

Abstract
Brain development and function are governed by precisely regulated protein expressions in different regions. To date, multiregional brain proteomes have been systematically analyzed only for adult human and mouse brains. To understand the underpinnings of brain development and function, we generated proteomes from six regions of the postnatal brain at three developmental stages of domestic dogs (Canis familiaris), which are special among animals in terms of their remarkable human-like social cognitive abilities. Quantitative analysis of the spatiotemporal proteomes identified region-enriched synapse types at different developmental stages and differential myelination progression in different brain regions. Through integrative analysis of inter-regional expression patterns of orthologous proteins and genome-wide cis-regulatory element frequencies, we found that proteins related with myelination and hippocampus were highly correlated between dog and human but not between mouse and human, although mouse is phylogenetically closer to human. Moreover, the global expression patterns of neurodegenerative disease and autism spectrum disorder–associated proteins in dog brain more resemble human brain than in mouse brain. The high similarity of myelination and hippocampus-related pathways in dog and human at both proteomic and genetic levels may contribute to their shared social cognitive abilities. The inter-regional expression patterns of disease-associated proteins in the brain of different species provide important information to guide mechanistic and translational study using appropriate animal models.
Keywords: brain proteome, cis-regulatory element, myelination, autism spectrum disorder, domestic dog
Abbreviations: AGC, automatic gain control; Amy, amygdala; ASD, autism spectrum disorder; Cere, cerebellum; CRE, cis-regulatory element; CREF, cis-regulatory element frequency; FA, formic acid; GO, gene ontology; Hippo, hippocampus; KEGG, Kyoto Encyclopedia of Genes and Genomes; MBP, myelin basic protein; MFOL, myelin-forming oligodendrocyte; MOL, mature oligodendrocyte; Olf, Olfactory bulb; OPC, oligodendrocyte precursor cell; PD, parkinson's disease; PFC, prefrontal cortex; RP, reversed phase; Scz, schizophrenia; Stri, striatum; TMT, tandem mass tag
Graphical Abstract
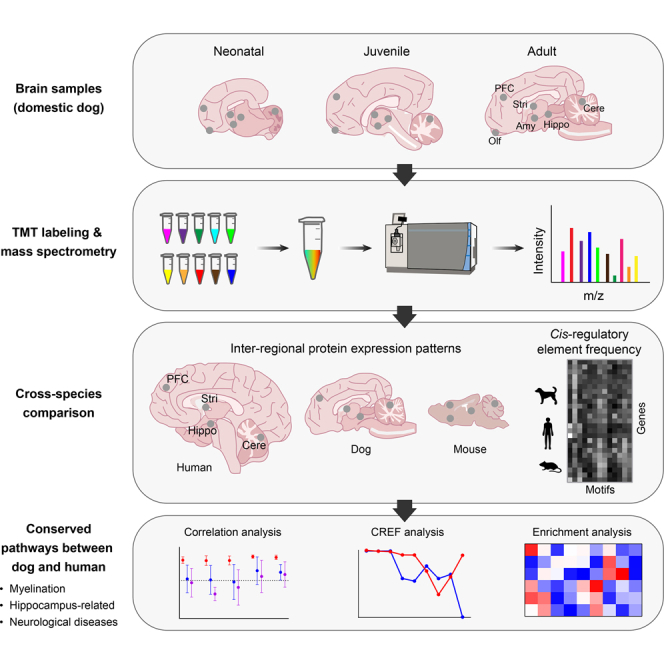
Highlights
-
•
First spatiotemporal brain proteomes of domestic dogs (Canis familiaris).
-
•
Defined developmental processes including myelination in different brain regions.
-
•
Myelin and neurological disease-related proteins express similarly in dog and human.
-
•
Cis-regulatory modules regulating specific pathways are conserved in dog and human.
In Brief
We generated the first spatiotemporal brain proteomes of domestic dogs. By comparison of brain proteomes together with genome-wide cis-regulatory elements frequency between dog, human, and mouse, we identified specific pathways involved in myelination, hippocampus, and neurological diseases conserved between dog and human; the conserved pathways between the two species may help understand their shared social cognitive abilities. The dog brain proteome we generated provides an invaluable resource for comparative studies of brain development, function, and disorder.
The complexity and inaccessibility of the human brain make it challenging to understand its development, function, and disorders. While genetic mouse models have contributed substantially to our current understanding of brain function and disease, the translational value of mouse models may be limited to certain aspects due to their diverged anatomic, behavioral, and cognitive properties from humans (1, 2). Although the brains of nonhuman primates such as macaques resemble human brains more closely in structure and development (3), the extremely high costs and slow reproduction rates render them inaccessible for most researchers. Despite the more distant evolutionary relationship with humans than mouse (human and mouse separated by ∼90 million years while human and dog separated by ∼96 million years) (4), domestic dogs (Canis familiaris) have a similar gyrencephalic brain structure to humans (5). Moreover, as a domesticated species with a long history of coevolution and coadaptation with humans, dogs have developed exquisite and complex dog-human heterospecific social and cognitive capabilities and are more skillful than chimpanzees and other primates in so called human unique social cognitive behaviors (6, 7). Thus, the domestic dog offers a unique opportunity to understand human brain development and function, especially social and cognitive functions.
The precise spatial and temporal regulation of the expression of proteins, the executors of biological function, is crucial for brain development and function. To date, multiregional brain proteomes have been systematically analyzed only for adult human and mouse brains (8, 9), while brain transcriptomes have been more widely studied in multiple species (3, 10, 11). The spatial patterning of gene expression has been shown to be closely linked with neuronal microcircuitry, inter-regional connectivity, and functional specialization of the brain (12). For example, the increased expression of genes regulating axonal and synaptic structure and function may distinguish connected from unconnected regions, whereas genes involved in oxidative synthesis and metabolism of ATP are highly expressed in brain network hubs such as frontal cortices (13, 14). Brain regions including prefrontal cortex (PFC), hippocampus (Hippo), and striatum (Stri) form neural circuits regulating different functions including social and cognition. However, the inter-regional expression patterns of various proteins remain unclear.
Multiple lines of evidence suggest that differences in protein expressions, which contribute to phenotypic divergence within or among species, are controlled by genetic alterations in cis-regulatory elements (CREs) for environmental adaption or selection during evolution (15, 16, 17). However, the gain and loss of CREs and their repetitive nature result in analytical complexities using conventional methods (18). Since change in the number of CREs is an important factor which affects transcription factor–binding sites and thus alter expression of proteins ultimately (17), Li et al. (19) developed a systematic analytical method based on cis-regulatory element frequency (CREF). Using the method, they analyzed the genome-wide frequency distribution of CREs in the proximal regulatory regions flanking transcription start sites of all protein-coding genes and successfully identified cis-regulatory modules regulating specific phenotypes in human compared with chimpanzee (19).
In this study, we generated the first multiregional proteomes of the postnatal dog brain using a high-throughput and accurate label-based approach. Cross-species comparative analysis of proteomes revealed highly correlated inter-regional expression patterns of orthologous proteins involved in myelin, Hippo, mitochondria, and brain disease-related pathways between dog and human but not between mouse and human. Genome-wide CREF analysis further confirmed conserved cis-regulatory modules regulating myelination and Hippo-related pathways between dog and human at the genetic level. Our findings provide insights into the molecular mechanism underlying similar or distinct brain region functions among different species. Our dog brain proteome provides a valuable resource for understanding brain development, function, and disorder.
Experimental Procedures
Dogs and Housing Conditions
Purebred healthy male Beagles at different postnatal ages (newborn, 3 months and adults) were obtained from Beijing Marshall Biotechnology Co. and raised at Sinogene Ltd. All dogs (3 dogs for each stage) were maintained in a near-natural 12-h light-dark cycle. All the experimental protocols were approved by the Institutional Animal Care Committee of the Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, and all procedures were carried out in accordance with the institutional policies for the Care and Use of Laboratory Animals.
Brain Dissection
Brains were dissected, weighed, and chilled on ice for 15 to 30 min prior to sectioning. Brains were placed ventral side up onto a chilled glass plate on ice. The whole brain was divided into left and right hemispheres by cutting along the midline using a long scalpel. The brainstem and cerebellum (Cere) were then removed from the cerebrum by making a transverse section at the junction between the diencephalon and midbrain. The Cere was separated from the brainstem by making a cut directly posterior to the brainstem, along the cerebellar peduncles. The regions of interest were dissected into 4 to 6 blocks using a scalpel blade. Each block was put into an EP tube and then frozen in liquid nitrogen immediately. Regions of interest were collected from fresh tissues and all specimens were stored at −80 °C after liquid nitrogen frozen. To ensure consistency, all dissections were performed by a brain anatomy expert Dr H. Z.
To precisely collect different brain regions of interest, we referred to the supplement of the book named the Beagle Brain in Stereotaxic Coordinates for detailed descriptions of each region (20). Briefly, the PFC was sampled between the anterior and middle third of the medial frontal gyrus. Olfactory bulb (Olf) was sampled near the anterior area of PFC. Hippo was sampled from the middle third of the retrocommissural hippocampal formation located on the medial side of the temporal lobe. Hippo included the dentate gyrus and the cornu ammonis. Amygdala (Amy) included the whole Amy. Stri included the head of the caudate nucleus and the putamen, separated by the internal capsule and ventrally connected to the nucleus accumbens. Cere was sampled from the lateral part of the posterior lobe. The sampled tissue of Cere at different stages contained all three layers of the cerebellar cortex and the underlying white matter but not the deep cerebellar nuclei. For consistency, all samples were dissected from the left hemisphere.
Protein Extraction
The protein extraction of dog brain proteome was performed following previous protocols (21). Briefly, the brain tissues of individual dogs were homogenized on ice in radioimmunoprecipitation assay buffer (5 times volume of 150 mg tissue weight) containing 50 mM Tris–HCl, 150 mM NaCl, 1% SDS, and 1× protease inhibitor mixture set I (Calbiochem). All samples were boiled at 99 °C for 10 min. After centrifugation at 4 °C for 10 min at 21,130g, the supernatant was recovered and the protein concentration was determined by bicinchoninic acid protein assay (Thermo Scientific).
Protein Digestion and TMT Labeling
Protein digestion was performed using the filter-aided sample preparation method. Briefly, 100 μg proteins were reduced by 100 mM DTT at 37 °C for 1 h and then transferred into the Microcon -30 kDa centrifugal filter units (EMD Millipore Corporation) to allow buffer exchange by centrifugation. After buffer exchange to urea (8M urea, 100 mM Tris–HCl, pH 8.5), the proteins were alkylated by 55 mM iodoacetamide (Sigma-Aldrich) for 1 h in the dark. A buffer containing 0.1 M triethylammonium bicarbonate (Sigma-Aldrich) was used to replace the denaturing buffer of the sample. Proteins were then digested with sequencing grade trypsin (Promega) at 37 °C overnight.
The resultant tryptic peptides were labeled with acetonitrile-dissolved 10plex tandem mass tag (TMT) reagents (Thermo Scientific) at room temperature in dark for 2 h. All 54 samples (3 stages × 6 regions × 3 replicates) were randomly assembled into six experimental groups for TMT 10plex labeling (supplemental Table S2). A pooled sample was made by pooling all 54 samples together and added in each group for inter-group control (supplemental Table S2). The labeling reaction was stopped by 5% hydroxylamine, and an equal amount of labeled samples was mixed together before prefractionation with high pH reversed phase (RP)-HPLC.
High pH Reversed-Phase Fractionation
Prefractionation of protein samples was performed using an offline basic RP-HPLC approach. The peptides were fractionated on a phenomenex gemini-NX 5μ C18 column (250 × 3.0 mm, 110 Å) (Torrance) using a Waters e2695 separations HPLC system. A 97 min basic RP-LC gradient with a flow rate of 400 μl/min was used for the entire LC separation as previously described (22). The separated samples were collected and combined into 15 fractions. All samples were dried with a Speed-Vac concentrator and stored at −20 °C before use.
LC-MS/MS/MS Analysis
The LC-MS/MS/MS analysis was performed using an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific) coupled online to an Easy-nLC 1200 in the data-dependent acquisition mode. The dried peptides were resuspended in 0.1% formic acid (FA), and about 1 μg of each sample was injected into a capillary self-packed analytic column (length: 25 cm, inner diameter: 150 μm) packed with C18 particles (Maisch GmbH, diameter: 1.9 μm). The LC was run with mobile phase containing buffer A (0.1% FA) and buffer B (80% acetonitrile, 0.1% FA). A 120-min nonlinear gradient with a flow rate of 600 nl/min was used for peptide separation. The positive ion mode was used for MS measurements, and the spectra were acquired across the mass range of 375 to 1500 m/z. For each cycle, one full MS scan was acquired in the Orbitrap at a resolution of 120,000 with an automatic gain control (AGC) target of 5 × 105. After a full scan, several ions were selected to collision-induced dissociation for fragmentation and then analyzed in the linear ion trap for peptide identification using an AGC target of 1 × 104. Up to 10 most intense ions from each MS2 were selected for additional fragmentation in the high-energy collision dissociation cell using an AGC of 1 × 105, 65% collision energy. The resultant fragment ions including TMT reporter ions were detected in the Orbitrap at a resolution of 50,000 at m/z 200, scan range 100 to 500 m/z, maximum injection time 85 ms, 2 m/z isolation window.
Protein Identification
The integrated dog (C. familiaris) protein database was interrogated using the software MaxQuant (version 1.6.3.4) for all raw MS files. The dog protein database with 76,225 entries was generated by combining Uniprot, Ensembl, and NCBI databases with redundancy removed. All searches were performed with oxidation of methionine and protein N-terminal acetylation as variable modifications and cysteine carbamidomethylation as fixed modification. The type of search was set to report ion MS3 and the 10plex TMT was chosen for isobaric labels. The TMT-labeled peptide N-termini and lysines were searched as fixed modifications. Trypsin was selected as protease allowing for up to two missed cleavages. The initial mass tolerance was 20 ppm for precursor ions and 20 ppm for fragment ions. Peptide spectrum match and protein false discovery rates were both applied at 1%. The minimum score for unmodified peptides was set to 15. Matching between runs of MS1 features is performed. In general, the values of parameters in MaxQuant have not been changed from their default values unless explicitly stated.
RNA Processing and RNA-seq
We performed tissue-level RNA extraction of six regions including Cere, Olf, Hippo, PFC, Stri, and Amy from juvenile dog brain using Trizol (Invitrogen, #15596-018) and purified with the RNeasy Micro kit (Qiagen, #74004) following the manufacturer’s protocol. After constructing the RNA library, RNA-seq was performed by Novogene using an Illumina NovaSeq 6000 platform by PE150 sequencing strategy. The paired-end reads were mapped to the Canis lupus familiaris reference genome (CanFam3.1) using Hisat2 (v2.1.0) (23), and then the transcriptome was reassembled and the expression level of genes and transcripts were quantified using StringTie (v2.1.4) (24).
Differential Protein Expression Analysis
The total reporter ion intensity of each sample was firstly normalized to the mean of total intensity of all samples, and then the reporter intensities of proteins in each sample were divided by that of the pool channel in each TMT group to obtain the ratios for further data analysis. The quality of TMT datasets was evaluated by box plot and clustering analysis which showed no obvious batch effect. Correlation analysis and principal component analysis were performed on the normalized reporter ion intensity data. To identify region-enriched and stage-specific differentially expressed proteins, we performed multiple t-tests between protein expression values in one region and the mean expression values of other five regions and between different stages (S2 versus S1 and S3 versus S2), respectively, employing the Benjamini-Hochberg p value adjustment method as well. Proteins with adjusted p value <0.05 and fold change >1.5 (the median coefficient of variation of TMT quantification between replicates = 0.08) were defined as differentially expressed proteins.
Functional Enrichment Analysis
Functional enrichment analysis (gene ontology (GO) terms and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways) was performed using the DAVID Functional Annotation tool with default parameters. Enrichments were computed using Fisher’s exact tests and p values were corrected for multiple comparisons using the Benjamini-Hochberg method. Significantly enriched terms were defined as those with a corrected p < 0.05.
Western Blotting Analysis
Equal amounts of total protein from different brain regions at different stages were loaded onto SDS/PAGE gels. Primary antibodies used were rat anti-MBP (1:5000; ab7349 from Abcam) and mouse anti-α-tubulin (1:20,000; B512 from Sigma-Aldrich). The primary antibodies were detected with horseradish peroxidase–coupled secondary antibodies.
ASD- and Schizophrenia (Scz)-Related Protein Expression Analysis
The autism spectrum disorder (ASD) set with 1003 risk genes was compiled from the online SFARI gene database (https://gene.sfari.org/database/). Three hundred twenty-one high-confidence Scz-associated genes were selected from an integrated analysis of schizophrenia genome-wide association study (PsychENCODE Integrative Analysis, http://resource.psychencode.org/) (25). The enrichment score of ASD- and Scz-related proteins in each region is defined by the ratio of actual percentage (region enriched/total ASD/Scz protein number)/theoretical percentage (region enriched/total protein number) using one tailed hypergeometric test. Region-enriched ASD proteins were identified by multiple t-tests as described above. The differentially expressed ASD- and non ASD-related proteins at different stages were used to determine the temporal expression patterns of ASD genes as previously described. “Complete linkage” hierarchical clustering was performed after “row” score normalization of the regulated dataset by Pheatmap R package. Functional enrichment analysis of GO/KEGG for region-enriched and temporally regulated ASD-related proteins was performed using DAVID as described above.
CREF Analysis
The CREF analysis was performed as previous study (19). The human genome (release 84) and mouse genomes (release 102) and gene annotation were downloaded from the Ensembl database using R package BiomaRt. The dog genome (release 102) was obtained by directly downloading the .gtf and .fa files from ftp.ensembl.org/pub/release −102/. We focused our analysis on protein-coding genes, including 15,746 dog genes, 19,763 human genes, and 21,733 mouse genes. The transcription starting sites of human and mouse genes were obtained using the principal transcripts defined by the APPRIS system (26). The proximal regulatory sequences between −1000 bp and +500 bp with respect to the transcription start sites were extracted from the human genome (GRCh38.p5) and the mouse genome (GRCm38.p6) through Ensembl Representational State Transfer Application Programming Interface. The proximal regulatory sequences of the dog (CanFam3.1) genes were obtained manually. A total of 1403 vertebrate motifs together with their position weight matrices from the TRANSFAC database were used in this study. Motif frequencies in the promoters were calculated using the MATCH program with the minimize false negative option.
Enrichment Analysis by the Wilcoxon Rank Sum Scoring Method
To find out the biological functions associated with each level of the CREF modules, we applied the rank-based Wilcoxon scoring method (27). The eigenvector of the CREF module at each level is polarized after its components are sorted in the ascending order. The most prominent genes at the two poles of the gene eigenvector were then analyzed by GO enrichment analysis. We used the Wilcoxon rank test to calculate p value (one-sided) for each comparison and rank these enriched pathway genes according to their significances.
Experimental Design and Statistical Rationale
Proteome analyses were performed on three biological replicates of brain tissues collected from six different brain regions at three different developmental stages. For data acquisition, we divided 54 samples into six experimental groups for 10plex LC-MS3 TMT isobaric labeling. A pool sample for inter-group control was made by pooling all 54 samples together. Each replicate was divided into 15 fractions for increasing proteome coverage, and extracted ion chromatograms were summed across the fractions for each peptide before statistical analysis. All statistical analyses were performed on three biological replicates for samples from a specific region at specific developmental stage, except Stri at S1 with one outlier (low Pearson correlation with the other two replicates) not included. To estimate the significance of difference in protein abundance, t test was used and p values were adjusted with the Benjamini-Hochberg FDR procedure. Statistical details are reported in each figure legend. Unpaired t test, two-sided Fisher’s exact test, and Kolmogorov-Smimov test were performed using Python.
Results
Spatiotemporal Proteomes of the Postnatal Dog Brain
To define dog brain development at the protein level, we quantitatively profiled the proteomes of six brain regions including cerebellar cortex (Cere), Olf, Hippo, PFC, Stri, and Amy across three postnatal developmental stages of Beagle dogs: neonatal (newborn, S1), juvenile (3-month-old, S2), and adult (3-year-old, S3) (Fig. 1A). Of those regions, PFC, Amy, Hippo, and Stri are involved in social cognition and often affected in ASD (28, 29). Proteomes were acquired using a TMT 10plex/MS3 strategy, which combines multiplexing capacity with high quantitative sensitivity and accuracy (30). To increase proteome coverage, each TMT 10plex sample was extensively fractionated by high-pH reversed-phase liquid chromatography to separate peptides by hydrophobicity and reduce sample complexity.
Fig. 1.

Spatiotemporal proteomic profiling of dog brain reveals specific synapse types enriched in different brain regions during development. A, the workflow of TMT 10plex- and MS3-based mass spectrometry quantitative proteomics. Six dog brain regions of cerebellar cortex (Cere), olfactory bulb (Olf), hippocampus (Hippo), prefrontal cortex (PFC), striatum (Stri), and amygdala (Amy) at three different stages were analyzed in the present study. For data acquisition, we divided 54 samples into 6 experimental groups for 10plex LC-MS3 TMT isobaric labeling. A pool sample for intergroup control was made by pooling all 54 samples together. B, heatmap of z-scored intensities of all 2461 region-enriched proteins at three different postnatal developmental stages after unsupervised hierarchical clustering (n = 2 for Stri at newborn stage; n = 3 for all others). Cere and Olf are apparently different from the other four regions at S2 and S3. C, heatmap of z-scored intensities of region-enriched synapse-related proteins across postnatal development. Red rectangles highlight synapse-related proteins enriched at a specific stage in a specific region. Different colors denote different types of synapses. D, heatmap of z-scored intensities of selected region-enriched proteins related with specific cell types or biological functions at different stages. Hippo, hippocampus; PFC, prefrontal cortex; TMT, tandem mass tag.
Mass spectra were searched against an integrated dog protein database (supplemental Table S1), which we constructed from UniProt, Ensembl, and NCBI databases after removing redundancy, using MaxQuant software with a peptide and protein false discovery rate of 1%. In total, 90,160 peptides corresponding to 8560 unique proteins with over 12 peptides/protein and 24% average amino acid sequence coverage were identified (supplemental Fig. S1A, and supplemental Table S2), and 6612 shared proteins were detected across all three stages (supplemental Fig. S1B). On average, more than 7000 proteins were quantifiable in each region at a specific stage (supplemental Table S2). The proteome data quality was supported by high reproducibility between biological replicates using Pearson correlation analysis (median R = 0.79; supplemental Fig. S1C). To evaluate the coverage of the proteomes, we carried out RNA-seq of samples from the same six brain regions at the juvenile stage using an Illumina platform, which resulted in 18,116 unique protein-coding genes covering 90% of annotated protein-coding genes (20,199 genes; CanFam3.1) in dog genome (supplemental Fig. S1D). The 7879 proteins identified in all six brain regions cover 43% of all protein-coding genes at the juvenile stage, indicating fewer proteins expressed than genes transcribed or many proteins were expressed at low abundance under the detection limit of mass spectrometry. Together, we generated a comprehensive proteomic resource for the postnatal dog brains.
Enrichment of Specific Synapse Types in Different Brain Regions
To identify enriched proteins for each brain region, we compared protein expression between each region and the other regions and identified 2461 region-enriched proteins in total with Cere and Olf the most different from the others (Fig. 1B and supplemental Table S3). By GO/KEGG enrichment analysis, we found that Cere and Olf were enriched with proteins involved in DNA replication and transcription regulation (Fisher’s exact test, Benjamini-Hochberg adj. p < 0.01) (supplemental Fig. S2), which may reflect a much higher density of neuronal soma in granule cell layer of Cere and Olf than the other brain regions. In addition, dopaminergic and several other types of synaptic proteins were enriched in Amy early at the newborn stage (Figs. 1C and S2). Proteins related to glutamatergic and cholinergic synapses were highly expressed in Stri while proteins associated with glutamatergic and GABAergic synapses were highly expressed in PFC from juvenile through adult stage (Figs. 1C and S2). The region-enriched synapse-related proteins at different stages are likely correlated with coordinated development of neural circuits in different brain regions. No significantly enriched pathways were identified in Hippo at any stages, suggesting that Hippo expresses various proteins at or below average levels of the other five brain regions. The top region-enriched proteins at a specific stage such as G-protein–coupled receptor family C group 5 member B (a Purkinje neuron marker) in Cere and synaptotagmin-like protein 2 (a synaptic vesicle associated protein) in PFC are related to region-specific cell types or functions (Fig. 1D). Thus, the region-enriched proteins revealed distinct cytoarchitecture and functions in different brain regions.
Timeline of Myelination in Different Regions During Postnatal Brain Development
To define temporal developmental features of the postnatal brain, we further characterized differentially expressed proteins across postnatal stages (S1 to S3) in different brain regions (supplemental Table S4). We found that there was a clear separation of proteomes between the newborn stage and the other two stages by principal component analysis (Fig. 2A). There were many more differentially expressed proteins from newborn to juvenile stages than from juvenile to adult stages (supplemental Fig. S3A). Functional enrichment analysis of all temporally regulated proteins showed that proteins involved in mitochondrial electron transport, myelin sheath, and synaptic vesicle cycle processes were upregulated in all brain regions from S1 to S2 (Figs. 2B and S3B). By contrast, the most enriched GO terms for downregulated proteins from S1 to S2 included mRNA splicing, translation initiation, and ribosome regulation in all regions except Amy (Figs. 2B and S3B). These findings suggest neural maturation and decreased neurogenesis during this period of development.
Fig. 2.

Similar trajectories of molecular events but differential progression of myelination in different brain regions. A, principal component analysis (PCA) of proteome shows a clear separation of S1 from the other two stages and Cere and Olf from the other 4 regions. Brain regions are color coded. Developmental stages are indicated by different shapes. B, bubble heatmap shows GO and KEGG enrichment of upregulated or downregulated proteins from newborn to juvenile or from juvenile to adult in six brain regions (Fisher’s exact test, fc > 1.5, adjusted p < 0.05). All six brain regions show similarly enriched pathways of upregulated proteins in S2 compared with S1. Size and color of dots indicate fold enrichment for that pathway and adjusted p value, respectively. Upregulated pathways in red and downregulated pathways in blue. C, heatmap shows the average levels of myelination stage-specific marker proteins in different brain regions during postnatal development. D, the diagram shows regional differences in the progression of myelination during postnatal development. The myelination progression at specific developmental stage is determined by the average protein levels of oligodendrocyte-lineage cell type markers indicated by colors in (C). The length of the progression bar is determined by the color intensity (above average level between white and red) shown in (C). Cere, cerebellum; GO, gene ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; Olf, olfactory bulb.
Myelination is essential for ensuring efficient connectivity within and among different brain regions (31). To better understand myelination progression in different brain regions across postnatal brain development, we analyzed the expression of 50 marker proteins that distinguish each oligodendrocyte-lineage cell type at five different stages of myelination (supplemental Table S5), including oligodendrocyte precursor cells (OPCs), differentiation-committed oligodendrocyte precursors, newly formed oligodendrocytes, myelin-forming oligodendrocytes (MFOLs), and mature oligodendrocytes (MOLs) (32). For each oligodendrocyte-lineage cell type in each region at a specific developmental stage, we calculated the average level of all expressed marker proteins for each cell type and labeled it with different color (Fig. 2C). The markers were considered substantially expressed if above the median level across the whole dataset (Fig. 2C). The myelination progression in one region at specific developmental stage is determined by the expression level of cell-type specific markers indicated by colors in Figure 2C (Fig. 2D). We note that if the number of a specific cell type is relatively low, the expression levels of marker proteins would be difficult to be detected or under the average level, while a high expression level of markers indicates more cells at a specific stage of OPC differentiation or oligodendrocyte maturation.
The results showed that Hippo reached mature MOL stage the earliest among all regions at S1 and expressed cell markers of both immature oligodendrocytes and MOLs throughout S3 (Fig. 2, C and D). By contrast, the mature MOL markers were expressed at the lowest level in Stri from S1 to S3 among all regions (Fig. 2C). In PFC, myelination stagnated at earlier MFOL stage at S3 (Fig. 2C). However, a substantial level of immature oligodendrocyte markers from OPC to MFOL expressed in PFC at S3 indicates a prolonged myelination through adulthood (Fig. 2C). Western blotting analysis confirmed an increasing level of myelin basic protein (MBP), an oligodendrocyte differentiation and maturation marker, in PFC, Hippo, and Stri from newborn to adult stages (supplemental Fig. S3C).
Together, these results revealed that the temporally regulated proteins are mostly associated with the completion of neurogenesis, formation of synapses, and myelination from S1 to S2. Moreover, our results demonstrate differential myelination progression in different brain regions with Hippo myelinated the earliest and strongest among all six brain regions.
Inter-Regional Expression Pattern of Mitochondria-Related Pathways in Dog Brain Resembles that in Human Brain
With a long history of coevolution and coadaptation with humans for over 30,000 years of domestication, dogs have developed unique human-like social cognitive capabilities (6, 33, 34, 35). Although dogs are phylogenetically more distant from humans than mice, they exhibit more complex social cognitive behaviors than mice (4). As a first step to understand the underpinnings of distinct brain functions among species, we compared the regional and inter-regional proteomes of dog brain with the corresponding proteomes of human and mouse brains which were published previously (8, 9) (Fig. 3A). Due to the unavailability of mouse and human brain proteomes at postnatal developing stages (lack of or a limited number of samples not enough for statistical analysis), we performed cross-species comparison only for adult brain proteomes. Moreover, direct comparison of expression levels for specific proteins in a brain region of different species is challenging because of the distinct protocols used in different studies. We thus examined correlations of proteomes in each region between two of the three different species and found no significant difference (supplemental Fig. S4). We then compared correlations of expression levels for each of all orthologs (2648 in total) in four matched brain regions (Cere, Hippo, PFC, and Stri) between adult dog, human, and mouse (Fig. 3B). The spatial protein expression patterns for all orthologs in the four regions of dog and human brains showed a significantly higher correlation than that between mouse and human (median Pearson correlation: 0.66 versus 0.24; Fig. 3B). We also compared the relative regional expression levels of orthologs in any two regions between dog (D)-human (H) and between mouse (M)-human (H) (Fig. 3, C and D, and supplemental Table S6). The comparisons revealed more proteins with similar relative expression levels in two regions (<2-fold in magnitude) in D-H than in M-H (Fig. 3, C and D, green). By contrast, there were more proteins exhibiting large differences (>2-fold in magnitude) or opposite changes in expression levels between two regions in M-H than in D-H (Fig. 3, C and D, magenta and blue).
Fig. 3.

Mitochondria-associated proteins are expressed in a more similar inter-regional pattern in dog and human brains compared with mouse. A, diagram of comparisons of regional and inter-regional expression patterns of orthologous proteins in four matched brain regions of dog, human, and mouse. B, the frequency distribution of Pearson correlations for each 1:1 ortholog (n = 2648) protein expression in four matched brain regions among dog (D), mouse (M), and human (H). The median correlation of human and dog proteome is higher than that between human and mouse proteome when all ortholog proteins are compared (0.66 versus 0.24). The box indicates orthologous proteins that are highly correlated between two of the three species (r > 0.67). C, protein abundance differences between two specific brain regions Stri and PFC in dog and human. Proteins are labeled as green if the magnitude of protein change in dog was <2-fold of that for human; red if the protein is changed between regions in opposite directions in dog and human; blue if the protein expression is consistently enriched in one region with a large difference of protein change magnitudes in two specific brain regions between dog and human (D/H or H/D > 2-fold), or the protein expression varies between regions only in human or in dog. D, bar plot of the percentage of protein numbers annotated in each of the color categories defined in (C) across all unique pairs of brain regions from the three species. The protein expression patterns in D-H were significantly different from that in M-H (Fisher’s exact test, ∗∗∗p < 0.001). Significantly, more orthologs are expressed similarly (green bar) in any two regions in dog and human brain than that in mouse and human brain. E, scatter plot shows GO and KEGG enrichment results of proteins with highly similar spatial expression patterns between dog and human brain (Pearson correlation coefficient D-H > 0.67; coefficient difference between D-H and M-H > 0.33, Fisher’s exact test, fc > 1.5, adjusted p < 0.05) or between mouse and human brain (D-H > 0.67; coefficient difference between M-H and D-H > 0.33). The proteins with highly similar spatial expression patterns in D-H (red) are enriched in mitochondria function, mitochondria-related disease, and myelin sheath, while the proteins with highly similar spatial expression patterns in M-H (blue) are not significantly enriched in any pathways. F, inter-regional expression patterns of mitochondria electron transport–related proteins similarly expressed in dog and human brain but different in mouse brain. The size of the circle indicates average expression level of all pathway-related proteins in each region. G, heatmap of z-scored intensities of representative proteins involved in mitochondria electron transport and myelin sheath with similar expression patterns in adult dog and human brain but different in mouse brain. KEGG, Kyoto Encyclopedia of Genes and Genomes; PFC, prefrontal cortex; Stri, striatum.
We further categorized the orthologous proteins into different groups which have similar inter-regional expression patterns across three species or between two species. Specifically, of the 2648 orthologs in all three species, 532 orthologs were expressed with similar patterns across the three species (Pearson correlation coefficient: D-H > 0.67, M-H > 0.67). Seven hundred and two and two hundred and two orthologs displayed high correlation coefficients of inter-regional expression patterns between D-H (D-H > 0.67; (D-H) - (M-H) > 0.33) and between M-H (M-H > 0.67; (M-H) - (D-H) > 0.33), respectively (Fig. 3E and supplemental Table S7). GO/KEGG enrichment analysis showed that orthologs expressed with similar patterns in the three species were enriched in mRNA splicing and synapse-related pathways (supplemental Table S7). While orthologs with high expression correlation in M-H but not in D-H were not significantly enriched in any pathways, orthologs with high expression correlation in D-H but not in M-H were enriched in mitochondrial function (mitochondria electron transport, oxidative phosphorylation), neurodegenerative diseases (Parkinson’s disease (PD), Huntington’s disease), and myelin sheath (Fig. 3E). Notably, mitochondria-related proteins are also involved in neurodegenerative diseases and myelin sheath pathway. Mitochondria electron transport–related proteins were expressed at the highest level in Stri of both dog and human, while they were expressed at the highest level in mouse Cere (Fig. 3F). Specifically, representative proteins such as NADH dehydrogenase (ubiquinone) flavoprotein 2 (Ndufv2, mitochondria and PD-related), ubiquitin C-terminal hydrolase L1 (Uchl1, PD-related), and proteolipid protein 1 (Plp1, myelin-related) exhibited more similar inter-regional expression patterns between dog and human than between mouse and human (Fig. 3G).
Together, these results showed that the inter-regional expression patterns of orthologous proteins, especially those involved in mitochondria-related pathways, were similar between dog and human but different between mouse and human.
ASD-Related Proteins Show Similar Inter-Regional Expression Patterns in Dog and Human with a High Expression Level in Cere
Many human psychiatric disorders, in which social cognition deficits are a core feature, have apparent canine analogs (36, 37). To reveal the similarities and differences in spatial expression pattern of psychiatric disease–related proteins among species, we compared the inter-regional expression patterns of ASD/Scz orthologous proteins between adult dog, human, and mouse brains. We interrogated the dog brain proteome using a set of 1003 ASD genetic risk-genes listed in the SFARI database and 321 high-confidence Scz-associated genes from the public database (PsychENCODE Integrative Analysis, http://resource.psychencode.org/) (supplemental Table S8). In total, we identified 328 ASD/Scz orthologs expressed in four brain regions of the three species and showed that the inter-regional ASD/Scz protein expression in dog brain was significantly more similar to that in human than in mouse brain (Fig. 4A, median Pearson correlation: 0.77 versus 0.27). Specifically, 86 ASD proteins with highly correlated inter-regional expression patterns in dog and human (D-H > 0.67; (D-H) - (M-H) > 0.33) were enriched in synapse, myelin sheath, and translation-related pathways (Fig. 4B). Moreover, many ASD proteins such as MeCP2 (MeCP2 mutations cause Rett Syndrome) with similar expression patterns in D-H were highly expressed in Cere in dog and human but in Hippo of mouse (Fig. 4, C and D). By contrast, a few ASD proteins such as ubiquitin protein ligase E3 component N-recognin 1 (UBR1) were highly expressed in Hippo of both mouse and human, but in Cere of dog (Fig. 4D). Our results together showed highly correlated inter-regional expression patterns of ASD/Scz proteins between dog and human, and the highly correlated ASD proteins in D-H were enriched in Cere.
Fig. 4.

Dog and human brains show a more similar inter-regional expression patterns of ASD/Scz-related proteins compared with mouse. A, the frequency distribution of Pearson correlations for each ASD/Scz ortholog protein (n = 328) among dog (D), mouse (M), and human (H) brain proteome. The spatial expression patterns of ASD/Scz orthologs in adult dog brain are more similar to that in human brain compared with mouse brain. The box indicates disease proteins that are highly correlated between two of the three species (r > 0.67). B, enriched GO terms for ASD-related proteins with similar inter-regional expression patterns between dog and human brains and between mouse and human brains. C, inter-regional expression patterns of ASD-related proteins with similar expression patterns in D-H but different in M. The size of the circle indicates average expression level (z scored) of all ASD-related proteins. D, heatmap of z-scored intensities of representative ASD proteins whose spatial expression patterns are similar in adult dog and human brain (upper panel) or similar in mouse and human brain (lower panel). E, the enrichment score of ASD/Scz-related proteins in each region is defined by the ratio of actual percentage (region-enriched/total ASD/Scz protein number)/theoretical percentage (region-enriched/total protein number). ASD/Scz proteins are mostly enriched in the Hippo and PFC at S1, and in the PFC, Stri, and Cere at S2 and S3 (Hypergeometric test, ∗p < 0.05, ∗∗∗p < 0.001). F, bubble heatmap shows GO or KEGG enrichment results of temporally regulated ASD or non-ASD proteins from newborn to juvenile stage (Fisher’s exact test, fc > 1.5, adjusted p < 0.05). Size and color of dots indicate fold enrichment for that pathway and adjusted p value, respectively. The upregulated ASD-related proteins were specifically involved in synapse-related functions in PFC, Stri, and Cere (indicated by red box) while the downregulated ASD-related proteins were specifically involved in transcription in all regions. ASD, autism spectrum disorder; Cere, cerebellum; GO, gene ontology; Hippo, hippocampus; KEGG, Kyoto Encyclopedia of Genes and Genomes; PFC, prefrontal cortex; Scz, schizophrenia; Stri, striatum.
The spatiotemporal expression of ASD/Scz-related proteins in human brain remains unclear. To characterize the brain regions that may be preferentially affected in diseases, we characterized the spatiotemporal expression features of ASD/Scz-related proteins in postnatal dog brain. In total, we identified 697 ASD-associated proteins and 158 Scz-risk proteins in the dog brain proteome (supplemental Table S8). Of all detected ASD/Scz proteins, 560 (66%) proteins were evenly distributed in different brain regions, while 295 (34%) proteins were enriched in one of the regions. Region-enriched ASD/Scz proteins were mostly enriched in Hippo at S1 and in PFC at S2 and S3 (Fig. 4E). Region-enriched ASD proteins were specifically involved in synapse-related pathways in Hippo (S1), PFC (S3), and Stri (S3) compared with non-ASD enriched proteins (supplemental Fig. S5 and supplemental Table S8). Temporally, most of ASD-related proteins were differentially expressed from S1 to S2 in each brain region (supplemental Fig. S6, A and B). GO/KEGG enrichment analysis of temporally regulated ASD proteins from S1 to S2 showed that upregulated ASD-related proteins were involved in glutamatergic and other types of synapses, calcium signaling, and postsynaptic density in the Cere, PFC, and Stri compared with non-ASD proteins (Fig. 4F and supplemental Table S8). Downregulated ASD-associated proteins from S1 to S2 stages were involved in transcription-related pathways in all six brain regions (Fig. 4F).
Conserved Myelination and Hippo-Related Pathways Between Dog and Human by CREF Analysis
Alterations in CREs, especially the gain and loss of CREs by insertion or deletion of transposon elements, have a great influence on the number of transcription factor–binding site and methylation site which alter gene expression and contribute to phenotypic diversity within and between species and thus constitute a key genetic basis for environmental adaption or selection (15, 16, 17, 38). However, study on the changes of the number of CREs has been rare due to analytical complexities and lack of appropriate methods until recently (19). We applied the recently developed CREF analysis method (19) to compare the genome-wide frequency distribution of cis-motifs for all protein-coding genes in dog, human, and mouse in order to understand the genetic basis accounting for protein expression similarities and differences in the three species.
The CREF matrix of all genes in a specific species was stratified by robust singular value decomposition into eigen modules of levels from high to low cis-motif frequencies for each species as described previously (19). The CREF module at each frequency level has a dual feature: one eigenvector for the motifs and another for protein-coding genes. Each CREF module can be compared across species by correlation analysis of their motifs, while the biological relevance of each CREF module can be interpreted by enrichment analysis of the regulated genes (Fig. 5A).
Fig. 5.

The cis-regulatory element modules regulating myelination and hippocampus-related pathways are conserved between dog and human. A, the cis-regulatory element frequency (CREF) matrix of all genes in a specific species was stratified by robust singular value decomposition into eigen modules of levels from high to low cis-motif frequencies for each species. The CREF module at each frequency level has a dual feature: one eigenvector for the motifs and another for protein-coding genes. Each CREF module can be compared across species by correlation analysis of their motifs, while the biological relevance of each CREF module can be interpreted by enrichment analysis of the regulated genes. The similarity of enriched functional pathways between species was verified at both genetic and proteomic levels. B, Pearson correlations of top nine motif eigenvectors in CREF modules between dog and human, and between mouse and human. The first three modules are highly correlated (Pearson correlation >0.97), suggesting these three modules are conserved among three species. In contrast, the fourth, fifth, and ninth modules are more correlated between dog and human. C, biological processes enriched in the fourth, fifth, and ninth modules for human, dog, and mouse, as evaluated by the Wilcoxon scoring method. GO terms of myelination, hippocampus development, and long-term memory are enriched in the fourth or ninth modules in dog, human, or both, but are not in mouse. D, the dot plots show correlations of the inter-regional expression patterns of proteins in the enriched biological processes as shown in (C) between two species of the three. The gene set of CNS myelin proteins were manually curated. The expression pattern of proteins involved in long-term synaptic plasticity was similar among the three species and thus not included in the panel. E, the average expression level (z scored) of CNS myelin and long-term memory-related proteins in different brain regions of three species. The size of the circle indicates average expression level of all pathway-related proteins in each region. The red box indicate region with the highest average expression levels of pathway-related proteins in all three species (left panel) or one specific species (right panel). F, the bar graph shows higher correlated expressions of proteins related with myelin and long-term memory in the hippocampus between dog and human than between mouse and human. G, the bar graph shows the summarized level of three major myelin proteins MBP, PLP1, and MAG in Hippo and PFC with reference to that in Cere in three species (estimates of ratios with 95% confidence intervals). Cere, cerebellum; GO, gene ontology; Hippo, hippocampus; MBP, myelin basic protein; PFC, prefrontal cortex.
We identified conserved as well as divergent CREF modules at different frequency levels in the three species of dog, human, and mouse (Fig. 5B). Specifically, CREF modules at levels 1, 2, and 3 of high motif frequencies, with all Pearson correlations larger than 0.97, were conserved among the three species. While the CREF modules at levels 4, 5, and 9 with Pearson correlations 0.93, 0.93, and 0.92, respectively, were more conserved between dog and human than between mouse and human (0.58, 0.54, and 0.01, respectively). Although CREF modules at level 7 were more similar between mouse and human, the correlation coefficient was low at 0.57 (Fig. 5B). The correlation coefficients of CREF modules at levels 6 and 8 were similar between D-H and M-H pairs (Fig. 5B).
We then analyzed the distribution of genes which encode 2648 orthologous proteins detected in the four brain regions of the three species in CREF modules at the top nine levels. The mouse orthologous genes were distributed at the highest level in modules at level 5 but distributed at the highest level at level 4 for both dog and human (supplemental Fig. S7), indicating that these orthologous genes in dog and human are under the control of similar CREF modules with a higher frequency level than that in mouse.
Cis-regulatory modules that are conserved among three species at the top three levels mainly regulate genes involved in basic biological pathways including reproduction, embryogenesis, fetal maturation, immune system, stress response, and mitosis as reported before for human and chimpanzee comparison (19). The genes regulated by CREF modules at level 4 were enriched in multiple pathways including CNS myelination, hippocampal CA3 synapse, synaptic plasticity, and long-term memory in dog or human (Fig. 5C). In mouse, however, genes enriched in CA3 synapse, synaptic plasticity, and long-term memory were regulated by CREF modules at level 5 (Fig. 5C).
Consistent with the conserved CREF modules, the inter-regional expression patterns of proteins involved in myelination and Hippo-related pathways as shown in Figure 4C were more similar between dog and human than between mouse and human (Fig. 5D). There were only a few CNS myelination-related proteins identified in the dog brain proteome which are not enough for correlation analysis, we thus manually curated a gene set of CNS myelin proteins (supplemental Table S9) based on previous studies of myelin proteome (39, 40). We found that although the average expression levels of CNS myelin proteins were the highest in Hippo of all three species (Fig. 5E, left panel), the specific proteins enriched in Hippo were similar in dog and human but different in mouse (supplemental Table S9). In addition, the average expression level of proteins involved in Hippo-related long-term memory was similar in different regions of dog and human but enriched in Hippo of mouse (Fig. 5E, right panel). The regional expression of CNS myelin and long-term memory proteins in Hippo was also highly correlated between dog and human (Fig. 5F). To verify if the expression level of specific orthologous proteins in different species are consistent with the CREF and proteomic correlation analysis, we estimated the relative levels of three major myelin proteins including MBP, PLP1, and myelin-associated glycoprotein, in Hippo and PFC with reference to Cere for each species, since direct comparison is challenging due to the different protocols used in different studies. The statistical results showed that the total level of myelin proteins in Hippo was ∼6-fold more abundant in human and 2.2-fold more abundant in dog, but slightly less than 1-fold in mouse, compared with the levels in Cere (Fig. 5G). A similar trend of myelin proteins between the three species was also observed in PFC (Fig. 5G).
These results suggest that although the mouse and human are more closely related phylogenetically (4), specific cis-regulatory modules regulating myelination and Hippo-related pathways in dog are more similar with that in humans, in agreement with the inter-regional expression patterns of those pathway-related proteins.
Discussion
The brain is a network consisting of spatially localized but functionally linked regions whose protein expressions are precisely regulated. Herein, we generated spatiotemporal brain proteome using a label-based approach for domestic dogs, which have gyrencephalic brain structure and remarkable social cognitive abilities similar to humans. We provided quantitative protein expression profiles in six brain regions from newborn to adult stages. Moreover, we performed comparison of adult brain proteomes together with genome-wide CREF analysis between dog, human, and mouse, which enabled us to identify evolutionarily conserved pathways underlying brain functions in different species.
We found similar inter-regional expression patterns of mitochondria-related proteins, with the highest expression in Stri, in both dog and human but not in mouse. Genes enriched in oxidative synthesis and metabolism of ATP have been reported to be highly expressed in brain network hubs (13). Human brain network hubs include areas of association cortex, putamen (part of dorsal Stri), and thalamus, regions that play a central role in higher-order cognition (41). The similar high expression level of mitochondria-related proteins in Stri supports Stri as a hub in brain network in both dog and human. By contrast, mitochondria-related proteins were highly expressed in Cere but not Stri of mouse, indicating that mouse Stri may have a lower energy demand than that in dog and human.
The inter-regional expression patterns of myelin sheath and myelination-related proteins were also highly correlated between dog and human but not between mouse and human. Specifically, myelin proteins were highly expressed in Hippo and PFC of dog and human. As similar expressions of genes, especially those regulating neuronal development and axonal guidance, in two regions partially predict mutual synaptic connectivity (13), the increased expression of proteins regulating myelination in Hippo and PFC may indicate stronger axonal connections and thus higher myelination between the two. Consistently, Hippo is structurally connected to medial PFC via white matter tracts in humans; Hippo and PFC are both involved in different circuits including anxiety, cognitive, and social behavior (42, 43). Whether myelin protein expressions between different regions in dog and human are correlated developmentally remains unknown due to the lack of postnatal multiregional brain proteomes of human.
In addition to mapping of physical locations, emerging evidence shows that Hippo also plays a critical role in social cognition by tracking and interpreting social relationships (44, 45, 46). Reduced white matter integrity of long-range fiber tracts linking Hippo with orbitofrontal cortex has been observed in ASD patients and is associated with impaired social functions (42, 47, 48, 49). In the present study, we showed that Hippo was myelinated at the highest level among all six regions early at the newborn stage and exhibited a long-lasting myelination through adulthood, which may allow the formation of efficient neural circuits and ensure its intra- and inter-regional connectivity with other brain regions. Similar long-lasting myelination may also occur in human brain, as neurogenesis was identified in the dentate gyrus of the adult Hippo (50). Moreover, we observed prolonged myelination in adult dog PFC, similar to that in human neocortex (11, 51). Prolonged myelination in human neocortex plays an important role in the development of cognitive abilities by providing a longer period for social learning to influence the establishment of neural circuits (51, 52). The similar high level and prolonged expression of myelin proteins in Hippo and PFC of dog and human may contribute to their similar social and cognitive abilities.
Consistent with the proteomic results, we identified specific cis-regulatory modules regulating myelination and Hippo-related pathways conserved between dog and human. It is worth noting that although the expression patterns of mitochondria-related proteins were also similar between dog and human, we did not identify CREF modules regulating mitochondria-related pathways conserved between dog and human, indicating that the conserved cis-regulatory modules between dog and human are limited to the regulation of specific pathways. Moreover, genes involved in the conserved myelination and Hippo-related pathways such as MBP, endophilin-A1, adenylate cyclase type 8, and major prion protein have also been identified as domestication-selected genes in dogs (34, 53, 54). We propose that specific cis-regulatory modules regulating myelination and Hippo-related pathways in dog might have undergone an adapted evolution during domestication. As wolf, which has not been domesticated, and dog share the common ancestor and have similar brain size and structure, it would be interesting to perform cross-species comparison between wolf, dog, and human to determine whether the conserved myelination and Hippo-related pathways are the result of domestication and coevolution of dogs with humans.
In contrast, we identified mouse-specific expression patterns of proteins involved in long-term memory, which were expressed the highest in Hippo only in mouse. The hippocampal longitudinal axis is oriented dorsoventrally in mice, while this axis is rotated into the posterior-anterior direction in primates and humans (55). Thus, the mouse Hippo might have different connectivities with other brain regions from that in human Hippo. Whether the enrichment of long-term memory-related proteins in mouse Hippo indicates different functions from that in dog and human needs further investigation.
We found that there are a large number of ASD-related proteins expressed with similar patterns, that is, the highest expression in Cere between dog and human but different between mouse and human. Compared with the extensive study of ASD in PFC, the literature for ASD study in Cere, which is primarily involved in motor control, is sparse. Structural abnormalities and volume differences have been detected in Cere of individuals with ASD (56). Mutations in a few genes with a high penetrance risk for ASD including chromodomain helicase DNA-binding protein 8 (CHD8) and Shank2 are reported to affect Purkinje cell function and cerebellar development (57, 58). These findings, together with our results support a previously underappreciated role of Cere in ASD.
In summary, our study delineated the spatial protein expression patterns during postnatal dog brain development. The comprehensive resource of dog brain proteome data we generated makes it possible to correlate spatial regulation of protein expression in different regions with neural circuit formation and functions. The similarity and difference of spatial expression patterns for each orthologous protein, including neurodegenerative and neuropsychiatric diseases–related proteins, among human, dog, and mouse provide important information for evaluation of the relevance of experimental findings in different animal models to that in human.
Data availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD033420 and PXD034449.
Supplemental data
This article contains supplemental data.
Conflict of interest
The authors declare no competing interests.
Acknowledgments
We thank Y. Li for advice on data analysis and generating the integrated dog proteome library.
Funding and additional information
This work was supported by grants from The National Key Research and Development Program (2019YFA0707100 and 2021ZD0203901 to Y. Q. Z.), the Strategic Priority Research Program B of the Chinese Academy of Sciences (XDBS1020100 to Y. Q. Z.), the National Science Foundation of China to Y. Q. Z. (31830036 and 31921002) and L. M. L. (32170679 and 11871462), and the Innovative Research Team (in Science and Technology) of Yunnan Province (201905E160019 to G.-D. W.), and Key Research Program of Frontier Sciences of the CAS (ZDBS-LY-SM011 to G.-D. W.).
Author contributions
Z. D., Y. Q. Z., and Y. X. conceptualization; Y. W. and Y. X. methodology; Z. Z. and Y. X. validation; H. H., Z. Z., C. G., G.-D. W., J. S., L. M. L., Z. D., and Y. X. formal analysis; H. H. and H. Z. investigation; J.-P. Z. resources; X. H. and Y. W. data curation; H. H., Y. Q. Z., and Y. X. writing–original draft; H. H., Y.-P. Z., Q.-F. W., Y.-H. J., L. M. L., Z. D., Y. Q. Z., and Y. X. writing–review and editing; H. H., Z. Z., and Y. X. visualization; Z. D. and Y. Q. Z. supervision; G.-D. W., L. M. L., and Y. Z. funding acquisition.
Footnotes
Present address for Zhiguang Zhao: Hebei Industrial Technology Research Institute of Genomics in Maternal & Child Health, Shijiazhuang 050000, China.
Contributor Information
Yong Q. Zhang, Email: yqzhang@genetics.ac.cn.
Ying Xiong, Email: yxiong@genetics.ac.cn.
Supplemental Data
References
- 1.Van Essen D.C., Donahue C.J., Coalson T.S., Kennedy H., Hayashi T., Glasser M.F. Cerebral cortical folding, parcellation, and connectivity in humans, nonhuman primates, and mice. Proc. Natl. Acad. Sci. U. S. A. 2019;116:26173–26180. doi: 10.1073/pnas.1902299116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nestler E.J., Hyman S.E. Animal models of neuropsychiatric disorders. Nat. Neurosci. 2010;13:1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bakken T.E., Miller J.A., Ding S.L., Sunkin S.M., Smith K.A., Ng L., et al. A comprehensive transcriptional map of primate brain development. Nature. 2016;535:367–375. doi: 10.1038/nature18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murphy W.J., Eizirik E., Johnson W.E., Zhang Y.P., Ryder O.A., O'Brien S.J. Molecular phylogenetics and the origins of placental mammals. Nature. 2001;409:614–618. doi: 10.1038/35054550. [DOI] [PubMed] [Google Scholar]
- 5.Johnson P.J., Luh W.M., Rivard B.C., Graham K.L., White A., FitzMaurice M., et al. Stereotactic cortical atlas of the domestic canine brain. Sci. Rep. 2020;10:4781. doi: 10.1038/s41598-020-61665-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hare B., Brown M., Williamson C., Tomasello M. The domestication of social cognition in dogs. Science. 2002;298:1634–1636. doi: 10.1126/science.1072702. [DOI] [PubMed] [Google Scholar]
- 7.MacLean E.L., Herrmann E., Suchindran S., Hare B. Individual differences in cooperative communicative skills are more similar between dogs and humans than chimpanzees. Anim. Behav. 2017;126:41–51. [Google Scholar]
- 8.Sharma K., Schmitt S., Bergner C.G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., et al. Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 2015;18:1819–1831. doi: 10.1038/nn.4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlyle B.C., Kitchen R.R., Kanyo J.E., Voss E.Z., Pletikos M., Sousa A.M.M., et al. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017;20:1787–1795. doi: 10.1038/s41593-017-0011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.He Z., Han D., Efimova O., Guijarro P., Yu Q., Oleksiak A., et al. Comprehensive transcriptome analysis of neocortical layers in humans, chimpanzees and macaques. Nat. Neurosci. 2017;20:886–895. doi: 10.1038/nn.4548. [DOI] [PubMed] [Google Scholar]
- 11.Zhu Y., Sousa A.M.M., Gao T., Skarica M., Li M., Santpere G., et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science. 2018;362 doi: 10.1126/science.aat8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fornito A., Arnatkeviciute A., Fulcher B.D. Bridging the gap between connectome and transcriptome. Trends Cogn. Sci. 2019;23:34–50. doi: 10.1016/j.tics.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 13.Bullmore E., Sporns O. The economy of brain network organization. Nat. Rev. Neurosci. 2012;13:336–349. doi: 10.1038/nrn3214. [DOI] [PubMed] [Google Scholar]
- 14.Collin G., Sporns O., Mandl R.C., van den Heuvel M.P. Structural and functional aspects relating to cost and benefit of rich club organization in the human cerebral cortex. Cereb. Cortex. 2014;24:2258–2267. doi: 10.1093/cercor/bht064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cordaux R., Batzer M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sahlen P., Yanhu L., Xu J., Kubinyi E., Wang G.D., Savolainen P. Variants that differentiate wolf and dog populations are enriched in regulatory elements. Genome Biol. Evol. 2021;13 doi: 10.1093/gbe/evab076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittkopp P.J., Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 2011;13:59–69. doi: 10.1038/nrg3095. [DOI] [PubMed] [Google Scholar]
- 18.Lanciano S., Cristofari G. Measuring and interpreting transposable element expression. Nat. Rev. Genet. 2020;21:721–736. doi: 10.1038/s41576-020-0251-y. [DOI] [PubMed] [Google Scholar]
- 19.Li L., Zhang S., Li L.M. Dual eigen-modules of cis-element regulation profiles and selection of cognition-language eigen-direction along evolution in hominidae. Mol. Biol. Evol. 2020;37:1679–1693. doi: 10.1093/molbev/msaa036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palazzi X. Springer-Verlag; NY: 2011. The Beagle Brain in Stereotaxic Coordinates. [Google Scholar]
- 21.Wisniewski J.R., Zougman A., Nagaraj N., Mann M. Universal sample preparation method for proteome analysis. Nat. Met. 2009;6:359–362. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 22.Udeshi N.D., Mertins P., Svinkina T., Carr S.A. Large-scale identification of ubiquitination sites by mass spectrometry. Nat. Protoc. 2013;8:1950–1960. doi: 10.1038/nprot.2013.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim D., Paggi J.M., Park C., Bennett C., Salzberg S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019;37:907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pertea M., Kim D., Pertea G.M., Leek J.T., Salzberg S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016;11:1650–1667. doi: 10.1038/nprot.2016.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang D., Liu S., Warrell J., Won H., Shi X., Navarro F.C.P., et al. Comprehensive functional genomic resource and integrative model for the human brain. Science. 2018;362 doi: 10.1126/science.aat8464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodriguez J.M., Maietta P., Ezkurdia I., Pietrelli A., Wesselink J.J., Lopez G., et al. Appris: annotation of principal and alternative splice isoforms. Nucl. Acids Res. 2013;41:D110–D117. doi: 10.1093/nar/gks1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng C., Fabrizio P., Ge H., Wei M., Longo V.D., Li L.M. Significant and systematic expression differentiation in long-lived yeast strains. PLoS One. 2007;2 doi: 10.1371/journal.pone.0001095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Varghese M., Keshav N., Jacot-Descombes S., Warda T., Wicinski B., Dickstein D.L., et al. Autism spectrum disorder: neuropathology and animal models. Acta Neuropathol. 2017;134:537–566. doi: 10.1007/s00401-017-1736-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amaral D.G., Schumann C.M., Nordahl C.W. Neuroanatomy of autism. Trends Neurosci. 2008;31:137–145. doi: 10.1016/j.tins.2007.12.005. [DOI] [PubMed] [Google Scholar]
- 30.McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wuhr M., Huttlin E.L., Erickson B.K., et al. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nave K.A., Werner H.B. Myelination of the nervous system: mechanisms and functions. Annu. Rev. Cell Dev. Biol. 2014;30:503–533. doi: 10.1146/annurev-cellbio-100913-013101. [DOI] [PubMed] [Google Scholar]
- 32.Marques S., Zeisel A., Codeluppi S., van Bruggen D., Mendanha Falcao A., Xiao L., et al. Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science. 2016;352:1326–1329. doi: 10.1126/science.aaf6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagasawa M., Mitsui S., En S., Ohtani N., Ohta M., Sakuma Y., et al. Social evolution. Oxytocin-gaze positive loop and the coevolution of human-dog bonds. Science. 2015;348:333–336. doi: 10.1126/science.1261022. [DOI] [PubMed] [Google Scholar]
- 34.Wang G.D., Zhai W., Yang H.C., Wang L., Zhong L., Liu Y.H., et al. Out of southern east asia: the natural history of domestic dogs across the world. Cell Res. 2016;26:21–33. doi: 10.1038/cr.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ostrander E.A., Wayne R.K., Freedman A.H., Davis B.W. Demographic history, selection and functional diversity of the canine genome. Nat. Rev. Genet. 2017;18:705–720. doi: 10.1038/nrg.2017.67. [DOI] [PubMed] [Google Scholar]
- 36.Ledford H. Dog DNA probed for clues to human psychiatric ills. Nature. 2016;529:446–447. doi: 10.1038/529446a. [DOI] [PubMed] [Google Scholar]
- 37.Overall K.L. Natural animal models of human psychiatric conditions: assessment of mechanism and validity. Prog. Neuropsychopharmacol. Biol. Psych. 2000;24:727–776. doi: 10.1016/s0278-5846(00)00104-4. [DOI] [PubMed] [Google Scholar]
- 38.Xu H., Sepulveda L.A., Figard L., Sokac A.M., Golding I. Combining protein and mRNA quantification to decipher transcriptional regulation. Nat. Met. 2015;12:739–742. doi: 10.1038/nmeth.3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jahn O., Siems S.B., Kusch K., Hesse D., Jung R.B., Liepold T., et al. The CNS myelin proteome: deep profile and persistence after post-mortem delay. Front. Cell Neurosci. 2020;14:239. doi: 10.3389/fncel.2020.00239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jahn O., Tenzer S., Werner H.B. Myelin proteomics: molecular anatomy of an insulating sheath. Mol. Neurobiol. 2009;40:55–72. doi: 10.1007/s12035-009-8071-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Heuvel M.P., Sporns O. Rich-club organization of the human connectome. J. Neurosci. 2011;31:15775–15786. doi: 10.1523/JNEUROSCI.3539-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ameis S.H., Catani M. Altered white matter connectivity as a neural substrate for social impairment in Autism Spectrum Disorder. Cortex. 2015;62:158–181. doi: 10.1016/j.cortex.2014.10.014. [DOI] [PubMed] [Google Scholar]
- 43.Benchenane K., Peyrache A., Khamassi M., Tierney P.L., Gioanni Y., Battaglia F.P., et al. Coherent theta oscillations and reorganization of spike timing in the hippocampal- prefrontal network upon learning. Neuron. 2010;66:921–936. doi: 10.1016/j.neuron.2010.05.013. [DOI] [PubMed] [Google Scholar]
- 44.Tavares R.M., Mendelsohn A., Grossman Y., Williams C.H., Shapiro M., Trope Y., et al. A map for social navigation in the human brain. Neuron. 2015;87:231–243. doi: 10.1016/j.neuron.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Smith A.S., Williams Avram S.K., Cymerblit-Sabba A., Song J., Young W.S. Targeted activation of the hippocampal CA2 area strongly enhances social memory. Mol. Psych. 2016;21:1137–1144. doi: 10.1038/mp.2015.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schafer M., Schiller D. Navigating social space. Neuron. 2018;100:476–489. doi: 10.1016/j.neuron.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Travers B.G., Adluru N., Ennis C., Tromp do P.M., Destiche D., Doran S., et al. Diffusion tensor imaging in autism spectrum disorder: a review. Autism Res. 2012;5:289–313. doi: 10.1002/aur.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Banker S.M., Gu X., Schiller D., Foss-Feig J.H. Hippocampal contributions to social and cognitive deficits in autism spectrum disorder. Trends Neurosci. 2021;44:793–807. doi: 10.1016/j.tins.2021.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y., Olson I.R. The original social network: white matter and social cognition. Trends Cogn. Sci. 2018;22:504–516. doi: 10.1016/j.tics.2018.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Denoth-Lippuner A., Jessberger S. Formation and integration of new neurons in the adult hippocampus. Nat. Rev. Neurosci. 2021;22:223–236. doi: 10.1038/s41583-021-00433-z. [DOI] [PubMed] [Google Scholar]
- 51.Miller D.J., Duka T., Stimpson C.D., Schapiro S.J., Baze W.B., McArthur M.J., et al. Prolonged myelination in human neocortical evolution. Proc. Natl. Acad. Sci. U. S. A. 2012;109:16480–16485. doi: 10.1073/pnas.1117943109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Monje M. Myelin plasticity and nervous system function. Annu. Rev. Neurosci. 2018;41:61–76. doi: 10.1146/annurev-neuro-080317-061853. [DOI] [PubMed] [Google Scholar]
- 53.Axelsson E., Ratnakumar A., Arendt M.L., Maqbool K., Webster M.T., Perloski M., et al. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature. 2013;495:360–364. doi: 10.1038/nature11837. [DOI] [PubMed] [Google Scholar]
- 54.Vonholdt B.M., Pollinger J.P., Lohmueller K.E., Han E., Parker H.G., Quignon P., et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature. 2010;464:898–902. doi: 10.1038/nature08837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Strange B.A., Witter M.P., Lein E.S., Moser E.I. Functional organization of the hippocampal longitudinal axis. Nat. Rev. Neurosci. 2014;15:655–669. doi: 10.1038/nrn3785. [DOI] [PubMed] [Google Scholar]
- 56.Wang S.S., Kloth A.D., Badura A. The cerebellum, sensitive periods, and autism. Neuron. 2014;83:518–532. doi: 10.1016/j.neuron.2014.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peter S., Ten Brinke M.M., Stedehouder J., Reinelt C.M., Wu B., Zhou H., et al. Dysfunctional cerebellar Purkinje cells contribute to autism-like behaviour in Shank2-deficient mice. Nat. Commun. 2016;7 doi: 10.1038/ncomms12627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kawamura A., Katayama Y., Kakegawa W., Ino D., Nishiyama M., Yuzaki M., et al. The autism-associated protein CHD8 is required for cerebellar development and motor function. Cell Rep. 2021;35 doi: 10.1016/j.celrep.2021.108932. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifiers PXD033420 and PXD034449.