

To the Editor,
Progressive familial intrahepatic cholestasis (PFIC) is a group of inherited liver diseases characterized by defects in the transporters of the biliary epithelia. With the development of genetic technologies, at least six major types of PFIC have been identified and are found to be attributed to disease‐causing genes (eg, ATP8B1, ABCB11, ABCB4, TJP2, NR1H4, MYO5B). 1 Autosomal recessive mutations of the ATP8B1 gene can cause progressive familial intrahepatic cholestasis type 1 (PFIC1), a severe form of hereditary cholestasis. If left untreated, PFIC1 may progress to liver fibrosis, cirrhosis and devastating end‐stage liver failure, eventually requiring liver transplantation in some cases. 2 , 3 , 4 , 5 , 6 The ATP8B1 gene is located on chromosome 18q21‐22 and encodes the familial intrahepatic cholestasis 1 (FIC1) protein, a P‐type ATPase in the P4 family that is involved in bile acid homeostasis and plays a critical role in bile secretion. 1 , 3 , 5 , 7 When the function of FIC1 is impaired due to ATP8B1 gene mutation, bile acids can accumulate, thus injure the hepatocytes and lead to cholestasis in PFIC1. 1 , 5 , 8 To date, the precise mechanisms by which FIC1 deficiency leads to the onset of cholestasis remain unclear. To our knowledge, there have been no evident results from clinical trials for identifying carriers. Thus, the genetic causes of PFIC1 and the carriers need to be identified for the early detection and prenatal diagnosis of PFIC1.
Although the spectrum of disease‐causing mutations of the ATP8B1 gene is expanding, such mutations have not yet been fully discovered. Currently available comprehensive treatment options for patients with PFIC1 include conservative management with medications, biliary bypass surgery, and liver transplantation. 2 To date, gene‐specific therapy has not been applied in clinical practice for FIC1 deficiency in patients with PFIC1. Here we reported a male patient with PFIC1 and tried to identify the genetic causes of PFIC1 in a male patient.
A 22‐year‐old man was admitted to our hospital due to progressive yellow discoloration of skin and sclera over a period of 2 months and pruritus that had lasted for 1 month. His serum liver biochemistry showed total bilirubin (TBIL) level of 667 μmol/L, direct bilirubin (DBIL) of 475 μmol/L, alanine aminotransferase (ALT) of 31 U/L, aspartate aminotransferase (AST) of 28 U/L, alkaline phosphatase (ALP) of 119 U/L, and gamma‐glutamyltransferase (GGT) of 28 U/L. It was noted that the patient's jaundice deteriorated following treatment with hydrocortisone, S‐adenosyl methionine, diammonium glycyrrhizinate, ursodeoxycholic acid and bilirubin adsorption. The patient had three first‐degree relatives, mother, father and sister, all of whom had a normal phenotype.
Upon his admission to our hospital, a clinical assessment was performed. Upon physical examination, the height and weight of the patient was 170 cm and 62 kg. He presented with severely yellowish skin and sclera, with scratches and right upper quadrant abdominal tenderness; no other abnormalities were noted. The laboratory test results at the time of his hospital admission were as follows: white blood cell (WBC) count 8.48 × 109/L, hemoglobin (Hb) 125 g/L, platelet (PLT) count 318 × 109/L, albumin (ALB) 35.4 g/L, globulin 18.6 g/L, TBIL 581.7 μmol/L, DBIL 428.1 μmol/L, ALT 24.5 U/L, AST 26.1 U/L, ALP 134.3 U/L, GGT 25.1 U/L, serum creatinine (Cr) 63.5 μmol/L, urea 5.1 mmol/L, uric acid 101.9 μmol/L, glucose 4.31 mmol/L, total cholesterol 3.18 mmol/L, triacylglycerol 3.17 mmol/L, NH3 18.1 μmol/L, α‐fetoprotein 1.1 μg/L, negativite hepatitis B surface antigen (HBsAg), hepatitis C virus (HCV), human immunodeficiency virus (HIV), Treponema pallidum antibody, Epstein‐Barr virus (EBV) DNA, cytomegalovirus (CMV) DNA, anti‐nuclear antibody (ANA) and anti‐mitochondrial antibody (AMA), normal serum immunoglobulin G (IgG) subtypes, ceruloplasmin 0.404 g/L, serum iron 29.16 μmol/L, and transferrin saturation 45.28%. Upper abdominal magnetic resonance imaging (MRI) and magnetic resonance cholangiopancreatography (MRCP) revealed that the surface of the liver was smooth with normal shape and size, in the absence of obvious expansion of the bile and pancreatic ducts (Figure 1). On the basis of these imaging findings, intrahepatic and extrahepatic obstructive jaundice were ruled out.
FIGURE 1.
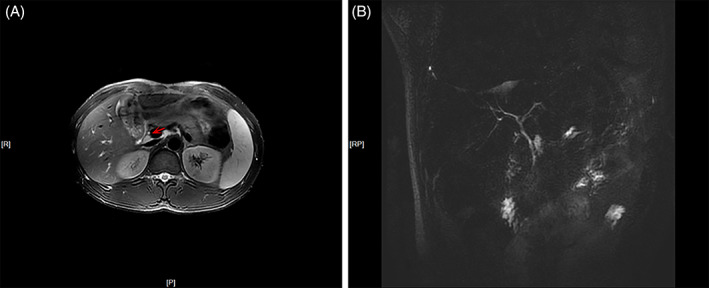
A, T2‐weighted cross‐sectional magnetic resonance imaging of the upper abdomen (arrow: bile duct) and B, magnetic resonance cholangiopancreatography revealed a smooth surface, normal shape and size of the liver with no obvious expansion of the bile duct and pancreatic duct [Color figure can be viewed at wileyonlinelibrary.com]
The patient underwent a liver biopsy, histopathological examination of which revealed an obvious dilatation of bile canaliculus, intrahepatic cholestasis, coarse granular bile plugs, blockage of bile flow in a portion of the small bile ducts and inflammation at the portal areas (Figure 2). Masson's trichrome staining of the portal tissues obtained by biopsy showed proliferation of fibrous connective tissues and fibrosis at the portal areas, whereas periodic acid–Schiff–diastase staining was negative. Reticular fiber staining of the liver tissue showed that the reticular scaffolds of liver cells collapsed focally. Prussian blue staining showed a small amount of iron particle deposition. Immunohistochemistry showed positive staining for CD19 in the bile duct epithelium, suggesting mild proliferation of the small bile ducts. In addition, CD68 staining revealed excessive proliferation and activation of Kupffer cells, and CD10 staining revealed a decreased bile canaliculus. It was of note that the copper staining and IgG4 test were both negative. While a small proportion of plasma cells presented MUM1 expression. These examinations led to an initial pathological diagnosis of cholestatic liver disease of mild to moderate activity and a Scheuer score of G2S2 in accordance with the modified Scheuer histological activity scoring system.
FIGURE 2.
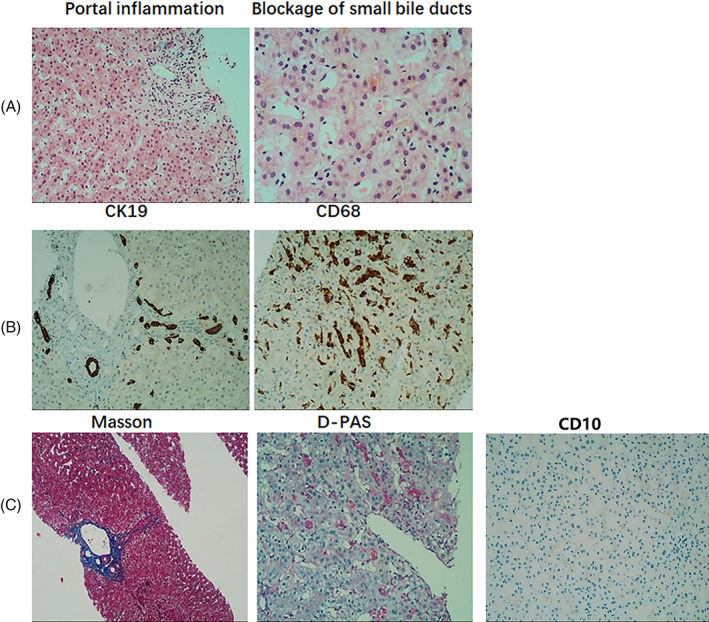
Histopathological examinations of the liver biopsy tissue revealed A, inflammation at the portal areas, blockage of small bile ducts, and biliary embolism (HE stain; magnification: portal inflammation, ×100; blockage of small bile ducts, ×200). B, Slight increase in CK19 positivity in the bile duct epithelium, suggesting a mild biliary duct hyperplasia with no visible bile duct disappearance, and overexpression of CD68, with an excessive proliferation and activation of Kupffer cells (magnification, ×100). C, The proliferation of fibrous connective tissue and fibrosis in the portal area (Masson's trichrome stain; magnification, ×40) and negative staining for periodic acid–Schiff–diastase (D‐PAS; magnification, ×100), together with decreased bile canaliculus in CD10 immunohistochemistry staining (magnification, ×100) [Color figure can be viewed at wileyonlinelibrary.com]
Notably, due to his normal serum GGT level, the patient was highly suspected to have PFIC1. Genetic study with whole exon sequencing (WES) was performed to identify potential pathogenic alterations in relation to the disease. Peripheral blood samples were obtained from the patient, his parents and sister. The results were further validated by direct Sanger sequencing of the polymerase chain reaction products, including the exons and flanking sequences of the ATP8B1 gene (Figure 3A). Two mutations of the ATP8B1 gene were identified in the patient and his sister, including 1375C>T mutation in exon 13 and 2025dupT mutation in exon 18 (Figures 3B). These two mutations formed a compound heterozygous mutation. Notably, these mutations were not listed in the Human Gene Mutation Database, ESP6500siv2‐ALL and DBsnp147, and had not been reported in patients diagnosed with PFIC1 so far. Furthermore, genetic studies of his parents showed that his father was a carrier of the 2025dupT heterozygous mutation in exon 18, while the 1375C>T heterozygous mutation in exon 13 was present in his mother. Although his sister carried the same gene mutations, up to the writing of this letter her liver function tests are normal with no clinical sign of PFIC1.
FIGURE 3.
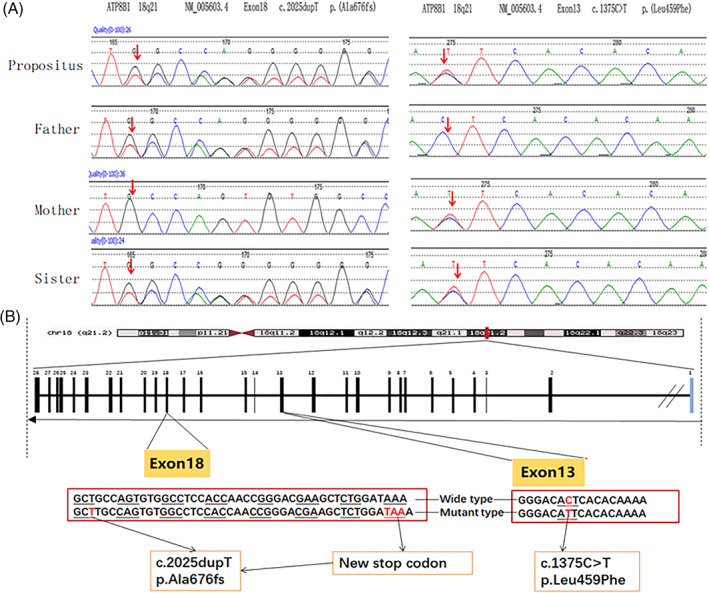
Sequencing results of exon 18 and exon 13 of the ATP8B1 gene in peripheral blood of the proband, his parents and sister. A, Exon 13 c.1375C>T (p.Leu459Phe) missense mutation and exon 18 c.2025dupT (p.Ala676fs) frame‐shift mutation identified in the proband and his sister. Exon 18 c.2025dupT (p.Ala676fs) frame‐shift mutation in the father and exon 13 c.1375C>T (p.Leu459Phe) missense mutation in the mother was identified, respectively. B, cDNA sequence (frame‐shift mutation and missense mutation) [Color figure can be viewed at wileyonlinelibrary.com]
Nonsense‐mediated mRNA decay is a RNA surveillance mechanism that detects the mRNA harboring premature termination codons and triggers their degradation to prevent the accumulation of truncated and potentially harmful proteins. Open reading frame introduced in the nonsense mutations have greatly accelerated the degradation of mRNA. 9 In our PFIC1 case, the nonsense‐mediated mRNA decay was triggered by the ATP8B1 mutation (2025dupT), which led to a reduced transcriptional ATP8B1 mRNA expression so that the corresponding FIC1 protein synthesis was insufficient and pathogenic. Intrigued by the genetic findings and with the expectation that three‐dimensional (3D) structure of the protein could be used to study PFIC1‐related mutations, we analyzed the 3D structures of the mutated FIC1 proteins and compared them with that of the wild‐type protein using the SWISS‐MODEL (https://www.swissmodel.expasy.org). The frame‐shift mutation (2025dupT in exon 18) introduced a stop codon, which caused an early termination of the protein translation process (Figure 4A). A comparative analysis of the predicted 3D structures revealed a truncated FIC1 protein with 688 amino acids, a portion of the 1251 amino acids in the wild‐type FIC1 protein. Furthermore, it was noted that the major domains (eg, P‐type ATPase ion transporting domains, calcium ATPase domains) 7 were missing in the truncated protein, profoundly affecting the structure and function of the FIC1 protein and making it non‐functional. The other heterozygous mutation (1375C>T in exon 13) was predicted to lead to a mutated FIC1 protein with a single alteration in the amino acid sequences (Leu459Phe) and local conformational changes (Figure 4B). The Leu459Phe mutation disrupted the hydrogen bonds, including breaking one original hydrogen bond (Thr460–Thr458) and forming three new hydrogen bonds (Lys455–Gly457, Lys455–Thr460 and Gly457–Thr460). Considering the important role of intramolecular hydrogen bonding in maintaining the secondary and tertiary structures of the protein, p.Leu459Phe could lead to a change in the protein conformation.
FIGURE 4.
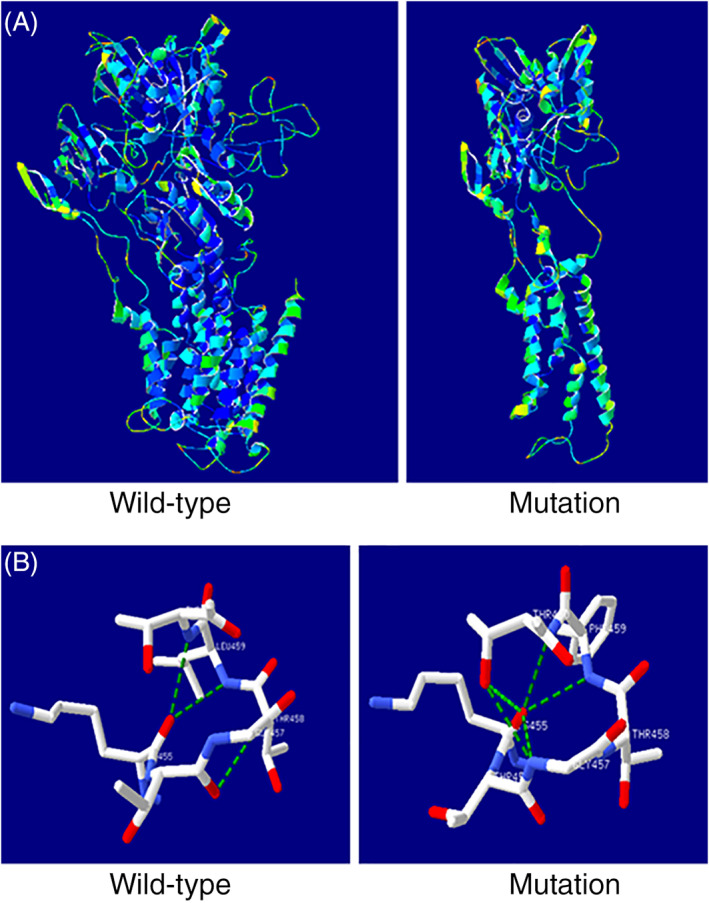
Models of the three‐dimensional (3D) structures of the wild‐type and mutated ATP8B1 proteins. A, A truncated ATP8B1 protein was predicted and identified as a result of the frame‐shift mutation (2025dupT) during homology modeling with the SWISS‐MODEL server. B, The missense mutation (1375C>T) led to a mutated ATP8B1 protein with change of a single amino acid (Leu459Phe). The number and position of hydrogen bonds have changed [Color figure can be viewed at wileyonlinelibrary.com]
It may merit attention that we newly identified two mutations (2025dupT and1375C>T) as forming the compound heterozygous mutation in PFIC1. PFIC1 is believed to result from ATP8B1 gene mutations,1, 5, 6hich markedly alter the protein structure and impair its function, such as frame‐shifts, large deletions and large insertions, and are generally thought to be associated with a phenotype change. In this study, the patient carried two mutations, including the 2025dupT mutation, which is predicted to cause a truncated FIC1 and to completely abrogate the FIC1 protein function. Therefore, we propose that 2025dupT mutation of the ATP8B1 gene may largely cause PFIC1 in our patient and could be pathogenic. The other mutation (1375C>T) of the ATP8B1 gene introduced a point mutation (Leu459Phe) and local conformational change of FIC1 and was likely to be pathogenic. It remains unclear why two family members with the same genetic alterations have variable phenotypes. We speculated that such difference might be due to the difference in their sex, patterns and levels of hormones, genetic mutation, epigenetic modifications and other environmental factors, or be associated with a single nucleotide polymorphism (SNP) within the key genes involved in bile formation. 10 We also postulate that the association between ATP8B1 gene mutations and clinical phenotype could be masked by other genetic or epigenetic modifying factors. The main gene has a great influence on the phenotype; however, modifying genes have subtle secondary effects. The modifier can influence the rates of expression and explicit of other alleles , ultimately change the phenotype. Further studies are needed to explain why two individuals carrying identical genetic mutations for the same disease (eg, PFIC1) have different levels of risk for disease development. It has been demonstrated that genetic factors increase the susceptibility to intrahepatic cholestasis of pregnancy (ICP), and that the FIC1 protein encoded by ATP8B1 gene is functionally interdependent with ABCB11 and MDR3 proteins, for which ATP8B1 is also considered a candidate gene for ICP susceptibility. 11 As the patient's sister has a high risk of developing ICP due to the two mutations of the ATP8B1 gene, regular follow‐up will be required when she is pregnant. 12
Rifampicin (RFP) has been used to treat cholestatic liver diseases via the enhancement of the hepatic efflux of organic anions, reduction of the bilirubin levels and amelioration of pruritus. As a potent pregnane X receptor (PXR) antagonist, RFP stimulates the expression of PXR‐sensitive genes, cytochrome P450 3A4 (CYP3A4), which is responsible for the hydroxylation of bile salts and drugs, and UDP‐glucuronosyltransferase 1A1 (UGT1A1). It enhances the expressions of MRP2 and OSTB. These mechanisms were considered to exclude toxic bile acid. 13 , 14 Our patient was treated with 300 mg RFP once daily and responded well to the medication. Notably, after being treated with RFP for 1 week, his serum TBIL level declined sharply (Figure 5). In addition, biochemical tests for drug‐related changes in the liver and kidney functions showed that his serum ALT and AST levels had increased temporarily to twice the upper limit of normal, but rapidly returned to normal without treatment, while his Cr level remained normal during RFP treatment (Figure 5). During the follow‐up since the patient was discharged from hospital, his liver function parameters remain normal and no recurrence of the disease has been observed (Table 1). There may be concerns about the use of RFP in cholestatic liver disease due to its initial recognition as an antituberculosis agent with potential hepatorenal toxicity, but its use in this patient with PFIC1 may provide additional clinical reference value.
FIGURE 5.
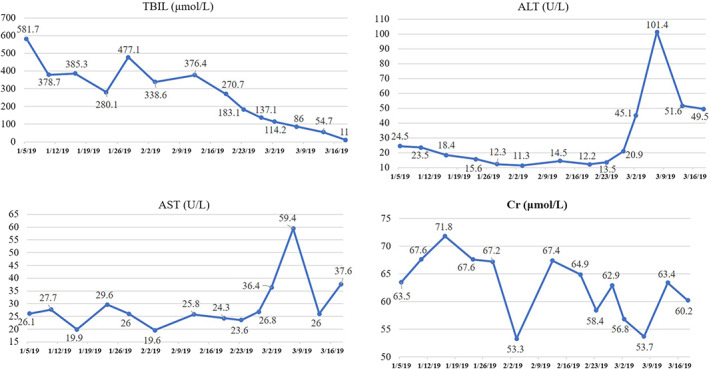
Changes in levels of total bilirubin (TBIL), alanine aminotransferase (ALT), aspartate aminotransferase (AST) and serum creatinine (Cr) at admission, during the hospitalization, and after treatment. Biochemical tests were performed in the patient at the time of hospital admission and after treatment. The date of patient's admission is January 4, 2019. He was discharged on March 14, 2019. After that, he continued to take 300mg RFP every day [Color figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Biochemical tests for the liver during the follow‐up of the patient after his discharge
| Follow‐up visits | TBIL (μmol/L) | DBIL (μmol/L) | IBIL (μmol/L) | ALT (U/L) | AST (U/L) | GGT (U/L) | ALP (U/L) |
|---|---|---|---|---|---|---|---|
| 18 March 2019 | 11 | 3.6 | 7.4 | 49.5 | 37.6 | 63 | 66.2 |
| 28 March 2019 | 34.1 | 28.7 | 5.4 | 18 | 19 | 26 | 100 |
| 5 April 2019 | 21 | 16.1 | 4.9 | 15 | 18 | 27 | 117 |
| 21 April 2019 | 10.7 | 9 | 1.7 | 16 | 18 | 26 | 111 |
| 21 May 2019 | 7.3 | 4.1 | 3.2 | 28 | 18 | 35 | 114 |
| 30 December 2019 | 12.8 | 8.9 | 3.9 | 12 | 19 | 32 | 125 |
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; DBIL, direct bilirubin; GGT, gamma‐glutamyltransferase; IBIL, indirect bilirubin; TBIL, total bilirubin.
In summary, we reported a rare case of PFIC1 with two novel mutations (2025dupT and1375C>T) of the ATP8B1 gene. This compound heterozygous mutation is a potentially disease‐causing mutation. RFP seems to be effective in the treatment of PFIC1 in this patient. These findings have expanded the known spectrum of disease‐causing mutations of the ATP8B1 gene and may benefit carrier testing in prenatal diagnosis and early detection of PFIC1.
CONFLICT OF INTEREST
All authors declare no conflicts of interest.
ACKNOWELDGMENTS
Written informed consent for the mutation analysis of hereditary human diseases and for publishing the current report were obtained from the patient and his family members.
REFERENCES
- 1. Bull LN, Thompson RJ. Progressive familial intrahepatic cholestasis. Clin Liver Dis. 2018;22(4):657‐669. [DOI] [PubMed] [Google Scholar]
- 2. Bull LN, Pawlikowska L, Strautnieks S, et al. Outcomes of surgical management of familial intrahepatic cholestasis 1 and bile salt export protein deficiencies. Hepatol Commun. 2018;2(5):515‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bull LN, van Eijk MJ, Pawlikowska L, et al. A gene encoding a P‐type ATPase mutated in two forms of hereditary cholestasis. Nat Genet. 1998;18(3):219‐224. [DOI] [PubMed] [Google Scholar]
- 4. Copeland E, Renault N, Renault M, et al. Novel splice‐site mutation in ATP8B1 results in atypical progressive familial intrahepatic cholestasis type 1. J Gastroenterol Hepatol. 2013;28(3):560‐564. [DOI] [PubMed] [Google Scholar]
- 5. Paulusma CC, de Waart DR, Kunne C, Mok KS, Elferink RPJO. Activity of the bile salt export pump (ABCB11) is critically dependent on canalicular membrane cholesterol content. J Biol Chem. 2009;284(15):9947‐9954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ujhazy P, Ortiz D, Misra S, et al. Familial intrahepatic cholestasis 1: studies of localization and function. Hepatology. 2001;34(4 Pt 1):768‐775. [DOI] [PubMed] [Google Scholar]
- 7. Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS. P4‐ATPases as phospholipid flippases—structure, function, and enigmas. Front Physiol. 2016;7:275. 10.3389/fphys.2016.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Paulusma CC, Groen A, Kunne C, et al. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 2006;44(1):195‐204. [DOI] [PubMed] [Google Scholar]
- 9. Hwang HJ, Park Y, Kim YK. UPF1: from mRNA surveillance to protein quality control. Biomedicines. 2021;9(8):995. 10.3390/biomedicines9080995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dröge C, Bonus M, Baumann U, et al. Sequencing of FIC1, BSEP and MDR3 in a large cohort of patients with cholestasis revealed a high number of different genetic variants. J Hepatol. 2017;67(6):1253‐1264. [DOI] [PubMed] [Google Scholar]
- 11. Chen F, Ghosh A, Shneider BL. Phospholipase D2 mediates signaling by ATPase class I type 8B membrane 1. J Lipid Res. 2013;54(2):379‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dixon PH, Sambrotta M, Chambers J, et al. An expanded role for heterozygous mutations of ABCB4, ABCB11, ATP8B1, ABCC2 and TJP2 in intrahepatic cholestasis of pregnancy. Sci Rep. 2017;7(1):11823. 10.1038/s41598-017-11626-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marschall HU, Wagner M, Zollner G, et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology. 2005;129(2):476‐485. [DOI] [PubMed] [Google Scholar]
- 14. Geenes V, Chambers J, Khurana R, et al. Rifampicin in the treatment of severe intrahepatic cholestasis of pregnancy. Eur J Obstet Gynecol Reprod Biol. 2015;189:59‐63. [DOI] [PubMed] [Google Scholar]