

Abstract
Background and Purpose
Inward rectifier potassium (KIR) channels are key effectors of vasodilatation in neurovascular coupling (NVC). KIR channels expressed in cerebral endothelial cells (ECs) have been confirmed as essential modulators of NVC. Alzheimer's disease (AD) and cerebrovascular disease (CVD) impact on EC‐KIR channel function, but whether oxidative stress or inflammation explains this impairment remains elusive.
Experimental Approach
We evaluated KIR channel function in intact and EC‐denuded pial arteries of wild‐type (WT) and transgenic mice overexpressing a mutated form of the human amyloid precursor protein (APP mice, recapitulating amyloid β‐induced oxidative stress seen in AD) or a constitutively active form of TGF‐β1 (TGF mice, recapitulating inflammation seen in cerebrovascular pathology). The benefits of antioxidant (catalase) or anti‐inflammatory (indomethacin) drugs also were investigated. Vascular and neuronal components of NVC were assessed in vivo.
Key Results
Our findings show that (i) KIR channel‐mediated maximal vasodilatation in APP and TGF mice reaches only 37% and 10%, respectively, of the response seen in WT mice; (ii) KIR channel dysfunction results from KIR2.1 subunit impairment; (iii) about 50% of K+‐induced artery dilatation is mediated by EC‐KIR channels; (iv) oxidative stress and inflammation impair KIR channel function, which can be restored by antioxidant and anti‐inflammatory drugs; and (v) inflammation induces KIR2.1 overexpression and impairs NVC in TGF mice.
Conclusion and Implications
Therapies targeting both oxidative stress and inflammation are necessary for full recovery of KIR2.1 channel function in cerebrovascular pathology caused by AD and CVD.
Keywords: cerebral blood flow, cerebrovascular reactivity, endothelium, inflammation, neuronal activity, neurovascular coupling, oxidative stress
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- CBF
cerebral blood flow
- CVD
cerebrovascular disease
- EC
endothelial cell
- HbO
oxyhaemoglobin
- HbR
deoxyhaemoglobin
- KIR
inward rectifier potassium channels
- ML133
1‐(4‐methoxyphenyl)‐N‐(naphthalen‐1‐ylmethyl)methanamine hydrochloride
- NVC
neurovascular coupling
- PAP
papaverine
- SMC
smooth muscle cell
What is already known
The interplay of smooth muscle and endothelial KIR channels is essential for optimal vasodilation.
Alzheimer's disease and cerebrovascular pathology impair endothelial KIR channel function.
What does this study add
Both oxidative stress and inflammation impair endothelial Kir2.1 subunit similarly.
Inflammation, but not oxidative stress, up‐regulates KIR2.1 protein expression in pial arteries.
What is the clinical significance
Antioxidant and anti‐inflammatory therapies can restore endothelial KIR2.1 channel function and K+‐dependent vasodilatation.
1. INTRODUCTION
The tight relationship between local neuronal activity and cerebral blood flow (CBF) regulation is known as neurovascular coupling (NVC). Different cell types participate in this process: neurons, astrocytes, smooth muscle cells (SMCs), pericytes and endothelial cells (ECs) (Iadecola, 2017). However, the specific signalling molecules underlying NVC in health and disease remain largely unknown.
Recent studies point to increased astrocytic potassium (K+) release, acting through vascular inward rectifier potassium (KIR) channels, namely, the KIR2.1 subunit, as a key mediator of NVC (reviewed in Longden & Nelson, 2015). These channels are present in SMCs and ECs, and the interplay of both populations contributes to optimal arterial wall electrical communication and vasodilation (Filosa et al., 2006; Sancho et al., 2017, 2019; Wu et al., 2007). In NVC, the activation of KIR2.1 channels in brain capillary ECs is known to propagate upstream to parenchymal arteries (Longden et al., 2017), confirming an active role for brain endothelium in vasodilation. Intriguingly, KIR channels are also present in pial arteries, despite no direct K+ release from astrocytic endfeet at this level (Bastide et al., 1999; Erdös et al., 2004; Marrelli et al., 1998; Mayhan et al., 2004), where EC‐KIR channels play a major role in tuning electrical communication (Sancho et al., 2017). Pial arteries are responsible for ensuring proper blood supply downstream during NVC (Faraci & Heistad, 1990), and K+‐induced hyperpolarization in parenchymal arteries is transmitted to pial arteries likely through EC–EC and EC–SMC gap junctions (Longden et al., 2017).
KIR channel dysfunction is present in several murine models of cerebrovascular pathology: stress (Longden et al., 2014), diabetes (Mayhan et al., 2004), ischaemia/reperfusion (I/R) (Bastide et al., 1999; Marrelli et al., 1998), ageing (Hakim, Chum, et al., 2020), familial forms of Alzheimer's disease (AD), 3xTgAD mice (Hakim, Behringer, et al., 2020) and 5xFAD mice (Mughal et al., 2021), and small vessel disease (Dabertrand et al., 2021). All the aforementioned pathologies involve oxidative stress and/or inflammation (Cohen et al., 2012; Murray et al., 2014; Takeda et al., 2010). While a role of oxidative stress in KIR2.1 dysfunction has been suggested in isolated brain arteries (Bastide et al., 1999; Marrelli et al., 1998), recent studies in endothelial tubes isolated from cerebral vessels point to a minimal role of free radicals (Hakim, Chum, et al., 2020). Moreover, whether inflammation can affect these channels remains largely unknown. Cerebrovascular pathology augments the risk of suffering from AD and vascular cognitive impairment and dementia (Cohen et al., 2012; Murray et al., 2014; Takeda et al., 2010); hence, a better understanding of whether oxidative stress and/or inflammation affect KIR2.1 activity under different pathological conditions is critical to finding therapeutic targets.
Here, we investigate the role of EC‐ and SMC‐KIR channels in K+‐induced dilatation of pial arteries and study whether antioxidant and/or anti‐inflammatory drugs could rescue KIR channel function in transgenic mouse models of AD (overexpressing a mutated form of the human amyloid precursor protein, APP mice) (Mucke et al., 2000) and cerebrovascular pathology (overexpressing a constitutively active form of TGF‐β1, TGF mice) (Wyss‐Coray et al., 2000). In APP mice, cerebrovascular dysfunction results primarily from amyloid‐β (Aβ)‐induced ROS (Park et al., 2004; Tong et al., 2005) whereas increased vascular inflammation has been identified as the main culprit in TGF mice (Zhang et al., 2013). We also investigate, in vivo, the impact of inflammation on NVC mediated by KIR channels.
2. METHODS
Experimental protocols were approved by the Animal Ethics Committee of the Montreal Neurological Institute and complied with the Canadian Council on Animal Care. Animal studies are reported in compliance with the ARRIVE guidelines (Percie du Sert et al., 2020) and with the recommendations made by the British Journal of Pharmacology (Lilley et al., 2020).
2.1. Mouse models
Heterozygous APP and TGF transgenic mice on a C57BL/6J background and their wild‐type (WT) littermates were bred in‐house. APP mice (line J20) overexpress the human APP carrying the Swedish (K670N, M671L) and Indiana (V717F) familial AD mutations under the PDGF β‐chain promoter (Mucke et al., 2000) and display Aβ‐induced cerebrovascular oxidative stress and impaired dilatory function (Tong et al., 2005). TGF mice (line T64) overexpress a constitutively active form of porcine TGF‐β1 under the glial fibrillary acidic protein (GFAP) promoter, leading to cerebrovascular fibrosis, inflammation and reduced dilatory function (Tong et al., 2005; Wyss‐Coray et al., 2000). Mice were housed (4–5 per cage) under controlled temperature (24 ± 1°C), humidity (50 ± 10%) and 12‐h light–dark cycle. Food and water were available ad libitum. Experiments were performed on adult mice (7–10 months old) with males and females used in approximately equal numbers. A group of young (age = 3–4 months) APP mice was studied to investigate plaque‐independent Aβ toxicity on KIR channel function in APP mice (Mucke et al., 2000). The same age range was selected in TGF mice to match experimental design for APP mice. Males and females were not studied independently, because a recent study has discarded sex effect on KIR channel function (Hakim, Behringer, et al., 2020).
2.2. Materials
Compounds for Krebs solution were purchased from Sigma‐Aldrich (Oakville, ON, Canada) or Fisher Chemical (Waltham, MA, USA). Catalase (from bovine liver, non‐PEG conjugated), KIR channel blockers (BaCl2, ML133), 5‐HT, ACh and papaverine (PAP) were purchased from Sigma‐Aldrich. Indomethacin was acquired from Tocris Bioscience (Oakville, ON, Canada). Ketamine (Narketan) and Xylazine (Rompun) were purchased respectively from Vetoquinol, Québec, Canada, and Bayer Inc., Ontario, Canada.
2.3. Cerebrovascular reactivity studies
Mice were killed by decapitation and the posterior (PCA) and middle (MCA) cerebral arteries were isolated and collected in cold (4°C) and oxygenated (95% O2 and 5% CO2) Krebs solution (pH 7.4) containing the following (in mM): 118 NaCl, 4.5 KCl, 2.5 CaCl2, 1 MgSO4, 1 KH2PO4, 25 NaHCO3, and 6 glucose, as described before (Tong et al., 2005). Vessel segments (~2 mm in length) were mounted in a superfusion chamber equipped with a pressure‐servo micropump (PS‐200) (Living Systems Instrumentation, Burlington, VT, USA) used for maintaining intraluminal pressure. Vessels were cannulated on a glass micropipette (~40‐μm diameter) at one end, sealed to another glass micropipette on the other end and slowly pressurized with oxygenated Krebs (37 ± 0.5°C, pH 7.4). Intraluminal pressure (in mmHg) and time required for vessel stabilization were adjusted depending on experiments (see below). Vessels were continuously superfused with oxygenated Krebs (6 ml·min−1) and vasomotor responses measured as intraluminal diameter changes, using a closed‐circuit video system (National Electronics, Taiwan) coupled with a video caliper (Image Instrumentation, Trenton, NJ, USA). All compounds were administered abluminally in the superfused solution, and only one experiment was performed per artery.
2.3.1. Characterization of pressure‐induced myogenic tone
Cannulated arteries were pressurized at 40 mmHg and allowed to stabilize for 1 h in Krebs solution. Pressure was then gradually increased over 5‐min increments to 60, 80, 100 and 120 mmHg. Vessels were maintained for 20 min at each pressure before measuring diameter. At the end of the experiments, vessels were gradually returned to 40 mmHg and incubated (15 min) in calcium (Ca2+)‐free Krebs containing 5‐mM EGTA. Arterial diameter was measured again at each pressure. Myogenic tones were calculated as percentage of change in diameter from regular Krebs to Ca2+‐free Krebs: [Diameter (Ca2+‐free) − Diameter (basal)/Diameter (Ca2+‐free)] × 100 (Scotland et al., 2001; Zaritsky et al., 2000).
2.3.2. Characterization of KIR channel‐mediated dilatation
In vessel preconstricted ~10%–15% by myogenic tone (at 80 mmHg), KIR channels were activated by increasing K+ concentrations (6 to 16 mM), substituting NaCl for KCl in circulating Krebs (Filosa et al., 2006). Krebs solutions were adjusted to ~300 mOsm, confirmed with a microosmometer (5004 micro OSMETTE™, Precision Systems Inc., Natick, MA, USA). Alternatively, K+‐induced dilatations were tested in the presence of the non‐selective KIR channel blocker barium (Ba2+ ) (Standen & Quayle, 1998). Preconstricted arteries were incubated with Ba2+ (100 μM, ~10 min) in 4‐mM K+ Krebs and arterial dilatations to increasing K+ concentrations (6 to 16 mM in 100‐μM Ba2+ Krebs) (Erdös et al., 2004) were measured.
Dilatations in control and Ba2+‐blocked arteries were calculated as percentage of change in diameter from basal diameter (4‐mM K+) to diameter at each K+ concentration (6 to 16 mM), relative to maximum diameter in Ca2+‐free solution (15‐min incubation at the end of the experiment, as described above): [Diameter (dilatated) − Diameter (basal)/(Diameter (Ca2+‐free) − Diameter (basal))] × 100 (Sonkusare et al., 2016).
2.3.3. Role of the KIR 2.1 subunit in KIR channel function
The contribution of KIR2.1 subunit to optimal KIR channel function was determined in myogenic tone‐preconstricted (80 mmHg) vessels incubated (20 min) in 4‐mM K+ Krebs in the presence of ML133 (20 μM), a selective KIR2.1 subunit blocker (Wang et al., 2011), followed by 5‐min incubation with 14‐mM K+ circulating Krebs containing 20‐μM ML133 (Sonkusare et al., 2016). Incremental changes in diameters in control and ML133‐blocked arteries were calculated as described above. Additionally, we investigated whether Ba2+ and ML133 can have synergistic effect on KIR2.1 blockade. To address this, the above experiment was reproduced by incubating WT arteries with either Ba2+ alone (100 μM) or Ba2+ (100 μM) + ML133 (20 μM).
2.3.4. Role of KIR channels in basal arterial tone maintenance
The role of KIR channels in basal arterial tone (Chrissobolis & Sobey, 2003) was investigated in arteries pressurized at 60 mmHg (physiological spontaneous tone) by incubating them for 3 min with increasing Ba2+ concentrations (20–120 μM). Ba2+ was pipetted in the superfusion chamber containing a physiological 4.5‐mM K+ Krebs solution. Ba2+‐induced vasoconstriction was measured as percentage change in diameter from basal condition to each Ba2+ concentration (Erdös et al., 2004).
2.3.5. Role of SMC and EC‐located KIR channels
K+‐induced vasodilation (6 to 16 mM) was investigated before and after EC denudation by passing an air bubble through the lumen (60 s) followed by Krebs solution. Control and denuded arteries were pressurized at 60 mmHg, allowed to stabilize for ~20 min and incubated with 5‐HT (2.10−7 M, 20–30 min) for submaximal preconstriction (~10% from basal tone) before measuring K+‐induced vasodilation. Due to the role of EC in myogenic tone (Scotland et al., 2001), we used 5‐HT as an alternative to myogenic tone to preconstrict vessels because it does not alter KIR channel function (Wu et al., 2007) and 5‐HT‐induced constriction is preserved in both APP and TGF mice (Tong et al., 2005).
At the end of the experiments, EC denudation was confirmed with the endothelium‐dependent dilator ACh (10−10–10−4 M) (Wu et al., 2007; Zhang et al., 2013), whereas SMC integrity was tested with the SMC relaxant PAP (10−10–10−4 M). Dilatations were measured as percentage change in diameter from 5‐HT‐preconstricted to dilated arteries in response to K+, ACh or PAP (Zhang et al., 2013).
2.3.6. Benefits of antioxidant and anti‐inflammatory treatments
Arterial dilatation to K+ (6 to 16 mM) and constriction to Ba2+ (20–120 μM) or ML133 (20 μM) (experiments described in previous sections) were studied before and after superfusion with a Krebs solution containing the antioxidant catalase (1000 units·ml−1, 60 min), because we have shown before that it rescues endothelial function in APP mice (Zhang et al., 2013), or containing the nonsteroidal anti‐inflammatory drug indomethacin (10 μM, 30 min) (Rubio‐Ruiz et al., 2014), which has been shown to restore other potassium channels in a model of COX‐dependent dysfunction (Armstead, 2001). We also used these treatments because neither catalase nor indomethacin has been reported to alter myogenic tone‐induced preconstriction (Erdös et al., 2004; Scotland et al., 2001). Vessels were washed with regular Krebs (4 mM of K+) for about 10 min before measuring changes in diameter, in order to avoid indomethacin‐mediated PKC activation (Kanno et al., 2012), which is known to inhibit KIR channel function when combined with Ba2+ (Vetri et al., 2012).
2.4. Western blotting
Major cerebral arteries and their pial branches, collected fresh at the time of reactivity experiments, were frozen on dry ice and kept in −80°C. Protein levels of the KIR2.1 subunit and oxidative stress marker SOD2 were measured in protein extracts from pial vessels from two cohorts of mice. Data were pooled for analysis (WT: n = 6, APP: n = 8 and TGF: n = 8). Vessels were dissolved in 30 μl of Laemmli buffer (62.5‐mM Tris, pH 7.5, 6‐M urea, 160‐mM 1,4‐DTT, 2% SDS and 0.001% bromophenol blue), boiled (5 min), protein assayed (assay kit; Bio‐Rad, Hercules, CA, USA), loaded (10 μg) onto a 10% acrylamide SDS‐PAGE gel and transferred to nitrocellulose membrane (Schleicher & Schuell, Keene, NH) (Tong et al., 2005). The membranes were blocked (1 h at room temperature) with 7% skim milk in Tris‐buffered saline‐Tween 20 (20‐mM Tris, 137‐mM NaCl and 0.1% Tween 20) and incubated overnight (4°C) with rabbit anti‐KIR2.1 (1:200 from Sigma‐Aldrich, RRID:AB_2040107), rabbit anti‐SOD2 (1:3000, MnSOD, Enzo Life Science, ON, Canada) or mouse anti‐β‐actin (1:16,000, Sigma‐Aldrich, RRID:AB_476744) to normalize for loading. Blots were further incubated for 1 h with HRP‐conjugated (goat anti‐mouse IgG, Fcγ fragment specific [RRID:AB_2313585] or goat anti‐rabbit IgG [H + L] [RRID: AB_2307391]) secondary antibody (1:2000; Jackson ImmunoResearch, PA, USA). Membranes were developed (5 min) using WesternSure Premium Chemiluminescent substrate (LI‐COR kit; Lincoln, NE, USA) and scanned using a C‐Digit Blot phosphor Imager (scanner STORM 860, GE Health Care, Piscataway, NJ, USA). Image densitometry was analysed with Image Studio Digit software, Version 5. The Immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology.
2.5. Sensory‐evoked changes in neuronal activity and haemodynamic responses of NVC
In contrast to APP mice which show both neuronal and cerebrovascular deficits in sensory‐evoked NVC responses (Tong et al., 2012), TGF mice are exempt from neuronal alterations. Particularly, TGF mice have intact glucose uptake in response to whisker stimulation whereas the haemodynamic response is severely impaired (Nicolakakis et al., 2011). Hence, they allow investigating the outcome of defective cerebrovascular KIR channels in the whisker‐evoked NVC response independently from changes in neuronal activity. Mice (WT, n = 9, TGF, n = 10, 7–10 months) were anaesthetized with Ketamine/Xylazine (85 mg·kg−1, i.m., half dose reinjected as needed), placed in ear bars on a physiological platform for monitoring of heart and respiratory rates and maintenance of body temperature (Harvard Apparatus, Saint‐Laurent QC, Canada). The barrel cortex was exposed by carefully drilling and removing the bone. A flexible 32‐channel micro‐electrocorticogram (ECoG) electrode array (Neuronexus, Ann Arbor, USA) was placed on the dura, covered with warm agarose gel (1%) and a coverglass (5‐mm diameter). A mini‐screw on the cerebellum was used as a reference. Whiskers were attached to a piezo actuator (piezo.com, Woburn, MA, USA) and stimulated at 1 and 4 Hz for 2 and 5 s. Upon completion of electrophysiological recording, the ECoG was slowly slid out the window for optical imaging of intrinsic signals by illuminating the cortical surface with three interleaved LEDs (530, 560 and 625 nm) and laser‐speckle contrast imaging of CBF with a 798‐nm laser (Labeotech.com). Ten trials were acquired (40‐s baseline, 20‐s whisker stimulation, 4 Hz). Agarose and the coverglass were carefully removed and a pool was formed around the barrel cortex for 20‐min superfusion of warm oxygenated Krebs (in mM) (124 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 4 glucose) containing 100‐μM Ba2+ (Longden et al., 2017). Electrophysiology and haemodynamic recordings were repeated post‐Ba2+ superfusion. A group of five WT mice were also recorded with a low dose of Ba2+ (10 μM) as a control for the procedure.
2.5.1. Analysis of electrophysiology and haemodynamic recordings
Electrophysiological data were acquired at a 30‐kHz sampling rate by an OpenEphys system and analysed using a custom‐made MATLAB (RRID:SCR_001622) script. P1‐N1 amplitude of the evoked responses was computed as the maximum and the minimum values over the initial 62.5 ms of each trigger. P1‐N1 amplitude was based on the first peaks of each stimulation train. Further investigation of the ECoG signal was carried out by analysis of the ECoG power in frequency bands (θ: 4–8 Hz, α: 8–12 Hz, β: 15–30 Hz, low γ: 40–60 Hz and high γ: 80–150 Hz), and evoked changes in power were normalized from baseline epoch. Haemodynamic data were analysed using MATLAB as described in Dubeau et al. (2011). Briefly, reflectance signals were converted to changes in absorption ΔA = log(R/R0), and a pseudo‐inverse and the modified Beer–Lambert law were used to extract relative changes in deoxygenated haemoglobin (HbR) and oxygenated haemoglobin (HbO). CBF was computed by quantifying the spatial contrast during laser illumination, defined as the ratio of the SD to the mean intensity in a given spatial area. Changes in total haemoglobin, HbR, HbO and CBF were computed from the manually delineated region of interest, based on the maximum response before Ba2+ superfusion.
2.6. Data analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). Mice were randomly distributed into experimental groups, and all experiments were performed blind to the identity of the mouse genotype. Responses to intraluminal pressure and to individual drugs were graphically represented as % of diameter change from baseline to facilitate visualization of group differences in diameter changes. Data are expressed as mean ± SEM and were analysed by Student's t‐test, one‐ or two‐way (genotype and treatment as factors) ANOVA followed by Newman–Keuls post hoc or Dunnett's multiple comparison tests. A post‐test was only performed if F achieved P < 0.05. For all experiments, n refers to the number of mice per group. Any variance in group sizes different from n = 5 was due to either exclusion of defective vessels (eventual loss of response or abrupt changes of pressure) or large intra‐group variability as shown in the young APP mice. Statistical analysis was undertaken only for studies where each group size was at least n = 5, with the exception of experiments where compounds (Ba2+ and ML133, Figure 2) were tested in different vessels under different conditions that showed comparable results. In these specific cases, we prioritized reducing the number of mice involved in the study and a justification has been included in the text. Two studies (pressure‐induced myogenic tone and the effect of age on KIR channel function) have been included as exploratory (n < 5) and no statistics are shown. Including these experiments was necessary to justify the age range selected for the study and the use of 80‐mmHg pressure to induce arterial preconstruction. Again, in order to reduce the number of mice involved in the study, for vascular reactivity studies and western blots, APP and TGF mice were compared to pooled WT mice from J20 and T64 lines, because both lines have identical backgrounds (C57BL/6J), and no differences were found during the study. Raw data for representative luminal diameters (µm) of pial arteries from each mouse group are shown in the supporting information (Tables S1‐S6). GraphPad Prism, Version 8.0.1 (RRID:SCR_002798; GraphPad Software, San Diego, CA, USA) was used for statistical analysis of in vitro and in vivo studies. A P < 0.05 was considered significant.
FIGURE 2.
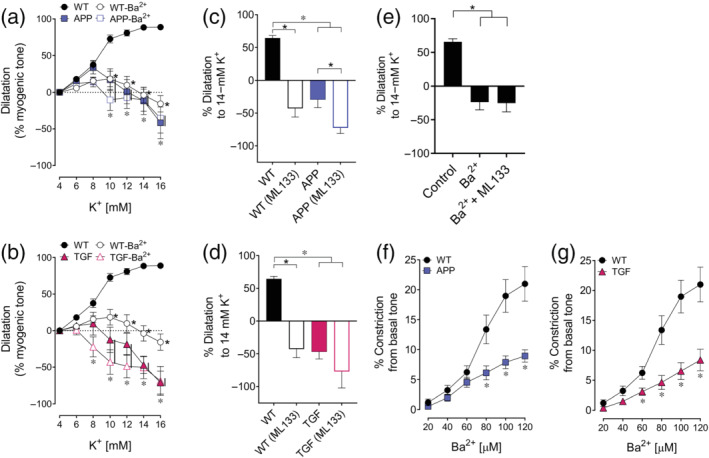
Dysfunctional dilatations to K+ ions and altered basal tone are mediated by impaired KIR2.1 subunit. (a,b) KIR channel blockade with Ba2+ significantly impaired K+‐induced dilatations in WT mice while it had no effect in APP or TGF mice. n = 5 for WT, APP and TGF. (c,d) At 14‐mM K+, selective blockade of the KIR2.1 subunit with ML133 exerted a deleterious effect in WT arteries, while having a smaller or no effect in APP or TGF arteries, highlighting the key role of the KIR2.1 subunit in both pathologies. n = 5 for WT and TGF; n = 4 for APP. (e) The combination of Ba2+ and ML133 did not have synergistic effect in WT mice (n = 5). *,* P < 0.05 for differences with WT and treated condition, respectively, in two‐way ANOVA followed by a Newman–Keuls post hoc comparison test. Error bars represent SEM. Vertical bars represent equal P‐values. (f,g) KIR channel blockade by increasing concentrations of Ba2+ at basal (4.5 mM) K+ concentration induced a greater vasoconstriction in WT than in APP or TGF mice, pointing to dysfunctional KIR channel at basal tone in APP and TGF mice. n = 6 for WT; n = 4 for APP and n = 5 for TGF. *P < 0.05 for Student's t‐test. Error bars represent SEM
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3. RESULTS
3.1. Myogenic tone is conserved in pial vessels of APP and TGF mice
We aimed to investigate whether diminished NO bioavailability in APP and TGF mice (Nicolakakis et al., 2011; Tong et al., 2005), or increased vessel stiffness in TGF mice (Gaertner et al., 2005), could be detrimental to myogenic tone maintenance. We found that both APP and TGF mice had intact myogenic tone in response to increasing intraluminal pressure (40–120 mmHg) when compared with WT mice (Figure 1a). At 80 mmHg, the intraluminal pressure that we used to induce arterial preconstriction in most vascular reactivity studies, myogenic tone was similar between all groups (WT: 11.6 ± 0.9%, APP:13.3 ± 2.6%, TGF: 16.2 ± 2.9%). Even at the highest intraluminal pressure tested (120 mmHg), APP and TGF vessels were still able to develop myogenic tone at levels comparable with WT mice (WT: 18.1 ± 3.3%, APP: 23.6 ± 3.7%, TGF: 23.1 ± 5.8%). Because only an n = 4 was used for this study, these results are considered exploratory, but, overall, the myogenic tone developed by arteries from all groups compared well with values previously reported in WT mice of the same background (Bai et al., 2004; Springo et al., 2015). Our results further support previous studies reporting that neither impaired NO bioavailability nor vessel stiffness compromise myogenic tone (Scotland et al., 2001; Springo et al., 2015), hence justifying the use of myogenic tone for preconstriction of pial arteries in vascular reactivity experiments. However, in all experiments where 80‐mmHg pressure was used for preconstricting vessels before testing K+‐induced dilatations, arteries from APP, and specially from TGF mice, reached pressure‐induced preconstriction faster (40–50 min) than WT (60–75 min). This observation may be due to the dysfunction of additional ion channels known to oppose pressure‐induced myogenic tone in resistance arteries (Tykocki et al., 2017).
FIGURE 1.
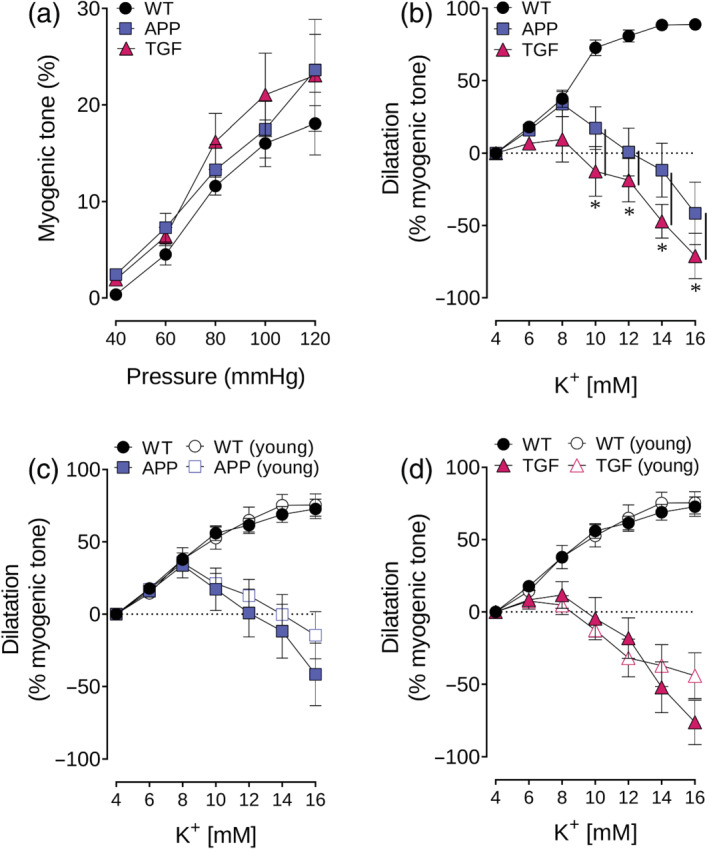
Conserved myogenic tone and impaired K+‐induced dilatations in APP and TGF mice. (a) Myogenic tone was conserved in pial arteries of APP and TGF mice for a range of pressure 40–120 mmHg. n = 4 for WT, APP and TGF. (b) K+‐induced dilatations were impaired in adult (7–10 months) APP and TGF mice when compared with WT vessels. n = 5 for WT, APP and TGF. *P < 0.05 for ANOVA followed by a Newman–Keuls post hoc comparison test. (c,d) Impaired dilatations were already present in young (3–4 months) APP and TGF mice. n = 3 for WT and TGF; n = 11 for APP. Error bars represent SEM. Vertical bars represent equal P‐values
3.2. K+‐induced dilatation is significantly impaired in APP and TGF mice
Compared with WT vessels, both APP and TGF mice showed markedly impaired dilatory responses to K+ (6 to 16 mM). WT vessels reached maximal dilatation at 16‐mM K+ (88.8 ± 2.5%), while in APP and TGF vessels, maximal dilatation occurred at 8‐mM K+ (APP: 33.7 ± 8.7%, TGF: 9.5 ± 15.7%), followed by prominent vasoconstriction at higher K+ concentrations (Figure 1b), thus showing a biphasic response. Preliminary results show that this response is similarly impaired in young APP and TGF mice (Figure 1c,d, respectively). However, young APP mice displayed a large variability possibly suggesting an inflection point in KIR channel dysfunction at this age.
3.3. Dysfunction of KIR channels and KIR2.1 subunit in APP and TGF mice
In WT vessels, the KIR channels blocker, Ba2+ (100 μM), drastically reduced the dilatory response to increasing K+ concentrations, reversing it to vasoconstriction at higher K+ concentrations (14–16 mM). In contrast, Ba2+ did not further worsen the already affected responses in APP and TGF vessels, pointing to dysfunctional cerebrovascular KIR channels in both groups (Figure 2a,b). Furthermore, when investigating the contribution of the KIR2.1 subunit, we found that, in WT mice, selective KIR2.1 subunit blockade with ML133 completely blocked arterial dilatation to 14‐mM K+ and was actually reversed to a constriction (64.5 ± 3.8% vs. −42.8 ± 13.1% of myogenic tone). In APP vessels, ML133 significantly enhanced the vasocontractile response to 14‐mM K+ (−29.3 ± 12.4% vs. −72.5 ± 8.7% of myogenic tone) (Figure 2c). In TGF mice, selective KIR2.1 subunit blockade exerted a similar potentiating effect on the contractile response, albeit not significant (−47.0 ± 10.5% before vs. −77.0 ± 25.2% after ML133) (Figure 2d). Our results indicate that the KIR2.1 subunit of KIR channels is impaired in APP and TGF mice. We did not find any synergistic blocking effect of concurrent treatment with Ba2+ and ML133 on KIR channel, tested in WT arteries: control = 65.6 ± 4.6%, Ba2+ = −23.8 ± 11.4, Ba2+ + ML133 = −25.3 ± 13.1 (Figure 2e). Even though the APP group has an n = 4 (Figure 2c,f), these results were repeatedly confirmed among different experiments showing no or virtually no effect of KIR blockers on arterial responses to K+.
3.4. KIR channels and cerebrovascular basal tone
At baseline, small diameter peripheral vessels and cerebral arteries constrict in response to KIR channel blockade with Ba2+, suggesting a modulatory role for KIR channels in arterial basal tone (reviewed in Chrissobolis & Sobey, 2003; Ko et al., 2008). In WT pial arteries, increasing Ba2+ concentrations (20–120 μM) induced a marked contractile response (21.0 ± 2.9% from baseline tone at 120 μM) whereas it had a much smaller effect in APP (8.9 ± 1.0%) and TGF (8.3 ± 1.8%) arteries (Figure 2f,g), indicating an impaired role of KIR channels in APP and TGF arteries at baseline.
3.5. ECs are essential for K+‐induced maximal dilatation of pial arteries
We aimed to identify the contribution of EC‐KIR channels to K+‐induced vasodilatation. Using 5‐HT‐preconstricted EC‐denuded arteries, we found that vasodilatation was diminished (~50%) compared with intact arteries in WT (Figure 3a). In APP mice, EC denudation only decreased (~50%) vasodilatation at 8‐mM K+ (Figure 3b) and had no effect on the highest K+ concentrations. In TGF mice, EC denudation did not worsen the already impaired K+‐induced vasomotor responses at any K+ concentration (Figure 3c). In all cases, EC denudation was confirmed by the loss of endothelium‐dependent dilation to ACh (<1%, data now shown), whereas SMC integrity was preserved as shown by similar dilatations to PAP in all groups (Figure 3d).
FIGURE 3.
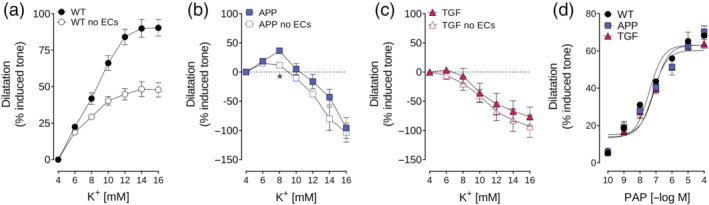
Endothelial cell denudation reduces K+‐dependent dilatation in WT, but not in APP or TGF, mice. (a,b,c) EC removal reduced maximal dilatations by about 50% in WT arteries but had little or no effect in vessels from APP or TGF mice. (d) SMC function was not impaired after in EC‐denuded arteries as shown by conserved dilatation to the SMC‐relaxant papaverine (PAP). *P < 0.05 represent differences in Student's t‐test. n = 4 for WT; n = 5 for APP and TGF
3.6. Antioxidant or anti‐inflammatory treatment restores KIR channel function in APP and TGF pial vessels
Incubation of pial artery segments with the antioxidant catalase did not affect KIR channel function in WT and TGF vessels, whereas it normalized vasodilatation in APP vessels (WT: 74.9 ± 5.8%, APP: 48.6 ± 11.7% of myogenic tone) (Figure 4a,d). In contrast, the anti‐inflammatory drug indomethacin did not have any effect on K+‐induced vasomotor responses in WT or APP vessels, but it restored that of TGF mice to a dilatation comparable with that of WT arteries (WT mice: 69.9 ± 5.5%, TGF mice: 53.2 ± 7.3% of myogenic tone) (Figure 4b,e). Interestingly, ML133 prevented the beneficial effects of catalase and indomethacin on APP and TGF arteries, respectively, indicating that functional recovery is, at least in part, due to rescue of the KIR2.1 subunit of KIR channels (Figure 4c,f).
FIGURE 4.
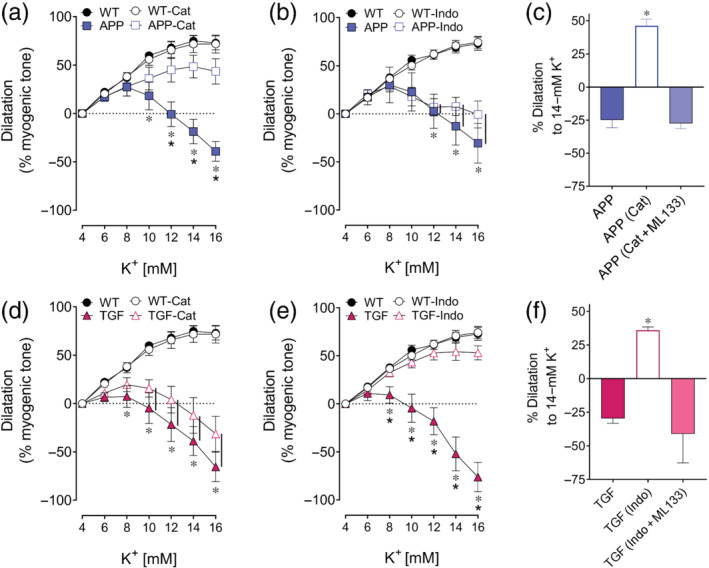
Catalase rescued KIR2.1 function in APP mice while indomethacin rescued KIR2.1 function in TGF mice. (a,b,d,e) K+ mediated dilatation was restored in APP mice and in TGF mice with catalase and indomethacin, respectively. *, * P < 0.05, for differences with WT and treated condition, respectively, in two‐way ANOVA followed by a Newman–Keuls post hoc comparison test. Error bars represent SEM. Vertical bars represent equal P‐values. n = 5 for WT, APP and TGF. (c,f) KIR2.1 subunit rescue could be blocked with ML133, which resulted in a vasoconstriction pattern similar to non‐treated vessels. *P < 0.05 for one‐way ANOVA followed by a Newman–Keuls post hoc comparison test. Error bars represent SEM. n = 5 for WT and TGF; n = 4 for APP
Similarly, preliminary results showed that catalase and indomethacin treatments fully restored basal tone in APP (Figure 5a,b) and TGF mice (Figure 5c,d), respectively. Indomethacin also exerted a small deleterious effect on WT arteries (Figure 5c,d) at high Ba2+ concentrations (>80 μM) at basal tone, likely due to a remaining effect of indomethacin‐driven PKC activation (Kanno et al., 2012), an effect previously reported and countered by washing off indomethacin (Filosa et al., 2006).
FIGURE 5.
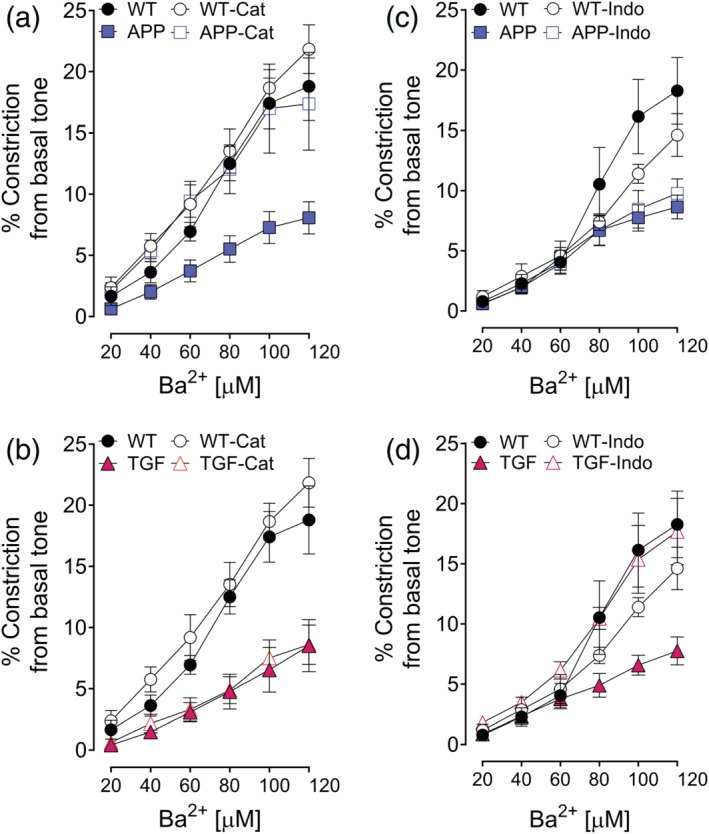
Catalase and indomethacin selectively restored basal tone. (a–d) Impaired vasoconstriction induced by Ba2+ in APP and TGF mice was restored by catalase and indomethacin, respectively. Error bars represent SEM. n = 5 for WT; n = 4 for APP and TGF
3.7. Inflammation but not oxidative stress modulates KIR2.1 channel expression in pial arteries
Whether the altered KIR channel function in APP and TGF mice is accompanied by changes in protein expression of the key KIR2.1 subunit was studied by western blotting. Pial arteries from TGF, but not APP, mice showed significantly increased protein levels of the KIR2.1 subunit compared with WT arteries (234.9 ± 34.2% from WT) (Figure 6a). As expected from previous studies (Tong et al., 2005), the oxidative stress marker SOD2 was increased only in APP arteries (228.8 ± 25.2% from WT), which suggests that inflammation can modify KIR2.1 protein expression in TGF mice (Figure 6b).
FIGURE 6.
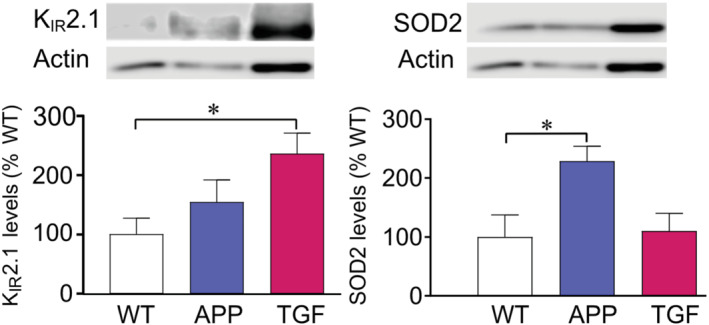
Inflammation but not oxidative stress up‐regulates KIR2.1 subunit protein levels in pial arteries. (a) KIR2.1 subunit was up‐regulated in TGF mice but not in APP mice compared with WT. (b) Only arteries from APP mice displayed increase in the oxidative stress marker SOD2. *P < 0.05 in one‐way ANOVA followed by Dunnett's test. Error bars represent SEM. n = 6 for WT; n = 8 for APP and TGF
3.8. Barium differently impairs in vivo NVC responses in WT and TGF mice
In WT mice, whisker stimulation led to increased CBF, enhanced supply of oxyhaemoglobin (HbO) and wash‐out of deoxyhaemoglobin (HbR) in the barrel cortex. This effect was significantly reduced by KIR channel blockade using 100‐μM Ba2+ (CBF: −43%, HbO: −61%, P < 0.05). In contrast, the low 10‐μM Ba2+ concentration did not affect haemodynamic function (CBF: +26%, HbO: +13%), confirming no effect on the procedure (Figure 7a,b). TGF mice displayed significantly reduced CBF and HbO whisker‐evoked responses compared with WT, but these were not further altered by 100‐μM Ba2+. However, in TGF, the HbR response was significantly reduced by Ba2+ ([HbR]: −0.1 to −0.6, P = 0.01); as CBF was not changed, this may suggest a decreased HbO demand (Figure 7b). Whereas the whisker‐evoked neuronal responses were not affected by Ba2+ in WT mice, as measured by the P1‐N1 amplitude and power of the ECoG signal, the latter was significantly lessened by Ba2+ across most frequency bands in TGF mice, pointing to neuronal susceptibility to KIR channel blockade in TGF mice (Figure 7c).
FIGURE 7.
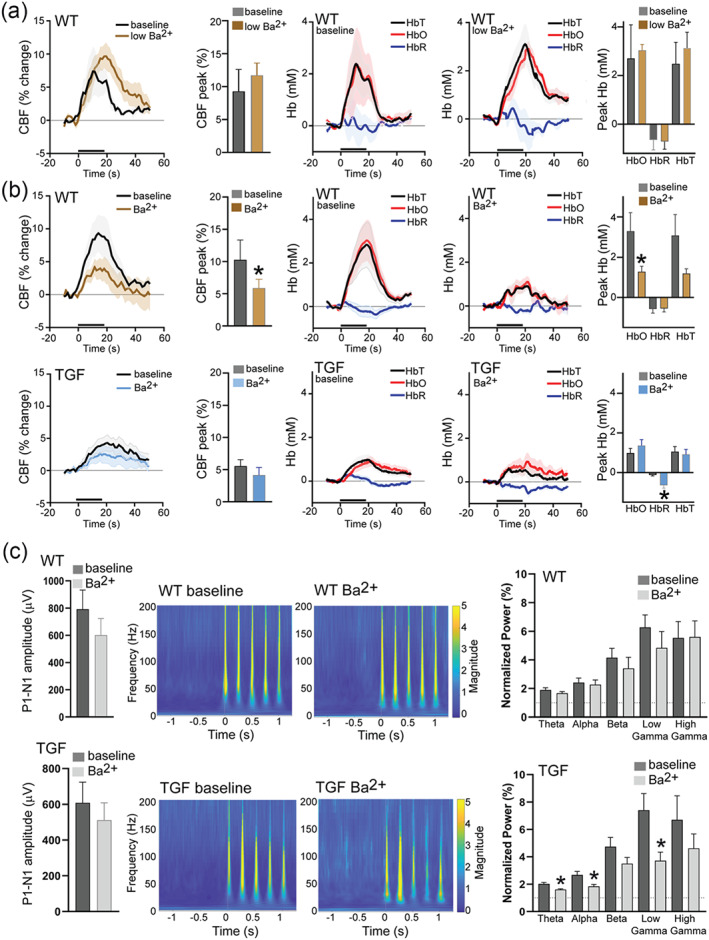
Inflammation alters KIR channel‐mediated haemodynamic responses and electrophysiology to whisker stimulation. (a) No effect of Ba2+ (10 μM) on haemodynamic responses to whisker stimulation in WT mice (n = 5). (b) Significantly reduced cerebral blood flow (CBF) and oxyhaemoglobin responses (peak HbO) in WT but not in TGF mice to whisker stimulation after superfusion with Ba2+ (100 μM). (c) Deoxyhaemoglobin (HbR) was reduced in TGF mice after Ba2+ superfusion. Conserved neuronal activity after superfusion with Ba2+ (100 μM) in WT mice but not in TGF mice (showing reduced θ, α and γ power). *P < 0.05 represent differences between baseline and treatment in Student's t‐test. Error bars represent SEM. n = 9 for WT; n = 10 for TGF. HbT; total haemoglobin [Correction added on 20 July 2022, after first online publication: Figure 7 has been corrected in this version.]
4. DISCUSSION
KIR channels are involved in CBF regulation and arterial basal tone maintenance (reviewed in Chrissobolis & Sobey, 2003; Ko et al., 2008; Tykocki et al., 2017), and the fact that they are dysfunctional in several pathologies associated with higher risk of dementia (Bastide et al., 1999; Longden et al., 2014; Marrelli et al., 1998; Mayhan et al., 2004; McCarron & Halpern, 1990a) makes these channels promising therapeutic targets. Thus, understanding the mechanisms triggering KIR channel dysfunction is essential in order to develop appropriate interventions.
We found that KIR channel dysfunction is an early event in APP and TGF mice (already present at 3–4 months), consistent with previous studies reporting early cerebrovascular deficits in both transgenic lines mainly characterized by reduced EC‐dependent dilatory responses (Tong et al., 2005). KIR channel function in APP and TGF mice was restored by catalase and indomethacin, respectively. These results not only show that KIR channels are sensitive to oxidative stress, in line with Aβ‐induced cerebrovascular ROS and SOD2 up‐regulation in APP mice (Park et al., 2004; Tong et al., 2005), but also to cerebrovascular inflammation characteristic of brain vessels in TGF mice (Tong & Hamel, 2015; Wyss‐Coray et al., 2000). We also confirm recent results reporting that a functional endothelium is necessary for maximal K+‐induced dilatation of pial arteries.
4.1. KIR channels in APP and TGF mice: Impact of oxidative stress and inflammation
Under normal conditions, low concentrations of extracellular K+ (<25‐mM K+) induce hyperpolarization of SMCs and vasodilatation, whereas higher concentrations induce SMC depolarization, leading to vasoconstriction. However, in pathological conditions, K+‐mediated vasodilatation is diminished and vasoconstriction can appear at concentrations lower than 25‐mM K+ (Longden et al., 2014). We found that in both APP and TGF mice, arterial dilatations were maintained at K+ concentrations lower than 8 mM, possibly explained by the contribution of the electrogenic sodium pump to dilatation at concentrations lower than 7‐mM K+ (McCarron & Halpern, 1990b), a pump presumably unaltered in cerebrovascular pathology associated with oxidative stress and inflammation (McCarron & Halpern, 1990a). K+‐induced vasodilatation, in APP mice, was significantly reduced at 10–12 mM K+, which corresponds to physiological K+ concentrations seen during NVC in vivo (Filosa et al., 2006). In TGF mice, dilatory impairments were already detectable at 8‐mM K+ and, in some experiments, vessels started to constrict at 10‐mM K+, suggesting a more severe impairment driven by inflammation. In healthy arteries, K+ concentrations above 25 mM change equilibrium potential (E K) to levels that depolarize pial arteries, resulting in constriction through voltage‐gated calcium channel activation (Longden & Nelson, 2015). Dysfunctional KIR channels in APP and TGF mice may alter E K and facilitate voltage‐gated calcium channel activation at lower K+ concentrations, similarly to that observed in WT arteries after blocking KIR channels with either Ba2+ or ML133. Although ML133 is supposedly more selective for KIR2.1 channels than Ba2+, its IC50 values at KIR 2.1 and KIR2.2 isoforms are relatively similar (1.8 and 2.9 μM, respectively) (Wang et al., 2011), which could explain the similar blocking effect exerted by Ba2+ or ML133 on vasoconstriction.
When further investigating the impact of altered KIR channel function on arterial basal tone, we found impairments in both APP and TGF mice. Impaired KIR channel‐mediated vasodilatation and basal tone maintenance could explain, at least in part, the reduced whisker‐evoked hyperaemic responses seen in APP (Nicolakakis et al., 2008) and TGF mice (Nicolakakis et al., 2011), and the chronic cerebral hypoperfusion previously reported in these mice (Gaertner et al., 2005; Hébert et al., 2013). However, we do not have patch clamp electrophysiological data to show that KIR channel currents are reduced in APP and TGF models, a possibility that would be worth further investigation.
Whereas a possible detrimental effect of oxidative stress on KIR channel function has been postulated (Bastide et al., 1999; Erdös et al., 2004; Vetri et al., 2012), the impact of inflammation has been poorly documented despite its known deleterious effects on large conductance calcium‐activated potassium (BKCa ) and ATP‐sensitive K+ channels (KATP ) (Wei et al., 1996). Cortical parenchymal arteriolar KIR channel function was altered in a rat model of preeclampsia with both inflammation and oxidative stress, but the contribution of each pathology was not elucidated (Johnson & Cipolla, 2018).
It was recently shown that Kir2.1 activity in brain ECs from 5xFAD mice depends on the binding of phosphatidylinositol 4,5‐bisphosphate (PIP2 ) to the channel (Mughal et al., 2021). Interestingly, oxidative stress and, specifically, hydrogen peroxide (H2O2) can decrease PIP2 levels by deactivating phosphatidylinositol‐4‐phosphate 5‐kinase β (PIP5Kβ) (Chen et al., 2009). Therefore, we hypothesize that, in APP mice, the increased levels of H2O2, a sub‐product of the ROS dismutation by SOD2, may impede the binding of PIP2 to KIR2.1 and, hence, its activity. Alternatively, H2O2‐induced PKC activation (Cosentino‐Gomes et al., 2012) could also explain the loss of response to K+ in APP mice, as PKC activation was found to result in loss of K+‐induced vasodilatation in a rat model of type 1 diabetes through modulation of KIR2.1 channel function (Vetri et al., 2012), a deficit restored by acute inhibition of PKC activity.
On the other hand, our findings indicate that inflammation impairs KIR channel function as severely as oxidative stress, if not more. Interestingly, we also found that brain arteries from TGF mice exhibited increased protein levels of the KIR2.1 subunit, whereas no significant changes were found in APP mice. Previous murine models of cerebrovascular pathology due to I/R or to type 1 diabetes have reported stable expression of the KIR2.1 subunit; however, these models did not cause KIR2.1 dysfunction or it was mild and undetectable (Erdös et al., 2004; Vetri et al., 2012). While cytokine‐driven brain KIR2.1 subunit overexpression could explain the increased protein levels seen in TGF vessels, as reported following systemic inflammation (Vicente et al., 2004), it cannot be discarded that KIR2.1 up‐regulation is a compensatory mechanism from chronic KIR channel dysfunction. Indeed, while APP arteries still partially dilated at K+ concentrations mimicking physiological levels in functional hyperaemia (10 mM), those from TGF mice constricted, which suggest that compensatory mechanisms may have emerged in response to a severe KIR2.1 dysfunction. The molecular mechanisms beyond this up‐regulation are worth further investigating, but up‐regulation of KIR2.1 protein levels has been reported before, namely, after pharmacological blockade of the cytoplasmic pore of the channel (Ji et al., 2017).
4.2. Role of endothelial KIR2.1 in K+‐induced vasodilatation of pial arteries
Traditionally, KIR channels have been exclusively associated with vascular SMCs, but more recent studies in mesenteric arteries (Sonkusare et al., 2016), pial arteries (Hakim, Behringer, et al., 2020; Hakim, Chum, et al., 2020; Sancho et al., 2017) and intraparenchymal brain vessels (Longden et al., 2017) have confirmed their presence in ECs and their active role in electrical communication and vasodilatation, driven by the KIR2.1 subunit. In our study, endothelial denudation of WT arteries reduced K+‐induced dilatation by ~50% and, while it also reduced dilatation in APP arteries at 8‐mM K+, TGF arterial responses were unaffected. These findings demonstrate the requirement of a healthy endothelium for optimal dilatation of pial arteries in response to K+ ion electrical signalling. Further, they indicate that EC‐KIR2.1 channels are impaired in both APP and TGF mice, but more severely in TGF arteries because removal of EC does not result in worsening of their already reduced dilatory capacity. These findings are in line with previous reports of impaired ACh‐mediated vasodilatation in pial arteries of APP and TGF mice (Tong et al., 2005; Tong & Hamel, 2015; Zhang et al., 2013), as ACh engages EC‐KIR channels to boost their signal strength (Sonkusare et al., 2016).
Endothelial KIR 2.1 channels are also essential for the upstream propagation of K+ signalling in intraparenchymal arteries (Longden et al., 2017), a role postulated to be maintained at the level of pial arteries to ensure adequate blood supply at baseline and during NVC (Iadecola, 2004). EC/EC gap junctions permit the fast transmission of the electrical signalling upstream that is then transmitted from ECs to SMCs through gap junctions of myoendothelial projections allowing vasomotor functions (Sancho et al., 2017; Segal, 2000). Some in vivo studies suggest that endothelial NO production may contribute to the maintenance of vasodilation induced by K+ (Dormanns et al., 2016), a role that was not tested in the present study but that cannot be discarded. Similarly, we did not investigate the respective contribution of EC‐ or SMC‐KIR channels in basal tone maintenance, but the interplay of both cell types appears necessary (Sancho et al., 2019).
4.3. KIR channels and whisker‐evoked NVC
When investigating NVC in WT mice, we confirmed that KIR channel blockade with Ba2+ impaired whisker‐evoked haemodynamic responses without affecting neuronal activity, pointing to a selective effect of Ba2+ on the vasculature in WT mice, as previously reported in Longden et al. (2017). To the contrary, in TGF mice, Ba2+ exerted a detrimental effect on whisker‐evoked neuronal activity but did not worsen the already impaired haemodynamic responses. Because the ECoG signal power was reduced across most frequency bands by Ba2+ in TGF, it suggests that Ba2+ acts through a broad mechanism rather than affecting selective neurons driving a specific cortical oscillation (Chen et al., 2017). Together, these findings indicate dysfunctional KIR channels in intraparenchymal microvessels of TGF mice and further reveal the existence of intact KIR channels that ensure proper neuronal function. We explain this increased susceptibility of neuronal activity to Ba2+ in TGF by the underlying neuroinflammation and severe astrogliosis (Ongali et al., 2018). Astrocytes play a key role in synapse formation and K+ homeostasis; hence, astrogliosis can have a detrimental effect on neuronal function (Kim et al., 2019). Reactive astrocytes, identified by GFAP marker, express the KIR2.1 subunit (Brasko & Butt, 2018; Kang et al., 2008). Thus, in TGF mice, blockade of astrocytic KIR2.1 subunit with Ba2+ could lead to altered astrocyte‐dependent K+ ion homeostasis and, hence, reduced changes in neuronal activity. The interplay between astrocytic and elevated endothelial KIR2.1 subunit in TGF mice deserves further investigation to elucidate possible compensatory mechanisms to preserve neuronal function under chronic neuroinflammatory conditions.
4.4. Benefits of antioxidant and anti‐inflammatory therapies on KIR channel function
Catalase restored KIR channel‐mediated dilatations through KIR2.1 activation in APP mice. Although there is no literature reporting the benefit of antioxidant therapies in restoring KIR function, combined catalase and SOD treatment rescued BKCa and KATP channels in a rat model of insulin resistance, both channels being sensitive to oxidative stress (Erdös et al., 2004). Here, in APP pial arteries, SOD2 protein levels were up‐regulated, presumably as a compensatory mechanism to trap Aβ‐generated free radicals and increase NO bioavailability (Tong et al., 2009; Tong & Hamel, 2007). When treated with catalase, cerebrovascular KIR channel functionality in APP mice virtually fully recovered, indicating that antioxidant mechanisms are beneficial to multiple types of K+ channels. These findings also agree with the reported benefits of antioxidant therapies on cerebrovascular function in APP mice (reviewed in Hamel, 2015). SOD2 is the main superoxide radical (O2 −) scavenger in mitochondria, where it catalyses O2 − dismutation into oxygen (O2) and H2O2. Free radical scavengers rescue H2O2‐induced perturbation of PIP2 homeostasis (Chen et al., 2009). It is thus possible that catalase restored KIR2.1 function in APP pial arteries throughout recovery of PIP2 homeostasis by reducing circulating H2O2. However, catalase failed to rescue KIR channel function in TGF mice, findings consistent with previous studies from our group showing no benefit of antioxidants (catalase, N‐acetylcysteine and SOD) on endothelium‐dependent dilatory function in TGF mice whose pathology has been attributed to perivascular inflammatory processes (Nicolakakis et al., 2011; Tong et al., 2005; Zhang et al., 2013). Hence, knowing that TGF‐β1 can induce COX‐2 expression in vascular cells (Rodríguez‐Barbero et al., 2006; Tong & Hamel, 2007), we tested the non‐selective COX inhibitor indomethacin (Lucas, 2016) and found that it completely restored KIR channel function in TGF brain arteries, providing the first evidence of indomethacin restoring cerebrovascular KIR channel function. These findings demonstrate that inflammation alters KIR channel function, consistent with in vivo studies in TGF mice with drugs displaying cerebrovascular anti‐inflammatory properties (Nicolakakis et al., 2011; Tong & Hamel, 2015). Our results also agree with previous studies reporting benefits of indomethacin on KATP and BKCa channels in a model of brain injury, although the benefits were attributed to reduced COX‐dependent ROS generation (Armstead, 2001). Here, in TGF arteries, inflammation‐mediated KIR channel impairment appears independent from COX‐induced ROS generation or, at least, not H2O2 mediated, because catalase did not display any benefits. Indomethacin is able to rescue function of other K+ channels such as BKCa (Armstead, 2001), the impairment of which being related to enhanced PKC activity in vascular pathology involving an inflammatory response such as type 1 diabetes (Vetri et al., 2012). Similar mechanisms could be at play regarding the recovery of the KIR2.1 function in TGF mice.
In summary, our results indicate that an intact endothelium is essential for optimal KIR channel‐induced cerebral vasodilatation and NVC, that endothelial KIR2.1 channels are sensitive to perivascular Aβ‐ or TGF‐β1‐induced oxidative stress or inflammation and that cerebrovascular KIR2.1 function can be fully restored by both antioxidant (APP mice) and anti‐inflammatory (TGF mice) treatments. Our results suggest that in pathologies with mixed cerebrovascular oxidative stress and inflammation, like cardiovascular diseases with a cerebrovascular pathology or neurodegenerative diseases like AD, and vascular cognitive impairment and dementia, combined therapies should have greater benefits on resting CBF and evoked haemodynamic responses.
AUTHOR CONTRIBUTIONS
All authors have made substantial contributions as follows: M. L.‐A., C. L. and E. H. conceived the study. M. L.‐A. and C. L. wrote the manuscript. L. T. and E. H. edited the manuscript. Vascular reactivity experiments were performed by M. L.‐A. western blots were performed by M. L.‐A. and L. T. In vivo imaging studies were performed by C. L., and M. B. contributed to optimization of in vivo imaging studies. M. L.‐C., L. T. and C. L. performed statistical analysis. All authors gave final approval for publication.
CONFLICT OF INTEREST
Nothing to disclose.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design and Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Table S1A. Myogenic tone in wild‐type (WT), APP or TGF mice in the presence and absence of calcium (Ca2+)
Table S1B. Vasodilatation to increasing concentrations of K+ in arteries from adult mice
Table S1C & D. Vasodilatation to increasing concentrations of K+ in arteries from adult vs young mice
Table S2A & B. Effect of Ba2+ on K+‐induced vasodilatation in WT, APP and TGF arteries.
Table S2C & D. Effect of ML133 on 14 mM K+‐induced vasodilatation in WT, APP and TGF arteries.
Table S2E. Comparative effect of Ba2+ and Ba2+ + ML133 on brain arteries from WT mice
Table S3A, B, C. Effect of endothelium denudation in WT arteries on K+‐induced dilatation
Table S4A, B, D & E. Effects of catalase (Cat) and Indomethacin (Indo) on K+‐mediated vasodilatations
Table S6A,B,C,D. Rescue of basal tone in APP and TGF mice with catalase (Cat) and indomethacin (Indo), respectively.
ACKNOWLEDGEMENTS
This work was supported by grants (to E. H.) from the Canadian Institute of Health Research (CIHR) (MOP‐126001), the Canadian Vascular Network‐Canadian Institutes of Health Research (CVN‐CIHR) and the Alzheimer Society of Canada, as well as by a CVN‐CIHR Scholar Award and a Jeanne Timmins Postdoctoral Fellowship (to M. L.‐A.). We would like to thank Dr. Lennart Mucke from the Gladstone Institute of Neurological Disease and Department of Neurology, University of California, San Francisco, CA, USA, for providing the APP and TGF mouse breeders. We would also like to thank Dr. Sujaya Neupane, from Department of Brain and Cognitive Sciences, McGovern Institute for Brain Research, Massachusetts Institute of Technology, Cambridge, MA, USA, for developing the custom‐made MATLAB script used for the analysis of electrophysiological and haemodynamic data.
Lacalle‐Aurioles, M. , Trigiani, L. J. , Bourourou, M. , Lecrux, C. , & Hamel, E. (2022). Alzheimer's disease and cerebrovascular pathology alter inward rectifier potassium (KIR2.1) channels in endothelium of mouse cerebral arteries. British Journal of Pharmacology, 179(10), 2259–2274. 10.1111/bph.15751
Funding information Alzheimer Society of Canada; Canadian Vascular Network‐Canadian Institutes of Health Research (CVN‐CIHR); Canadian Institute of Health Research (CIHR), Grant/Award Number: MOP‐126001
Contributor Information
María Lacalle‐Aurioles, Email: maria.lacalleaurioles@mcgill.ca.
Edith Hamel, Email: edith.hamel@mcgill.ca.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Boison, D. , Burns, K. E. , Dessauer, C. , Gertsch, J. , Helsby, N. A. , Izzo, A. A. , Koesling, D. , … Wong, S. S. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Enzymes. British Journal of Pharmacology, 178, S313–S411. [DOI] [PubMed] [Google Scholar]
- Armstead, W. M. (2001). Vasopressin induced cyclooxygenase dependent superoxide generation contributes to K(+) channel function impairment after brain injury. Brain Research, 910, 19–28. 10.1016/S0006-8993(01)02716-0 [DOI] [PubMed] [Google Scholar]
- Bai, N. , Moien‐Afshari, F. , Washio, H. , Min, A. , & Laher, I. (2004). Pharmacology of the mouse‐isolated cerebral artery. Vascular Pharmacology, 41, 97–106. 10.1016/j.vph.2004.07.001 [DOI] [PubMed] [Google Scholar]
- Bastide, M. , Bordet, R. , Pu, Q. , Robin, E. , Puisieux, F. , & Dupuis, B. (1999). Relationship between inward rectifier potassium current impairment and brain injury after cerebral ischemia/reperfusion. Journal of Cerebral Blood Flow and Metabolism, 19, 1309–1315. 10.1097/00004647-199912000-00003 [DOI] [PubMed] [Google Scholar]
- Brasko, C. , & Butt, A. (2018). Expression of Kir2.1 inward rectifying potassium channels in optic nerve glia: Evidence for heteromeric association with Kir4.1 and Kir5.1. Neuroglia, 1, 176–187. 10.3390/neuroglia1010012 [DOI] [Google Scholar]
- Chen, G. , Zhang, Y. , Li, X. , Zhao, X. , Ye, Q. , Lin, Y. , Tao, H. W. , Rasch, M. J. , & Zhang, X. (2017). Distinct inhibitory circuits orchestrate cortical beta and gamma band oscillations. Neuron, 96, 1403–1418.e6. 10.1016/j.neuron.2017.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, M. , Zhu, X. , Sun, H. , Mao, Y. , Wei, Y. , Yamamoto, M. , & Yin, H. L. (2009). Oxidative stress decreases phosphatidylinositol 4,5‐bisphosphate levels by deactivating phosphatidylinositol‐4‐phosphate 5‐kinase beta in a Syk‐dependent manner. The Journal of Biological Chemistry, 284, 23743–23753. 10.1074/jbc.M109.036509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrissobolis, S. , & Sobey, C. G. (2003). Inwardly rectifying potassium channels in the regulation of vascular tone. Current Drug Targets, 4, 281–289. 10.2174/1389450033491046 [DOI] [PubMed] [Google Scholar]
- Cohen, S. , Janicki‐Deverts, D. , Doyle, W. J. , Miller, G. E. , Frank, E. , Rabin, B. S. , & Turner, R. B. (2012). Chronic stress, glucocorticoid receptor resistance, inflammation, and disease risk. Proceedings of the National Academy of Sciences of the United States of America, 109, 5995–5999. 10.1073/pnas.1118355109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosentino‐Gomes, D. , Rocco‐Machado, N. , & Meyer‐Fernandes, J. R. (2012). Cell signaling through protein kinase C oxidation and activation. International Journal of Molecular Sciences, 13, 10697–10721. 10.3390/ijms130910697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabertrand, F. , Harraz, O. F. , Koide, M. , Longden, T. A. , Rosehart, A. C. , Hill‐Eubanks, D. C. , Joutel, A. , & Nelson, M. T. (2021). PIP 2 corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity. Proceedings of the National Academy of Sciences of the United States of America, 118, e2025998118. 10.1073/pnas.2025998118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormanns, K. , Brown, R. G. , & David, T. (2016). The role of nitric oxide in neurovascular coupling. Journal of Theoretical Biology, 394, 1–17. 10.1016/j.jtbi.2016.01.009 [DOI] [PubMed] [Google Scholar]
- Dubeau, S. , Desjardins, M. , Pouliot, P. , Beaumont, E. , Gaudreau, P. , Ferland, G. , & Lesage, F. (2011). Biophysical model estimation of neurovascular parameters in a rat model of healthy aging. NeuroImage, 57, 1480–1491. 10.1016/j.neuroimage.2011.04.030 [DOI] [PubMed] [Google Scholar]
- Erdös, B. , Simandle, S. A. , Snipes, J. A. , Miller, A. W. , & Busija, D. W. (2004). Potassium channel dysfunction in cerebral arteries of insulin‐resistant rats is mediated by reactive oxygen species. Stroke, 35, 964–969. 10.1161/01.STR.0000119753.05670.F1 [DOI] [PubMed] [Google Scholar]
- Faraci, F. M. , & Heistad, D. D. (1990). Regulation of large cerebral arteries and cerebral microvascular pressure. Circulation Research, 66, 8–17. 10.1161/01.RES.66.1.8 [DOI] [PubMed] [Google Scholar]
- Filosa, J. A. , Bonev, A. D. , Straub, S. V. , Meredith, A. L. , Wilkerson, M. K. , Aldrich, R. W. , & Nelson, M. T. (2006). Local potassium signaling couples neuronal activity to vasodilation in the brain. Nature Neuroscience, 9, 1397–1403. 10.1038/nn1779 [DOI] [PubMed] [Google Scholar]
- Gaertner, R. F. , Wyss‐Coray, T. , Von Euw, D. , Lesné, S. , Vivien, D. , & Lacombe, P. (2005). Reduced brain tissue perfusion in TGF‐β1 transgenic mice showing Alzheimer's disease‐like cerebrovascular abnormalities. Neurobiology of Disease, 19, 38–46. 10.1016/j.nbd.2004.11.008 [DOI] [PubMed] [Google Scholar]
- Hakim, M. A. , Behringer, E. J. , & Aguayo, L. (2020). Development of Alzheimer's disease progressively alters sex‐dependent K Ca and sex‐independent K IR channel function in cerebrovascular endothelium. Journal of Alzheimer's Disease, 76, 1423–1442. 10.3233/JAD-200085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hakim, M. A. , Chum, P. P. , Buchholz, J. N. , & Behringer, E. J. (2020). Aging alters cerebrovascular endothelial GPCR and K+ channel function: Divergent role of biological sex. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences, 75, 2064–2073. 10.1093/gerona/glz275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel, E. (2015). Cerebral circulation: Function and dysfunction in Alzheimer's disease. Journal of Cardiovascular Pharmacology, 65, 317–324. 10.1097/FJC.0000000000000177 [DOI] [PubMed] [Google Scholar]
- Hébert, F. , Grand'Maison, M. , Ho, M.‐K. , Lerch, J. P. , Hamel, E. , & Bedell, B. J. (2013). Cortical atrophy and hypoperfusion in a transgenic mouse model of Alzheimer's disease. Neurobiology of Aging, 34, 1644–1652. 10.1016/j.neurobiolaging.2012.11.022 [DOI] [PubMed] [Google Scholar]
- Iadecola, C. (2004). Neurovascular regulation in the normal brain and in Alzheimer's disease. Nature Reviews. Neuroscience, 5, 347–360. 10.1038/nrn1387 [DOI] [PubMed] [Google Scholar]
- Iadecola, C. (2017). The neurovascular unit coming of age: A journey through neurovascular coupling in health and disease. Neuron, 96, 17–42. 10.1016/j.neuron.2017.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji, Y. , Veldhuis, M. G. , Zandvoort, J. , Romunde, F. L. , Houtman, M. J. C. , Duran, K. , van Haaften, G. , Zangerl‐Plessl, E. M. , Takanari, H. , Stary‐Weinzinger, A. , & van der Heyden, M. A. G. (2017). PA‐6 inhibits inward rectifier currents carried by V93I and D172N gain‐of‐function KIR2.1 channels, but increases channel protein expression. Journal of Biomedical Science, 24, 44. 10.1186/s12929-017-0352-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, A. C. , & Cipolla, M. J. (2018). Impaired function of cerebral parenchymal arterioles in experimental preeclampsia. Microvascular Research, 119, 64–72. 10.1016/j.mvr.2018.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, S. J. , Cho, S. H. , Park, K. , Yi, J. , Yoo, S. J. , & Shin, K. S. (2008). Expression of Kir2.1 channels in astrocytes under pathophysiological conditions. Molecules and Cells, 25, 124–130. [PubMed] [Google Scholar]
- Kanno, T. , Yaguchi, T. , Nagata, T. , & Nishizaki, T. (2012). Indomethacin activates protein kinase C and potentiates α7 ACh receptor responses. Cellular Physiology and Biochemistry, 29, 189–196. 10.1159/000337600 [DOI] [PubMed] [Google Scholar]
- Kim, Y. , Park, J. , & Choi, Y. K. (2019). The role of astrocytes in the central nervous system focused on BK channel and heme oxygenase metabolites: A review. Antioxidants (Basel, Switzerland), 8, 121. 10.3390/antiox8050121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko, E. A. , Han, J. , Jung, I. D. , & Park, W. S. (2008). Physiological roles of K+ channels in vascular smooth muscle cells. Journal of Smooth Muscle Research, 44, 65–81. 10.1540/jsmr.44.65 [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/bph.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden, T. A. , Dabertrand, F. , Hill‐Eubanks, D. C. , Hammack, S. E. , & Nelson, M. T. (2014). Stress‐induced glucocorticoid signaling remodels neurovascular coupling through impairment of cerebrovascular inwardly rectifying K+ channel function. Proceedings of the National Academy of Sciences of the United States of America, 111, 7462–7467. 10.1073/pnas.1401811111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden, T. A. , Dabertrand, F. , Koide, M. , Gonzales, A. L. , Tykocki, N. R. , Brayden, J. E. , Hill‐Eubanks, D. , & Nelson, M. T. (2017). Capillary K(+)‐sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nature Neuroscience, 20, 717–726. 10.1038/nn.4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longden, T. A. , & Nelson, M. T. (2015). Vascular inward rectifier K+ channels as external K+ sensors in the control of cerebral blood flow. Microcirculation, 22, 183–196. 10.1111/micc.12190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas, S. (2016). The pharmacology of indomethacin. Journal of Head and Face Pain, 56, 436–446. 10.1111/head.12769 [DOI] [PubMed] [Google Scholar]
- Marrelli, S. P. , Johnson, T. D. , Khorovets, A. , Childres, W. F. , & Bryan, R. M. (1998). Altered function of inward rectifier potassium channels in cerebrovascular smooth muscle after ischemia/reperfusion. Stroke, 29, 1469–1474. 10.1161/01.STR.29.7.1469 [DOI] [PubMed] [Google Scholar]
- Mayhan, W. G. , Mayhan, J. F. , Sun, H. , & Patel, K. P. (2004). In vivo properties of potassium channels in cerebral blood vessels during diabetes mellitus. Microcirculation, 11, 605–613. 10.1080/10739680490503410 [DOI] [PubMed] [Google Scholar]
- McCarron, J. G. , & Halpern, W. (1990a). Impaired potassium‐induced dilation in hypertensive rat cerebral arteries does not reflect altered Na+,K(+)‐ATPase dilation. Circulation Research, 67, 1035–1039. 10.1161/01.RES.67.4.1035 [DOI] [PubMed] [Google Scholar]
- McCarron, J. G. , & Halpern, W. (1990b). Potassium dilates rat cerebral arteries by two independent mechanisms. American Journal of Physiology‐Heart and Circulatory Physiology, 259, H902–H908. 10.1152/ajpheart.1990.259.3.H902 [DOI] [PubMed] [Google Scholar]
- Mucke, L. , Masliah, E. , Yu, G. Q. , Mallory, M. , Rockenstein, E. M. , Tatsuno, G. , Hu, K. , Kholodenko, D. , Johnson‐Wood, K. , & McConlogue, L. (2000). High‐level neuronal expression of Aβ1–42 in wild‐type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. The Journal of Neuroscience, 20, 4050–4058. 10.1523/JNEUROSCI.20-11-04050.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughal, A. , Harraz, O. F. , Gonzales, A. L. , Hill‐Eubanks, D. , & Nelson, M. T. (2021). PIP2 improves cerebral blood flow in a mouse model of Alzheimer's disease. Function (Oxford, England), 2, zqab010. 10.1093/function/zqab010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, K. N. , Girard, S. , Holmes, W. M. , Parkes, L. M. , Williams, S. R. , Parry‐Jones, A. R. , & Allan, S. M. (2014). Systemic inflammation impairs tissue reperfusion through endothelin‐dependent mechanisms in cerebral ischemia. Stroke, 45, 3412–3419. 10.1161/STROKEAHA.114.006613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis, N. , Aboulkassim, T. , Aliaga, A. , Tong, X.‐K. , Rosa‐Neto, P. , & Hamel, E. (2011). Intact memory in TGF‐β1 transgenic mice featuring chronic cerebrovascular deficit: Recovery with pioglitazone. Journal of Cerebral Blood Flow and Metabolism, 31, 200–211. 10.1038/jcbfm.2010.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolakakis, N. , Aboulkassim, T. , Ongali, B. , Lecrux, C. , Fernandes, P. , Rosa‐Neto, P. , Tong, X. K. , & Hamel, E. (2008). Complete rescue of cerebrovascular function in aged Alzheimer's disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator‐activated receptor gamma agonist. The Journal of Neuroscience, 28, 9287–9296. 10.1523/JNEUROSCI.3348-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongali, B. , Nicolakakis, N. , Tong, X. K. , Lecrux, C. , Imboden, H. , & Hamel, E. (2018). Transforming growth factor‐β1 induces cerebrovascular dysfunction and astrogliosis through angiotensin II type 1 receptor‐mediated signaling pathways. Canadian Journal of Physiology and Pharmacology, 96, 527–534. 10.1139/cjpp-2017-0640 [DOI] [PubMed] [Google Scholar]
- Park, L. , Anrather, J. , Forster, C. , Kazama, K. , Carlson, G. A. , & Iadecola, C. (2004). Aβ‐induced vascular oxidative stress and attenuation of functional hyperemia in mouse somatosensory cortex. Journal of Cerebral Blood Flow and Metabolism, 24, 334–342. 10.1097/01.WCB.0000105800.49957.1E [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, W. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Würbel, H. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLoS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez‐Barbero, A. , Dorado, F. , Velasco, S. , Pandiella, A. , Banas, B. , & López‐Novoa, J. M. (2006). TGF‐β1 induces COX‐2 expression and PGE2 synthesis through MAPK and PI3K pathways in human mesangial cells. Kidney International, 70, 901–909. 10.1038/sj.ki.5001626 [DOI] [PubMed] [Google Scholar]
- Rubio‐Ruiz, M. E. , Pérez‐Torres, I. , Diaz‐Diaz, E. , Pavón, N. , & Guarner‐Lans, V. (2014). Non‐steroidal anti‐inflammatory drugs attenuate the vascular responses in aging metabolic syndrome rats. Acta Pharmacologica Sinica, 35, 1364–1374. 10.1038/aps.2014.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho, M. , Fabris, S. , Hald, B. O. , Brett, S. E. , Sandow, S. L. , Poepping, T. L. , & Welsh, D. G. (2019). Membrane lipid‐KIR2.x channel interactions enable hemodynamic sensing in cerebral arteries. Arteriosclerosis, Thrombosis, and Vascular Biology, 39, 1072–1087. 10.1161/ATVBAHA.119.312493 [DOI] [PubMed] [Google Scholar]
- Sancho, M. , Samson, N. C. , Hald, B. O. , Hashad, A. M. , Marrelli, S. P. , Brett, S. E. , & Welsh, D. G. (2017). KIR channels tune electrical communication in cerebral arteries. Journal of Cerebral Blood Flow and Metabolism, 37, 2171–2184. 10.1177/0271678X16662041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotland, R. S. , Chauhan, S. , Vallance, P. J. , & Ahluwalia, A. (2001). An endothelium‐derived hyperpolarizing factor‐like factor moderates myogenic constriction of mesenteric resistance arteries in the absence of endothelial nitric oxide synthase‐derived nitric oxide. Hypertension (Dallas, Tex. 1979), 38, 833–839. [DOI] [PubMed] [Google Scholar]
- Segal, S. S. (2000). Integration of blood flow control to skeletal muscle: Key role of feed arteries. Acta Physiologica Scandinavica, 168, 511–518. 10.1046/j.1365-201x.2000.00703.x [DOI] [PubMed] [Google Scholar]
- Sonkusare, S. K. , Dalsgaard, T. , Bonev, A. D. , & Nelson, M. T. (2016). Inward rectifier potassium (Kir2.1) channels as end‐stage boosters of endothelium‐dependent vasodilators. The Journal of Physiology, 594, 3271–3285. 10.1113/JP271652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springo, Z. , Toth, P. , Tarantini, S. , Ashpole, N. M. , Tucsek, Z. , Sonntag, W. E. , Csiszar, A. , Koller, A. , & Ungvari, Z. I. (2015). Aging impairs myogenic adaptation to pulsatile pressure in mouse cerebral arteries. Journal of Cerebral Blood Flow and Metabolism, 35, 527–530. 10.1038/jcbfm.2014.256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standen, N. B. , & Quayle, J. M. (1998). K+ channel modulation in arterial smooth muscle. Acta Physiologica Scandinavica, 164, 549–557. 10.1046/j.1365-201X.1998.00433.x [DOI] [PubMed] [Google Scholar]
- Takeda, S. , Sato, N. , Uchio‐Yamada, K. , Sawada, K. , Kunieda, T. , Takeuchi, D. , Kurinami, H. , Shinohara, M. , Rakugi, H. , & Morishita, R. (2010). Diabetes‐accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proceedings of the National Academy of Sciences of the United States of America, 107, 7036–7041. 10.1073/pnas.1000645107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, X.‐K. , & Hamel, E. (2007). Transforming growth factor‐β1 impairs endothelin‐1‐mediated contraction of brain vessels by inducing mitogen‐activated protein (MAP) kinase phosphatase‐1 and inhibiting p38 MAP kinase. Molecular Pharmacology, 72, 1476–1483. 10.1124/mol.107.039602 [DOI] [PubMed] [Google Scholar]
- Tong, X.‐K. , & Hamel, E. (2015). Simvastatin restored vascular reactivity, endothelial function and reduced string vessel pathology in a mouse model of cerebrovascular disease. Journal of Cerebral Blood Flow and Metabolism, 35, 512–520. 10.1038/jcbfm.2014.226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, X.‐K. , Lecrux, C. , Rosa‐Neto, P. , & Hamel, E. (2012). Age‐dependent rescue by simvastatin of Alzheimer's disease cerebrovascular and memory deficits. The Journal of Neuroscience, 32, 4705–4715. 10.1523/JNEUROSCI.0169-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong, X.‐K. , Nicolakakis, N. , Fernandes, P. , Ongali, B. , Brouillette, J. , Quirion, R. , & Hamel, E. (2009). Simvastatin improves cerebrovascular function and counters soluble amyloid‐beta, inflammation and oxidative stress in aged APP mice. Neurobiology of Disease, 35, 406–414. 10.1016/j.nbd.2009.06.003 [DOI] [PubMed] [Google Scholar]
- Tong, X.‐K. , Nicolakakis, N. , Kocharyan, A. , & Hamel, E. (2005). Vascular remodeling versus amyloid beta‐induced oxidative stress in the cerebrovascular dysfunctions associated with Alzheimer's disease. The Journal of Neuroscience, 25, 11165–11174. 10.1523/JNEUROSCI.4031-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykocki, N. R. , Boerman, E. M. , & Jackson, W. F. (2017). Smooth muscle ion channels and regulation of vascular tone in resistance arteries and arterioles. Comprehensive Physiology, 7, 485–581. 10.1002/cphy.c160011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetri, F. , Xu, H. , Paisansathan, C. , & Pelligrino, D. A. (2012). Impairment of neurovascular coupling in type 1 diabetes mellitus in rats is linked to PKC modulation of BKCa and Kir channels. American Journal of Physiology‐Heart and Circulatory Physiology, 302, H1274–H1284. 10.1152/ajpheart.01067.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente, R. , Coma, M. , Busquets, S. , Moore‐Carrasco, R. , López‐Soriano, F. J. , Argilés, J. M. , & Felipe, A. (2004). The systemic inflammatory response is involved in the regulation of K(+) channel expression in brain via TNF‐α‐dependent and ‐independent pathways. FEBS Letters, 572, 189–194. 10.1016/j.febslet.2004.07.030 [DOI] [PubMed] [Google Scholar]
- Wang, H.‐R. , Wu, M. , Yu, H. , Long, S. , Stevens, A. , Engers, D. W. , Sackin, H. , Daniels, J. S. , Dawson, E. S. , Hopkins, C. R. , Lindsley, C. W. , Li, M. , & McManus, O. B. (2011). Selective inhibition of the Kir2 family of inward rectifier potassium channels by a small molecule probe: The discovery, SAR, and pharmacological characterization of ML133. ACS Chemical Biology, 6, 845–856. 10.1021/cb200146a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, E. P. , Kontos, H. A. , & Beckman, J. S. (1996). Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am. J. Physiol. Circ. Physiol., 271, H1262–H1266. 10.1152/ajpheart.1996.271.3.H1262 [DOI] [PubMed] [Google Scholar]
- Wu, B.‐N. , Luykenaar, K. D. , Brayden, J. E. , Giles, W. R. , Corteling, R. L. , Wiehler, W. B. , & Welsh, D. G. (2007). Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. American Journal of Physiology. Heart and Circulatory Physiology, 292, H1085–H1094. 10.1152/ajpheart.00926.2006 [DOI] [PubMed] [Google Scholar]
- Wyss‐Coray, T. , Lin, C. , Sanan, D. A. , Mucke, L. , & Masliah, E. (2000). Chronic overproduction of transforming growth factor‐β1 by astrocytes promotes Alzheimer's disease‐like microvascular degeneration in transgenic mice. The American Journal of Pathology, 156, 139–150. 10.1016/S0002-9440(10)64713-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaritsky, J. J. , Eckman, D. M. , Wellman, G. C. , Nelson, M. T. , & Schwarz, T. L. (2000). Targeted disruption of Kir2.1 and Kir2.2 genes reveals the essential role of the inwardly rectifying K(+) current in K(+)‐mediated vasodilation. Circulation Research, 87, 160–166. 10.1161/01.RES.87.2.160 [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Papadopoulos, P. , & Hamel, E. (2013). Endothelial TRPV4 channels mediate dilation of cerebral arteries: Impairment and recovery in cerebrovascular pathologies related to Alzheimer's disease. British Journal of Pharmacology, 170, 661–670. 10.1111/bph.12315 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1A. Myogenic tone in wild‐type (WT), APP or TGF mice in the presence and absence of calcium (Ca2+)
Table S1B. Vasodilatation to increasing concentrations of K+ in arteries from adult mice
Table S1C & D. Vasodilatation to increasing concentrations of K+ in arteries from adult vs young mice
Table S2A & B. Effect of Ba2+ on K+‐induced vasodilatation in WT, APP and TGF arteries.
Table S2C & D. Effect of ML133 on 14 mM K+‐induced vasodilatation in WT, APP and TGF arteries.
Table S2E. Comparative effect of Ba2+ and Ba2+ + ML133 on brain arteries from WT mice
Table S3A, B, C. Effect of endothelium denudation in WT arteries on K+‐induced dilatation
Table S4A, B, D & E. Effects of catalase (Cat) and Indomethacin (Indo) on K+‐mediated vasodilatations
Table S6A,B,C,D. Rescue of basal tone in APP and TGF mice with catalase (Cat) and indomethacin (Indo), respectively.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.