

Abstract
Colony stimulating factor 1 receptor (CSF1R)‐related leukoencephalopathy is a rare, genetic disease caused by heterozygous mutations in the CSF1R gene with rapidly progressive neurodegeneration, behavioral, cognitive, motor disturbances.
Objective
To describe four cases of CSF1R‐related leukoencephalopathy from three families with two different pathogenic mutations in the tyrosine kinase domain of CSF1R and to develop an integrated presentation of inter‐individual diversity of clinical presentations.
Methods
This is an observational study of a case series. Patients diagnosed with CSF1R encephalopathy were evaluated with standardized functional estimation scores and subject to analysis of cerebrospinal fluid biomarkers. Brain computed tomography (CT) and magnetic resonance imaging (MRI) were evaluated. We performed a functional phosphorylation assay to confirm the dysfunction of mutated CSF1R protein.
Results
Two heterozygous missense mutations in the CSF1R gene were identified, c.2344C>T; p.Arg777Trp and c.2329C>T; p.Arg782Cys. A phosphorylation assay in vitro showed markedly reduced autophosphorylation in cells expressing mutations. According to ACMG criteria, both mutations were pathogenic. A radiological investigation revealed typical white matter lesions in all cases. There was inter‐individual diversity in the loss of cognitive, motor‐neuronal, and extrapyramidal functions.
Conclusions
Including the present cases, currently three CSF1R mutations are known in Sweden. We present a visualization tool to describe the clinical diversity, with potential use for longitudinal follow‐up for this and other leukoencephalopathies.
Keywords: adult‐onset leukoencephalopathy with spheroids and pigmented glia, biomarkers, colony stimulating factor 1 receptor, CSF1R gene, neurodegeneration, primary microgliopathy
1. INTRODUCTION
The CSF1R gene on chromosome 5q32 encodes a receptor for colony stimulating factor 1, which regulates the function and maturation of the monocyte/macrophage lineage, including microglia. 1 The receptor is also expressed in osteoclasts and plays an important role in bone mineralization. 2 Heterozygous mutations in this gene have been identified in patients with autosomal dominant adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP), which includes hereditary diffuse leukoencephalopathy with spheroids (HDLS) and pigmentary orthochromatic leukodystrophy (POLD). 3 Recent genetic data and common neuropathology confirm that HDLS and POLD constitute a single nosological unit, 4 now termed CSF1R‐related leukoencephalopathy. It is characterized by rapidly progressive neurodegeneration and variable behavioral, cognitive, and motor disturbances as well as seizures. In the last stage of the disease, affected individuals become bedridden with spasticity and progress to a vegetative state. 5 Mean age at onset is in the fourth decade, and the disease course ranges from 2 to 30 years. 5 Brain magnetic resonance imaging (MRI) typically shows progressive bifrontal and biparietal cerebral white matter abnormalities in the subcortical and periventricular regions. 6 At least 71 different CSF1R mutations (56 missense mutations, eight splice‐site mutations, three frameshift mutations, two nonsense mutations, and two small deletions) have been described, 7 but the true prevalence and incidence of the disease are still unknown.
In this study, we describe four cases from three families with two different pathogenic mutations in the tyrosine kinase domain of the CSF1R gene. We capture the range of clinical presentations using several standardized scoring scales to facilitate a description of multifaceted, individual disease trajectories.
2. PARTICIPANTS AND METHODS
2.1. Patients
Patients were initially assessed at their local hospitals according to each clinic's routine. All patients were diagnosed either with definitive ALSP (patients 1, 3, and 4) or probable ALSP (patient 2) using ALSP diagnostic criteria 8 (Table 1). A comprehensive assessment was performed by the study team using several standardized clinical rating scales to capture the diverging clinical phenotypes: Unified Parkinson's Disease Rating Scale (UPDRS) parts I–VI, 9 Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS‐R), 10 Hospital Anxiety and Depression Scale [HADS, 11 ], and Expanded Disability Status Scale [EDSS, 12 ]. Cognitive status was evaluated with Mini‐Mental State Examination [MMSE, 13 ], Montreal Cognitive Assessment [MOCA, 14 ], and the Symbol Digit Modalities Test [SDMT, 15 ]. To estimate the quality of life, the Multiple Sclerosis Impact Scale [MSIS‐29, 16 ] and EuroQol‐dimension [EQ5D/VAS, 17 ] scales were used. Computed tomography (CT) and MRI were performed according to clinical routine with different standard scanners and protocols. We systematically re‐evaluated all available imaging. Patient 2 died in 1991 at 45 years of age, while her daughter, patient 3, died 30 years later, 44 years of age.
TABLE 1.
Demographics and clinical characteristics of patients included in this study
| Patient | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Variable | ||||
| Age at symptom onset | 41 | 42 | 42 | 48 |
| Presenting symptoms | Frontal | Frontal | Hemiparesis | Bulbar |
| Sex | Male | Female | Female | Male |
| Ethno‐regional descent | Nordic | Nordic | Nordic | Nordic |
| Mutation position | 5:149435880 | N/A | 5:149435895 | 5:149435895 |
| Mutation | Heterozygous c.2344C>T | N/A | Heterozygous c.2329C>T | Heterozygous c.2329C>T |
| Amino acid exchange | p. Arg782Cys | N/A | p. Arg777Trp | p. Arg777Trp |
| Diagnosis according to ALSP diagnostic criteria 5 , 8 |
Definitive 2a, 2b, 3, 4a |
Possible 2a, 3, 4a |
Definitive 2a, 2b, 3, 4a |
Definitive 2a, 2b, 2c, 3, 4a |
| Mutation pathogenicity criteria 18 | PS1 and PS3 | N/A | PS1 and PS3 | PS1 and PS3 |
Abbreviations: ALSP, adult leukoencephalopathy with spheroids and pigmented glia; Arg, arginine; CSF1R, colony stimulating factor 1 receptor (NM_005211.3); Cys, cysteine; N/A, not available; PS1, same amino acid change as a previously established pathogenic variant regardless of nucleotide change; PS3, well‐established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product; Trp, tryptophan.
2.2. Genetic analysis
Sequence analysis was performed by Blueprint© Genetics using the Leukodystrophy and Leukoencephalopathy panel (Version 3, Mar 01, 2018, https://blueprintgenetics.com/tests/panels/neurology/leukodystrophy‐and‐leukoencephalopathy‐panel/) for patient 1 and patient 3; for patient 4, the CSF1R gene (NM_005211.3) Single Gene Test analysis (Version 1, Mar 01, 2018; https://blueprintgenetics.com/tests/single‐gene‐tests/csf1r‐single‐gene‐test‐2/) was used. Both for the Leukodystrophy /Leukoencephalopathy panel and for the Single Gene Test, the reads were aligned to the human reference genome (GRC37/hg19) by using the Burrows–Wheeler Aligner, and the GATK algorithms was used for variant calling. Interpretation of variant was performed by using the Blueprint Genetic Variant Classification Schemes (https://blueprintgenetics.com/variant‐classification/).
The identified variants were classified using the ACMG criteria. 18
2.3. Neuropathological analysis
A neuropathologic examination was performed at autopsy of patient 2 in 1991, and sectioning and staining were renewed in 2020 for this study. Selected blocks were sectioned at 6 and 3 micrometers for large and small blocks, respectively, and the sections were stained with hematoxylin–eosin, Luxol Fast Blue‐Cresyl violet, and Gallyas silver.
2.4. Phosphorylation assay
To assess the pathogenicity of the CSF1R p. Arg777Trp and p. Arg782Cys mutants, we performed a functional assay of these mutants as previously described. 19 HEK293T cells were transfected with cDNA encoding p. Arg777Trp and p. Arg782Cys. After 20 min of ligand (CSF1) stimulation, autophosphorylation of CSF1R at residues of Tyr546, Tyr708, and Tyr723 was examined using antibodies against specific phosphorylated CSF1Rs. Statistical analysis was performed with one‐way ANOVA using a Tukey multiple comparison test.
2.5. Graphical visualization
Graphical visualization of functional estimate scores (ALSP‐FES) was performed using Microsoft Excel (2016). Details of calculations and Excel sheets with formulas are included in Supplements 4, 5, and 6.
2.6. Ethics
The Swedish Research Ethics Committee (Ref. 2019–05742, March 19, 2020) approved the study. Patients 1, 3, and 4 signed written informed consent. Patient 2 was deceased before the study initiation. Permission was granted by Patient 2’s family to publish both clinical history and histopathological findings. All procedures were performed in accordance with the principles of the Declaration of Helsinki.
3. RESULTS
Table 1 summarizes the demographic and clinical characteristics of patients 1–4.
3.1. Individual description of cases
3.1.1. Patient 1
A 41‐year‐old previously healthy man of Nordic ethnicity was admitted to a local hospital for sepsis and erysipelas of the arm. Hospital staff noted abnormal social behavior, reduced speech fluency, and verbal response latency. Brain CT revealed moderate periventricular and frontal white matter lesions and small frontal calcifications bilaterally (Figure 1A). Family members confirmed progressive cognitive and neuropsychiatric decline over the past year, including personality changes with agitation, apathy, anxiety, and emotional lability. There was no history of substance abuse. The neurological assessment revealed moderate dysarthria and reduced speech fluency but no other neurological deficits. At clinical 10‐month follow‐up, speech modalities had declined. The patient had an increased tonus in all extremities and a spastic gait. On a 2‐year follow‐up, the patient lived in a nursing home. Neurological examination revealed significant deterioration with dementia, mutism, choreoathetoid movements, and a complex motor pyramidal–extrapyramidal syndrome. The neuropsychiatric assessment showed reduced attention, information processing, verbal and visuospatial abilities, verbal and visual memory, and executive skills. Relatives reported reduced school performance. The patient was suspected to have had a neuropsychiatric disturbance long before a full‐blown neurodegenerative process developed (Neuropsychiatric assessment summary is given in Supplements 1). MRI demonstrated confluent, symmetric, bifrontoparietal white matter changes (WMC) with a clear U‐fiber sparing (Figure 1B,F). Axial T2‐weighted (Figure 1A,E) and T2‐weighted fluid‐attenuated inversion recovery (FLAIR) images (Figure 1B,F) showed progression of confluent white matter changes with a subtle left‐sided predominance between the baseline scan at 42 years of age (Figure 1A‐D) and follow‐up scan 10 months later (Figure 1E‐H). The lesions showed a pronounced low T1 signal, indicative of marked demyelination. There was also central atrophy with enlarged ventricles and thinning of the corpus callosum. At baseline, T1‐weighted imaging (Figure 1C‐D) demonstrated very subtle peripheral foci of contrast enhancement at the leading edge (arrows). Small calcifications were seen frontally on the right side on CT (Figure 1G‐H). Small foci of persistent diffusion restriction were also seen (not shown). A follow‐up brain MRI 10 months later revealed progression of WMC and global cortical atrophy (GCA) scale 20 rating of 2. There was no history of neurological hereditary diseases. Both parents were alive and healthy at the time of presentation. Genetic analysis was performed at Blueprint© Genetics using the Leukodystrophy and Leukoencephalopathy panel (Version 3, Mar 01, 2018) and revealed a heterozygous missense variant c.2344C>T; p. Arg782Cys in the CSF1R gene.
FIGURE 1.
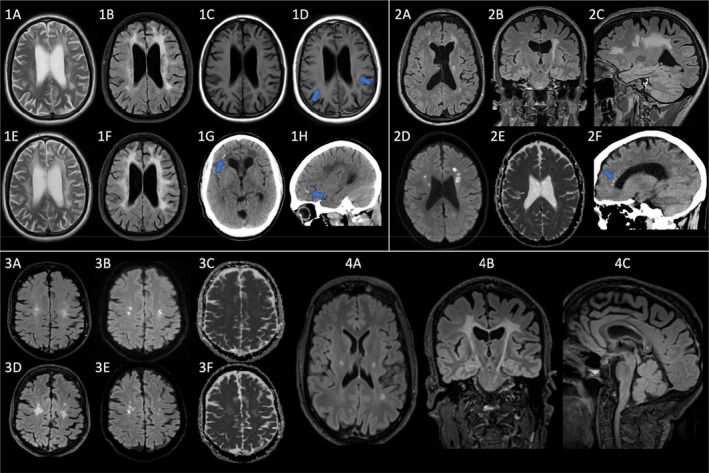
Brain MRI and CT scan images. Progressive white matter changes with calcifications (arrows 1G‐H, 2F), persistent diffusion restriction (2D‐E, 3B‐C, 3E‐C), and contrast enhancement (arrows D). 1: Patient 1 (age 42, 10 months later); 2: Patient 3 (age 41); 3: Patient 4 (age 48, 2.5 years later); 4: Patient 4 (age 51)
3.1.2. Patient 2
A 42‐year‐old woman of Nordic ethnicity was admitted to a psychiatric clinic with a few months history of depersonalization and cognitive dysfunction. Both parents were alive without any known neurological diseases. She had psoriasis but no other medical history. Neurological examination was normal except for positive glabellar reflex and muscular jerkiness. She was initially diagnosed with depression. Testing revealed lack of emotional contact, confabulation tendency, symptom denial, reduced concentration and endurance, and dysphasia. The proposed diagnosis was frontal lobe dementia. Brain CT showed atrophy, most pronounced in the frontal lobes. MRI showed atrophy and widespread, mostly frontal WMCs, mainly in the left hemisphere. Electroencephalography showed bilateral, generalized slowing. Sensory evoked potentials were normal. She deteriorated gradually and spent her last years in a nursing home. She died three years after the onset of symptoms.
Postmortem brain autopsy on macroscopic examination revealed a general and slight gyral atrophy accentuated in the frontal lobe, while the posterior regions showed no surface atrophy. The white matter showed severe frontal and central demyelination, while the parietal–occipital and temporal regions were markedly less deteriorated. In a coronal section of the anterior frontal lobes, the cortical ribbon was well‐delineated, while the subcortical white matter was severely attenuated and atrophied, resulting in shrinkage of the entire frontal lobes (Figure 2A). In the posterior frontal lobes, the hematoxylin–eosin staining showed an intact cortical tissue (left) and a relatively preserved subcortical white matter (center), while the deep white matter was markedly attenuated (right) (Figure 2B). There was an extreme scarcity of myelin sheaths and axonal and myelin sheath swelling in one of the few remaining long structures (Figure 2C). In the white matter area from the posterior frontal lobes (behind the region of maximal damage, as in Figure 2B), many reactive astrocytes and a few mildly pigmented macrophages were visualized (Figure 2D). In the area with more severe damage, pleomorphic reactive cells (astrocytes and macrophages) and a more attenuated matrix can be seen (Figure 2E). Figure 2F shows severely attenuated white matter, where Gallyas silver staining reveals rudimental fragments of axons and two mildly pigmented macrophages with granular intracellular debris. Axonal thickenings, occasionally resembling spheroids, were seen, but they were relatively scarce.
FIGURE 2.
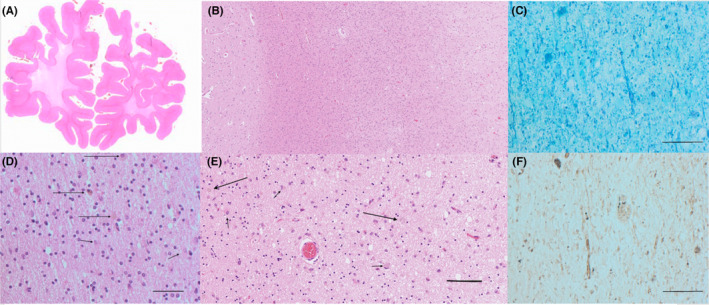
Histopathology of patient 2 postmortem brain tissue sections. (A) Macrophotograph anterior frontal lobes, hematoxylin–eosin staining. Note the well‐delineated cortical ribbon, which is of normal width and the subjacent white matter, which is severely attenuated, with shrinkage of the entire frontal lobes (B) Microphotograph posterior frontal lobe, hematoxylin–eosin. Note the intact cortical tissue with dense and preserved matrix (left) and the relatively preserved subcortical white matter (center), while deeper white matter is markedly attenuated (right) (C) Microphotograph anterior frontal lobe, Luxol Fast Blue‐Cresyl violet. Note the extreme scarcity of myelin sheaths and the axonal and myelin sheath swelling in one of the few remaining long structures. D: White matter of the posterior frontal lobes, hematoxylin–eosin. Note the attenuated tissue with many reactive astrocytes and a few mildly pigmented macrophages. (E) Pleomorphic reactive astrocytes and macrophages. (F) Attenuated white matter from the anterior frontal lobe, as in A and C, Gallyas silver staining for identification of axons. Note the rudimental fragments of axons and the two macrophages (pigmented) with granular intracellular debris. Bar indicates 0.05 mm
To sum up, myelin and silver stains revealed massive axonal breakdown as well as near‐total demyelination and near‐complete loss of axonal structures. We attempted to extract DNA from paraffin blocks archived at autopsy; however, repeated attempts were unsuccessful due to DNA degradation, possibly due to long formalin fixation time or a long time since tissue sampling in 2001.
3.1.3. Patient 3
At 42 years of age, the daughter of patient 2 was admitted to a regional hospital with the right arm and leg weakness that had progressed over the course of a month. She had no prior significant medical history. There were no signs of a neuropsychiatric or cognitive dysfunction. At our 18‐month follow‐up, she was confined to a wheelchair and needed help with most daily activities. She was oriented to time and space. She had severe dysarthria and dysphasia. She exhibited pseudobulbar syndrome and a marked combined pyramidal–extrapyramidal syndrome. Brain CT showed multiple punctate calcifications in a stepping‐stone pattern along the corpus callosum (arrow in Figures 1, 2F). CT angiography was normal. MRI showed symmetric bilateral atrophy and widespread confluent WMCs. Axial (Figures 1, 2A), coronal (Figures 1, 2B), and sagittal (Figures 1, 2C) T2‐weighted FLAIR images at 41 years of age showed symmetric, confluent white matter hyperintensities sparing the subcortical U fibers with prominent involvement of the corticospinal tracts and a left‐sided predominance. Diffusion‐weighted b1000 (Figures 1, 2D) and apparent diffusion coefficient (Figures 1, 2E) images demonstrated foci of restricted diffusion that persisted four months later at a follow‐up scan (not shown). U fibers were mostly spared, and the MRI revealed no contrast enhancement. The lesions were essentially unchanged four months later. A spinal cord MRI was normal. Testing for NOTCH3 gene mutations was negative. Genetic analysis was performed at Blueprint© Genetics using the Leukodystrophy and Leukoencephalopathy panel (Version 3, Mar 01, 2018) and revealed a heterozygous missense variant c.2329C>T, p. Arg777Trp in the CSF1R gene.
3.1.4. Patient 4
A 48‐year‐old man was referred to a private neurological outpatient department due to dysarthria. Following symptom progression, the patient was referred to a university clinic a year later. Neurological examination revealed dysarthria, dysphagia, and numbness of the right arm and left leg as well as subjective breathing difficulty. At a 1‐year follow‐up, the patient reported spastic paraparesis, urine incontinence, and reduced balance. Due to dysarthria that progressed to anarthria, he used a speech‐generating tablet. At our 2‐year follow‐up, a severe bulbar syndrome with hypersalivation and a slight pseudobulbar component had developed, as well as discrete pyramidal–extrapyramidal and deep sensory symptoms. Brain MRI at baseline and follow‐up revealed confluent WMCs with relatively symmetrical frontoparietal distribution (Figures 1, 3). T2‐weighted FLAIR images at 51 years of age showed symmetric confluent white matter hyperintensities sparing the subcortical U fibers with prominent involvement of the corticospinal tracts and the central portion of the splenium of corpus callosum: axial (Figures 1, 4A), coronal (Figures 1, 4B), and sagittal (Figures 1, 4C). Axial T2‐weighted FLAIR (Figures 1, 3A, D), diffusion‐weighted (Figures 1, 3B, E), and apparent diffusion coefficient (Figures 1, 3C, F) images at 48 years of age (Figures 1, 3A‐C) and 2.5 years later (Figures 1, 3D‐F) show progression of relatively symmetric bifrontal white matter hyperintensities with foci of persistent diffusion restriction at the leading edge. The WMCs affected corticospinal tracts down to the decussation in the pons. The corpus callosum was affected in its isthmus and centrally in the splenium region. The patient had developed moderate frontotemporal atrophy as well as slight to moderate central atrophy. Brain CT did not show any calcifications. The Blueprint Genetics© CSF1R single gene test (Version 1, Mar 01, 2018) Plus analysis identified a heterozygous missense variant c.2329C>T; p. Arg777Trp.
3.2. Quantitative outcomes in present case series
3.2.1. Autophosphorylation assay
Ligand‐dependent autophosphorylation of CSF1R was examined in cells transfected with wild‐type or variant CSF1Rs. The mutation of CSF1R p. Ile794Thr has been frequently identified in patients with ALSP 5 and is known to be pathogenic, as has been previously reported. 19 Although phosphorylation of CSF1R at Tyr546, Tyr708, and Tyr723 was observed in cells expressing wild‐type CSF1R after ligand (CSF1) stimulation, neither of the CSF1R mutants underwent autophosphorylation (Figure 3A).
FIGURE 3.
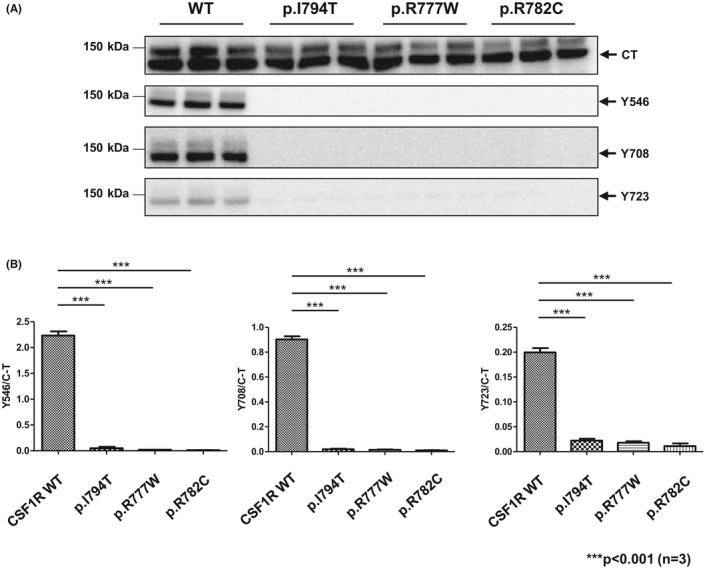
In vitro functional assay of mutant CSF1Rs. (A) Ligand‐dependent autophosphorylation of CSF1R in cells transfected with wild‐type or variant CSF1Rs. (B) Semi‐quantitative assessment of immunoblot signal intensity between wild‐type, positive control, and mutant CSF1R proteins. Data are presented as mean ± SEM
The signal intensity was assessed using semi‐quantitative immunoblot analysis. Each autophosphorylation signal of CSF1R was normalized by the signal from the total amount of CSF1R. Horizontal bars indicate the difference between wild‐type and the positive control as well as the mutations in the patients presented in this publication (Figure 3B).
3.2.2. CSF biomarkers
CSF tau was significantly elevated in Patient 1, while CSF beta‐amyloid and CSF phosphorylated tau were normal in all cases (Table S2). The tau/phospho‐tau ratio in Patient 1 was roughly 28, which suggests relatively fast progression. Glial fibrillary protein (GFAP) in patient 1 was markedly elevated, suggestive of astrocytic damage or activation. CSF‐NFL levels were significantly elevated, indicating ongoing severe axonal damage, especially in patient 1.
3.2.3. Function estimation scores
Table 2 presents the results of the clinical assessment scales that were used to rate patients described in this case series. Figure 4 shows a composite graphic representation of the ALSP‐FES: a radar chart.
TABLE 2.
Function estimation scores
| Patient | Patient 1 | Patient 2 | Patient 3 | Patient 4 |
|---|---|---|---|---|
| Estimation instrument, time | ||||
| Rating time point, years after onset | 2 years | N/A | 1 year | 3 years |
| UPDRS part I | 9 | N/A | 4 | 4 |
| UPDRS part II | 33 | N/A | 24 | 15 |
| UPDRS part III | 35 | N/A | 32 | 8 |
| UPDRS part IV | N/A | N/A | N/A | N/A |
| UPDRS part V | Stage 5 | N/A | Stage 5 | Stage 1 |
| UPDRS part VI | 10% | N/A | 25% | 100% |
| ALSFRS‐R | 24 | N/A | 17 | 31 |
| HADS Depression/Anxiety | N/A | N/A | 9/9 | 13/9 |
| EQ5D/VAS | 10/N/A | N/A | 14/10 | 10/4 |
| EDSS | 8.0 | N/A | 7.5–8.0 | 5.5 |
| MMSE | N/A | N/A | 15 | 24 |
| MSIS−29 | N/A | N/A | N/A | N/A |
| SDMT | N/A | N/A | N/A | 31 |
Abbreviations: ALSFRS, ALS Functional Rating Scale; EDSS, Expanded Disability Status Scale; EQ5D, EuroQol‐5D standardized instrument for measuring generic health status; HADS, Hospital Anxiety Depression Rating Scale; MMSE, Mini‐Mental State Examination; MSIS‐29, Multiple Sclerosis Impact Scale; N/A, not available; SDMT, Symbol Digits Modalities Test; UPDRS, Unified Parkinson's Disease Rating Scale; VAS, Visual Assessment Scale.
FIGURE 4.
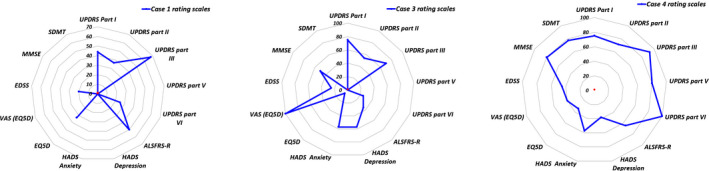
Radar chart visualizing function evaluation by several rating scales. Blue dots localized at the outer point on the specific evaluation scale axis signify 100% performance. Patient's performance was evaluated between 0 to 100% on specific rating scale. If patient was unable to perform the activity of rating scale due to disability, performance was evaluated 0%
3.3. Determination of pathogenicity of mutations
The genetic analysis revealed two heterozygous missense variants, c.2329C>T; p. Arg777Trp and c.2344C>T; p. Arg782Cys, and both of them were classified as pathogenic according to ACMG guidelines (details are presented in Table 1). 18
4. DISCUSSION
In this case series, we describe four cases of CSF1R‐related leukoencephalopathy caused by CSF1R gene mutations found in members of three families in Sweden. Patients 3 and 4, who were not related, had a previously described mutation in the CSF1R gene (c.2329C>T; p. Arg777Trp). For the other identified mutation in this study (c.2344C>T; p. Arg782Cys) identified in patient 1, there are no reports describing the clinico‐pathological or pathogenicity for the variant in detail, although the review by Konno et al. 7 briefly mentioned this mutation. In this paper, we report that these CSF1R mutants had lost ligand‐induced autophosphorylation of CSF1R, which confirms the pathogenicity of these heterozygous missense mutations. The postmortem tissue biobank at Lund University gave us the opportunity to connect a clinical case with definitive CSF1R‐related leukoencephalopathy to autopsy findings. Despite paraffin‐embedded brain tissue specimen was stored for 30 years, we were able to create workable histopathological images with findings typical for CSF1R‐related leukoencephalopathy: axonal spheroids (Figure 2C) and pigmented macrophages (Figure 2D). 21 , 22
Table S3 summarizes previously reported cases and families with identified mutations in CSF1R affecting the amino acid arginine at positions 777 and 782. The amino acid at position 782 seems to be a hot spot for missense changes, and this arginine has been described to be exchanged either with glycine, cysteine, leucine, or histidine all described with clinical outcome. 5 , 7 , 23 , 24 , 25 A change to cysteine at position 782 as in patient 1 may have a stronger physiochemical impact (Grantham score: 180 [0–215] indicating a greater evolutionary distance) than the change to histidine (Grantham score: 29 [0–215]), while a change to glycine may have an intermediate impact (Grantham score: 125 [0–215]). Thus, the mutation in our patient 1—c.2344C>T (p. Arg782Cys) of the CSF1R gene—may predict more severe disease. Nevertheless, a rapid course was observed in a case with a change to glycine, c.2344C>G; p. Arg782Gly in CSF1R with a disease duration of only 2 years and 2 months. 25 The cases with the CSF1R p. Arg777Trp mutations—one likely familiar and the other arguably sporadic—have no family relationship, although we did not explore the patients’ ancestral lines. A CSF1R c.2329C>T; p. Arg777Trp point substitution was previously described as a novel mutation in a Japanese patient with alcoholism, personality changes, and dementia. 26 Our autophosphorylation test confirmed the pathogenicity of this specific mutation. Another missense alteration at this residue, Arg777Gln, was described in three individuals from a French family who exhibited progressive frontal dementia, dysarthria, and apraxia 21 and in a family with early onset and rapid disease progression of HDLS. 27
We explored the clinical heterogeneity of CSF1R‐related encephalopathy, integrating scoring systems for extrapyramidal, motor‐neuron, and neuropsychiatric/cognitive functions into a radar chart. Patients with CSF1R‐related leukoencephalopathy present with a wide range of adult‐onset focal or systemic neurological and cognitive symptoms. Previous reports present a spectrum of phenotypes, 22 protracted asymptomatic courses, 28 a phenotype with features of progressive multiple sclerosis (MS), 29 and patients with parkinsonian features, 30 as well as dominant bulbar symptoms, 31 as in our patient 4. We scored our current cases with a composite of several established function estimation scores (FESs) used to diagnose specific neurological diseases, which we termed ALSP‐FES. We propose our novel radar chart as an integrated representation of the ALSP‐FES as a candidate for assessment of CSF1R‐related encephalopathy and similar leukodystrophies.
CSF‐NFL correlates well with disease activity and disability progression in multiple sclerosis (MS) and is a diagnostic and prognostic biomarker in MS. 32 , 33 In ALS, CSF‐NFL levels correlate with disease progression. 34 Hayer et al. reported NFL levels to be considerably elevated in the serum and CSF in CSF1R‐related leukoencephalopathy patients 35 with a lack of overlap between patients and, therefore, proposed NFL as a valid biomarker. This is consistent with the NFL level in patient 1. However, patients 3 and 4 indicate that CSF‐NFL may be only moderately elevated, as commonly seen in MS (relapsing and progressive) and atypical parkinsonism (Supplement 2, Table S2). 36 , 37 , 38
Recently, Gelfand et al. published a case report in which they describe delayed clinical stabilization of two symptomatic patients with genetically verified CSF1R‐related leukoencephalopathy after allogeneic hematopoietic stem cell transplantation (HSCT). 5 , 39 Furthermore, some researchers have reported that CSF2 expression is increased in patients with mutant CSF1R, making it an attractive therapeutic target. 40 In a recent report, the authors proposed that a protracted, nearly asymptomatic course in a patient with a documented CSF1R pathogenic mutation was explained by her constant medication with corticosteroids for another disease and the inhibitory effect of the corticosteroids on CSF2’s pro‐inflammatory effects. 41 The recent advancement in genetic engineering techniques, such as CRISPR gene editing and therapies focused on microglial modulation, 42 could potentially offer new treatment avenues for patients with genetic leukoencephalopathies or pre‐symptomatic carriers. Because the disease is autosomal dominant with a high degree of penetrance, 5 offspring's risk of being affected is close to 50%. Hypothetically, it could be meaningful to identify such asymptomatic carriers and consider allogeneic HSCT at an earlier stage before a full‐blown leukoencephalopathy ensues. 21 , 22
A weakness of our study is that CSF biomarkers were analyzed in different laboratories that use slightly different methods, which could result in minor discrepancies in values between patients. MRI protocols were not harmonized between different centers.
5. CONCLUSION
A precondition for implementing therapy that is still experimental is long‐term clinical monitoring with standardized, validated tools to evaluate clinical outcomes and endpoints in these disorders. In this paper, we highlight the variability of CSF1R‐related leukoencephalopathy semi‐quantitatively along the axes of frontal, motor‐neuron, and extrapyramidal disease. We suspect that adult‐onset leukodystrophies in general, and CSF1R‐related leukoencephalopathies in particular, are considerably underdiagnosed. 43 We intend to increase the awareness among neurologists and psychiatrists of CSF1R‐related encephalopathies and similar leukodystrophies including AARS‐ and AARS2‐dependent diseases. 44
AUTHOR CONTRIBUTIONS
OA, IR, and VDK designed and conceptualized the study. TG, EE, AY, IT, OA, IR, VDK, KJ, and VD involved in interpretation of results, design of the work, and manuscript drafting and revision. EE (Histopathology images); TG (MRI images); AY, IT (In vitro phosphorylations assay); VDK (Radar chart); and CH‐O (attempted DNA extraction and revised manuscript) involved in collection of specific data and interpretation. Patients were examined by neurologists, OA, IR, VDK, KJ, and VD. All authors read and approved the final manuscript.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the Regional Ethical Committee in Stockholm, ethical permit Dnr 2009/2017‐31/2 “STOPMS‐II” (patient 4), and the Swedish Research Ethics Authority, ethical permit Dnr 651–05, “HDLS” (patients 1, 3, and 4). Patients signed informed consent to participate in these studies.
CONSENT FOR PUBLICATION
Patients signed consent to publish MRI images and clinical data in this publication.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/ane.13589.
Supporting information
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the patients and their relatives for their participation in the study and consent for this publication. We appreciate Antanas Romas at Vattenfall AB, KSU, Sweden, for help with radar chart diagrams and calculations. We appreciate the biomedical technicians at Lund University, Sweden, for making brain sections and stainings.
Rosenstein I, Andersen O, Victor D, et al. Four Swedish cases of CSF1R‐related leukoencephalopathy: Visualization of clinical phenotypes. Acta Neurol Scand. 2022;145:599–609. doi: 10.1111/ane.13589
Funding information
TI was funded by AMED JP21dk0207045. VDK has received financial support from Stockholm County Council (grant ALF 20160457); Biogen (recipient of grant and scholarship, PI for project sponsored by); Novartis (Scientific Advisory board member, recipient of scholarship and lecture honoraria); Merc (Scientific Advisory Board member, recipient of lecture honoraria); and Vigil Neuro (Scientific Advisory Board member)
Contributor Information
Oluf Andersen, Email: oluf.andersen@neuro.gu.se.
Virginija Danylaité Karrenbauer, Email: virginija.karrenbauer@ki.se.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Chitu V, Gokhan Ş, Nandi S, Mehler MF, Stanley ER. Emerging roles for CSF‐1 receptor and its ligands in the nervous system. Trends Neurosci. 2016;39(6):378‐393. doi: 10.1016/j.tins.2016.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Guo L, Bertola DR, Takanohashi A, et al. Bi‐allelic CSF1R mutations cause skeletal dysplasia of dysosteosclerosis‐pyle disease spectrum and degenerative encephalopathy with brain malformation. Am J Hum Genet. 2019;104(5):925‐935. doi: 10.1016/j.ajhg.2019.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rademakers R, Baker M, Nicholson AM, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet. 2011;44(2):200‐205. doi: 10.1038/ng.1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nicholson AM, Baker MC, Finch NA, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology. 2013;80(11):1033‐1040. doi: 10.1212/WNL.0b013e31828726a7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Konno T, Yoshida K, Mizuno T, et al. Clinical and genetic characterization of adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia associated with CSF1R mutation. Eur J Neurol. 2017;24(1):37‐45. doi: 10.1111/ene.13125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sundal C, Van Gerpen JA, Nicholson AM, et al. MRI characteristics and scoring in HDLS due to CSF1R gene mutations. Neurology. 2012;79(6):566‐574. doi: 10.1212/WNL.0b013e318263575a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Konno T, Kasanuki K, Ikeuchi T, Dickson DW, Wszolek ZK. CSF1R‐related leukoencephalopathy: a major player in primary microgliopathies. Neurology. 2018;91(24):1092‐1104. doi: 10.1212/wnl.0000000000006642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Konno T, Yoshida K, Mizuta I, et al. Diagnostic criteria for adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia due to CSF1R mutation. Eur J Neurol. 2018;25(1):142‐147. doi: 10.1111/ene.13464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goetz CG, Fahn S, Martinez‐Martin P, et al. Movement Disorder Society‐sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): process, format, and clinimetric testing plan. Mov Disord. 2007;22(1):41‐47. doi: 10.1002/mds.21198 [DOI] [PubMed] [Google Scholar]
- 10. Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS Study Group (Phase III). J Neurol Sci. 1999;169(1–2):13‐21. doi: 10.1016/s0022-510x(99)00210-5 [DOI] [PubMed] [Google Scholar]
- 11. Spinhoven P, Ormel J, Sloekers PP, Kempen GI, Speckens AE, Van Hemert AM. A validation study of the Hospital Anxiety and Depression Scale (HADS) in different groups of Dutch subjects. Psychol Med. 1997;27(2):363‐370. doi: 10.1017/s0033291796004382 [DOI] [PubMed] [Google Scholar]
- 12. Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444‐1452. doi: 10.1212/wnl.33.11.1444 [DOI] [PubMed] [Google Scholar]
- 13. Tombaugh TN, McIntyre NJ. The mini‐mental state examination: a comprehensive review. J Am Geriatr Soc. 1992;40(9):922‐935. doi: 10.1111/j.1532-5415.1992.tb01992.x [DOI] [PubMed] [Google Scholar]
- 14. Shi D, Chen X, Li Z. Diagnostic test accuracy of the Montreal Cognitive Assessment in the detection of post‐stroke cognitive impairment under different stages and cutoffs: a systematic review and meta‐analysis. Neurol Sci. 2018;39(4):705‐716. doi: 10.1007/s10072-018-3254-0 [DOI] [PubMed] [Google Scholar]
- 15. Strober L, DeLuca J, Benedict RH, et al. Symbol Digit Modalities Test: a valid clinical trial endpoint for measuring cognition in multiple sclerosis. Mult Scler J. 2019;25(13):1781‐1790. doi: 10.1177/1352458518808204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hobart J, Lamping D, Fitzpatrick R, Riazi A, Thompson A. The Multiple Sclerosis Impact Scale (MSIS‐29): a new patient‐based outcome measure. Brain. 2001;124(Pt 5):962‐973. doi: 10.1093/brain/124.5.962 [DOI] [PubMed] [Google Scholar]
- 17. Balestroni G, Bertolotti G. EuroQol‐5D (EQ‐5D): an instrument for measuring quality of life. Monaldi Arch Chest Dis. 2012;78(3): doi: 10.4081/monaldi.2012.121 [DOI] [PubMed] [Google Scholar]
- 18. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405‐424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miura T, Mezaki N, Konno T, et al. Identification and functional characterization of novel mutations including frameshift mutation in exon 4 of CSF1R in patients with adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. J Neurol. 2018;265(10):2415‐2424. doi: 10.1007/s00415-018-9017-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasquier F, Leys D, Weerts JG, Mounier‐Vehier F, Barkhof F, Scheltens P. Inter‐ and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol. 1996;36(5):268‐272. doi: 10.1159/000117270 [DOI] [PubMed] [Google Scholar]
- 21. Guerreiro R, Kara E, Le Ber I, et al. Genetic analysis of inherited leukodystrophies: genotype‐phenotype correlations in the CSF1R gene. JAMA Neurology. 2013;70(7):875‐882. doi: 10.1001/jamaneurol.2013.698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lynch DS, Jaunmuktane Z, Sheerin UM, et al. Hereditary leukoencephalopathy with axonal spheroids: a spectrum of phenotypes from CNS vasculitis to parkinsonism in an adult onset leukodystrophy series. J Neurol Neurosurg Psychiatry. 2016;87(5):512‐519. doi: 10.1136/jnnp-2015-310788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kinoshita M, Yoshida K, Oyanagi K, Hashimoto T, Ikeda S. Hereditary diffuse leukoencephalopathy with axonal spheroids caused by R782H mutation in CSF1R: case report. J Neurol Sci. 2012;318(1–2):115‐118. doi: 10.1016/j.jns.2012.03.012 [DOI] [PubMed] [Google Scholar]
- 24. Robinson JL, Suh E, Wood EM, et al. Common neuropathological features underlie distinct clinical presentations in three siblings with hereditary diffuse leukoencephalopathy with spheroids caused by CSF1R p.Arg782His. Acta Neuropathol Commun. 2015;3:42. doi: 10.1186/s40478-015-0219-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Foulds N, Pengelly RJ, Hammans SR, et al. Adult‐Onset Leukoencephalopathy with Axonal Spheroids and Pigmented Glia Caused by a Novel R782G Mutation in CSF1R. Sci Rep. 2015;5:10042. doi: 10.1038/srep10042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mitsui J, Matsukawa T, Ishiura H, et al. CSF1R mutations identified in three families with autosomal dominantly inherited leukoencephalopathy. Am J Med Genet. 2012;159b(8):951‐957. doi: 10.1002/ajmg.b.32100 [DOI] [PubMed] [Google Scholar]
- 27. Hoffmann S, Murrell J, Harms L, et al. Enlarging the nosological spectrum of hereditary diffuse leukoencephalopathy with axonal spheroids (HDLS). Brain Pathology (Zurich, Switzerland). 2014;24(5):452‐458. doi: 10.1111/bpa.12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. La Piana R, Webber A, Guiot MC, Del Pilar CM, Brais B. A novel mutation in the CSF1R gene causes a variable leukoencephalopathy with spheroids. Neurogenetics. 2014;15(4):289‐294. doi: 10.1007/s10048-014-0413-1 [DOI] [PubMed] [Google Scholar]
- 29. Granberg T, Hashim F, Andersen O, Sundal C, Karrenbauer VD. Hereditary diffuse leukoencephalopathy with spheroids ‐ a volumetric and radiological comparison with multiple sclerosis patients and healthy controls. Eur J Neurol. 2016;23(4):817‐822. doi: 10.1111/ene.12948 [DOI] [PubMed] [Google Scholar]
- 30. Sundal C, Fujioka S, Van Gerpen JA, et al. Parkinsonian features in hereditary diffuse leukoencephalopathy with spheroids (HDLS) and CSF1R mutations. Parkinsonism Relat Disord. 2013;19(10):869‐877. doi: 10.1016/j.parkreldis.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Inui T, Kawarai T, Fujita K, et al. A new CSF1R mutation presenting with an extensive white matter lesion mimicking primary progressive multiple sclerosis. J Neurol Sci. 2013;334(1–2):192‐195. doi: 10.1016/j.jns.2013.08.020 [DOI] [PubMed] [Google Scholar]
- 32. Comabella M, Sastre‐Garriga J, Montalban X. Precision medicine in multiple sclerosis: biomarkers for diagnosis, prognosis, and treatment response. Curr Opin Neurol. 2016;29(3):254‐262. doi: 10.1097/wco.0000000000000336 [DOI] [PubMed] [Google Scholar]
- 33. Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577‐589. doi: 10.1038/s41582-018-0058-z [DOI] [PubMed] [Google Scholar]
- 34. Poesen K, Van Damme P. Diagnostic and Prognostic Performance of Neurofilaments in ALS. Front Neurol. 2018;9:1167. doi: 10.3389/fneur.2018.01167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hayer SN, Krey I, Barro C, et al. NfL is a biomarker for adult‐onset leukoencephalopathy with axonal spheroids and pigmented glia. Neurology. 2018;91(16):755‐757. doi: 10.1212/wnl.0000000000006357 [DOI] [PubMed] [Google Scholar]
- 36. Novakova L, Axelsson M, Khademi M, et al. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing‐remitting multiple sclerosis. J Neurochem. 2017;141(2):296‐304. doi: 10.1111/jnc.13881 [DOI] [PubMed] [Google Scholar]
- 37. Williams T, Zetterberg H, Chataway J. Neurofilaments in progressive multiple sclerosis: a systematic review. J Neurol. 2021;268:3212–3222. doi: 10.1007/s00415-020-09917-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Parnetti L, Gaetani L, Eusebi P, et al. CSF and blood biomarkers for Parkinson's disease. Lancet Neurol. 2019;18(6):573‐586. doi: 10.1016/s1474-4422(19)30024-9 [DOI] [PubMed] [Google Scholar]
- 39. Gelfand JM, Greenfield AL, Barkovich M, et al. Allogeneic HSCT for adult‐onset leukoencephalopathy with spheroids and pigmented glia. Brain. 2020;143(2):503‐511. doi: 10.1093/brain/awz390 [DOI] [PubMed] [Google Scholar]
- 40. Chitu V, Biundo F, Shlager GGL, et al. Microglial Homeostasis Requires Balanced CSF‐1/CSF‐2 Receptor Signaling. Cell Rep. 2020;30(9):3004‐3019.e5. doi: 10.1016/j.celrep.2020.02.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tipton PW, Stanley ER, Chitu V, Wszolek ZK. Is Pre‐Symptomatic Immunosuppression Protective in CSF1R‐Related Leukoencephalopathy? Mov Disord. 2021;36(4):852‐856. doi: 10.1002/mds.28515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Han J, Sarlus H, Wszolek ZK, Karrenbauer VD, Harris RA. Microglial replacement therapy: a potential therapeutic strategy for incurable CSF1R‐related leukoencephalopathy. Acta Neuropathol Commun. 2020;8(1):217. doi: 10.1186/s40478-020-01093-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hillert J, Stawiarz L. The Swedish MS registry – clinical support tool and scientific resource. Acta Neurol Scand. 2015;132(199):11‐19. doi: 10.1111/ane.12425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Sundal C, Carmona S, Yhr M, et al. An AARS variant as the likely cause of Swedish type hereditary diffuse leukoencephalopathy with spheroids. Acta Neuropathol Commun. 2019;7(1):188. doi: 10.1186/s40478-019-0843-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Supplementary Material
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.