OMIM | FD | VUS (recently reclassified) | Functional polymorphism and not disease causing | not provided | not provided |
| ACMG classification according to VarSome (date of query) |
LP (2019‐12‐05) VUS because highest ethnic frequency = 0.10% (2020/01/20) P because a user has reported this variant is classified LP in one article (Spada et al. 5 ) and that is confirmed by a functional study (2020‐08‐03) |
VUS (2019‐12‐05) B because highest ethnic frequency = 0.69% (2020‐01‐20) LP because alternative variant (Asp313Asn) is classified P by UniProt Variants (and confirmed using ACMG) (2020‐08‐04) |
VUS (2019‐12‐05) B because highest ethnic frequency = 0.15% (2020‐01‐20) LP because highest ethnic frequency no longer takes into account again (2020‐08‐04) |
LB (2019‐12‐05) B (2020‐01‐20) LB (2020‐08‐04) |
VUS (2019‐12‐05) B because highest ethnic frequency = 0.074% (2020‐01‐20) VUS because highest ethnic frequency no longer taken into account (2020‐08‐04) |
| Polyphen‐2 | Probably damaging (1) | Probably damaging (0.996) | Probably damaging (0.996) | Probably damaging (0.993) | Benign (0.043) |
| Provean | Deleterious (−3.119) | Deleterious (−3.183) | Deleterious (−2.754) | Deleterious (−4.667) | Deleterious (−2.823) |
| SIFT | Damaging (0.004) | Damaging (0.001) | Damaging (0.002) | Damaging (0.001) | Tolerated (0.060) |
| Mutation taster | Disease causing | Polymorphism | Disease causing | Polymorphism | Disease causing |
Note: Last accessed 2020‐08‐04. [ ]: The number of times referenced.
Abbreviations: AF, allele frequency; B, benign; dbFGP, International Fabry Disease Genotype–Phenotype Database; FD, Fabry disease; gnomAD, Genome Aggregation Database; LB, likely benign; LOVD, Leiden Open (source) Variation Database; LP, likely pathogenic; np, not provided; P, pathogenic; VUS, variant of unknown significance.
The high residual enzyme activity associated with p.(Ala143Thr) is in favor of a pseudo‐deficiency rather than a disease‐causing allele per se. 35 , 56 Of note, the dbFGP database, which includes inputs from FD experts and researchers, clearly states p.(Ala143Thr) as benign (Table 3).
4.1.2. c.937G>T; p.(Asp313Tyr)/p.(D313Y)
A 67‐year‐old female had undergone chronic hemodialysis since age 26, due to biopsy‐confirmed membranoproliferative glomerulonephritis with unclear etiology. DBS analysis demonstrated reduced α‐Gal A activity (0.99 μmol/l/h; normal: 1.5–2 μmol/l/h) and normal lyso‐Gb3 (1.18 ng/ml; normal: 0.0–3.5 ng/ml). The GLA variant c.937G>T; p.(Asp313Tyr) was identified, but an expert geneticist (D.P.G.) advised to exclude FD, since there was no other clinical sign of FD or relevant family history, apart from slightly decreased α‐Gal A activity (57% of normal). Renal biopsy was not suggestive of FD.
A 50‐year‐old male consulted due to a family history of HCM and GLA variant p.(Asp313Tyr)/Tyr313, also identified in his two sisters affected with familial cardiomyopathy and his asymptomatic mother. Notably, the patient's father had died of a cardiac arrest, which raises suspicion of an autosomal dominant transmission. Extensive investigations did not reveal any FD feature. In contrast, additional molecular studies using a panel of five sarcomeric genes identified a pathogenic variant in the TNNT2 troponin T gene. This variant was likely responsible for the familial cardiomyopathy, inherited from the patient's father whereas the p.(Asp313Tyr) GLA variant inherited from patient's mother was coincidental.
The missense p.(Asp313Tyr) variant was historically considered pathogenic for a while. 57 It was first described in 1993 in a classic FD male patient with only partial sequencing of the GLA gene, though clinical description was not provided. However, another group subsequently published a case with the additional presence of another GLA variant [c.1232G>A; p.(Gly411Asp)], creating an uncertainty regarding pathogenicity of p.(Asp313Tyr). 58 Functional studies showed that p.(Asp313Tyr) is a sequence variant associated with 75% of normal α‐Gal A activity (pseudo‐deficiency). 58 Similarly, cells transfected with p.(Asp313Tyr) showed even higher residual α‐Gal A levels (59%) than cells transfected with other non‐pathogenic GLA variants, that is, c.352C>T; p.(Arg118Cys)/p.(R118C) (24.5%), 59 and c.196G>C; p.(Glu66Gln)/(p.(E66Q) (47.6%). 35 , 60 , 61 While in vitro prediction tools give conflicting predictions for this variant (Table 3), p.(Asp313Tyr) prevalence in the non‐Finnish European population is 0.45% in the GnomAD database, much higher than the global FD prevalence. Such high allelic frequency supports the classification of p.(Asp313Tyr) as benign. Three major databases (dbFGP, ClinVar and LOVD) agree on classifying p.(Asp313Tyr) as benign/likely benign (Table 3). Several recent reports on p.(Asp313Tyr) demonstrated clinical and biochemical absence of FD features and concluded that p.(Asp313Tyr) per se does not cause FD. 35 , 62
4.1.3. c.376A>G; p.(Ser126Gly)/p.(S126G)
A 45‐year‐old Italian male was referred to the French Referral Center for Fabry disease for a suspicion of familial FD in association to the p.(Ser126Gly) GLA variant. His sister, an asymptomatic p.(Ser126Gly) GLA carrier, was already considered for FD‐specific treatment in Italy. Extensive investigations did not reveal any sign or symptom of FD; α‐Gal A levels were 57% of normal in two independent assays. Brain MRI, glomerular filtration rate and lyso‐Gb3 were normal and FD was consequently excluded.
p.(Ser126Gly) has been found in 74 cases (exomes frequency of 0.033%, n = 60; genomes frequency of 0.063%, n = 14) in GnomAD, above the global frequency of FD (Table 3). While several publications claimed p.(Ser126Gly) to be pathogenic when associated with a given haplotype (though no convincing clinical evidence was provided), 63 this expert opinion authors considers p.(Ser126Gly) as likely benign, in line with all major databases which do not associate p.(Ser126Gly) with FD (Table 3).
4.2. Novel GLA variants resulting in FD diagnosis
4.2.1. c.931dupC; p.(Leu311Profs*4)
A 48‐year‐old Algerian female was referred for evaluation of isolated HCM. The patient reported acroparesthesia during childhood. NGS sequencing of genes involved in hereditary cardiomyopathies identified a novel, previously undescribed frameshift GLA variant, c.931dupC; p.(Leu311Profs*4), which was categorized as pathogenic. Patient had elevated lyso‐Gb3 (10.8 ng/ml, normal: <1.8 ng/ml). Investigations found no renal/neurological involvement, angiokeratoma, or cornea verticillata. Her two sisters, (both with HCM) and rest of family were unavailable for further evaluation. FD was confirmed and disease‐specific treatment initiated. The frameshift associated with this variant was predicted to result in classic FD even though patient's symptoms were mostly limited to the heart, illustrating the phenotypic variability of many genetic diseases.
4.2.2. c.1281_1282insCTTA; p.(Leu429Ilefs*22)
A 54‐year‐old male was hospitalized for syncope, due to complete atrioventricular block; a dual chamber pacemaker was implanted. He had no FD relevant medical history (except for clustered angiokeratomas). His mother had died from sudden cardiac arrest at age 69. Investigations revealed markedly reduced leukocyte α‐Gal A activity (1 nmol/h/mg; normal: 16–39 nmol/h/mg) and elevated plasma lyso‐Gb3 levels (10.1 ng/ml; normal: <5 ng/ml). Fabry disease diagnosis was confirmed, and disease‐specific treatment initiated, together with close clinical and biochemical follow‐up. Additional tests showed microalbuminuria, presence of T2/FLAIR signal alterations in the periventricular white matter on brain MRI, normal audiogram, and absence of cornea verticillata. Family screening is ongoing for his two sisters (aged 57 and 59), his brother (aged 42), and his two daughters (aged 26 and 27).
GLA genotyping revealed a novel hemizygous 4‐base insertion (c.1281_1282insCTTA) predicted to preserve the entire coding sequence of α‐galactosidase A, with the exception of a conservative change of the last amino‐acid (leucine to isoleucine) at codon 429, together with the addition of a 22‐amino‐acid tail. A frameshift variant is theoretically predicted to cause a severe full‐blown classic phenotype in a 54‐year‐old male patient, which was not the case in this hemizygote with predominantly cardiac symptoms. This may be due to the unique nature of the genetic variant preserving the coding sequence until the last codon of the protein.
4.3. Databases and in‐silico prediction softwares
Genetic databases and in silico prediction softwares are useful resources (Table 2), but physicians with limited FD experience may face conflicting results of pathogenicity depending on the database queried and the time of the query. For example, the classification of the p.(Ala143Thr) variant was changed from “likely pathogenic” (on December 5th, 2019) to VUS (on January 20th, 2020) in Varsome (https://varsome.com) with the addition of “BS1” (Benign Strong criterion number 1, AF higher than expected for the disorder). It changed again from “VUS” to “pathogenic” on August 3rd, 2020, with the addition of the “PS3” (Pathogenic Strong criterion number 3), as a Varsome user reported this variant as “likely pathogenic” in a paper supported by a functional expression study, with additional removal of both criteria BS1 and BS2 criteria. Furthermore, p.(Ala143Thr) is classified as benign in dbFGP, with conflicting results in other databases (ClinVar, LOVD, the Japanese Fabry Database) (Table 3).
TABLE 2.
Available databases and in silico prediction softwares
| Name | Description |
|---|---|
| Databases | |
| Genome Aggregation Database (gnomAD) https://gnomad.broadinstitute.org/ | Includes aggregated exome and genome sequencing data. Useful to compare the frequency of a variant in the general population or in an ethnic group against the disease prevalence or against the frequency of the most common pathogenic variant. A GLA variant is not considered disease‐causing if its frequency is higher than FD prevalence or the reported most frequent variant (Popmax Filtering AF). |
| ClinVar https://www.ncbi.nlm.nih.gov/clinvar | Collects evidence‐supported interpretations of clinical significance of variants for FD submitted by clinical testing laboratories, researchers, other databases. |
| Leiden Open (source) Variation Database (LOVD) https://www.lovd.nl/ | Displays gene variants. |
| OMIM® (Online Mendelian Inheritance in Man) https://omim.org/ | Contains information on all known Mendelian disorders and over 15,000 genes, focusing on the relationship between phenotype and genotype. |
| International Fabry Disease Genotype‐Phenotype Database (dbFGP) http://dbfgp.org/dbFgp/fabry | Combines data from databases such as the Human Gene Mutation Database (HGMD) and The Japanese Fabry Database, diagnostic and clinical evaluations of patients with data from peer reviewed publications and input from expert FD researchers and care teams. The database provides information of the associated phenotype for a given variant. |
| The Japanese Fabry Database http://fabry‐database.org/mutants/ | Created by Meiji Pharmaceutical University and led by H. Sakuraba, this database lists GLA variants and the reported clinical phenotypes with references. |
| In silico prediction softwares | |
| VarSome https://varsome.com/ | Annotation tool that classifies the genetic variant according to ACMG criteria. The verdict arises from a vast quantity of accurate curated data such as coding impact of the variant, in vitro functional studies, allele frequency, previous publications and other databases analyses. |
| PolyPhen‐2 http://genetics.bwh.harvard.edu/pph2/ | A tool which predicts whether an amino acid substitution or indel has an impact on the biological function of a protein. |
| Provean (Protein Variation Effect Analyzer) http://provean.jcvi.org/index.php | A tool which predicts whether an amino acid substitution or indel has an impact on the biological function of a protein. |
| SIFT https://sift.bii.a‐star.edu.sg/ | A tool which predicts whether an amino acid substitution or indel has an impact on the biological function of a protein. |
| Mutation taster http://www.mutationtaster.org/ | A tool which predicts whether an amino acid substitution, insertion or deletion has an impact on the biological function of a protein. |
While in silico prediction softwares (e.g., PolyPhen‐2, PROVEAN, SIFT, Mutation Taster) can help in determining the pathogenicity of missense variants, their partial reliability is well‐known as confirmed with/for the five most common (likely) benign GLA variants (Table 3).
With respect to databases, those specific for FD and curated by FD experts, appear to be most valuable for classifying GLA variant pathogenicity (Table 2).
4.4. Literature search
A thorough literature search can also help to determine the pathogenicity of allelic variants. In a recent paper, a group of FD experts, including senior medical geneticists, re‐examined information (clinical, biochemical, and histopathological) from 22 individuals with the p.(Arg118Cys) GLA variant. This variant had been originally considered pathogenic, based on poorly documented cases, low AF and prediction of the consequence of amino acid substitution (Table 3). 59 Careful reassessment of patients and their families ruled out FD, identifying alternative causes for renal failure (e.g., HIV‐associated nephropathy, old age) and stroke (multiple major cardiovascular risk factors). 59 In agreement, dbFGP and ACMG criteria categorize p.(Arg118Cys) as benign and likely benign, respectively (Table 3).
While the p.(Glu66Gln) GLA variant was first suggested to cause FD over 20 years ago, 64 re‐analysis by Japanese 60 and Korean 65 expert groups have subsequently excluded its pathogenicity. Several studies have reported that patients with p.(Glu66Gln) had α‐Gal A levels about 56% (24%–65%) of normal, normal lyso‐Gb3 and normal tissue biopsies. Frequency of this variant in Asian populations may reach 0.83% in Japan and 1% in South Korea, which is much higher than FD global prevalence. 60 , 65 , 66 , 67 It should be noted that literature search yields conflicting results when interpreting p.(Glu66Gly) pathogenicity with data from recognized expert groups in favor of benignity of this GLA variant. 60 , 65 , 67
5. LESSONS FROM FD FOR MORE ACCURATE VARIANT INTERPRETATION
Distinguishing disease‐causing variants from benign bystanders is probably one of the biggest challenges in contemporary clinical genetics. Below, the authors provide practical recommendations for accurate diagnosis of FD (Table 4, Figure 3), which may be extended to some other genetic conditions.
TABLE 4.
Practical recommendations for a more accurate diagnosis of Fabry disease
|
FIGURE 3.
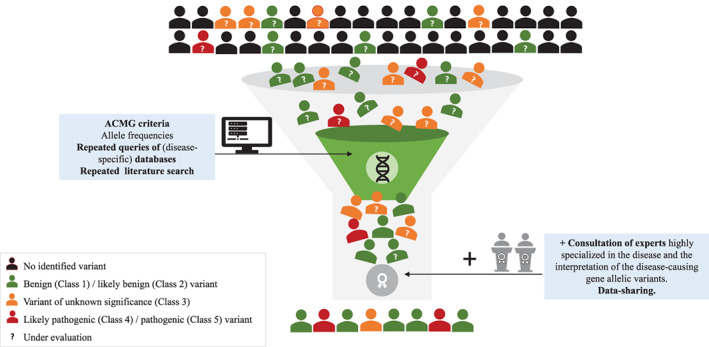
Interpreting pathogenicity of genetic variants when facing uncertainty: lessons from Fabry disease (FD)
5.1. Insights from disease specific biomarkers
In FD and other genetic diseases of metabolism, surrogate biomarkers are available, which, when properly used, can optimize interpretation of the clinical relevance of a genetic variant. In male patients, α‐Gal A levels are key to confirm or exclude pathogenicity of a variant. In both genders, plasma lyso‐Gb3 levels may help in the interpretation, although the possibility of normal lyso‐Gb3 levels in heterozygotes affected with the later‐onset phenotype should be considered.
In the case of FD, where a “pharmacogenetic” specific treatment exists, it should be noted that “amenability” of a given GLA variant does not necessarily imply pathogenicity, since the available amenability table includes proven non‐pathogenic variants such as p.(Arg118Cys), p.(Ala143Thr) and p.(Asp313Tyr) (www.galafoldamenabilitytable.com, last accessed on November 24th, 2021).
5.2. Insights from disease knowledge and expert recommendations to weight ACMG criteria
GLA sequencing is conducted in two contexts: clinical suspicion of FD, or screening at‐risk populations (i.e., HCM, end‐stage renal disease, cryptogenic stroke/TIA). Screening of large at‐risk populations will therefore disclose GLA VUS with relatively high prevalence. In patients with GLA VUS and a non‐specific phenotype, other common genetic causes should be systematically searched (e.g., sarcomeric genes in HCM as illustrated with both aforementioned cases). Erroneous classification of benign variants as pathogenic delays correct diagnosis and can prompt expensive unnecessary treatments. In contrast, detection of a likely benign GLA variant in a patient with very strong clinical suspicion of FD, should prompt search for a second GLA variant. 58 , 67 Biopsy to check for sphingolipids deposits may be considered, 34 although histological confirmation of VUS pathogenicity may be challenging. Indeed, documentation of only a few Gb3 inclusions in some cells does not necessarily imply pathogenicity. Electron microscopy can better assess the amount of stored material, although the sampled tissue and the cellular type (terminally‐differentiated vs. others) may yield different results and interpretation should consider phenocopies of FD (e.g., amiodarone intake).
5.3. Compare allele frequencies of unknown GLA variants with prevalence of FD, and with frequency of the most common pathogenic allele
Traditionally, an AF < 1% supports pathogenicity and >1% benignity. The most frequent pathogenic GLA variants (p.(Asn215Ser) in Caucasians and IVS4 + 919G>A in Asians) occur indeed at a frequency <1%. However, this rule does not always apply, as exemplified by FD, where many GLA variants considered or proven as benign (p.(Glu66Gln), p.(Arg118Cys), p.(Ser126Gly), p.(Ala143Thr), p.(Asp313Tyr)) do not meet the >1% frequency threshold.
When considering the pathogenicity of a GLA variant, if the variant's AF is higher than 0.0125% (FD prevalence: 1/8000) and higher than the frequency of the most common pathogenic allele (indicated as the “Popmax Filtering AF” in GnomAD), the variant should be considered as “likely benign.” 13 , 66 , 68 Furthermore, the variant's AF may vary in different populations and the highest one should be used to determine the likelihood of pathogenicity. Since FD is considered underdiagnosed, ruling out a variant as non‐pathogenic, due to a high AF could be questionable. However, statistical models (http://cardiodb.org/allelefrequencyapp/) can help compute the maximum expected AF and provide better estimates of disease‐causing variants in the general population (maximum credible population AF). 68
Once pathogenicity of a missense variant has been verified, the next step is to determine its association with a specific phenotype. Whether the “classic” or “later‐onset” phenotype of FD is more prevalent is still unclear; several arguments exist for both hypotheses. The total number of genetic variants responsible for classic FD overwhelms the number of genetic variants associated with later‐onset FD (Figure 2). 69 However, a few later‐onset variants have been found at relatively higher frequency. Later‐onset phenotype with single‐organ impact and lower morbidity has minimal effects on genetic fitness, with undetected but higher segregation within populations and consequent higher prevalence. 51 Familial segregation analysis of a GLA VUS can be performed for further information. Absence of co‐segregation strongly suggests benignity while presence of co‐segregation is in favor pathogenicity. 70 However, identifying relatives who can effectively contribute to pathogenicity classification of a VUS, is not always feasible, especially when no male relative exists, thereby limiting the relevance of α‐Gal A assay. 71
5.4. Periodically review the literature and specific databases
It is important to periodically review the literature and perform a specific database search for additional evidence on pathogenicity/benignity of variants, since with growing evidence over time, some variants originally designated VUS may be reclassified as either likely pathogenic/pathogenic or in contrast likely benign/benign and in the latter case, no longer considered to be FD‐causing variants. 35 , 54 , 58 , 59 , 62 , 72 However, careful consideration is necessary when using the variants classification from these databases, as they allow inclusion of different levels of evidence without a hierarchical ranking, such as an in vitro expression study with no clinical data or a report of a few poorly detailed clinical cases 73 which may bias the final interpretation of pathogenicity of a variant under study.
6. CONCLUSION
Since the publication of ACMG‐AMP, 13 the approach to interpreting unknown/novel GLA variants integrates information from in vitro prediction softwares, AF and gene databases. 13 However, this traditional approach is not universally applicable for rare diseases. We recommend that physicians should be aware of potential errors in interpreting GLA variants. Misdiagnosis prolongs patient diagnostic odyssey, resulting in higher morbidity and adding unnecessary financial burden to the healthcare system. 69 Accurate interpretation benefits from the input of rare disease expert clinicians and geneticists (Table 4; Figure 3). We call for more data sharing by referral centers worldwide to increase the robustness of disease specific databases. These lessons obtained from Fabry disease contribute, with practical concepts, to better interpret the pathogenicity of allelic variants in genetic diseases for a more accurate diagnosis and effective management.
CONFLICT OF INTEREST
D.P.G.: Consultant for Sanofi‐Genzyme, Idorsia, Takeda; speaker's honoraria from Takeda, Amicus, Sanofi‐Genzyme. T.L.: Hotel/travel grants from Sanofi‐Genzyme, Takeda, BioMarin, Enzyvant, Orphan. E.H.: Speaker's honoraria and/or travel grants from Sanofi‐ Genzyme, GSK, Actelion, Sobi; B.K.: Speaker's honoraria from Travere, Sanofi, Alnylam, Reata; hotel/travel grants from Sanofi, Travere, Reata; D.L.: Speaker's honoraria and hotel/travel expenses from Amicus, Sanofi‐Genzyme. V.L.S.: Speaker's honoraria from Sanofi‐Genzyme; hotel/travel grants from Amicus, Orphan, Takeda, Sanofi‐Genzyme; K.N.: Speaker's honoraria, travel grants from Sanofi Genzyme; grants from Amicus therapeutics; E.N.: Speaker's honoraria and/or travel grants from Amicus Therapeutics, Sanofi‐Genzyme. J.P.R.: Consultant for Sanofi‐Genzyme; hotel/travel expenses from Amicus Therapeutics; speaker's honoraria from Amgen.
AUTHOR CONTRIBUTIONS
Each author participated in the meetings, analyzing literature, revising, and interpreting the presented cases with current data, establishing recommendations and approved the manuscript. Dominique P. Germain designed and supervised the study, substantially contributed to developing the manuscript and wrote the revised version.
ACKNOWLEDGMENTS
The authors received editorial/writing support in the preparation of this manuscript that was founded by Sanofi Genzyme, but no payment for writing this publication. The authors are responsible for the content of this manuscript and the decision to submit the manuscript for publication.
Germain DP, Levade T, Hachulla E, et al. Challenging the traditional approach for interpreting genetic variants: Lessons from Fabry disease. Clinical Genetics. 2022;101(4):390-402. doi: 10.1111/cge.14102
DATA AVAILABILITY STATEMENT
Data are not shared due to patient confidentiality and ethical restrictions.
REFERENCES
- 1. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under‐recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338‐346. [DOI] [PubMed] [Google Scholar]
- 3. Elleder M, Bradová V, Smíd F, et al. Cardiocyte storage and hypertrophy as a sole manifestation of Fabry's disease. Report on a case simulating hypertrophic non‐obstructive cardiomyopathy. Virchows Arch A Pathol Anat Histopathol. 1990;417(5):449‐455. [DOI] [PubMed] [Google Scholar]
- 4. von Scheidt W, Eng CM, Fitzmaurice TF, et al. An atypical variant of Fabry's disease with manifestations confined to the myocardium. N Engl J Med. 1991;324(6):395‐399. [DOI] [PubMed] [Google Scholar]
- 5. Spada M, Pagliardini S, Yasuda M, et al. High incidence of later‐onset fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Linthorst GE, Bouwman MG, Wijburg FA, Aerts JMFG, Poorthuis BJHM, Hollak CEM. Screening for Fabry disease in high‐risk populations: a systematic review. J Med Genet. 2010;47(4):217‐222. [DOI] [PubMed] [Google Scholar]
- 7. Gragnaniello V, Burlina AP, Polo G, et al. Newborn screening for Fabry disease in northeastern Italy: results of five years of experience. Biomolecules. 2021;11(7):951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Doheny D, Srinivasan R, Pagant S, Chen B, Yasuda M, Desnick RJ. Fabry disease: prevalence of affected males and heterozygotes with pathogenic GLA mutations identified by screening renal, cardiac and stroke clinics, 1995‐2017. J Med Genet. 2018;55(4):261‐268. [DOI] [PubMed] [Google Scholar]
- 9. Germain DP, Biasotto M, Tosi M, Meo T, Kahn A, Poenaru L. Fluorescence‐assisted mismatch analysis (FAMA) for exhaustive screening of the alpha‐galactosidase A gene and detection of carriers in Fabry disease. Hum Genet. 1996;98(6):719‐726. [DOI] [PubMed] [Google Scholar]
- 10. Germain DP, Poenaru L. Fabry disease: identification of novel alpha‐galactosidase A mutations and molecular carrier detection by use of fluorescent chemical cleavage of mismatches. Biochem Biophys Res Commun. 1999;257(3):708‐713. [DOI] [PubMed] [Google Scholar]
- 11. Eng CM, Desnick RJ. Molecular basis of Fabry disease: mutations and polymorphisms in the human alpha‐galactosidase A gene. Hum Mutat. 1994;3(2):103‐111. [DOI] [PubMed] [Google Scholar]
- 12. The Human Gene Mutation Database . 2021. Accessed September 3, 2021. http://www.hgmd.cf.ac.uk/ac/gene.php?gene=GLA.
- 13. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. den Dunnen JT, Dalgleish R, Maglott DR, et al. HGVS recommendations for the description of sequence variants: 2016 update. Hum Mutat. 2016;37(6):564‐569. [DOI] [PubMed] [Google Scholar]
- 15. Germain DP, Fouilhoux A, Decramer S, et al. Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clin Genet. 2019;96(2):107‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lavalle L, Thomas AS, Beaton B, et al. Phenotype and biochemical heterogeneity in late onset Fabry disease defined by N215S mutation. PLoS One. 2018;13(4):e0193550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018;123(4):416‐427. [DOI] [PubMed] [Google Scholar]
- 18. Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. J Am Soc Nephrol. 2017;28(5):1631‐1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wasserstein MP, Caggana M, Bailey SM, et al. The New York pilot newborn screening program for lysosomal storage diseases: report of the first 65,000 infants. Genet Med. 2019;21(3):631‐640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Polo G, Burlina AP, Ranieri E, et al. Plasma and dried blood spot lysosphingolipids for the diagnosis of different sphingolipidoses: a comparative study. Clin Chem Lab Med. 2019;57(12):1863‐1874. [DOI] [PubMed] [Google Scholar]
- 21. Echevarria L, Benistan K, Toussaint A, et al. X‐chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89(1):44‐54. [DOI] [PubMed] [Google Scholar]
- 22. Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008;105(8):2812‐2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rozenfeld P, Feriozzi S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol Genet Metab. 2017;122(3):19‐27. [DOI] [PubMed] [Google Scholar]
- 24. Demuth K, Germain DP. Endothelial markers and homocysteine in patients with classic Fabry disease. Acta Paediatr Suppl. 2002;91(439):57‐61. [DOI] [PubMed] [Google Scholar]
- 25. Kaissarian N, Kang J, Shu L, Ferraz MJ, Aerts JM, Shayman JA. Dissociation of globotriaosylceramide and impaired endothelial function in α‐galactosidase‐A deficient EA.hy926 cells. Mol Genet Metab. 2018;125(4):338‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sanchez‐Niño MD, Carpio D, Sanz AB, Ruiz‐Ortega M, Mezzano S, Ortiz A. Lyso‐Gb3 activates Notch1 in human podocytes. Hum Mol Genet. 2015;24(20):5720‐5732. [DOI] [PubMed] [Google Scholar]
- 27. Pieroni M, Moon JC, Arbustini E, et al. Cardiac involvement in Fabry disease: JACC review topic of the week. J Am Coll Cardiol. 2021;77(7):922‐936. [DOI] [PubMed] [Google Scholar]
- 28. Togawa T, Kodama T, Suzuki T, et al. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol Genet Metab. 2010;100(3):257‐261. [DOI] [PubMed] [Google Scholar]
- 29. Rombach SM, Dekker N, Bouwman MG, et al. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta. 2010;1802(9):741‐748. [DOI] [PubMed] [Google Scholar]
- 30. Smid BE, van der Tol L, Biegstraaten M, Linthorst GE, Hollak CE, Poorthuis BJ. Plasma globotriaosylsphingosine in relation to phenotypes of Fabry disease. J Med Genet. 2015;52(4):262‐268. [DOI] [PubMed] [Google Scholar]
- 31. Pettazzoni M, Froissart R, Pagan C, et al. LC‐MS/MS multiplex analysis of lysosphingolipids in plasma and amniotic fluid: a novel tool for the screening of sphingolipidoses and Niemann‐Pick type C disease. PLoS One. 2017;12(7):e0181700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Duro G, Zizzo C, Cammarata G, et al. Mutations in the GLA gene and LysoGb3: is it really Anderson‐Fabry disease? Int J Mol Sci. 2018;19(12):3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nowak A, Mechtler TP, Hornemann T, et al. Genotype, phenotype and disease severity reflected by serum LysoGb3 levels in patients with Fabry disease. Mol Genet Metab. 2018;123(2):148‐153. [DOI] [PubMed] [Google Scholar]
- 34. van der Tol L, Cassiman D, Houge G, et al. Uncertain diagnosis of fabry disease in patients with neuropathic pain, angiokeratoma or cornea verticillata: consensus on the approach to diagnosis and follow‐up. JIMD Rep. 2014;17:83‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lenders M, Weidemann F, Kurschat C, et al. Alpha‐Galactosidase a p.A143T, a non‐Fabry disease‐causing variant. Orphanet J Rare Dis. 2016;11(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Talbot A, Nicholls K, Fletcher JM, Fuller M. A simple method for quantification of plasma globotriaosylsphingosine: utility for Fabry disease. Mol Genet Metab. 2017;122(1–2):121‐125. [DOI] [PubMed] [Google Scholar]
- 37. Nowak A, Beuschlein F, Sivasubramaniam V, Kasper D, Warnock DG. Lyso‐Gb3 associates with adverse long‐term outcome in patients with Fabry disease. J Med Genet. 2021;0:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Auray‐Blais C, Ntwari A, Clarke JT, et al. How well does urinary lyso‐Gb3 function as a biomarker in Fabry disease? Clin Chim Acta. 2010;411(23–24):1906‐1914. [DOI] [PubMed] [Google Scholar]
- 39. Alharbi FJ, Baig S, Rambhatla SB, et al. The clinical utility of total concentration of urinary globotriaosylsphingosine plus its analogues in the diagnosis of Fabry disease. Clin Chim Acta. 2020;500:120‐127. [DOI] [PubMed] [Google Scholar]
- 40. van Breemen MJ, Rombach SM, Dekker N, et al. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta. 2011;1812(1):70‐76. [DOI] [PubMed] [Google Scholar]
- 41. Sakuraba H, Togawa T, Tsukimura T, Kato H. Plasma lyso‐Gb3: a biomarker for monitoring fabry patients during enzyme replacement therapy. Clin Exp Nephrol. 2018;22(4):843‐849. [DOI] [PubMed] [Google Scholar]
- 42. Arends M, Wijburg FA, Wanner C, et al. Favourable effect of early versus late start of enzyme replacement therapy on plasma globotriaosylsphingosine levels in men with classical Fabry disease. Mol Genet Metab. 2017;121(2):157‐161. [DOI] [PubMed] [Google Scholar]
- 43. Kritzer A, Siddharth A, Leestma K, Bodamer O. Early initiation of enzyme replacement therapy in classical Fabry disease normalizes biomarkers in clinically asymptomatic pediatric patients. Mol Genet Metab Rep. 2019;21:100530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Germain DP, Nicholls K, Giugliani R, et al. Efficacy of the pharmacologic chaperone migalastat in a subset of male patients with the classic phenotype of Fabry disease and migalastat‐amenable variants: data from the phase 3 randomized, multicenter, double‐blind clinical trial and extension study. Genet Med. 2019;21(9):1987‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Müntze J, Gensler D, Maniuc O, et al. Oral chaperone therapy migalastat for treating Fabry disease: enzymatic response and serum biomarker changes after 1 year. Clin Pharmacol Ther. 2019;105(5):1224‐1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lenders M, Nordbeck P, Kurschat C, et al. Treatment of Fabry's disease with migalastat: outcome from a prospective observational multicenter study (FAMOUS). Clin Pharmacol Ther. 2020;108(2):326‐337. [DOI] [PubMed] [Google Scholar]
- 47. Schiffmann R, Fuller M, Clarke LA, Aerts JMFG. Is it Fabry disease? Genet Med. 2016;18(12):1181‐1185. [DOI] [PubMed] [Google Scholar]
- 48. Cimmaruta C, Citro V, Andreotti G, Liguori L, Cubellis MV, Hay MB. Challenging popular tools for the annotation of genetic variations with a real case, pathogenic mutations of lysosomal alpha‐galactosidase. BMC Bioinformatics. 2018;19(15):433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93(2):112‐128. [DOI] [PubMed] [Google Scholar]
- 50. Oliveira JP, Nowak A, Barbey F, et al. Fabry disease caused by the GLA p.Phe113Leu (p.F113L) variant: natural history in males. Eur J Med Genet. 2020;63(2):103703. [DOI] [PubMed] [Google Scholar]
- 51. Germain DP, Brand E, Burlina A, et al. Phenotypic characteristics of the p.Asn215Ser (p.N215S) GLA mutation in male and female patients with Fabry disease: a multicenter Fabry Registry study. Mol Genet Genom Med. 2018;6(4):492‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Germain DP, Jurca‐Simina IE. Principles of human genetics and Mendelian inheritance. In: Burlina AP, ed. Neurometabolic Hereditary Diseases of Adults. Springer International Publishing; 2018:1‐28. [Google Scholar]
- 53. Eng CM, Ashley GA, Burgert TS, Enriquez AL, D'Souza M, Desnick RJ. Fabry disease: thirty‐five mutations in the alpha‐galactosidase A gene in patients with classic and variant phenotypes. Mol Med. 1997;3(3):174‐182. [PMC free article] [PubMed] [Google Scholar]
- 54. Terryn W, Vanholder R, Hemelsoet D, et al. Questioning the pathogenic role of the GLA p.Ala143Thr “Mutation” in Fabry disease: implications for screening studies and ERT. JIMD Rep. 2013;8:101‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. ClinVar . p.Ala143Thr AND p.A143T. Accessed 3 May. https://www.ncbi.nlm.nih.gov/clinvar/?term=p.Ala143Thr%20AND%20p.A143T.
- 56. Conzelmann E, Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6(1):58‐71. [DOI] [PubMed] [Google Scholar]
- 57. Eng CM, Resnick‐Silverman LA, Niehaus DJ, Astrin KH, Desnick RJ. Nature and frequency of mutations in the alpha‐galactosidase A gene that cause Fabry disease. Am J Hum Genet. 1993;53(6):1186‐1197. [PMC free article] [PubMed] [Google Scholar]
- 58. Froissart R, Guffon N, Vanier MT, Desnick RJ, Maire I. Fabry disease: D313Y is an alpha‐galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol Genet Metab. 2003;80(3):307‐314. [DOI] [PubMed] [Google Scholar]
- 59. Ferreira S, Ortiz A, Germain DP, et al. The alpha‐galactosidase A p.Arg118Cys variant does not cause a Fabry disease phenotype: data from individual patients and family studies. Mol Genet Metab. 2015;114(2):248‐258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sakuraba H, Tsukimura T, Togawa T, et al. Fabry disease in a Japanese population‐molecular and biochemical characteristics. Mol Genet Metab Rep. 2018;17:73‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Benjamin ER, Della Valle MC, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Niemann M, Rolfs A, Giese A, et al. Lyso‐Gb3 indicates that the alpha‐galactosidase a mutation D313Y is not clinically relevant for Fabry disease. JIMD Rep. 2013;7:99‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Capuano I, Garofalo C, Buonanno P, et al. Identifying Fabry patients in dialysis population: prevalence of GLA mutations by renal clinic screening, 1995‐2019. J Nephrol. 2020;33(3):569‐581. [DOI] [PubMed] [Google Scholar]
- 64. Okumiya T, Ishii S, Takenaka T, et al. Galactose stabilizes various missense mutants of alpha‐galactosidase in Fabry disease. Biochem Biophys Res Commun. 1995;214(3):1219‐1224. [DOI] [PubMed] [Google Scholar]
- 65. Lee BH, Heo SH, Kim GH, et al. Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J Hum Genet. 2010;55(8):512‐517. [DOI] [PubMed] [Google Scholar]
- 66. GnoMAD . 2021. Accessed 3 September 2021. https://gnomad.broadinstitute.org/variant/X-100658972-C-G?dataset=gnomad_r2_1.
- 67. Ishii S, Sakuraba H, Suzuki Y. Point mutations in the upstream region of the alpha‐galactosidase A gene exon 6 in an atypical variant of Fabry disease. Hum Genet. 1992;89(1):29‐32. [DOI] [PubMed] [Google Scholar]
- 68. Whiffin N, Minikel E, Walsh R, et al. Using high‐resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19(10):1151‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Germain DP, Oliveira JP, Bichet DG, et al. Use of a rare disease registry for establishing phenotypic classification of previously unassigned GLA variants: a consensus classification system by a multispecialty Fabry disease genotype‐phenotype workgroup. J Med Genet. 2020;57(8):542‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Silva CA, Barreto FC, Dos Reis MA, Moura Junior JA, Cruz CM. Targeted screening of Fabry disease in male hemodialysis patients in Brazil highlights importance of family screening. Nephron. 2016;134(4):221‐230. [DOI] [PubMed] [Google Scholar]
- 71. Germain DP, Moiseev S, Suárez‐Obando F, et al. The benefits and challenges of family genetic testing in rare genetic diseases‐lessons from Fabry disease. Mol Genet Genom Med. 2021;9(5):e1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Yasuda M, Shabbeer J, Benson SD, Maire I, Burnett RM, Desnick RJ. Fabry disease: characterization of alpha‐galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22(6):486‐492. [DOI] [PubMed] [Google Scholar]
- 73. du Moulin M, Koehn AF, Golsari A, et al. The mutation p.D313Y is associated with organ manifestation in Fabry disease. Clin Genet. 2017;92(5):528‐533. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are not shared due to patient confidentiality and ethical restrictions.