

Summary
Identification of protein interactors is fundamental to understanding their functions. Here, we describe a modified protocol for tandem affinity purification coupled with mass spectrometry (TAP/MS), which includes two-step purification. We detail the S-, 2×FLAG-, and Streptavidin-Binding Peptide (SBP)- tandem tags (SFB-tag) system for protein purification. This protocol can be used to identify protein interactors and establish a high-confidence protein–protein interaction network based on computational models. This is particularly useful for identifying bona fide interacting proteins for subsequent functional studies.
For complete details on the use and execution of this protocol, please refer to Bian et al. (2021).
Subject areas: Bioinformatics, Molecular Biology, Protein Biochemistry, Proteomics, Mass Spectrometry
Graphical abstract
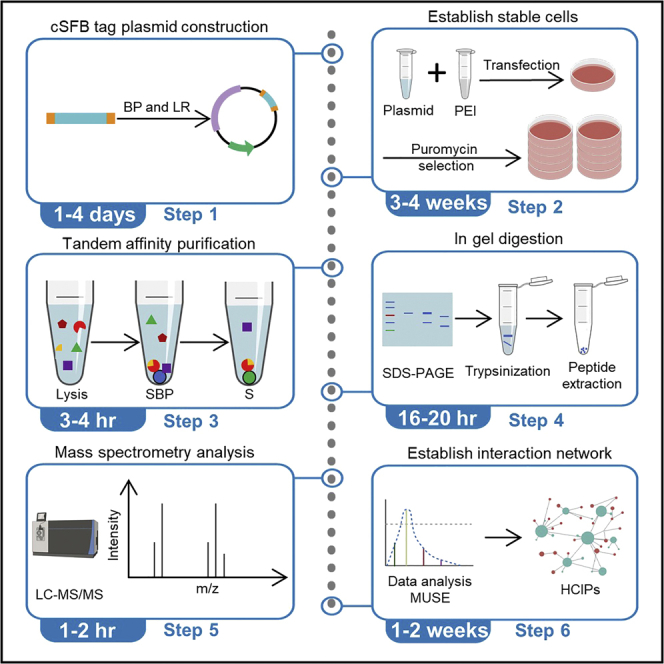
Highlights
-
•
An optimized protocol for establishing SFB-tag-based TAP/MS system
-
•
Streptavidin-biotin purification system enables denaturing washing conditions
-
•
Tandem-affinity purification to acquire specific interactors
-
•
MS data and bioinformatics analysis to establish a protein–protein interaction network
Publisher’s note: Undertaking any experimental protocol requires adherence to local institutional guidelines for laboratory safety and ethics.
Identification of protein interactors is fundamental to understanding their functions. Here, we describe a modified protocol for tandem affinity purification coupled with mass spectrometry (TAP/MS), which includes two-step purification. We detail the S-, 2×FLAG-, and Streptavidin-Binding Peptide (SBP)- tandem tags (SFB-tag) system for protein purification. This protocol can be used to identify protein interactors and establish a high-confidence protein–protein interaction network based on computational models. This is particularly useful for identifying bona fide interacting proteins for subsequent functional studies.
Before you begin
Affinity purification coupled with mass spectrometry (AP/MS) is a powerful tool in systematically identifying protein–protein interactions (Aebersold and Mann, 2003). However, significant efforts have been wasted on repeatedly identifying the same abundant peptides or peptides from “sticky proteins” that bind to many baits in AP/MS analysis (Listgarten and Emili, 2005). Tandem affinity purification coupled with mass spectrometry (TAP/MS) (Puig et al., 2001; Rigaut et al., 1999), which eliminates many nonspecific binding interactions during the two-step purification process, could be more suitable for identifying bona fide interactions for follow-up in-depth functional studies (Dunham et al., 2012; Li, 2011). In 2007, we first used S-, 2×FLAG-, and Streptavidin-Binding Peptide (SBP)- tandem tags (SFB-tag) as TAP labels to identify protein interactions (Kim et al., 2007a, 2007b) (Figure 1E). The FLAG-tag in this system is mainly used to detect protein expression by western blotting. The SBP-tag has been proven to have the advantages of high yield and high purity in one-step protein purification (Keefe et al., 2001). The S-tag has the characteristics of a small tag (15 amino acids), antibody-like ligand binding specificity, and high-capacity matrices (Kim and Raines, 1993). The SFB-tag-based TAP/MS system can be used in most mammalian cells to study protein–protein interactions under physiological or pathological conditions, helping researchers understand the molecular basis for diseases and therefore shedding light on disease prevention, diagnosis, and treatment (Figure 1E). In the past decade, many laboratories have successfully used this technique to discover the connections between important proteins and have made outstanding achievements in the studies of DNA damage and cancer signaling (Bian et al., 2021; Hussain et al., 2018; Kim et al., 2007a, 2008, 2021; Li et al., 2015; Liu et al., 2022; Wang et al., 2021; Yuan et al., 2012; Zhang et al., 2014, 2018; Zhou et al., 2020).
Figure 1.

Map of plasmids used in this protocol
(A and B) pDONR201 (A) and pDEST-cSFB (B) are shown.
We summarized the main difference of different AP/MS approaches, as well as their strengths and limitations in Table 1. Comparing with other AP/MS techniques, our method does not require additional enzyme digestion or protein recovery with high efficacy and specificity and has high-capacity matrices. The affinity tags are small, which may not affect protein folding and interactions. The elution conditions for biotin are mild and do not cause protein denaturation or require optimization.
Table 1.
Summary of comparison between various AP/MS approaches
| Type | Tag | Length | Binding matrix | Elution | Strengths | Limitations |
|---|---|---|---|---|---|---|
| Antibody immunoprecipitation | Endogenous protein (Bontinck et al., 2018) | N/A | Antibodies recognizing specific proteins | Low pH | Recover endogenous protein complex; no interference of tag. | Antibody cross-reactivity is random; limited by antibody availability; optimization of purification required. |
| FLAG, Myc, HA (Brizzard et al., 1994; Evan et al., 1985; Field et al., 1988; Hopp et al., 1988) | 8–10 aa | Antibodies recognizing epitope tags | Peptide or low pH | Small size tags cause minimal impacts to protein folding/functions; can use generic antibodies and protocols. | Binding matrix is not very stable; relatively high background. | |
| One-step AP | High molecular weight tags, such as MBP, GST (Kellermann and Ferenci, 1982; Smith and Johnson, 1988) | 346 aa, 218 aa |
MBP beads, glutathione beads | Maltose, reduced glutathione | Enhance protein solubility and shield toxic proteins. | Large tags may affect protein structure and function |
| His tag (Janknecht et al., 1991) | 6 aa | Ni2+–NTA | Imidazole | Small tags cause minimal impacts to protein folding/functions; using generic antibodies and protocols; high yield of bait proteins. | Relatively high background; not suitable for EDTA, DTT environment. | |
| Staphylococcus protein A (ProtA) (Rigaut et al., 1999), Streptococcus protein G (ProtG) (Burckstummer et al., 2006) | 58 aa, 60 aa |
IgG | TEV cleavage | Tolerate harsh washes. | The TEV-protease cleavage causes significant loss of yield. | |
| SBP (Keefe et al., 2001) | 38 aa | Streptavidin | Biotin | Purified with high yield and purity; tolerate harsh washes; high elution efficiency. | Cross-reactivity with endogenous biotinylated proteins. | |
| Proximity-based labeling | BioID (Roux et al., 2012) | 310 aa | Streptavidin | Biotin | Nontoxic labeling conditions and extensively applied across many studies. | Poor temporal resolution and limited application in vivo due to the low catalytic activity. |
| APEX/APEX2 (Lam et al., 2015; Rhee et al., 2013) | 250 aa | Streptavidin | Biotin | High temporal resolution and high activity in most cellular compartments. | Limited application in vivo due to H2O2 toxicity and low BP permeability. | |
| TurboID (Branon et al., 2018; Cho et al., 2020) | 319 aa | Streptavidin | Biotin | Highest activity promiscuous biotin ligase. | Labeling window is narrow due to the high biotin affinity; potential toxicity in long-term experiments | |
| TAP | Original TAP (ProtA and CBP) (Janknecht et al., 1991) | ∼100 aa | IgG and calmodulin | TEV cleavage, EGTA | The protein purity is greatly improved compared with that of one-step AP; relatively high recovery efficiency. | TEV-protease cleavage causes significant loss of yield; heavy cross-reactivity of calmodulin. |
| GS TAP (ProtG and SBP) (Burckstummer et al., 2006) | ∼100 aa | IgG and streptavidin | TEV cleavage, biotin | Marked improvement in protein yield compared with the original TAP system. | TEV-protease cleavage causes significant loss of the yield; IgG binding recovers less than 40% of the bait. |
|
| SFB-TAP (S, 2×FLAG, SBP) (Kim et al., 2007a, 2007b) | 84 aa | Streptavidin and S protein beads | Biotin | Does not require additional enzyme digestion; mild washing condition; high elution efficiency; high yield. | May lose weakly interacting proteins. |
The main difference of different AP/MS approaches, as well as their strengths and limitations are listed in detail.
This protocol describes the use of TAP followed by MS, using cell lines stably expressing proteins of interest (“baits”) to identify the interacting proteins (“preys”) with high confidence. For each bait protein, at least two biological repeats were required. We outline the steps to prepare plasmids encoding C-terminal S protein tag-2 × FLAG tag-SBP tag (cSFB)-tagged bait proteins, establish HEK293T cells stably expressing the plasmids, and perform TAP and MS identification. However, we also applied similar protocols to other cell lines, such as HepG2 and Sh-SY5Y cells, with relatively high transfection efficiency. For cells with low plasmid transfection efficiency, such as MCF10A, JURKAT and CEM cells, this protocol can be modified at the plasmid preparation step by using a lentiviral vector containing the SFB tag instead.
Prepare plasmid of cSFB-tagged protein of interest
Timing: 1 week
This step mainly focuses on the preparation of plasmid.
-
1.cSFB-tagged plasmid construction.Note: Both N- and C-terminal SFB tags are available for use, and the choice can be made based on validation of correct bait protein localization.Note: The signal peptide responsible for protein subcellular localization is usually located at the N- or C- terminal of the protein. When the purification tags are close to the signal peptide, it may interfere with the correct subcellular localization of the bait protein. Incorrect subcellular localization may result in the loss of natural complexes of bait and interacting proteins.
-
a.Amplifying the gene from cDNA by Phusion DNA polymerase. For the Gateway cloning system, the attB1 and attB2 homologous sequences are included in the forward and reverse primers, respectively (Figure 1A).
-
i.The PCR reaction system is as follows:
Reagent Final concentration Amount 5× Phusion HF or GC Buffer 1× 10 μL 10 mM dNTPs 200 μM 1 μL 10 μM Forward Primer 0.5 μM 2.5 μL 10 μM Reverse Primer 0.5 μM 2.5 μL Template DNA < 500 ng variable DMSO (optional) 3% (1.5 μL) Phusion DNA Polymerase 1.0 units/50 μL PCR 0.5 μL Nuclease-free water n/a Up to 50 μL Total n/a 50 μL -
ii.PCR conditions:
PCR cycling conditions
Step Temperature Time Cycles Initial Denaturation 98°C 1 min 1 Denaturation 98°C 10 s 25–35 cycles Annealing 50°C–65°C 20 s Extension 72°C 30 s per kb Final extension 72°C 10 min 1 Hold 4°C forever -
iii.Extract PCR products from the DNA gel with the FastPure® Gel DNA Extraction Mini Kit.
-
i.
-
b.Set BP reaction for entry plasmid.
-
i.The BP reaction system is as follows:
Component Amount BP Clonase™ II enzyme mix 2 μL attB-PCR product (≥10 ng/μL; final amount ∼15–150 ng) 1–7 μL pDonor201 vector (150 ng/μL) 1 μL TE buffer, pH 8.0 to 10 μL -
ii.Perform BP reaction at 25°C for 1 h.Optional: The BP reaction can be performed for 12 h, as this can improve BP reaction efficiency.
-
iii.Add 1 μL of the Proteinase K solution to each sample to terminate the reaction. Vortex briefly. Incubate samples at 37°C for 10 min.
-
iv.Transform the BP reaction product into DH5α competent cells, and select the positive clones on Kanamycin LB plates. Extract entry plasmid containing the gene of interest and confirmed by sequencing. The sequence information of sequencing primers is as follows: DNF: 5′-TCGCGTTAACGCTAGCATGGA-3′, DNR: 5′-GTAACATCAGAGATTTTGAGA-3′.
-
i.
-
c.Transfer the gene of interest into the cSFB destination vector by the LR reaction (Figure 1B).
-
i.The LR reaction system is as follows:
Component Amount LR Clonase™ II enzyme mix 2 μL entry clone (50–150 ng) 1–7 μL destination vector (cSFB 150 ng/μL) 1 μL TE buffer, pH 8.0 Up to 10 μL -
ii.Perform LR reaction at 25°C for 1 h.
-
iii.Add 1 μL of the Proteinase K solution to each sample to terminate the reaction. Vortex briefly. Incubate samples at 37°C for 10 min.
-
iv.Transform the LR reaction product into DH5α competent cells, and select positive clones on Ampicillin LB plates. Extract plasmid encoding the cSFB-tagged gene of interest and confirmed by sequencing, and protein expression is detected via western blotting. The sequencing primer information is as follows: CTF: 5′-CACTATAGAATAACATCCACTTT-3′, CTR: 5′-GAATTTAGCAGCAGCGGTTTCT-3′.
-
i.
-
a.
Establishment of a stable cell line expressing the target gene
During this step, the stable cell expressing the target protein is established and validated for expression and localization.
-
2.Cell transfection.
-
a.HEK293T cells (passage number < 15) are seeded in a 6 well plate and incubated for less than 24 h (37°C, 5% CO2). The cells should be 70%–80% confluent at moment of transfection.
-
b.Transfect HEK293T cells:
-
i.Prepare a mixture of the transfection plasmids (2 μg) in OptiMEM.
-
ii.Prepare OptiMEM and polyethylenimine (PEI) dilutions for a 3:1 ratio of PEI: DNA. Incubate for a maximum 5 min at 25°C.
-
iii.Following 5 min of incubation, drop the PEI dilution into the plasmid mixture and mix by gently flicking the tube.
-
iv.Incubate the transfection mix for 20 min.
 CRITICAL: Ensure that the plasmid DNA is endotoxin free and concentrated to >500 ng/μL. Do not syringe filter the transfection master mix or the final transfection mix after the DNA and PEI have been added as this will reduce the transfection efficiency.
CRITICAL: Ensure that the plasmid DNA is endotoxin free and concentrated to >500 ng/μL. Do not syringe filter the transfection master mix or the final transfection mix after the DNA and PEI have been added as this will reduce the transfection efficiency.
-
i.
-
c.Carefully transfer the transfection mix to cells by adding drop-wise with a circular motion.
-
d.Incubate cells for 24 h (37°C, 5% CO2).
-
a.
-
3.Puromycin selection.
-
a.Passage the transfected cells into a 10 cm dish containing the media with puromycin at the required concentration for the cells. For HEK293T cells, 2 μg/mL puromycin is used.
-
b.Forty-eight hours after puromycin addition (all untransfected cells should be killed), remove the medium and gently wash the plate with warm PBS to dislodge the remaining dead cells.
-
c.Trypsinize and collect cells into a single-cell suspension. The cells are then serially diluted and plated into a 96-well plate for single-cell colony formation.CRITICAL: Using multichannel pipettes can greatly reduce the time cost.
-
d.Incubate the plate undisturbed at 37°C in a humidified CO2 incubator.
-
a.
-
4.Validation of stable cells expressing target protein.
-
a.Detect the clones under microscope after 7 days; check each well, mark the ones with only a single colony, and transfer cells from these wells into large vessels (e.g., 24 well plate).
-
b.The isolated stable cell colonies are checked for the expression of cSFB-tagged bait proteins by western blotting using anti-FLAG antibody.CRITICAL: In one well of the 24-well plate, half of the cells are removed for expression analysis after trypsin digestion, and the remaining cells are cultured further.
-
c.Following expansion of the positive cell clones, confirm the cSFB-tagged bait protein expression by western blotting again, and examine its subcellular localization by immunofluorescence to ensure its correct subcellular localization. After that, the stable cells are expanded for the subsequent TAP.CRITICAL: After picking out cell colonies, make sure that almost all cells express the tagged bait protein (if not, subcloning should be conducted) with correct subcellular localization. Incorrect subcellular localizations of the tagged proteins were observed occasionally, while changing tags to the other end of the proteins solved the problem.
-
a.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| ANTI-FLAG® M2 antibody (1:5000) | Sigma-Aldrich | Cat#B3111 |
| THE™ beta Actin Antibody, mAb, Mouse (1:5000) | Genscript | Cat#A00702 RRID: AB_914102 |
| Bacterial and virus strains | ||
| Trans5α Chemically Competent Cell | TransGen Biotech | Cat#CD201-01 |
| TransDB3.1 Chemically Competent Cell | TransGen Biotech | Cat#CD531-01 |
| Chemicals, peptides, and recombinant proteins | ||
| Trizol | Thermo Fisher Scientific | Cat#15596026 |
| Phusion® High-Fidelity DNA Polymerase | New England Biolabs | Cat#M0530L |
| BP Clonase™ II enzyme mix | Thermo Fisher Scientific | Cat#11789020 |
| LR Clonase™ II enzyme mix | Thermo Fisher Scientific | Cat#11791020 |
| FBS | Gibco | Cat#C11875500CP |
| Penicillin and streptomycin | Thermo Fisher Scientific | Cat#15140163 |
| DMEM | Basalmedia | Cat#L110KJ |
| Opti-MEM™ I Reduced Serum Medium | Gibco | Cat#31985070 |
| PEI | Polysciences | Cat#23966-1 |
| Puromycin | Sangon | Cat#A610593 |
| Aprotinin | Sigma-Aldrich | Cat#A1153-25MG |
| Pepstatin A | Sigma-Aldrich | Cat#P5318-25MG |
| TurboNuclease | Accelagen | Cat#N0103M |
| Coomassie brilliant blue R-250 | Sangon | Cat#A100472 |
| Biotin | Sigma-Aldrich | Cat#B4501 |
| Streptavidin-conjugated beads | GE | Cat#17-5113-01 |
| Trypsin | Hualishi Scientific | Cat#HLS TRY001C |
| S-protein beads | Millipore | Cat#69704 |
| NP40 | Sigma-Aldrich | Cat#74385 |
| EDTA | Sigma-Aldrich | Cat#V900106 |
| Acetic acid | Sangon | Cat#A501931 |
| Formic acid | Thermo Fisher Scientific | Cat#85178 |
| Acetonitrile | Aladdin | Cat# A104442 |
| Critical commercial assays | ||
| HiScript II 1st Strand cDNA Synthesis Kit | Vazyme | Cat#R212-02 |
| FastPure Gel DNA Extraction Mini Kit | Vazyme | Cat#DC301-01 |
| FastPure Plasmid Mini Kit | Vazyme | Cat#DC201-01 |
| Experimental models: Cell lines | ||
| HEK293T | ATCC | Cat# CRL-11268 RRID: CVCL_0063 |
| Oligonucleotides | ||
| PDONR-F: GGGGACAAGTTTGTACAAAAAAGCAGGCTCC |
This paper | N/A |
| PDONR-R: GGGGACCACTTTGTACAAGAAAGCTGGGTT |
This paper | N/A |
| Recombinant DNA | ||
| pDONR201 | Invitrogen | N/A |
| pDEST-cSFB | This paper | N/A |
| Software and algorithms | ||
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo Fisher Scientific | Lumos |
| Mascot software program | Matrix Science | RRID: SCR_014322 |
| Ingenuity Pathway Analysis | QIAGEN | RRID: SCR_008653 |
| Cytoscape (Version 3.7.2) | Institute for Systems Biology |
https://cytoscape.org RRID: SCR_003032 |
| cBioPortal | Memorial Sloan Kettering Cancer Center | https://www.cbioportal.org/ |
| GEPIA | Zhang Lab, Peking University | http://gepia.cancer-pku.cn/ |
| R (Version 4.2.1) | Free Software Foundation’s GNU project | https://www.r-project.org/ |
| Uniport | European Bioinformatics Institute (EMBL-EBI); Swiss Institute of Bioinformatics (SIB); Protein Information Resource (PIR) | https://www.uniprot.org |
| CRAPome | Alexey Nesvizhskii and Anne-Claude Gingras Labs | http://www.crapome.org |
| STRING | Swiss Institute of Bioinformatics (SIB); Novo Nordisk Foundation Center Protein Research (CPR); European Molecular Biology Laboratory (EMBL) | https://string-db.org/ |
| ImmPort | The Northrop Grumman Information Technology Health Solutions team | https://www.immport.org/shared/home |
| Xcalibur 4.1 software | Thermo Fisher Scientific | Cat#OPTON-30967 |
| Proteome Discoverer 2.5 software | Thermo Fisher Scientific | Cat#OPTON-30957 |
| Deposited data | ||
| Mass spectrometry proteomics data | This study; deposited to the ProteomeXchange Consortium via the PRIDE partner repository | Dataset identifier: PXD031772 and 10.6019/ PXD031772. |
| Other | ||
| EASY-nLC1200 | Thermo Fisher Scientific | Cat#LC140 |
| Silica capillary column | Genetec | Cat#SL20S05-15E8RU |
| C-18 resin | Thermo Fisher Scientific | Cat#060263 |
| Orbitrap Fusion Lumos Tribrid Mass Spectrometer | Thermo Fisher Scientific | Cat#IQLAAEGAAPFADBMBHQ |
Materials and equipment
NETN lysis buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| NP-40 | 0.5% | 20 mL |
| 1 M Tris.HCl, pH8.0 | 20 mM | 80 mL |
| 5 M NaCl | 100 mM | 80 mL |
| 0.5 M EDTA | 1 mM | 8 mL |
| ddH2O | n/a | Up to 4 L |
| Total | n/a | 4 L |
Storage: Store for up to 6 months at 25°C.
Aprotinin (1000×)
| Reagent | Final concentration | Amount |
|---|---|---|
| Aprotinin | 1 mg/mL | 10 mg |
| ddH2O | n/a | Up to 10 mL |
| Total | n/a | 10 mL |
Storage: aliquot 500 μL into 20 1.5 mL micro centrifuge tubes and store at −20°C for several months.
Pepstatin A (1000×)
| Reagent | Final concentration | Amount |
|---|---|---|
| Pepstatin A | 0.5 mg/mL | 25 mg |
| Ethanol | n/a | Up to 50 mL |
| Total | n/a | 50 mL |
Storage: aliquot 1.5 mL in 35 1.5 mL micro centrifuge tubes and store at −20°C for several months.
Turbonuclease buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| 1 M Tris.HCl, pH8.0 | 50 mM | 0.5 mL |
| 1 M MgCl2 | 1 mM | 10 μL |
| ddH2O | n/a | Up to 10 mL |
| Total | n/a | 10 mL |
Storage: fresh prepared with addition of proteinase inhibitors.
Coomassie brilliant blue solution
| Reagent | Final concentration | Amount |
|---|---|---|
| Coomassie brilliant blue R-250 | 0.025% | 0.025 g |
| MeOH | 40% | 0.4 L |
| Acetic acid | 7% | 0.07 L |
| ddH2O | n/a | Up to 1 L |
| Total | n/a | 1 L |
Storage: Store for up to 6 months at 25°C.
Destain I solution
| Reagent | Final concentration | Amount |
|---|---|---|
| MeOH | 40% | 0.4 L |
| Acetic acid | 7% | 0.07 L |
| ddH2O | n/a | Up to 1 L |
| Total | n/a | 1 L |
Storage: Store for up to 6 months at 25°C.
Destain II solution
| Reagent | Final concentration | Amount |
|---|---|---|
| MeOH | 50% | 0.5 L |
| Acetic acid | 7% | 0.07 L |
| ddH2O | n/a | Up to 1 L |
| Total | n/a | 1 L |
Storage: Store for up to 6 months at 25°C.
Step-by-step method details
Preparation of cell lysate
During this step, cells are collected and lysed into soluble and chromatin fractions.
Note: All the following steps are performed using 1 × 108 cells (Figure 2A).
-
1.
Lyse the cells in 5 mL of lysis buffer (NETN with aprotinin and pepstatin A) for 30 min on shaker in a cold room.
Note: The lysis volume depends on how many cells you collect (normally 10 times the pellet volume).
Pause point: If suspension is required, we recommend freezing the collected cells at −80°C rather than freezing the lysates to reduce protein loss.
-
2.
Spin down the 15 mL tubes at 2,500 ×g and 4°C for 20 min.
-
3.Transfer the supernatant to fresh tubes (1), and add 1.5 mL of TurboNuclease buffer containing 2.5 μL of TurboNuclease to the pellets.
-
a.Heat in a 37°C water bath and vortex for 10 s every 5 min till the solution turn white (this usually takes 30–40 min).
-
b.Spin down the tubes at 2,500 ×g and 4°C for 20 min and collect the supernatant (2).CRITICAL: Please ensure that the chromatin is interrupted and the precipitate is milky white (Figure 2C).
-
c.Combine (1) and (2) and spin down at 2,500 ×g and 4°C for an additional 20 min.CRITICAL: During all steps except TurboNuclease treatment, samples should be kept on ice or in a cold room (i.e., 4°C). Proteinase inhibitors should be added to the solutions to prevent protein degradation.Note: Combination of two supernatants is not necessary for studying chromatin or soluble protein interactions, separating these two parts for subsequent TAP.
-
a.
Figure 2.

The macroscopic view of cell pellet in different conditions
(A–E) The cell state in collection (A), NETN lysis (B), TurboNuclease digestion (C), Streptavidin agarose enrichment (D), and S-bead enrichment (E) are shown in Eppendorf tubes.
Tandem affinity purification
This step describes how the purification is accomplished using streptavidin-sepharose (also called SBP beads) and S beads.
-
4.
Add streptavidin-sepharose to the combined solution (total 150 μL beads, depending upon how well your protein is expressed and how many plates are used). Incubate on a shaker in a cold room for 1 h (Figure 2D). Troubleshooting 1.
-
5.Spin down at 2,500 ×g and 4°C for 1 min and aspirate the buffer out of the tube without disturbing the beads.
-
a.Resuspend the beads in 0.8 mL of lysis buffer and transfer them into an Eppendorf tube.
-
b.Wash out the tube with 0.5 mL of lysis buffer, and transfer the buffer into the previous Eppendorf tube to recover the remaining beads.
-
c.Spin and aspirate the supernatant.
-
a.
Note: The transfer of beads to a new tube prevents contamination by any proteins that may precipitate on the sidewall of the previous tube during incubation.
-
6.
Wash beads twice with 1 mL of lysis buffer by centrifuging the Eppendorf tube at 1,500 ×g and 4°C for 1 min.
-
7.Remove all remaining trapped lysis buffer by pipetting using a thin gel-loading tip firmly pressed to the bottom of the tube.
-
a.Elute bound proteins with 0.9 mL of lysis buffer containing 2 mg/mL biotin with proteinase inhibitors.
-
b.Rotate the tube in a cold room for 30 min. Spin briefly to pellet the beads, and carefully transfer the supernatant to a new Eppendorf tube without disturbing the beads.
-
c.Repeat the elution step using 0.5 mL of lysis buffer containing biotin for another 30 min. Troubleshooting 2.
-
a.
-
8.
Combine both eluates. Spin at maximum speed, and transfer the supernatant to a new tube.
Note: Ensure that there are no streptavidin-sepharose beads remaining in the 2nd purification.
-
9.
Add 30 μL of S beads to the eluates. Rotate the tube in a cold room for 1 h (Figure 2E).
-
10.
Spin down the S beads at 1,500 ×g for 1 min, aspirate the supernatant, and wash the S beads 2–3 times with lysis buffer (1 mL each time).
-
11.
Add 35 μL 2× SDS sample buffer to the S beads, briefly vortex the tube, and boil for 15 min.
Note: Once you have performed this protocol a few times, you do not need to check by western blotting each time. Instead, run all of the control samples on a gel, and perform Coomassie blue staining for analysis. There should be a difference in band pattern between the eluate from streptavidin beads and that from S beads.
-
12.
Subject the protein sample to SDS–PAGE until it runs into the separation gel for a short distance (i.e., 1 cm). Stain the gel with clean Coomassie blue solution for 3–10 min. Troubleshooting 3.
Note: SDS–PAGE is performed until the dye runs less than 1 cm into the separation gel. The time of Coomassie blue staining is carefully controlled, and staining is not performed for more than 15 min.
In gel digestion
During this step, the purified protein is digested by trypsin in gel and extracted for mass spectrometry detection.
-
13.
Destain the gel with Destain I solution for 30 min and repeat this step for several times (Figure 3A).
-
14.
Cut the gel band into two pieces from top to bottom and transferred to a clean Eppendorf tube for further complete destaining using Destain II solution. Troubleshooting 4.
-
15.
Incubate the gel pieces in water for 1 h twice.
Optional: Reduction and Alkylation: Dehydrate the gel pieces with 100% acetonitrile (ACN) for 5 min. Soak the gel pieces with 50 μL of 10 mM dithiothreitol (DTT) in 10 mM ammonium bicarbonate (ABC) at 55°C for 45 min. Then alkylate the gel pieces with 50 μL of 100 mM iodoacetamide in 10 mM ABC at 25°C for 30 min in the dark. (The reduction and alkylation steps facilitate peptide identification, especially Cysteine-containing peptides. However, the reaction incompletion and side reactions may compromise the experimental results.).
-
16.
Dehydrate the gel pieces with 75% ACN for 1 h. Change the pH of the gel to basic pH with 50 mM ABC for 30 min twice. Remove the ABC solution and add 300 ng of trypsin (200 ng/μL, 1.5 μL) in 30 μL of 50 mM ABC solution to each tube, grind mildly, and incubate at 37°C 14–16 h.
-
17.
Acidify the digest by adding 30 μL of 2% formic acid (FA) for 5 min, extract the peptides from the gel pieces by adding 400 μL of 100% ACN, shake the solution on a rocker platform at 25°C for 15 min, centrifuge the tubes at 16,000 ×g for 1 min, and transfer the supernatant into new tubes.
-
18.
Vacuum-dry the samples.
Figure 3.

Example data of SFB-TAP/MS
(A) Coomassie brilliant blue staining of SDS-PAGE showing the protein bands after purification.
(B) Data summary for the human Notch pathway interaction network study. Total numbers of peptides and proteins identified in the MS analysis are shown. A MUSE score greater than 0.85 was used as the cut-off to identify HCIPs.
LC-MS analysis
During this step, the extracted peptide is analyzed on mass spectrometer.
-
19.Separate the peptides by a 65 min gradient elution (from 1% to 40% buffer B) at a flow rate 0.3 μL/min with the Thermo EASY-nLC1200 integrated nano-HPLC system which is directly interfaced with the MS.
-
a.The analytical column was a home-made fused silica capillary column packed with C-18 resin.
-
b.Mobile phase A consists of 0.1% formic acid in water, and mobile phase B consists of 100% acetonitrile and 0.1% formic acid.
-
a.
-
20.As the peptides are eluted, they are subjected to electrospray ionization and then analyzed by an Orbitrap Fusion Lumos Tribrid Mass Spectrometer.
-
a.The source is operated at 1.9 kV, with no sheath gas flow and with the ion transfer tube at 350°C.
-
b.The mass spectrometer is operated in the data-dependent acquisition mode using the Xcalibur 4.1 software and there is a single full-scan mass spectrum in the Orbitrap (350–1,500 m/z, 60,000 resolution at m/z 200) followed by 20 data-dependent MS/MS scans at 30% normalized collision energy.
-
c.The selection window of the Quadrupole is 300–1,500 m/z.
-
d.Set the AGC target as 5e4, and the maximum injection time is 50 ms.
-
e.Set the intensity threshold at 1,000.
-
f.MS2 spectra are acquired with a resolution of 15,000.
-
g.The peptides are detected, isolated, and fragmented to produce a tandem mass spectrum of specific fragment ions for each peptide.
-
h.Analyze each mass spectrum using the Thermo Xcalibur Qual Browser. Troubleshooting 5.
-
a.
-
21.Determine peptide sequences (protein identity) by matching fragmentation patterns in the Swiss-Prot protein database (Homo sapiens) using the Mascot software program (Matrix Science, USA) in Proteome Discoverer 2.5 software (Thermo Fisher Scientific, San Jose, CA) (Figure 3B).
-
a.The mass tolerance is set to 20 ppm for both precursor ions and fragment ions.
-
b.Enzyme specificity is set to partially tryptic with two missed cleavages.
-
a.
Note: No fixed modification is set. The variable modifications included oxidation on Methionine, acetylation on peptide N-terminus, and Carbamidomethylation on Cysteine. Spectral matches are filtered to include a false-discovery rate of less than 1% at the peptide level.
MS data analysis and bioinformatics analysis
This step mainly focuses on the analysis and visualization of MS data.
-
22.
Perform a quality control step to ensure a reasonable number of prey proteins with correctly distributed cellular localization and high data reproducibility across the replicates (Figure 4A).
-
23.
Evaluate the confidence scores of bait-prey binary interactions using computational models, such as CompPASS (Sowa et al., 2009), SAINT (Choi et al., 2011) and MUSE (Li et al., 2016) algorithms. Here, for example, we set the MUSE score > 0.85 as the cutoff for identifying high-confidence interacting proteins (HCIPs) (Figure 4B). Troubleshooting 6.
-
24.
High-confidence interactions can be further validated by knowledge databases, including BioGRID (Chatr-Aryamontri et al., 2017), STRING (Szklarczyk et al., 2019), IntAct (Orchard et al., 2014), MINT (Licata et al., 2012), HI-union/HuRI/Lit-BM (Luck et al., 2020) and HI-II-14 (Rolland et al., 2014) (Figure 4C).
Note: The results could also be obtained from the CRAPome database, a repository of prey frequently found in AP/MS experiments (Mellacheruvu et al., 2013) (Figure 4C).
-
25.Visualize protein–protein interaction networks composed of HCIP in Cytoscape (Smoot et al., 2011) (Figures 4C and 5).
-
a.In detail, the identified bait-prey interactions and their corresponding peptide-spectrum match (PSM) values are integrated into excel or .csv format and imported into Cytoscape software for interaction network visualization.
-
a.
Note: For better annotation of the interaction network, properties of each node and edge could be added via additional table format. Other “R”-based visualization methods are also available. The R framework (R-project.org) and Bioconductor (Gentleman et al., 2004) are also frequently used for data analysis, clustering, and visualization. Troubleshooting 7.
-
26.
Perform GO/KEGG analysis, disease enrichment and other bioinformatic analyses for analyzing biological functions of HCIPs (Ashburner et al., 2000) (Figure 4C).
-
27.Perform integrated analysis of proteomic data with genomic data, such as phenotypical data in model organisms, correlation with diseases, expression alterations and mutations in diseases, and correlation with survival and prognosis (Figure 4D).
-
a.For instance, the identified HCIPs can be used for the overlapping analyses with other regulators involved in the detailed biological process according to the users’ research purpose. Troubleshooting 8.
-
a.
-
28.
Conduct in-depth mechanistic studies for several potentially functionally relevant proteins to produce biologically meaningful results (Li et al., 2017), and eventually establish a functional PPI network (Figure 4E).
Note: For data validation, a co-IP assay using antibodies against endogenous proteins or epitope tags is the most frequently used approach (Couzens et al., 2013; Hauri et al., 2013; Kwon et al., 2013; Wang et al., 2014). Other approaches include two-hybrid- and flow cytometry-based interaction validations, such as fluorescence resonance energy transfer and bimolecular fluorescence complementation assays (Fields and Song, 1989; Piehler, 2005; Yan and Marriott, 2003).
Figure 4.

Schematic diagram showing the workflow of bioinformatics analysis in TAP/MS study
(A) Quality control of MS data.
(B) HCIPs are defined by MUSE algorithm.
(C) Validation of HCIPs utilizing knowledge databases, GO term enrichment analysis and visualization of protein–protein interaction networks composed of HCIP by Cytoscape.
(D) Functional analysis of proteomic data by integrating with genomic or model organism data.
(E) Establishing a functional PPI network.
Figure 5.

Example interaction network identified in human Notch pathway established by TAP/MS
The interaction network was constructed using HCIPs identified in SFB-TAP/MS data of our human Notch pathway study.
Expected outcomes
A successful outcome of the TAP/MS experiment is shown in Figures 3 and 5. First, a clear bait protein band was observed after Coomassie brilliant blue staining. Second, PSM resulted in relatively high bait enrichment (in HEK293T cells, PSM usually >100) and a reasonable number of prey (in HEK293T cells, unique prey numbers are usually >100). Third, the confidence scores of bait-prey interactions were generated using computational models and used for establishing the protein-protein interaction network.
Limitations
Transiently transfected cells can be directly subjected to AP/MS, but this usually leads to the bait being expressed at relatively high levels and cells under stress conditions. Since overexpressed proteins often bind to proteins that they normally do not interact with under physiological conditions, the transient transfection followed by the AP/MS approach may lead to considerable false-positive findings. A better approach may be to establish stable pools or cell lines with an expression level of bait protein similar to that of the endogenous protein (Gingras et al., 2007). The inducible system and knock-in strategy can also help with purification of the protein complex at the endogenous level and overcome proliferation defects caused by overexpression of some toxic baits.
The protein–protein interaction network establishment protocol described here depends mainly on MS-based whole-protein complex analysis and thus cannot distinguish between direct and indirect protein interactions. To confirm direct binding between proteins, additional assays, such as in vitro pull-down analysis, are required.
The analysis step in this protocol usually requires a group of control TAP/MS experiments with bait proteins that are not associated with the proteins of interest. Moreover, several medium- to large-scale SFB-based TAP/MS results in HEK293T and MCF10A cells are available for use as controls (Bian et al., 2021; Li et al., 2015, 2016; Wang et al., 2014; Xu et al., 2014).
Troubleshooting
Problem 1
During the streptavidin-sepharose and S-bead incubation steps, proteins are precipitated on the beads (white flocculent pellets was observed on the beads after the 1000×g centrifugation). (step-by-step method details step 4).
Potential solution
Reduce the initial cell number or increase the cell lysis volume accordingly.
Problem 2
The elution of streptavidin-sepharose precipitated samples is insufficient. (step-by-step method details step 7).
Potential solution
Prepare fresh biotin solution in NETN lysis buffer and completely dissolved by warming in a water bath. Modify the biotin concentration if needed.
Problem 3
No protein band is observed after gel staining. (step-by-step method details step 12).
Potential solution
First, check the protein expression level just before collecting cells for purification; second, during purification, collect samples for each IP step, e.g., samples before addition of streptavidin-sepharose, after incubation, and eluate, to identify the step that failed.
Problem 4
The gel cannot be destained completely. (step-by-step method details step 14).
Potential solution
Ensure that R-250 is used. Carefully control the staining time to no more than 15 min, usually. Watch the gel while staining, and immediately change to the destaining solution once the band becomes visible.
Problem 5
Poor quality LC-MS data and hard to interpret. (step-by-step method details step 20).
Potential solution
If the LC-MS data peaks are too small to confidently interpret, inject more sample into the LC-MS. If the LC-MS data has a large contaminating peak, it is likely to be from tubes used in purification process. Therefore, it is important to use high quality and clean tubes to avoid contamination.
Problem 6
Low score of bait-prey binary interactions. (step-by-step method details step 23).
Potential solution
If the scores of most bait-prey binary interactions evaluated using computational models are extremely low, confirm whether a correct control group is chosen, or whether there is problem in the purification process.
Problem 7
Too redundant interaction network visualized by cytoscape software. (step-by-step method details step 25).
Potential solution
In integrating the data ready for importing into cytoscape, users need to eliminate the duplicated binary interaction. For example, NOTCH1-NOTCH2 /NOTCH2-NOTCH1.
Problem 8
Low overlap for function studies when integrated with genomic data or other resource. (step-by-step method details step 27).
Potential solution
In integrating the HCIPs data with other resource data, gene ID and protein ID conversion, as well as ortholog among different species need to be noticed carefully.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Xu Li (lixu@westlake.edu.cn).
Materials availability
All unique reagents generated in this study are listed in the key resources table and available upon reasonable request from the lead contact.
Acknowledgments
X.L. and W.W. thank J.C. for his mentoring and continuous support. We thank colleagues in the Xu Li, Wenqi Wang, Jun Qin, and Junjie Chen laboratories for the inputs to this protocol. We thank the Westlake University Biomedical Research Core Facilities for assistance with the MS analysis. This work was supported by grants from Natural Science Foundation of China 91954103 and an Institutional Startup Grant from the Westlake Education Foundation to X.L. This work was also supported in part by an NIH grant (R01 GM126048) and an American Cancer Society Research Scholar Award grant (RSG-18-009-01-CCG) to W.W.
Author contributions
J.C., W.W., and X.L. designed the protocol, analyzed the data, revised the manuscript, and supervised the whole study. W.B., H.J., and S.F. helped design the protocol, performed the experiments, and wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Junjie Chen, Email: jchen8@mdanderson.org.
Wenqi Wang, Email: wenqiw6@uci.edu.
Xu Li, Email: lixu@westlake.edu.cn.
Data and code availability
The published article includes all data sets generated or analyzed during this study. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD031772 and 10.6019/ PXD031772. All other data are available from the authors upon reasonable request.
References
- Aebersold R., Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T., et al. Gene ontology: tool for the unification of biology. Nat. Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bian W., Tang M., Jiang H., Xu W., Hao W., Sui Y., Hou Y., Nie L., Zhang H., Wang C., et al. Low-density-lipoprotein-receptor-related protein 1 mediates Notch pathway activation. Dev. Cell. 2021;56:2902–2919.e8. doi: 10.1016/j.devcel.2021.09.015. [DOI] [PubMed] [Google Scholar]
- Bontinck M., Van Leene J., Gadeyne A., De Rybel B., Eeckhout D., Nelissen H., De Jaeger G. Recent trends in plant protein complex analysis in a developmental context. Front. Plant Sci. 2018;9:640. doi: 10.3389/fpls.2018.00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branon T.C., Bosch J.A., Sanchez A.D., Udeshi N.D., Svinkina T., Carr S.A., Feldman J.L., Perrimon N., Ting A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018;36:880–887. doi: 10.1038/nbt.4201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzard B.L., Chubet R.G., Vizard D.L. Immunoaffinity purification of FLAG epitope-tagged bacterial alkaline phosphatase using a novel monoclonal antibody and peptide elution. Biotechniques. 1994;16:730–735. [PubMed] [Google Scholar]
- Burckstummer T., Bennett K.L., Preradovic A., Schütze G., Hantschel O., Superti-Furga G., Bauch A. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat. Methods. 2006;3:1013–1019. doi: 10.1038/nmeth968. [DOI] [PubMed] [Google Scholar]
- Chatr-Aryamontri A., Oughtred R., Boucher L., Rust J., Chang C., Kolas N.K., O'Donnell L., Oster S., Theesfeld C., Sellam A., et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res. 2017;45:D369–D379. doi: 10.1093/nar/gkw1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho K.F., Branon T.C., Udeshi N.D., Myers S.A., Carr S.A., Ting A.Y. Proximity labeling in mammalian cells with TurboID and split-TurboID. Nat. Protoc. 2020;15:3971–3999. doi: 10.1038/s41596-020-0399-0. [DOI] [PubMed] [Google Scholar]
- Choi H., Larsen B., Lin Z.Y., Breitkreutz A., Mellacheruvu D., Fermin D., Qin Z.S., Tyers M., Gingras A.C., Nesvizhskii A.I. SAINT: probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods. 2011;8:70–73. doi: 10.1038/nmeth.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzens A.L., Knight J.D.R., Kean M.J., Teo G., Weiss A., Dunham W.H., Lin Z.Y., Bagshaw R.D., Sicheri F., Pawson T., et al. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]
- Dunham W.H., Mullin M., Gingras A.C. Affinity-purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 2012;12:1576–1590. doi: 10.1002/pmic.201100523. [DOI] [PubMed] [Google Scholar]
- Evan G.I., Lewis G.K., Ramsay G., Bishop J.M. Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell Biol. 1985;5:3610–3616. doi: 10.1128/mcb.5.12.3610-3616.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field J., Nikawa J., Broek D., MacDonald B., Rodgers L., Wilson I.A., Lerner R.A., Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol. Cell Biol. 1988;8:2159–2165. doi: 10.1128/mcb.8.5.2159-2165.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields S., Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- Gentleman R.C., Carey V.J., Bates D.M., Bolstad B., Dettling M., Dudoit S., Ellis B., Gautier L., Ge Y., Gentry J., et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras A.C., Gstaiger M., Raught B., Aebersold R. Analysis of protein complexes using mass spectrometry. Nat. Rev. Mol. Cell Biol. 2007;8:645–654. doi: 10.1038/nrm2208. [DOI] [PubMed] [Google Scholar]
- Hauri S., Wepf A., Drogen A., Varjosalo M., Tapon N., Aebersold R., Gstaiger M. Interaction proteome of human Hippo signaling: modular control of the co-activator YAP1. Mol. Syst. Biol. 2013;9:713. doi: 10.1002/msb.201304750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopp T.P., Prickett K.S., Price V.L., Libby R.T., March C.J., Pat Cerretti D., Urdal D.L., Conlon P.J. A short polypeptide marker sequence useful for recombinant protein identification and purification. Bio Technol. 1988;6:1204–1210. doi: 10.1038/nbt1088-1204. [DOI] [Google Scholar]
- Hussain T., Lee J., Abba M.C., Chen J., Aldaz C.M. Delineating WWOX protein interactome by tandem affinity purification-mass spectrometry: identification of top interactors and key metabolic pathways involved. Front. Oncol. 2018;8:591. doi: 10.3389/fonc.2018.00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R., de Martynoff G., Lou J., Hipskind R.A., Nordheim A., Stunnenberg H.G. Rapid and efficient purification of native histidine-tagged protein expressed by recombinant vaccinia virus. Proc. Natl. Acad. Sci. USA. 1991;88:8972–8976. doi: 10.1073/pnas.88.20.8972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe A.D., Wilson D.S., Seelig B., Szostak J.W. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag. Protein Expr. Purif. 2001;23:440–446. doi: 10.1006/prep.2001.1515. [DOI] [PubMed] [Google Scholar]
- Kellermann O.K., Ferenci T. Maltose-binding protein from Escherichia coli. Methods Enzymol. 1982;90:459–463. doi: 10.1016/s0076-6879(82)90171-9. [DOI] [PubMed] [Google Scholar]
- Kim H., Chen J., Yu X. Ubiquitin-binding protein RAP80 mediates BRCA1-dependent DNA damage response. Science. 2007;316:1202–1205. doi: 10.1126/science.1139621. [DOI] [PubMed] [Google Scholar]
- Kim H., Huang J., Chen J. CCDC98 is a BRCA1-BRCT domain-binding protein involved in the DNA damage response. Nat. Struct. Mol. Biol. 2007;14:710–715. doi: 10.1038/nsmb1277. [DOI] [PubMed] [Google Scholar]
- Kim J.E., Chen J., Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- Kim J.S., Raines R.T. Ribonuclease S-peptide as a carrier in fusion proteins. Protein Sci. 1993;2:348–356. doi: 10.1002/pro.5560020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.J., Cervantes C., Jung Y.S., Zhang X., Zhang J., Lee S.H., Jun S., Litovchick L., Wang W., Chen J., et al. PAF remodels the DREAM complex to bypass cell quiescence and promote lung tumorigenesis. Mol. Cell. 2021;81:1698–1714.e6. doi: 10.1016/j.molcel.2021.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon Y., Vinayagam A., Sun X., Dephoure N., Gygi S.P., Hong P., Perrimon N. The Hippo signaling pathway interactome. Science. 2013;342:737–740. doi: 10.1126/science.1243971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam S.S., Martell J.D., Kamer K.J., Deerinck T.J., Ellisman M.H., Mootha V.K., Ting A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods. 2015;12:51–54. doi: 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Tran K.M., Aziz K.E., Sorokin A.V., Chen J., Wang W. Defining the protein-protein interaction network of the human protein tyrosine phosphatase family. Mol. Cell. Proteomics. 2016;15:3030–3044. doi: 10.1074/mcp.m116.060277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Wang W., Chen J. Recent progress in mass spectrometry proteomics for biomedical research. Sci. China Life Sci. 2017;60:1093–1113. doi: 10.1007/s11427-017-9175-2. [DOI] [PubMed] [Google Scholar]
- Li X., Wang W., Wang J., Malovannaya A., Xi Y., Li W., Guerra R., Hawke D.H., Qin J., Chen J. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 2015;11:775. doi: 10.15252/msb.20145504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y. The tandem affinity purification technology: an overview. Biotechnol. Lett. 2011;33:1487–1499. doi: 10.1007/s10529-011-0592-x. [DOI] [PubMed] [Google Scholar]
- Licata L., Briganti L., Peluso D., Perfetto L., Iannuccelli M., Galeota E., Sacco F., Palma A., Nardozza A.P., Santonico E., et al. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012;40:D857–D861. doi: 10.1093/nar/gkr930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten J., Emili A. Statistical and computational methods for comparative proteomic profiling using liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics. 2005;4:419–434. doi: 10.1074/mcp.R500005-MCP200. [DOI] [PubMed] [Google Scholar]
- Liu Y., Luo Y., Yan S., Lian Y.F., Wu S., Xu M., Feng L., Zhang X., Li R., Zhang X., et al. CRL2(KLHDC3) mediates p14ARF N-terminal ubiquitylation degradation to promote non-small cell lung carcinoma progression. Oncogene. 2022;41:3104–3117. doi: 10.1038/s41388-022-02318-6. [DOI] [PubMed] [Google Scholar]
- Luck K., Kim D.K., Lambourne L., Spirohn K., Begg B.E., Bian W., Brignall R., Cafarelli T., Campos-Laborie F.J., Charloteaux B., et al. A reference map of the human binary protein interactome. Nature. 2020;580:402–408. doi: 10.1038/s41586-020-2188-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellacheruvu D., Wright Z., Couzens A.L., Lambert J.P., St-Denis N.A., Li T., Miteva Y.V., Hauri S., Sardiu M.E., Low T.Y., et al. The CRAPome: a contaminant repository for affinity purification-mass spectrometry data. Nat. Methods. 2013;10:730–736. doi: 10.1038/nmeth.2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orchard S., Ammari M., Aranda B., Breuza L., Briganti L., Broackes-Carter F., Campbell N.H., Chavali G., Chen C., del-Toro N., et al. The MIntAct project--IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014;42:D358–D363. doi: 10.1093/nar/gkt1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piehler J. New methodologies for measuring protein interactions in vivo and in vitro. Curr. Opin. Struct. Biol. 2005;15:4–14. doi: 10.1016/j.sbi.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Puig O., Caspary F., Rigaut G., Rutz B., Bouveret E., Bragado-Nilsson E., Wilm M., Séraphin B. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24:218–229. doi: 10.1006/meth.2001.1183. [DOI] [PubMed] [Google Scholar]
- Rhee H.W., Zou P., Udeshi N.D., Martell J.D., Mootha V.K., Carr S.A., Ting A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science. 2013;339:1328–1331. doi: 10.1126/science.1230593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G., Shevchenko A., Rutz B., Wilm M., Mann M., Séraphin B. A generic protein purification method for protein complex characterization and proteome exploration. Nat. Biotechnol. 1999;17:1030–1032. doi: 10.1038/13732. [DOI] [PubMed] [Google Scholar]
- Rolland T., Taşan M., Charloteaux B., Pevzner S., Zhong Q., Sahni N., Yi S., Lemmens I., Fontanillo C., Mosca R., et al. A proteome-scale map of the human interactome network. Cell. 2014;159:1212–1226. doi: 10.1016/j.cell.2014.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux K.J., Kim D.I., Raida M., Burke B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012;196:801–810. doi: 10.1083/jcb.201112098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D.B., Johnson K.S. Single-step purification of polypeptides expressed in Escherichia coli as fusions with glutathione S-transferase. Gene. 1988;67:31–40. doi: 10.1016/0378-1119(88)90005-4. [DOI] [PubMed] [Google Scholar]
- Smoot M.E., Ono K., Ruscheinski J., Wang P.L., Ideker T. Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics. 2011;27:431–432. doi: 10.1093/bioinformatics/btq675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowa M.E., Bennett E.J., Gygi S.P., Harper J.W. Defining the human deubiquitinating enzyme interaction landscape. Cell. 2009;138:389–403. doi: 10.1016/j.cell.2009.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk D., Gable A.L., Lyon D., Junge A., Wyder S., Huerta-Cepas J., Simonovic M., Doncheva N.T., Morris J.H., Bork P., et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Qiu Z., Hou Y., Deng X., Xu W., Zheng T., Wu P., Xie S., Bian W., Zhang C., et al. AXL is a candidate receptor for SARS-CoV-2 that promotes infection of pulmonary and bronchial epithelial cells. Cell Res. 2021;31:126–140. doi: 10.1038/s41422-020-00460-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Li X., Huang J., Feng L., Dolinta K.G., Chen J. Defining the protein-protein interaction network of the human hippo pathway. Mol. Cell. Proteomics. 2014;13:119–131. doi: 10.1074/mcp.m113.030049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S., Li X., Gong Z., Wang W., Li Y., Nair B.C., Piao H., Yang K., Wu G., Chen J. Proteomic analysis of the human cyclin-dependent kinase family reveals a novel CDK5 complex involved in cell growth and migration. Mol. Cell. Proteomics. 2014;13:2986–3000. doi: 10.1074/mcp.m113.036699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y., Marriott G. Analysis of protein interactions using fluorescence technologies. Curr. Opin. Chem. Biol. 2003;7:635–640. doi: 10.1016/j.cbpa.2003.08.017. [DOI] [PubMed] [Google Scholar]
- Yuan J., Ghosal G., Chen J. The HARP-like domain-containing protein AH2/ZRANB3 binds to PCNA and participates in cellular response to replication stress. Mol. Cell. 2012;47:410–421. doi: 10.1016/j.molcel.2012.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P., Wei Y., Wang L., Debeb B.G., Yuan Y., Zhang J., Yuan J., Wang M., Chen D., Sun Y., et al. ATM-mediated stabilization of ZEB1 promotes DNA damage response and radioresistance through CHK1. Nat. Cell Biol. 2014;16:864–875. doi: 10.1038/ncb3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Shi J., Liu X., Feng L., Gong Z., Koppula P., Sirohi K., Li X., Wei Y., Lee H., et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 2018;20:1181–1192. doi: 10.1038/s41556-018-0178-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.A., Zhou J., Zhao L., Yu G., Zhan J., Shi C., Yuan R., Wang Y., Chen C., Zhang W., et al. KLHL22 maintains PD-1 homeostasis and prevents excessive T cell suppression. Proc. Natl. Acad. Sci. USA. 2020;117:28239–28250. doi: 10.1073/pnas.2004570117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The published article includes all data sets generated or analyzed during this study. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD031772 and 10.6019/ PXD031772. All other data are available from the authors upon reasonable request.