

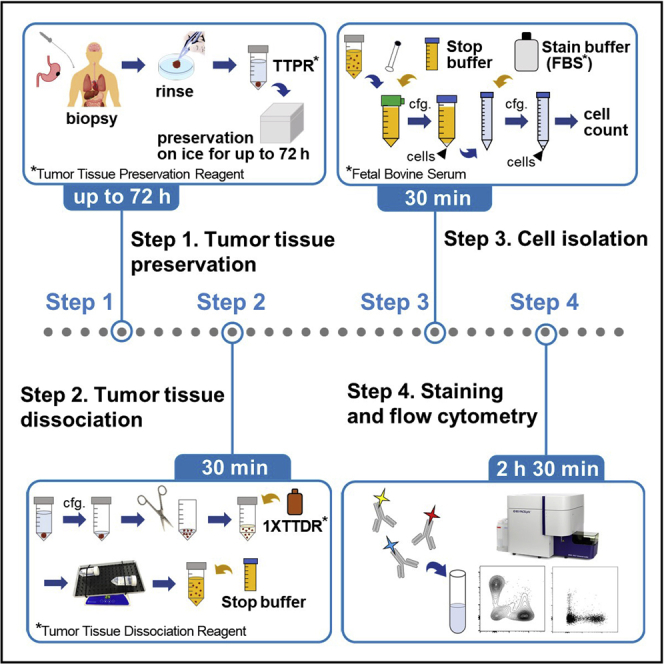
Summary
Immunophenotyping of tumor-infiltrating lymphocytes (TILs) by flow cytometry can predict clinical efficacy of immunotherapy. However, several obstacles need to be overcome for developing a flow cytometry assay starting from solid tumor specimens. Here, we show a detailed enzyme-based protocol to isolate TILs from human tumor tissues. The protocol was optimized to obtain enough viable TILs from a biopsy tissue specimen for flow cytometry-based TIL immunophenotyping. Additionally, tissue samples could be preserved for up to 72 h for subsequent characterization.
For complete details on the use and execution of this protocol, please refer to Kumagai et al. (2020, 2022).
Subject areas: Cancer, Clinical Protocol, Immunology
Graphical abstract
Highlights
-
•
Long-term preservation of live immune cells derived from tumor tissues
-
•
Optimized enzyme-based protocol for isolating immune cells from tumor tissues
-
•
Applicable for phenotypic analyses with high yields of tumor-infiltrating lymphocytes
-
•
Applicable for multiple immune monitoring in clinical settings
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Immunophenotyping of tumor-infiltrating lymphocytes (TILs) by flow cytometry can predict clinical efficacy of immunotherapy. However, several obstacles need to be overcome for developing a flow cytometry assay starting from solid tumor specimens. Here, we show a detailed enzyme-based protocol to isolate TILs from human tumor tissues. The protocol was optimized to obtain enough viable TILs from a biopsy tissue specimen for flow cytometry-based TIL immunophenotyping. Additionally, tissue samples could be preserved for up to 72 h for subsequent characterization.
Before you begin
The following protocol describes steps for isolation of cells from tumor tissue samples for flow cytometry analysis. Here, the protocol focuses on analyzing immune cells in the tumor microenvironment although cells which comprise the tumor microenvironment such as tumor cells, fibroblasts and epithelial and endothelial cells are also isolated. To preserve cell viability and activation status, tissue samples are maintained in a Tumor Tissue Preservation Reagent (TTPR) we developed. With TTPR, tissues can be preserved for up to 72 h for subsequent cell analyses. Tissue samples were subjected to an enzyme-based protocol to isolate tumor infiltrating lymphocytes (TILs) using Tumor Tissue Dissociation Reagent (TTDR), which can enzymatically dissociate tissues for single-cell suspension. The procedure was optimized for biopsy and surgical specimens of human tumor tissues as little as 5 mg. The procedure can be also used for murine tissue samples (Nishikawa, 2019). Refer to the key resources table for a complete list of reagents and tools.
All participants provided written informed consent, and this study was approved by the institutional review boards of the National Cancer Center (2015-048) and was conducted in accordance with ethics guidelines, including the Declaration of Helsinki.
Preparation of TTDR reconstitution and stop buffer
Timing: 20 min (on the day prior to cell isolation)
Refer to materials and equipment for buffer recipes.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD3 V500-C (SK7) (dilution 1:20) | BD | Cat#647454 |
| PerCP-Cy5.5 Mouse Anti-Human CD4 (SK3) (dilution 1:5) | BD | Cat#341654 |
| FITC Mouse Anti-Human CD8 (SK1) (dilution 1:5) | BD | Cat#347313 |
| PE-Cy7 Mouse Anti-Human CD45RA (L48) (dilution 1:20) | BD | Cat#337167 |
| APC Mouse Anti-Human CD152 (CTLA-4) (BNI3) (dilution 1:5) | BD | Cat#555855 |
| BV421 Mouse Anti-Human CD279 (PD-1) (MIH4) (dilution 1:20) | BD | Cat#564323 |
| PE Mouse Anti-Human FoxP3 (236A/E7) (dilution 1:20) | BD | Cat#560852 |
| FITC Mouse Anti-Human HLA-DR, DP, DQ (Tu39) (dilution 1:10) | BD | Cat#555558 |
| BV421 Mouse Anti-Human CD11b (ICRF44) (dilution 1.5:100) | BD | Cat#562632 |
| PE Mouse Anti-Human CD11c (B-ly6) (dilution 1:10) | BD | Cat#555392 |
| CD14 Monoclonal Antibody (61D3), PerCP-Cyanine5.5 (dilution 1.5:100) | Thermo Fisher Scientific | Cat#45-0149-42 |
| Brilliant Violet 605™ Anti-Human CD33 Antibody (P67.6) (dilution 1:40) | BioLegend | Cat#366612 |
| BV510 Mouse Anti-Human CD163 (GHI/61) (dilution 1:40) | BD | Cat#744921 |
| Alexa Fluor® 700 Mouse Anti-Human CD3 (UCHT1) (dilution 1.5:100) | BD | Cat#641406 |
| VENTANA® CONFIRM CD3 rabbit monoclonal (2GV6) | Roche | Cat#790-4341 |
| Biological samples | ||
| Human colorectal cancer tissue (aged 34–87, 14 males, 7 females) | National Cancer Center Hospital East | https://www.ncc.go.jp/en/ncce/index.html |
| Human gastric cancer tissue (aged 43–93, 26 males, 13 females) | National Cancer Center Hospital East | https://www.ncc.go.jp/en/ncce/index.html |
| Human non-small-cell lung cancer (NSCLC) tissue (aged 54–83, 15 males, 8 females) | National Cancer Center Hospital East | https://www.ncc.go.jp/en/ncce/index.html |
| Chemicals, peptides, and recombinant proteins | ||
| DPBS, no calcium, no magnesium | Thermo Fisher Scientific | Cat#14190136 |
| Bovine Serum Albumin (BSA) | Sigma-Aldrich | Cat#A7906 |
| UltraPure™ 0.5 M EDTA, pH 8.0 | Thermo Fisher Scientific | Cat#15575020 |
| Dimethyl Sulfoxide (DMSO) | FUJIFILM | Cat#031-24051 |
| Stain Buffer (FBS: fetal bovine serum) | BD | Cat#554656 |
| Lysing Buffer | BD | Cat#555899 |
| Otsuka Normal Saline | Otsuka | Cat#3311401A2026 |
| Transcription Factor Buffer Set (Components: Fix/Perm Buffer (4×), Diluent Buffer, Perm/Wash Buffer (5×)) | BD | Cat#562725 |
| Human BD Fc Block™ | BD | Cat#564220 |
| Brilliant Stain Buffer (BSB) Plus | BD | Cat#566385 |
| BD Horizon™ Fixable Viability Stain 780 (FVS780) | BD | Cat#565388 |
| 10% Buffered Neutral Formalin Solution | MUTO PURE CHEMICALS | Cat#20215 |
| Paraffin wax (Parabett 60 GR) | MUTO PURE CHEMICALS | Cat#43257 |
| Xylene (Xylene MS) | MUTO PURE CHEMICALS | Cat#43131 |
| Ethanol 100 | MUTO PURE CHEMICALS | Cat#43102 |
| Xylene (Xylene S) | FUJIFILM | Cat#244-00081 |
| I-VIEW DAB universal kit | Roche | Cat#518-100032 |
| non-aqueous mounting medium for microscopy | Merck | Cat#100579 |
| Critical commercial assays | ||
| BD® Tumor & Tissue Preservation Reagent (TTPR) 20 mL/tube | BD | Cat#664648 |
| BD Horizon™ Dri Tumor & Tissue Dissociation Reagent (TTDR) 15 vials/box | BD | Cat#661563 |
| Deposited data | ||
| Raw and analyzed data | This paper | n/a |
| Tumor Tissue Preservation Reagent (TTPR) | PCT/JP2020/005991 | n/a |
| Software and algorithms | ||
| FlowJoTM v10 Software | BD | https://www.bdbiosciences.com/en-us/products/software/flowjo-v10-software |
| BD FACSuiteTM Software v1.3 or higher | BD | https://www.bdbiosciences.com/en-us/products/software/instrument-software/bd-facsuite-application# |
| NDP.view2 | Hamamatsu | https://www.hamamatsu.com/us/en/product/life-science-and-medical-systems/digital-slide-scanner/U12388-01.html |
| Fiji Image J v1.53c | n/a | https://imagej.net/software/fiji/ |
| Other | ||
| EASYstrainer for 50 mL Tubes, 40 μm | Greiner | Cat#542040 |
| Terumo® Syringe | Terumo | Cat#SS-02LZ |
| Nutating Mixer | Fisher Scientific | Cat#05-450-213 |
| gentleMACS™ Dissociator | Miltenyi Biotec | Cat#130-093-235 |
| gentleMACS™ C Tubes | Miltenyi Biotec | Cat#130-093-237 |
| Multipurpose- and Micro-centrifuge | n/a | n/a |
| BD FACSLyric™ Flow Cytometry | BD | https://www.bdbiosciences.com/en-us/products/instruments/flow-cytometers/clinical-cell-analyzers/facslyric |
| BD Trucount™ Absolute Counting Tubes | BD | Cat#340334 |
| 0.22 μm filtration filter | n/a | n/a |
| Napox Scissors | Natsume | Cat#B-5 |
| TC20™ Automated Cell Counter | Bio-Rad | Cat#1450101J1 |
| Cell Counting Slides | Bio-Rad | Cat#145-0011 |
| Trypan Blue | Bio-Rad | Cat#145-0013 |
| Falcon® 5 mL Round Bottom Polystyrene Test Tube | Corning | Cat#352008 |
| Falcon® 5 mL Round Bottom Polystyrene Test Tube, with Cell Strainer Snap Cap | Corning | Cat#352235 |
| Falcon® 15 mL High Clarity PP Centrifuge Tube | Corning | Cat#352096 |
| Falcon® 50 mL High Clarity PP Centrifuge Tube | Corning | Cat#352070 |
| Eppendorf Conical Tube 25 mL | Eppendorf | Cat#0030-122-437 |
| Retoratome | Yamato | Cat#REM-710 |
| NanoZoomer® S360 | Hamamatsu | Cat#C13220-01 |
Materials and equipment
TTDR reconstitution buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 0.5% (w/v) | 0.25 g |
| DPBS | n/a | Adjust to 50 mL |
| Total | n/a | 50 mL |
Note: Adjust volume to 50 mL and sterilize using a 0.22 μm filter. Aliquot into smaller tubes if needed. Keep refrigerated at 4°C for up to 1 week, unopened.
Stop buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| BSA | 1% (w/v) | 2.5 g |
| 0.5 M EDTA | 2 mM | 1 mL |
| DPBS | n/a | Adjust to 250 mL |
| Total | n/a | 250 mL |
Note: Adjust volume to 250 mL and sterilize using a 0.22 μm filter. Aliquot into smaller tubes if needed. Keep refrigerated at 4°C for up to 1 week, unopened.
FVS780 stock solution
| Reagent | Final concentration | Amount |
|---|---|---|
| FVS780 (dry powder) | n/a | 1 vial |
| DMSO | n/a | 0.18 mL |
Note: Vortex the solution well and aliquot 2 μL into tubes. Keep refrigerated at –20°C for up to 90 days. Do not reuse once it is thawed.
Alternatives: Equipment and reagent alternatives.
-
•
Nutating Mixer (Fisher Scientific Cat#05-450-213) can be replaced by an equivalent product with gentle three-dimensional shaking motion.
-
•
TC20™ Automated Cell Counter (Bio-Rad Cat#1450101J1) and its accessories (Cell Counting Slides and Trypan Blue) can be replaced by any other cell counters with accessories.
-
•
The BD FACSLyric™ Flow Cytometer was used to perform flow cytometry analysis. Any other flow cytometer with similar performance characteristics can be used for cell analysis.
-
•
In the key resources table, the listed antibodies for cell staining were used to validate this protocol. Any other staining panels of interest can be used.
Step-by-step method details
Tumor tissue preservation
This step describes our approach to preserve tissue specimens including human tumor tissues for up to 72 h before the dissociation step. This method for tissue preservation overcomes the limitation of specimen transportation time and enables wider application of the TIL analysis method.
-
1.Tumor tissue preservation.
-
a.Gently rinse the fresh specimen with saline to remove blood from tissue and place into a TTPR tube.
-
b.Make sure the sample is completely immersed in the TTPR solution, and then close the cap tightly.
-
c.Place the TTPR tube with specimen in a styrofoam box filled with crushed ice and store the box at 2°C–8°C for up to 72 h.
-
a.
Note: Rinsing step with saline can be skipped unless an obvious blood contamination is seen. If using surgically resected samples instead of biopsy, the tissue samples should be cut into small pieces of biopsy size (approximately 5 mm3 as shown in Figure 8) before placing sample into TTPR for preservation. Up to 100 mg of tumor tissue can be preserved in a TTPR tube (20 mL).
Note: A styrofoam box or the substitute should be filled with as much ice as possible, and the lid should be sealed with plastic tape (Figure 1). Size of the box should be large enough to keep ice during the storage time.
Figure 8.

The number of viable cells based on tumor tissue size
(A) Surgically resected tumor tissues of NSCLC (n=13) and endoscopic biopsy specimens of gastric cancer (n=39) were prepared. Tumor tissue specimens were cut and divided into 5, 10, 20, and 30 mg pieces. Due to the limited amount of each specimen, 5, 10, 20, and 30 mg in NSCLC were obtained from 9, 10, 10, and 7 out of 13 specimens, respectively. And 5, 10, and 20 mg in gastric were obtained from 20, 26, and 12 out of 39 specimens, respectively. Horizontal lines indicate means. The decreased number of CD8+ T cells (at 20 mg) in gastric cancer samples was partly due to the low immunogenicity and high heterogeneity in gastric cancer.
(B) Representative image (5 mg) of NSCLC tissue is shown. One-way ANOVA was used (∗p < 0.05).
Figure 1.

Box preparation for TTPR preservation
(A) TTPR tubes are placed in a styrofoam box filled with ice.
(B) The lid is sealed with plastic tape.
Tumor tissue dissociation and isolation
This step describes the approach to dissociate tumor tissue to obtain single-cell suspensions. The expected outcome is to extract a sufficient number of various types of immune cells although other types of cells which comprise the tumor microenvironment including tumor cells, fibroblasts, and epithelial and endothelial cells are also isolated simultaneously. We show data of isolated TILs with this method in comparison with the conventional mechanical dissociation method, which was reported previously (Sugiyama et al., 2013; Saito et al., 2016). The cell number and the immune phenotype of cells isolated from multiple types of tumors, various tissue sizes and different preservation time are presented.
Note: All reagents are stored at 4°C or on ice unless otherwise specified. Sample incubations and centrifugation are performed at 4°C or on ice unless otherwise specified.
-
2.Tumor tissue dissociation.
-
a.Prepare 1× TTDR by adding 5 mL of TTDR reconstitution buffer at 20°C–25°C to each TTDR vial and incubate for 5–10 min at 20°C–25°C. Mix well by pipetting and transfer reconstituted TTDR to a 15 mL conical tube containing additional 5 mL of TTDR reconstitution buffer. Keep it aside at 20°C–25°C while preparing the sample. Do not leave the reconstituted TTDR at 20°C–25°C longer than 30 min prior to use.Note: Make sure that dried reagent at the bottom of the vial is completely reconstituted.
-
b.Centrifuge the TTPR tube containing the tissue at 300 × g for 2 min at 4°C. Troubleshooting 1.
-
c.Aspirate the supernatant down to approximately 0.3 mL in the tube.
-
d.Cut tissue into small pieces (around 0.5–1.0 mm per side) using scissors (Figure 2).
 CRITICAL: Excessive cutting with scissors may increase cell death. Cutting time should be usually about 20–40 s and no longer than 90 s when the scissor stroke is about 100 times per 60 s. In the case of hard tissues and/or large tissue size, place tube on ice for about 1 min to cool down and resume cutting.
CRITICAL: Excessive cutting with scissors may increase cell death. Cutting time should be usually about 20–40 s and no longer than 90 s when the scissor stroke is about 100 times per 60 s. In the case of hard tissues and/or large tissue size, place tube on ice for about 1 min to cool down and resume cutting. -
e.Place the tube at 20°C–25°C and add 5–10 mL of 1× TTDR at 20°C–25°C into the tube, rinsing the scissors while adding it.Note: We recommend 5 mL of 1× TTDR for up to 50 mg and 10 mL for up to 100 mg. Insufficient volume of 1× TTDR may cause poor cell recovery.
-
f.Close the cap tightly and incubate at 20°C–25°C for 15 min on the nutator. Gently tilt and check every 5 min that the tissue pieces are not stuck to the walls of the tube without touching the solution.
-
g.During the incubation, place a 50 mL conical tube with a 40 μm cell strainer directly on ice and add 10 mL cold stop buffer to equilibrate the filter membrane.CRITICAL: Incubation time with TTDR should not exceed 20 min.Note: The use of a 40 μm cell strainer is recommended to eliminate cell aggregates and/or tissue debris as much as possible, as the purpose of this protocol is mainly to isolate immune cells for flow cytometry analysis. Please choose a larger pore size strainer if you need to isolate larger cells as well.
-
h.After the incubation, place the incubation tube at 20°C–25°C and immediately add 5 mL cold stop buffer to the tube and the cap.
-
a.
-
3.Cell isolation.
-
a.Immediately decant the solution in the cap to the tube, and then transfer the solution into the strainer.Note: Pour the solution into the strainer slowly while avoiding overflowing (Figure 3). Exercise caution with mucosal samples (e.g., colorectal tissues) since they may be stickier and often disrupt the flow of the solution from the strainer.
-
b.Add an additional 10 mL cold stop buffer in the TTDR incubation tube for rinsing and decant the solution into the strainer.
-
c.Lightly grind the pieces of tissue on the strainer using the top of the syringe plunger for 20–30 s (Figure 4).
-
d.Add an additional 8 mL cold stop buffer in the strainer, rinsing the tip of the plunger.
-
e.Centrifuge the tube with the strainer at 400 × g for 2 min at 4°C to drop the remaining solution into the tube.
-
f.Remove the strainer and close the cap tightly.
-
g.Mix by inverting of the tube 10 times.CRITICAL: The mixing step is indispensable to achieve optimal cell recovery.
-
h.Centrifuge at 400 × g for 12 min at 4°C.
-
i.Aspirate the supernatant cautiously, leaving approximately 5 mL in the tube. Troubleshooting 2.
-
j.Resuspend the pellet and transfer to a 15 mL conical tube.
-
k.Rinse the original tube with 4–5 mL of Stain Buffer (FBS) and transfer to the 15 mL tube. Repeat this step; total volume is 13–15 mL.
-
l.Close the cap tightly and invert the tube at least twice.
-
m.Centrifuge at 400 × g for 5 min at 4°C.
-
n.Aspirate the supernatant cautiously, leaving approximately 100 μL in the tube.
-
o.Resuspend the pellet in a total of 2–5 mL Stain Buffer (FBS), and pipette appropriate volume of the cell suspension for cell count.Note: We recommend hemolysis of cell counting portion if the suspension looks reddish or pinkish. Up to 5 × 106 viable cells can be stained in a tube in this protocol. Optimization of antibodies and reagents are required for your staining panel.
-
p.Centrifuge at 400 × g for 5 min at 4°C.
-
q.Aspirate the supernatant cautiously, leaving 100 μL in the tube for cell staining. Pipette the cell suspension carefully and transfer the cell suspension to a 5 mL round bottom tube. The volume can be adjusted depending on the assay needs.Note: Leaving 50–100 μL in the tube is recommended for cell surface staining. Different types of tubes can be used although a 5 mL round bottom tube is recommended here. For isotype control staining, cell suspension has to be divided into two tubes.
-
a.
Figure 2.

Tissue for the treatment with TTDR
Tissue specimens were cut into small pieces.
(A and B) Before (A) and after (B) cutting tissue.
Figure 3.

Preparation of single-cell suspensions
The solution poured in strainer was filtered by gravity flow.
Figure 4.

Before and after grinding of tissues with strainer
(A and B) Before (A) and after (B) grinding tissue specimens by the tip of the plunger.
Cell staining for flow cytometry analysis
In this part, we describe the staining protocol for TILs isolated in the previous step. The protocol is optimized for flow cytometry analysis. The panel shown here is an example. You may set your own staining panel according to your study design.
Note: All steps including centrifugation are performed at 4°C or on ice unless otherwise specified. Reagents are kept at 4°C or on ice unless otherwise specified. All incubation steps should be performed at 4°C or on ice and protected from light. We recommend using a steel tube stand for incubation on ice as it can cool faster and keep stable temperature during the incubation (Figure 5).
-
4.Staining for dead cell exclusion and blocking.
-
a.Prepare FVS780 working solution by adding 28 μL DPBS(-) to a 2 μL FVS780 stock solution (dilute it 1:15).Note: Discard the remaining FVS780 stock solution once thawed.
-
b.Add 3 μL of FVS780 working solution to 100 μL TIL suspension, mix thoroughly by pipetting, and incubate for 10 min, protected from light.
-
c.Add 5 μL of human Fc Block to the tube, mix thoroughly by pipetting, and incubate for 10 min, protected from light.
-
d.During the incubation, prepare the following mixture for staining cell surface molecules.
-
i.Add 10 μL of Brilliant Stain Buffer (BSB) Plus to a 1.5 mL microtube.
-
ii.Add antibodies one by one in the tube containing BSB Plus. Lightly vortex the tube (∼ 1 s) each time an antibody is added. Spin the tube down when needed.
-
iii.Vortex lightly and spin tube down after the addition of the last antibody. Keep tube on ice or at 4°C, protected from light.Mixture for staining cell surface molecules (TIL panel)
Reagent or marker (clone) Fluorophore Final dilution Volume (for 100 μL cell suspension) Brilliant Stain Buffer (BSB) Plus 1:10 10 μL CD3 (SK7) V500-C 1:20 5 μL CD4 (SK3) PerCP-Cy5.5 1:5 20 μL CD8 (SK1) FITC 1:5 20 μL CD45RA (L48) PE-Cy7 1:20 5 μL CD279/PD-1 (MIH4) BV421 1:20 5 μL CD152/CTLA-4 (BNI3) APC 1:5 20 μL Mixture for staining cell surface molecules (Myeloid panel)Reagent or marker (clone) Fluorophore Final dilution Volume (for 100 μL cell suspension) Brilliant Stain Buffer (BSB) Plus 1:10 10 μL HLA-DR, DP, DQ (Tu39) FITC 1:10 10 μL CD11b (ICRF44) BV421 1.5:100 1.5 μL CD11c (B-ly6) PE 1:10 10 μL CD14 (61D3) PerCP-Cy5.5 1.5:100 1.5 μL CD33 (P67.6) BV605 1:40 2.5 μL CD163 (GHI/61) BV510 1:40 2.5 μL CD3 (UCHT1) AF700 1.5:100 1.5 μL
-
i.
-
a.
-
5.Staining of cell surface markers.
-
a.After Fc block incubation (step 4c), add antibody mixture to the tube and mix thoroughly by pipetting 10 times, and incubate for 30 min, protected from light.
-
b.Add 3 mL of Stain Buffer (FBS) to the tube and pipette it several times gently.
-
c.Centrifuge at 400 × g for 5 min at 4°C.
-
d.Aspirate supernatant carefully down to approximately 100 μL in the tube.
-
e.Repeat step b and c.
-
f.Aspirate supernatant, leaving 100 μL in the tube.
-
a.
-
6.Intracellular staining.
-
a.Add 1 mL of 1× Fix/Perm Buffer and mix gently by pipetting several times.
-
b.Incubate for 50–60 min, protected from light.
-
c.Add 3 mL of 1× Perm/Wash Buffer and mix gently by pipetting several times.Note: Use cold ddH2O when preparing 1× Perm/Wash Buffer.
-
d.Centrifuge at 800 × g for 5 min at 4°C.
-
e.Aspirate supernatant carefully down to approximately 100 μL in the tube.
-
f.Repeat step c and d.
-
g.Aspirate supernatant, leaving 100 μL in the tube.
-
h.Add anti-human FoxP3 antibody (5 μL) and mix well by pipetting several times.
-
i.Incubate for 30 min, protected from light.
-
j.Add 3 mL of 1× Perm/Wash Buffer vigorously to mix.
-
k.Centrifuge at 800 × g for 5 min at 4°C.
-
l.Aspirate supernatant carefully down to approximately 100 μL in the tube.
-
m.Add 3 mL Stain Buffer (FBS) and mix gently by pipetting several times.
-
n.Centrifuge at 800 × g for 5 min at 4°C.
-
o.Aspirate supernatant carefully, leaving approximately 100 μL in the tube.Note: Addition and incubation of anti-human FoxP3 antibody (steps 6h–6l) can be skipped for the myeloid panel.
-
a.
-
7.Flow cytometry analysis.
-
a.Resuspend cell pellets in an appropriate volume of Stain Buffer (FBS), usually in 0.5 mL, and transfer it into a 5 mL round bottom tube with cell strainer snap cap.
-
b.Centrifuge at 400 × g for around 30 s at 4°C to drop the solution in the strainer.
-
c.Set the tube on the flow cytometer after pipetting the sample in the tube.
- d.
-
a.
Note: Keep stained samples on ice or at 2°C–8°C protected from light until acquisition. Data acquisition should be executed within 2 h of staining step.
Figure 5.

Sample tubes on ice during the incubation
Sample tubes were kept on a steel tube stand.
Expected outcomes
We expect to obtain a sufficient number of TILs for immunophenotyping by flow cytometry from tissue specimens. This protocol allows for up to 72 h of preservation of tissue specimens for subsequent isolation of immune cells.
There is no impact on the frequency and immune phenotype of different subpopulations as demonstrated by analyses of mean fluorescence intensity (MFI) and % positive parameters of each cell population in both dissociation methods (Figure 6).
Figure 6.

The number and frequency of each immune cell population prepared by TTDR and mechanical dissociation methods
(A) Representative dot plots and gating for each T cell population are shown. Fresh NSCLC tissues (15 mg for each) were used for the isolation of immune cells by TTDR and mechanical dissociation (MD) and subjected to flow cytometry using BD Trucount™ Absolute Counting Tubes.
(B) Summaries for the number and frequency of each T cell population (NSCLC: n=3, colorectal: n=10). Cell number per 10 mg tumor tissue is shown here. One-way ANOVA was used (∗p < 0.05).
To validate the isolation method with TTDR, we also examined the number of CD3-positive cells with immunohistochemistry (IHC) using the same tumor tissues (n=6). The tumor tissues were divided into two pieces for IHC and flow cytometry analysis. The number of CD3-positive cells analyzed with flow cytometry using isolated cells with this protocol correlates with the IHC results (Figure 7).
Figure 7.

Confirmation of flow cytometry data with IHC
(A and B) Tumor tissues from six patients with NSCLC or colorectal cancer were cut into two pieces and divided for flow cytometry and IHC analyses. One piece of tumor tissue was examined with flow cytometry. The second piece was analyzed with IHC with anti-CD3 (2GV6) rabbit mAb by DAB-HRP method. All images were acquired using the NanoZoomer S360 and analyzed with Image J to count CD3-positive cells. (A) A correlation plot with linear regression in the number of CD3-positive cells between flow cytometry and IHC. For flow cytometry, 10 mg of each tumor tissue was examined. For IHC, 100 mm2 area of each image was examined. In this analysis, the live-dead discrimination was skipped in flow cytometry in accordance with IHC analysis. (B) Representative IHC images of the a), b) and c) points in (A) are shown. Scale bar; 500 μm and 100 μm for 100× (left) and 500× (right) magnification, respectively.
We examined the number of viable immune cells isolated from different sizes (mg) of tumor tissues (Figure 8A). Tumor tissues were cut and divided into 5, 10, 20, and 30 mg pieces in TTPR. They were treated according to this protocol and analyzed with flow cytometry. The recovery of each immune cell population correlated with the size of tumor tissues.
We examined the impact of tissue preservation on the cell number and frequency of immune cells that were isolated from tumor tissues preserved in TTPR at different time points for up to 72 h. Colorectal and lung tumor tissues were used for this experiment (Figures 9A and 9B). In addition, myeloid cell populations (Filippo et al., 2018) were also assessed (Figures 9C and 9D). We did not observe any significant changes in the cell number and frequency of immune cells during the preservation.
Figure 9.

Flow cytometry analysis of tumor tissues preserved at different time points in TTPR
(A–D) Tumor tissues from colorectal cancer (n=10) and NSCLC (n=8) were cut into small pieces (around 5 mg) and were equally divided for three conditions [Fresh(F), TTPR preservation, 48 or 72 h]. Each sample consists of 2–6 pieces (10–30 mg in total). Fresh; treated within 1–2 h after collection, 48 h; treated after 43–50 h preservation, 72 h; treated after 70–76 h preservation.
(A) Representative dot plots and gating for each T cell population in colorectal cancer (F, 48 and 72 h) are shown.
(B) The changes of each cell population in the number and frequency are shown. The relative changes compared to fresh samples are calculated.
(C) Representative dot plots and gating for myeloid cell population in colorectal cancer (F, 48 and 72 h) are shown.
(D) The changes of each cell population in the number and frequency are shown. The relative changes compared to fresh samples are calculated. Data in (B, D) are shown as mean ± SD. One-way ANOVA was used. ns: not significant.
Limitations
This protocol has been optimized for the isolation and characterization of immune cells from multiple types of human tumor tissues. These procedures can be also applied to isolate immune cells from murine tumor tissues. The number of isolated viable cells depends on the tumor microenvironment such as immunogenic vs non-immunogenic tumors and may be impacted by tumor heterogeneity. If specimens contain necrotic areas, cell recovery may be reduced.
Troubleshooting
Problem 1
Specimens preserved in TTPR solution apparently look red. This is most likely a contamination with whole blood (step 2b).
Potential solution
To exclude lymphocytes derived from peripheral blood as much as possible, carefully transfer the specimen to a new TTPR tube or a 25 mL conical tube (Eppendorf Cat#0030-122-437) containing 20 mL cold DPBS(-) with tweezers.
Problem 2
Before removing the supernatant, you may find some floating matters in the solution above the 5 mL line in the tube (Figure 10) (step 3i).
Figure 10.

Floating matters in the centrifuged tube
The red arrows indicate floating matters in the tube.
Potential solution
To increase cell yield, decant the supernatant to a new 50 mL tube, mix it by inversion several times, and centrifuge at 400 × g for 10 min at 4°C. After centrifugation, aspirate the supernatant and mix the cell pellet in the original 50 mL tube. This may occur in mucous membrane rich tissues such as colorectal tumor specimens.
Problem 3
The number of isolated immune cells is low (step 7d).
Potential solution
This problem is most likely due to low levels of infiltrating immune cells in the tumor specimen. Several steps in the sample preparation protocols are critical to optimize TIL recovery from the sample: 1) tumor cutting: make sure tumor tissue is cut into appropriate size (Figure 2) (step 2d), 2) tumor dissociation in TTDR: it is possible to increase the volume of 1× TTDR up to 10 mL to ensure efficient dissociation (step 2e), 3) sample incubation in TTDR: ensure that sample is submerged in TTDR in the incubation tube during the nutation (step 2f), or 4) ensure that cell pellet is maintained during wash and centrifugation steps (steps 6d–6o) after Fix/Perm Buffer treatment.
Problem 4
Low viability of isolated immune cells (step 7d).
Potential solution
This problem may represent the state of the tumor microenvironment. If specimen contains necrotic areas, viable cell recovery may be impaired as previously mentioned. To avoid cell damage during the execution of sample protocol, several steps may affect cell viability; 1) cut large tumor specimen into appropriate size pieces before immersing into TTPR (Figures 8B), 2) make sure specimen in TTPR tube is always kept on ice during the preservation period (step 1), 3) avoid excessive cutting with scissors (step 2d), or 4) execute Fix/Perm Buffer treatment immediately after aspirating supernatant (steps 5f–6a).
Problem 5
Weak or no fluorescent signal is detected (step 7d).
Potential solution
This problem could have several reasons including sample staining and flow cytometry instrument settings. We recommend titration of each conjugate including negative control to determine final concentration in the panel and development of optimal photomultiplier tubes (PMT) assay settings for flow cytometry instrument. Follow practices recommended in H62 guideline (Clinical and Laboratory Standards Institute, 2021).
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Hiroyoshi Nishikawa (hnishika@ncc.go.jp).
Materials availability
Tumor Tissue Preservation Reagent (TTPR) is available for purchase from BD as a custom product. Please contact Customer Service to order, catalog number is provided in the key resources table.
Acknowledgments
This study was supported by Grants-in-Aid for Scientific Research [S grant no. 17H06162 (H.N.), Challenging Exploratory Research Grant no. 16K15551 (H.N.)] from the Ministry of Education, Culture, Sports, Science and Technology of Japan; by Projects for Cancer Research by Therapeutic Evolution [P-CREATE, no. 16cm0106301h0001 (H.N.)], by Development of Technology for Patient Stratification Biomarker Discovery grant [no. 19ae0101074s0401 (H.N.)] from Japan Agency for Medical Research and Development (AMED); by National Cancer Center Research and Development Fund [no. 28-A-7 and 31-A-7 (H.N.)]. This study was executed in part as a research program supported by BD Japan.
Author contributions
T.K. and H.N. designed the research. T.K., R.D., E.A., and H.N. performed the experiments and analyzed the data. S. Koyama, S. Kumagai, and H.N. collected the clinical specimens and conceived the project. T.K., E.A., and H.N. wrote the manuscript.
Declaration of interests
H.N. received research funding from BD Japan for this study and received research funding and honoraria from Ono Pharmaceutical, Chugai Pharmaceutical, Bristol-Myers Squibb, and MSD, and research funding from Taiho Pharmaceutical, Daiichi-Sankyo, Kyowa Kirin, Zenyaku Kogyo, Oncolys BioPharma, Debiopharma, Asahi-Kasei, Sysmex, FUJIFILM, SRL, Eisai, Astellas Pharmaceutical, and Sumitomo Dainippon Pharma outside of this study. H.N. is the primary inventor on pending patents PCT/JP2020/0059919 belonging to the National Cancer Center Japan and BD Biosciences. S. Koyama received honoraria from Bristol-Myers Squibb, Ono Pharmaceutical, Chugai Pharmaceutical, and MSD, and received research funding outside the scope of this work from Bristol-Myers Squibb, Ono Pharmaceutical, and Otsuka Pharmaceutical. S. Kumagai declares no competing interests. T.K., R.D., and E.A. are employees of Becton, Dickinson and Company, which is in the business of selling flow cytometers and flow cytometry reagents.
Contributor Information
Tamiyo Kobayashi, Email: tamiyo.kobayashi@bd.com.
Hiroyoshi Nishikawa, Email: hnishika@ncc.go.jp.
Data and code availability
This study did not generate/analyze datasets/code.
References
- Clinical and Laboratory Standards Institute . First Edition. 2021. H62 Validation of Assays Performed by Flow Cytometry. [Google Scholar]
- Kumagai S., Togashi Y., Kamada T., Sugiyama E., Nishinakamura H., Takeuchi Y., Vitaly K., Itahashi K., Maeda Y., Matsui S., et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 2020;21:1346–1358. doi: 10.1038/s41590-020-0769-3. [DOI] [PubMed] [Google Scholar]
- Kumagai S., Koyama S., Itahashi K., Tanegashima T., Lin Y., Togashi Y., Kamada T., Irie T., Okumura G., Kono H., et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer Cell. 2022;40:201–218.e9. doi: 10.1016/j.ccell.2022.01.001. [DOI] [PubMed] [Google Scholar]
- Nishikawa H. BD Life Science; 2019. Isolation of tumor infiltrating lymphocytes (TIL) using TTDR.https://www.bdbiosciences.com/content/dam/bdb/marketing-documents/Isolation_of_TILs.pdf [Google Scholar]
- Saito T., Nishikawa H., Wada H., Nagano Y., Sugiyama D., Atarashi K., Maeda Y., Hamaguchi M., Ohkura N., Sato E., et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat. Med. 2016;22:679–684. doi: 10.1038/nm.4086. [DOI] [PubMed] [Google Scholar]
- Sugiyama D., Nishikawa H., Maeda Y., Nishioka M., Tanemura A., Katayama I., Ezoe S., Kanakura Y., Sato E., Fukumori Y., et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA. 2013;110:17945–17950. doi: 10.1073/pnas.1316796110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippo V., Perego M., Gabrilovich D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018;19:108–119. doi: 10.1038/s41590-017-0022-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate/analyze datasets/code.