

Summary
In‐depth knowledge about spatial and temporal variation in microbial diversity and function is needed for a better understanding of ecological and evolutionary responses to global change. In particular, the study of microbial ancient DNA preserved in sediment archives from lakes and oceans can help us to evaluate the responses of aquatic microbes in the past and make predictions about future biodiversity change in those ecosystems. Recent advances in molecular genetic methods applied to the analysis of historically deposited DNA in sediments have not only allowed the taxonomic identification of past aquatic microbial communities but also enabled tracing their evolution and adaptation to episodic disturbances and gradual environmental change. Nevertheless, some challenges remain for scientists to take full advantage of the rapidly developing field of paleo‐genetics, including the limited ability to detect rare taxa and reconstruct complete genomes for evolutionary studies. Here, we provide a brief review of some of the recent advances in the field of environmental paleomicrobiology and discuss remaining challenges related to the application of molecular genetic methods to study microbial diversity, ecology, and evolution in sediment archives. We anticipate that, in the near future, environmental paleomicrobiology will shed new light on the processes of microbial genome evolution and microbial ecosystem responses to quaternary environmental changes at an unprecedented level of detail. This information can, for example, aid geological reconstructions of biogeochemical cycles and predict ecosystem responses to environmental perturbations, including in the context of human‐induced global changes.
Introduction
Bacteria, archaea and microbial eukaryotes are central components of aquatic ecosystems through their contribution to food web dynamics and global biogeochemical processes, including oxygen and biomass production and the cycling of carbon. Sequencing of the DNA present in the environment has greatly increased our understanding of microbial communities inhabiting aquatic systems and their variable and dynamic roles in biogeochemical processes (Grossart et al., 2020). Still, the question ‘How conserved are microbial functions across different spatial and temporal scales?’ was recently highlighted as a sustained research priority (Antwis et al., 2017). Because contemporary water monitoring projects based on molecular techniques typically only span a few decades (e.g. Fuhrman et al., 2015), our ability to tackle questions related to changes in biota over longer evolutionary time scales has so far been limited. Fortunately, ancient DNA time‐series offer an expanded temporal window to retrieve information from several decades to potentially 100s of thousands of years ago, which can be used for describing the natural history of aquatic ecosystems and their responses to environmental changes (Coolen et al., 2013; Domaizon et al., 2017; Armbrecht, 2020).
Paleomicrobiology research based on the study of ancient DNA preserved in fossils and other remains has already led to many important discoveries related to human health, including evolutionary patterns of microbial pathogens, e.g., those involved in tuberculosis (Donoghue, 2016) and plague (Rascovan et al., 2019) as well as the long‐term changes in human and Neanderthal oral microbiomes (Warinner et al., 2015; Weyrich et al., 2017). Additionally, it is possible to directly recover and sequence DNA molecules preserved in aquatic environmental archives (a.k.a. sediment ancient DNA, sedaDNA). Despite methodological limitations primarily due to DNA degradation over time and the difficulty to authenticate ancient DNA signals, the application of molecular genetic tools to sediment records has proven to be an extremely promising approach (Fig. 1) to reveal changes in past biota, from microorganisms to macro‐fauna, and from individuals to complex communities (Capo et al., 2021).
Fig. 1.
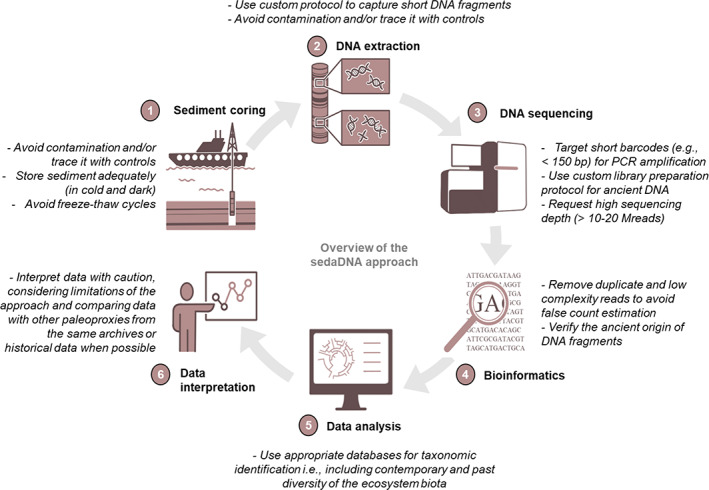
Conceptual workflow of the molecular palaeoecological approach applied to DNA preserved in marine and freshwater sediments. For each step of the workflow, main considerations related to the specificities of targeting ancient DNA are described briefly.
The current success in the analysis of historically deposited DNA in marine and freshwater sediments is linked to the recent advances in the applications of various molecular methods such as amplicon sequencing (metabarcoding) and quantitative amplification methods (quantitative PCR, droplet digital PCR). The field of sedaDNA has so far provided new knowledge about, for example, (i) compositional rearrangements within microbial assemblages in lakes subjected to various levels of human impacts, (ii) the recent regional homogenization of microbial diversity across lakes, and (iii) the microbial taxa favoured by recent changes in environmental conditions due to anthropogenic pressures (cyanobacteria; Monchamp et al., 2016, 2018, eukaryotic plankton; Capo et al., 2017; Keck et al., 2020). Further, the occurrence of algal blooms during the Holocene (Konkel et al., 2020) and the long‐term tracking of microbial interactions, such as parasitic or mutualistic interactions (Kyle et al., 2015; More et al., 2018), have also been investigated using sedaDNA‐based approaches.
Here, we provide a brief overview of the current applications and perspective on potential new sedaDNA research directions in aquatic environmental paleomicrobiology. Guidelines for this relatively young research field have been described (see, e.g., Armbrecht et al., 2019; Capo et al., 2021 for synthesis); thus, we here focus on the identification and discussion of current challenges and suggest possible solutions to overcome them. Specifically, we address three key research challenges: (i) the limitations in interpreting the ancient DNA signals recovered from sediments (ancient vs modern, dead vs alive) to infer ecological changes; (ii) the limited mechanistic understanding of the past evolutionary processes that have led to the genetic makeup of contemporary microorganisms; and (iii) the difficulty to reconstruct historical trophic networks in aquatic systems to better understand complex interspecific interactions (e.g., bacterial–eukaryotic host–parasite interactions) within these ecosystems.
Disentangling ancient and modern signals: dead or alive?
The pool of microbial DNA preserved in aquatic sediments is composed of two main fractions which are (i) the ancient molecules – DNA within dead cells or extracellular DNA either in free form or bound to particles – and (ii) DNA within viable/living cells (cysts, spores, pollen, and eggs) that are either actively growing in the sediments or dormant but able to regenerate under suitable environmental conditions (Fig. 2). Some microorganisms can remain viable for extended periods (>100s of thousands of years) following sediment burial; this is possible via various mechanisms that include switching to fermentation (Orsi et al., 2017), sporulation and/or formation of other resistant resting stages (Jörgensen, 2011; Bradley et al., 2019). For instance, in a recent study of the Dead Sea sediments, Thomas and Ariztegui (2019) illustrated a new pathway of carbon transformation at the subsurface and demonstrated how life can be maintained in extreme environments characterized by long‐term isolation and minimal energetic resources. Typically, to be considered alive, a cell must be intact, maintain an electrochemical potential across the cell membrane and be capable of growth and reproduction. A caveat is that based on this definition, it is most likely not possible to differentiate between very slow growing and dormant cells as both would be considered ‘alive’. However, besides revival and cultivation of the living subset of the community or methods based on metabolic probing, several indirect approaches have been used to test for microbial viability and/or activity (Emerson et al., 2017).
Fig. 2.
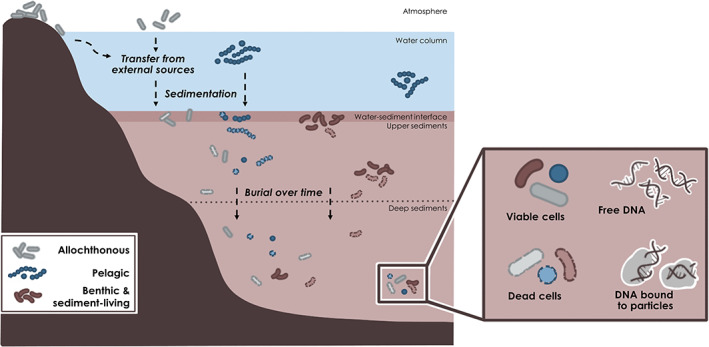
Composition of the microbial pool in aquatic sediments. The different sources of microbial cells (from external sources, the water column, and the sediments) are depicted with cells with different shapes and colours (see caption in the bottom left part of the figure). The different forms of microbial cells and DNA that can be found in deep sediments are shown at the bottom right part of the figure.
First, independent extraction of intracellular vs. extracellular DNA and subsequent amplicon or shotgun sequencing can reveal and contrast the taxonomic diversity and metabolic potential of living vs. dead subsurface bacteria cells (Vuillemin et al., 2017). Second, ancient RNA has the potential to help identify the active fraction of the microbiome. Because RNA is generally found to degrade rapidly, sequences of reverse transcribed sedimentary RNA are typically assumed to reflect the activity of contemporary sediment‐dwelling microorganisms (Vuillemin et al., 2020; Pearman et al., 2021). However, studies have shown that RNA can be recovered from ancient cells and that it can remain active even in soft tissues for at least several decades (reviewed by Smith and Gilbert, 2018). This means that the ancient RNA signal in sediment records might reflect the ecology of both contemporary and ancient microbes. Further exploration of the decay mechanisms of RNA in sediments is needed to use this approach to its highest potential. A third promising approach for distinguishing between living and dead organisms in sediment is viability PCR via propidium monoazide, a nucleic acid intercalating dye binding to extracellular DNA and DNA inside damaged cells making it unavailable for PCR and sequencing (Heise et al., 2016; Emerson et al., 2017). Additionally, certain DNA extraction protocols are best suited to the recovery of short DNA fragments, which are characteristic for ancient DNA (Dabney et al., 2013; Slon et al., 2017). Finally, bioinformatics strategies can be applied during post‐sequencing data processing to retrieve short sequences (Armbrecht et al., 2020). For metagenomic libraries specifically, bioinformatic methods (see Table 1 for adequate tools) can be applied to assess DNA damage patterns, a key feature to demonstrate ancient DNA authenticity and control for modern DNA contamination (Pedersen et al., 2016; Armbrecht et al., 2021b).
Table 1.
List of open‐source bioinformatics programmes and databases currently used in the field of sedaDNA to perform the main bioinformatics steps of post‐sequencing read processing. Although this list is not exhaustive, it provides some guidance for scientists wishing to apply sedaDNA approaches.
| Bioinformatic step/task |
|---|
| Post sequencing read processing |
|
‐Sequence quality check ‐Adapter removal and quality filtering ‐Removal of duplicate sequences and low‐complexity sequences ‐Identification and removal of contaminant sequences |
| Authentication and assessment of present‐day contamination in ancient DNA | ||
|---|---|---|
| Suggested tools | References | |
| ‐Verification of ancient DNA molecular damage | mapDamage2.0 | Jónsson et al. (2013) |
| AuthentiCT | Peyrégne and Peter (2020) | |
| HOPS | Hübler et al. (2019) | |
| PyDamage | Borry et al. (2021) | |
| Taxonomic assignment | ||
|---|---|---|
| Suggested tools | References | |
| ‐Read mapping against reference molecular databases (for shotgun data) | Holi pipeline | https://github.com/ancient‐eDNA/Holi |
| PIA | Cribdon et al. (2020) | |
| MALT | Herbig et al. (2016) | |
| BWA | Li and Durbin (2009) | |
| ‐Publicly available reference databases for read mapping | NCBI GenBank (all organisms) | Benson et al. (2018) |
| SILVA [16S and 18S (SSU), 23S&28S (LSU) rRNA sequences for bacteria, archaea and eukaryotes] | Quast et al. (2013) | |
| BOLD (COI gene, eukaryotes) | Ratnasingham and Hebert (2007) | |
| PR2, V9‐PR2 (18S rRNA sequences, eukaryotes, especially protists) | Guillou et al. (2012) | |
Both ancient and modern molecular signals recovered from aquatic sediments provide information about past environmental conditions. Indeed, in addition to taxonomic profiles, the molecular signal from dead aquatic organisms provides information on the environmental/water conditions prevailing at the time of deposition/burial of those taxa. In contrast, microbial communities living in sediments (notably facultative and obligate anaerobic microorganisms) are generally thought to be structured through in situ environmental conditions, such as the availability of electron acceptors and donors, porosity, and sediment lithology and chemistry (e.g., Parkes et al., 2000; Kallmeyer et al., 2012). However, recent studies suggest that the active subsurface microbial communities are also shaped by the conditions prevailing at the time when water column organisms were deposited in the superficial sediments (More et al., 2019; Orsi et al., 2017; Starnawski et al., 2017; Vuillemin et al., 2018). Accordingly, the molecular signal representing the taxonomic diversity from both dead and living microorganisms can provide useful and complementary information on past environmental conditions and how these have influenced and shaped microbial communities in the studied ecosystems. It will nevertheless be important to consider that apparent functional diversity seen in such environmental archives essentially represents both the metabolic footprint of dead microorganisms and that of populations growing and reproducing in the sediments. As a consequence of this, growth‐related shifts in the quantitative representation of taxa and functions may therefore happen with time and obscure the interpretation of sedaDNA archives. Hence, there is a need for further development and validation of efficient and precise analytical strategies for discriminating between contemporary DNA from living sediment microorganisms and the ancient DNA from those that died a long time ago.
Unravelling the evolution of aquatic microbial genomes
In this new era where sedaDNA approaches have been integrated in paleoecology research, one of the most evident and tractable challenges is to successfully recover (parts of) ancient genomes for a side‐by‐side comparison with their modern equivalents. This new information can then be used to reconstruct the occurrence and timings of past evolutionary events that led up to present day taxonomic and genetic diversity and do so at an unprecedented level of detail. There still remain some challenges before we can fully explore this avenue of sedaDNA research. Access to relevant reference genomes was until recently a major bottleneck, and while global initiatives now cover a large portion of the prokaryotic diversity (Thompson et al., 2017; Nayfach et al., 2021; Parks et al., 2022), this is still a challenge to be solved for eukaryotes (Lewin et al., 2018). Another remaining challenge lies in adequately assigning short and fragmented DNA sequences to their actual host populations that have persisted across the sedimentary records as either intact cells or detrital DNA. Even for prokaryotes and despite the dramatic increase in published reference genomes in recent years, there are still gaps in the databases due to the vast microbial diversity in freshwater and marine environments, the majority of which have not been cultured or sequenced. So far, there have only been a few attempts to tackle evolutionary questions from sedaDNA; for instance, Lammers et al. (2021) successfully reconstructed chloroplast and mitochondrial genomes from Nannochloropsis limnetica haplotypes from 20 000‐year‐old sediments. However, with continuously advancing technologies and the strong interest in pursuing evolutionary studies to address a broad range of microbial ecology questions, these types of works are expected to become an increasingly popular research direction in the near future.
While highly resolved sedimentary records of taxonomic and functional marker genes can be informative for understanding the ecological niches and dynamics of microbial populations and their functional associations, a genome‐centric view is needed for understanding the actual genomic changes that underscore evolutionary processes such as adaptation, micro‐diversification (intraspecific diversity in metabolic pathways), and selective sweeps (new beneficial mutations). Recent advances in sequencing methods and bioinformatic binning approaches now provide an unsurpassed ability to use direct shotgun metagenomic data for the reconstruction of nearly complete population genomes for at least the more abundant community members (Albertsen et al., 2013; Lui et al., 2021) and partition core genomes from the adaptable accessory genomes that vary across taxonomic groups (Buck et al., 2021). However, this is a much more challenging endeavour when dealing with the highly complex microbial communities found in sediments and, particularly, the fragmented and incomplete genomes typically occurring in ancient sediment records. The accumulation of DNA damage (including the substitution of nucleotides) and associated fragmentation of molecules can prevent the assembly of reads into contigs and the subsequent binning of contigs into metagenome‐assembled genomes. To obtain ancient genomic information for the purpose of exploring adaptive signals in the genome, single‐nucleotide polymorphism (SNP) signatures, and gene gain/loss, we need more relevant reference genomes as scaffolds for mapping short reads (Starnawski et al., 2017).
Following the production of more reference genomes, we might be able to reconstruct their overall genomic features and link any related changes to biotic and abiotic drivers acting over longer timescales. This can be achieved in a few different ways: (i) by ultra‐deep sequencing of multiple related samples to attempt a bioinformatically demanding assembly and binning based on genomic signatures and differential abundance patterns (Lammers et al., 2021); (ii) by applying hybridization capture methods (described in the section ‘Providing a holistic reconstruction of past aquatic ecosystems biota’) to selectively enrich the molecular signal from specific lineages (Armbrecht et al., 2021b) and reconstruct their genomic information; (iii) by generating reference genomes from isolates or metagenome‐assembled genomes from contemporary databases or adjacent ‘active’ habitats, such as the surface sediments or the overlying water column (Garner et al., 2020); (iv) by sequencing the genomes of single intact cells recovered in ancient sediments (Starnawski et al., 2017), or finally the perhaps most spectacular approach: (v) by reviving dormant cells from ancient sediments for further cultivation, genome sequencing and possible downstream experimentation (Ellegaard and Ribeiro, 2018; Morono et al., 2020).
While the use of the aforementioned approaches for addressing evolutionary processes is still in its infancy, they have now been benchmarked for some microbial groups, enabling future exploratory research of the evolutionary history of lineages, which still live, or have lived, in the studied ecosystems.
Providing a holistic reconstruction of past aquatic ecosystems biota
Although the short‐ and long‐term responses of aquatic biota to environmental change have long been investigated via classical paleoecology (e.g., Smol, 2009), such research typically focused on a single or a limited number of proxies and have so far not successfully captured the full diversity of organisms, their co‐occurrences and their potential interactions. Improving our understanding of the complex relationships between micro‐ and macro‐organisms is the key to pinpoint the impacts of environmental change on aquatic ecosystems. The sedaDNA approach is a unique opportunity to achieve this, as it theoretically allows the reconstruction and characterization of the ecosystem biodiversity across all domains of life. Of relevance here is the metagenomic shotgun approach, which can simultaneously capture the DNA signal of the full range of organisms that jointly make up the ecosystem (More et al., 2019; Orsi et al., 2017; Armbrecht, 2020; Capo et al., 2021). In addition, metagenomic datasets can be collected without the need for laborious and error‐prone amplicon sequencing of multiple genetic markers. Compared to more targeted molecular approaches (e.g., qPCR, ddPCR, amplicon sequencing), shotgun metagenomics has the potential to provide a more holistic view of biotic interactions and ecosystem functioning (Garner et al., 2020; Moguel et al., 2020; More et al., 2019; Lammers et al., 2021; Armbrecht et al., 2021a).
Although shotgun metagenomics is theoretically suitable to investigate ‘whole’ communities, it has mostly been used to study microbial and planktonic diversity (Grossart et al., 2020). For both modern and ancient DNA analysis, it can prove challenging to capture the signal of larger organisms which are generally present at lower abundances – and with a much patchier distribution – compared with microorganisms; hence, very deep sequencing is required to allow assessments of the distribution of such species, which can be costly. The major fraction of a sediment metagenome originates from bacterial or archaeal DNA, with eukaryotic DNA representing only a minute fraction of the total sedaDNA pool (<1.5% of the total sedaDNA based on small subunit (SSU) rRNA alignments in marine environments; Armbrecht et al., 2020). However, with shotgun sequencing becoming increasingly affordable, this technology is now more accessible than ever to obtain high read counts for rare species by increasing sequencing depth and improving reference databases to compare metagenomic and metabarcoding sequence against (detailed in the section ‘Unravelling the evolution of aquatic microbial genomes’).
Searching for a combination of taxonomic marker genes in metagenomic sedaDNA datasets (e.g. SSU rRNA and LSU rRNA genes for eukaryotes; Armbrecht, 2020) can provide a powerful framework to reconstruct entire trophic networks of ecosystems. To do so, there is a need for ultra‐deep sequencing to allow the detection of the DNA signal from rare or macro‐organisms from sediment metagenomes. Additionally, as a complement to the rapidly developing sequencing capacity, one emerging approach in molecular‐based paleoecology is the application of hybridization capture techniques where DNA molecules from specific biological groups (e.g. taxonomic marker genes or genes encoding for certain target functions) are enriched to enhance their representation in the metagenomic sequence libraries (Armbrecht et al., 2021b). These approaches rely on user‐defined oligonucleotide probes tethered to some solid support to capture and enrich target DNA fragments for subsequent sequencing (Horn, 2012). By designing and applying oligonucleotide probes for ~15 000 18S rRNA gene sequences (V9 region), and a combination of marker genes (18S rRNA, 28S rRNA, ITS, COI) to distinguish harmful microalgae, (Armbrecht et al., 2021b) were able to characterize marine phyto‐ and zoo‐plankton, as well as commercially important harmful microalgae, over a period of ~9000 years in Tasmania, Australia. Hybridization capture applied to the simultaneous investigation of key organisms at various trophic levels represents an economical and promising strategy for improving paleo‐reconstruction of aquatic food webs.
Conclusion
The sequencing of DNA preserved in sedimentary archives offers a unique way to uncover the role of microorganisms in past aquatic ecosystems and their responses to environmental perturbations. This information can greatly improve our knowledge of contemporary ecosystems and their future under ongoing climate change (Cavicchioli et al., 2019). Recent improvements in sedaDNA sampling and analysis protocols, as well as sequencing capacity and cost efficiency, allow us to use this new approach over very long timescales, i.e., up to 100s of thousands of years. Analyses of metagenomic data originating from ancient sediments have shown that the recovered reads are typically very short due to the accumulation of damage in DNA molecules, which poses difficulties for achieving robust assemblies into longer contigs, and binning into metagenome‐assembled genomes. To move beyond mapping of highly conserved taxonomic and functional marker genes over historical timescale and in essence use the DNA preserved in sediment records to its full potential to address burning eco‐evolutionary questions, there will first be a need for gathering relevant and highly curated reference genomes to guide the functional and taxonomic annotation and grouping of sediment DNA sequences. Additionally, the hybridization capture approach has emerged as a powerful tool to enrich DNA of underrepresented organisms in ancient sedimentary archives with the potential to contribute to a better characterization of past communities and keystone species. Altogether, the sedaDNA approach enables detailed studies of genes, genomes, populations, and communities over extended timescales and to reconstruct past evolutionary events that have led up to the contemporary biosphere. We envision that the upscaling of sedaDNA research from studies of targeted specific groups of organisms and genes to the investigation of long‐term microbial genomic evolution and reconstruction of whole trophic networks will provide new knowledge to fully comprehend the responses of aquatic microbiomes current and future responses to global change.
Acknowledgements
E.C. was supported by a postdoctoral fellowship from the Swedish Research Council VR (grant 2017‐04422) and the Swedish Research Council Formas (grant 2018‐01031). M.‐E.M. was supported by a postdoctoral fellowship from the Groupe de recherche interuniversitaire en limnologie (GRIL) and McGill University. L.A. is supported by an ARC DECRA Fellowship (DE210100929). The authors are very grateful for the work of Dr Marine Vandewalle‐Capo in producing the figures presented in this manuscript. The authors warmly thank two anonymous reviewers for their feedback. The authors also thank the members of the sedaDNA scientific society for fruitful discussions about the use of DNA preserved in sedimentary archives.
Contributor Information
Eric Capo, Email: eric.capo@hotmail.fr.
Marie‐Eve Monchamp, Email: me.monchamp@gmail.com.
References
- Albertsen, M. , Hugenholtz, P. , Skarshewski, A. , Nielsen, K.L. , Tyson, G.W. , and Nielsen, P.H. (2013) Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat Biotechnol 31: 533–538. [DOI] [PubMed] [Google Scholar]
- Antwis, R.E. , Griffiths, S.M. , Harrison, X.A. , Aranega‐Bou, P. , Arce, A. , Bettridge, A.S. , et al. (2017) Fifty important research questions in microbial ecology. FEMS Microbiol Ecol 93: 1–10. [DOI] [PubMed] [Google Scholar]
- Armbrecht, L. (2020) The potential of sedimentary ancient DNA to reconstruct past ocean ecosystems. Oceanography 33: 116–123. [Google Scholar]
- Armbrecht, L. , Eisenhofer, R. , Utge, J. , Sibert, E.C. , Rocha, F. , Ward, R. , et al. (2021a) Paleo‐diatom composition from Santa Barbara Basin deep‐sea sediments: a comparison of 18S‐V9 and diat‐rbcL metabarcoding vs shotgun metagenomics. ISME Commun 1: 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrecht, L. , Hallegraeff, G. , Bolch, C.J.S. , Woodward, C. , and Cooper, A. (2021b) Hybridisation capture allows DNA damage analysis of ancient marine eukaryotes. Sci Rep 11: 3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armbrecht, L. , Herrando‐Pérez, S. , Eisenhofer, R. , Hallegraeff, G.M. , Bolch, C.J.S. , and Cooper, A. (2020) An optimized method for the extraction of ancient eukaryote DNA from marine sediments. Mol Ecol Resour 20: 906–919. [DOI] [PubMed] [Google Scholar]
- Armbrecht, L.H. , Coolen, M.J.L. , Lejzerowicz, F. , George, S.C. , Negandhi, K. , Suzuki, Y. , et al. (2019) Ancient DNA from marine sediments: precautions and considerations for seafloor coring, sample handling and data generation. Earth‐Science Rev 196: 102887. [Google Scholar]
- Benson, D.A. , Cavanaugh, M. , Clark, K. , Karsch‐Mizrachi, I. , Ostell, J. , Pruitt, K.D. , and Sayers, E.W. (2018) GenBank. Nucleic Acids Res 46: D41–D47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borry, M. , Hübner, A. , Rohrlach, A.B. , and Warinner, C. (2021) PyDamage: automated ancient damage identification and estimation for contigs in ancient DNA de novo assembly. PeerJ 9: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, J.A. , Amend, J.P. , and LaRowe, D.E. (2019) Survival of the fewest: microbial dormancy and maintenance in marine sediments through deep time. Geobiology 17: 43–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck, M. , Mehrshad, M. , and Bertilsson, S. (2021) mOTUpan: a robust Bayesian approach to leverage metagenome assembled genomes for core‐genome estimation. bioRxiv 1–10. 10.1101/2021.06.25.449606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capo, E. , Debroas, D. , Arnaud, F. , Perga, M.‐E. , Chardon, C. , and Domaizon, I. (2017) Tracking a century of changes in microbial eukaryotic diversity in lakes driven by nutrient enrichment and climate warming. Environ Microbiol 19: 2873–2892. [DOI] [PubMed] [Google Scholar]
- Capo, E. , Giguet‐Covex, C. , Rouillard, A. , Nota, K. , Heintzman, P.D. , et al. (2021) Lake sedimentary DNA research on past terrestrial and aquatic biodiversity: overview and recommendations. Quaternary 4: 6. 10.3390/quat4010006 [DOI] [Google Scholar]
- Cavicchioli, R. , Ripple, W.J. , Timmis, K.N. , Azam, F. , Bakken, L.R. , Baylis, M. , et al. (2019) Scientists' warning to humanity: microorganisms and climate change. Nat Rev Microbiol 17: 569–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolen, M.J.L. , Orsi, W.D. , Balkema, C. , Quince, C. , Harris, K. , Sylva, S.P. , et al. (2013) Evolution of the plankton paleome in the Black Sea from the Deglacial to Anthropocene. Proc Natl Acad Sci 110: 8609–8614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribdon, B. , Ware, R. , Smith, O. , Gaffney, V. , and Allaby, R.G. (2020) PIA: more accurate taxonomic assignment of metagenomic data demonstrated on sedaDNA from the North Sea. Front Ecol Evol 8: 84. [Google Scholar]
- Dabney, J. , Knapp, M. , Glocke, I. , Gansauge, M.‐T. , Weihmann, A. , Nickel, B. , et al. (2013) Complete mitochondrial genome sequence of a middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci U S A 110: 15758–15763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domaizon, I. , Winegardner, A. , Capo, E. , Gauthier, J. , and Gregory‐Eaves, I. (2017) DNA‐based methods in paleolimnology: new opportunities for investigating long‐term dynamics of lacustrine biodiversity. J Paleolimnol 58: 1–21. [Google Scholar]
- Donoghue, H.D. (2016) Paleomicrobiology of human tuberculosis. Microbiol Spectr 4: 1–14. [DOI] [PubMed] [Google Scholar]
- Ellegaard, M. , and Ribeiro, S. (2018) The long‐term persistence of phytoplankton resting stages in aquatic ‘seed banks’. Biol Rev 93: 166–183. [DOI] [PubMed] [Google Scholar]
- Emerson, J.B. , Adams, R.I. , Román, C.M.B. , Brooks, B. , Coil, D.A. , Dahlhausen, K. , et al. (2017) Schrödinger's microbes: tools for distinguishing the living from the dead in microbial ecosystems. Microbiome 5: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman, J. , Cram, J. , and Needham, D. (2015) Marine microbial community dynamics and their ecological interpretation. Nat Rev Microbiol 13: 133–146. [DOI] [PubMed] [Google Scholar]
- Garner, R.E. , Gregory‐Eaves, I. , and Walsh, D.A. (2020) Sediment metagenomes as time capsules of Lake microbiomes. mSphere 5: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossart, H.P. , Massana, R. , McMahon, K.D. , and Walsh, D.A. (2020) Linking metagenomics to aquatic microbial ecology and biogeochemical cycles. Limnol Oceanogr 65: S2–S20. [Google Scholar]
- Guillou, L. , Bachar, D. , Audic, S. , Bass, D. , Berney, C. , Bittner, L. , et al. (2012) The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote Small Sub‐Unit rRNA sequences with curated taxonomy. Nucleic Acids Res 41: D597–D604. 10.1093/nar/gks1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise, J. , Nega, M. , Alawi, M. , and Wagner, D. (2016) Propidium monoazide treatment to distinguish between live and dead methanogens in pure cultures and environmental samples. J Microbiol Methods 121: 11–23. [DOI] [PubMed] [Google Scholar]
- Herbig, A. , Maixner, F. , Bos, K.I. , Zink, A. , Krause, J. , and Huson, D.H. (2016) MALT: fast alignment and analysis of metagenomic DNA sequence data applied to the Tyrolean Iceman. bioRxiv 050559. 10.1101/050559 [DOI] [Google Scholar]
- Horn, S. (2012) Target enrichment via DNA hybridization capture. Methods Mol Biol 840: 177–188. [DOI] [PubMed] [Google Scholar]
- Hübler, R. , Key, F.M. , Warinner, C. , Bos, K.I. , Krause, J. , and Herbig, A. (2019) HOPS: automated detection and authentication of pathogen DNA in archaeological remains. Genome Biol 20: 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jónsson, H. , Ginolhac, A. , Schubert, M. , Johnson, P.L.F. , and Orlando, L. (2013) mapDamage2. 0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 29: 1682–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jørgensen, B.B. (2011) Deep subseafloor microbial cells on physiological standby. Proc Natl Acad Sci U S A 108: 18193–18194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallmeyer, J. , Pockalny, R. , Adhikari, R. , Smith, D. , and D'Hondt, S. (2012) Global distribution of microbial abundance and biomass in subseafloor sediment. Proc Natl Acad Sci 109: 16213–16216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck, F. , Millet, L. , Debroas, D. , Etienne, D. , Galop, D. , Rius, D. , and Domaizon, I. (2020) Assessing the response of micro‐eukaryotic diversity to the great acceleration using lake sedimentary DNA. Nat Commun 11: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konkel, R. , Toruńska‐Sitarz, A. , Cegłowska, M. , Ežerinskis, Ž. , Šapolaitė, J. , Mažeika, J. , and Mazur‐Marzec, H. (2020) Blooms of toxic cyanobacterium Nodularia spumigena in norwegian fjords during Holocene warm periods. Toxins 12: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle, M. , Haande, S. , Ostermaier, V. , and Rohrlack, T. (2015) The red queen race between parasitic chytrids and their host, Planktothrix: a test using a time series reconstructed from sediment DNA. PLoS One 10: e0118738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lammers, Y. , Heintzman, P.D. , and Alsos, I.G. (2021) Environmental palaeogenomic reconstruction of an ice age algal population. Commun Biol 4: 220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin, H.A. , Robinson, G.E. , Kress, W.J. , Baker, W.J. , Coddington, J. , Crandall, K.A. , et al. (2018) Earth BioGenome project: sequencing life for the future of life. Proc Natl Acad Sci 115: 4325–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , and Durbin, R. (2009) Fast and accurate short read alignment with burrows‐wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui, L.M. , Nielsen, T.N. , and Arkin, A.P. (2021) A method for achieving complete microbial genomes and improving bins from metagenomics data. PLoS Comput Biol 17: e1008972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moguel, B. , Pérez, L. , Alcaraz, L.D. , Blaz, J. , Caballero, M. , Muñoz‐Velasco, I. , et al. (2020) Holocene life and microbiome profiling in ancient tropical Lake Chalco, Mexico. Sci Rep 11: 13848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monchamp, M.E. , Spaak, P. , Domaizon, I. , Dubois, N. , Bouffard, D. , and Pomati, F. (2018) Homogenization of lake cyanobacterial communities over a century of climate change and eutrophication. Nat Ecol Evol 2: 317–324. [DOI] [PubMed] [Google Scholar]
- Monchamp, M.E. , Walser, J.C. , Pomati, F. , and Spaak, P. (2016) Sedimentary DNA reveals cyanobacterial community diversity over 200 years in two perialpine lakes. Appl Environ Microbiol 82: 6472–6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- More, K.D. , Orsi, W.D. , Galy, V. , Giosan, L. , He, L. , Grice, K. , and Coolen, M.J.L. (2018) A 43 kyr record of protist communities and their response to oxygen minimum zone variability in the northeastern Arabian Sea. Earth Planet Sci Lett 496: 248–256. [Google Scholar]
- More, K.D. , Giosan, L. , Grice, K. , and Coolen, M.J.L. (2019) Holocene paleodepositional changes reflected in the sedimentary microbiome of the Black Sea. Geobiology 17: 436–448. 10.1111/gbi.12338 [DOI] [PubMed] [Google Scholar]
- Morono, Y. , Ito, M. , Hoshino, T. , Terada, T. , Hori, T. , Ikehara, M. , et al. (2020) Aerobic microbial life persists in oxic marine sediment as old as 101.5 million years. Nat Commun 11: 3626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach, S. , Roux, S. , Seshadri, R. , Udwary, D. , Varghese, N. , Schulz, F. , et al. (2021) A genomic catalog of Earth's microbiomes. Nat Biotechnol 39: 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi, W.D. , Coolen, M.J.L. , Wuchter, C. , He, L. , More, K.D. , Irigoien, X. , et al. (2017) Climate oscillations reflected within the microbiome of Arabian Sea sediments. Sci Rep 7: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkes, R. , Cragg, B. , and Wellsbury, P. (2000) Recent studies on bacterial populations and processes in subseafloor sediments: a review. Hydrgeol J 8: 11–28. [Google Scholar]
- Parks, D.H. , Chuvochina, M. , Rinke, C. , Mussig, A.J. , Chaumeil, P.‐A. , and Hugenholtz, P. (2022) GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome‐based taxonomy. Nucleic Acids Res 50: D785–D794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearman, J.K. , Biessy, L. , Howarth, J.D. , Vandergoes, M.J. , Rees, A. , and Wood, S.A. (2021) Deciphering the molecular signal from past and alive bacterial communities in aquatic sedimentary archives. Mol Ecol Resour. 10.1111/1755-0998.13515. [DOI] [PubMed] [Google Scholar]
- Pedersen, M.W. , Ruter, A. , Schweger, C. , Friebe, H. , Staff, R.A. , Kjeldsen, K.K. , et al. (2016) Postglacial viability and colonization in North America's ice‐free corridor. Nature 537: 45–49. [DOI] [PubMed] [Google Scholar]
- Peyrégne, S. , and Peter, B.M. (2020) AuthentiCT: a model of ancient DNA damage to estimate the proportion of present‐day DNA contamination. Genome Biol 21: 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C. , Pruesse, E. , Yilmaz, P. , Gerken, J. , Schweer, T. , Yarza, P. , et al. (2013) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rascovan, N. , Sjögren, K.G. , Kristiansen, K. , Nielsen, R. , Willerslev, E. , Desnues, C. , and Rasmussen, S. (2019) Emergence and spread of basal lineages of Yersinia pestis during the Neolithic decline. Cell 176: 295–305.e10. [DOI] [PubMed] [Google Scholar]
- Ratnasingham, S. , and Hebert, P.D.N. (2007) BOLD: the barcode of life data system. Mol Ecol Notes 7: 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slon, V. , Hopfe, C. , Weiß, C.L. , Mafessoni, F. , De Rasilla, M. , Lalueza‐Fox, C. , et al. (2017) Neandertal and Denisovan DNA from Pleistocene sediments. Science 608: 605–608. [DOI] [PubMed] [Google Scholar]
- Smith, O. , and Gilbert, M.T.P. (2018) Ancient RNA, Paleogenomics. In Population Genomics, Lindqvist, C. , and Rajora, O. (eds). Cham, Switzerland: Springer. [Google Scholar]
- Smol, J. (2009) Pollution of Lakes and Rivers a Paleoenvironmental Perspective, 2nd ed. Hoboken, NJ, USA: Blackwell Pub. [Google Scholar]
- Starnawski, P. , Bataillon, T. , Ettema, T.J.G. , Jochum, L.M. , Schreiber, L. , Chen, X. , et al. (2017) Microbial community assembly and evolution in subseafloor sediment. Proc Natl Acad Sci U S A 114: 2940–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, C. , and Ariztegui, D. (2019) Fluid inclusions from the deep Dead Sea sediment provide new insights on Holocene extreme microbial life. Quat Sci Rev 212: 18–27. [Google Scholar]
- Thompson, L.R. , Sanders, J.G. , McDonald, D. , Amir, A. , Ladau, J. , Locey, K.J. , et al. (2017) A communal catalogue reveals Earth's multiscale microbial diversity. Nature 551: 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Ariztegui, D. , Horn, F. , Kallmeyer, J. , Orsi, W.D. , Anselmetti, F. , et al. (2018) Microbial community composition along a 50 000‐year lacustrine sediment sequence. FEMS Microbiol Ecol 94: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Horn, F. , Alawi, M. , Henny, C. , Wagner, D. , Crowe, S.A. , and Kallmeyer, J. (2017) Preservation and significance of extracellular DNA in ferruginous sediments from Lake Towuti, Indonesia. Front Microbiol 8: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuillemin, A. , Vargas, S. , Coskun, Ö. , Pockalny, R. , Murray, R.W. , Smith, D.C. , et al. (2020) Atribacteria reproducing over millions of years in the Atlantic abyssal subseafloor. MBio 11: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warinner, C. , Speller, C. , and Collins, M.J. (2015) A new era in palaeomicrobiology: Prospects for ancient dental calculus as a long‐term record of the human oral microbiome. Philos Trans R Soc B Biol Sci 370: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyrich, L.S. , Duchene, S. , Soubrier, J. , Arriola, L. , Llamas, B. , Breen, J. , et al. (2017) Neanderthal behaviour, diet, and disease inferred from ancient DNA in dental calculus. Nature 544: 357–361. [DOI] [PubMed] [Google Scholar]