

Karnes and colleagues report on the results of a genome-wide association study to find predictors of heparin-induced thrombocytopenia, comparing patients with a positive functional assay for platelet activation, patients with anti-PF4 antibodies, and antibody-negative controls. A positive functional assay is highly associated with ABO O blood group, and a single nucleotide polymorphism is identified for future exploration of clinical utility in risk prediction.
Key Points
We identify the ABO O blood group as a novel risk factor for platelet reactivity and HIT.
Our results clarify the biology underlying HIT pathogenesis with ramifications for HIT prediction and implications for related conditions.
Visual Abstract

Abstract
Heparin-induced thrombocytopenia (HIT) is an unpredictable, potentially catastrophic adverse effect resulting from an immune response to platelet factor 4 (PF4)/heparin complexes. We performed a genome-wide association study (GWAS) with positive functional assay as the outcome in a large discovery cohort of patients divided into 3 groups: (1) functional assay-positive cases (n = 1269), (2) antibody-positive (functional assay-negative) controls (n = 1131), and (3) antibody-negative controls (n = 1766). Significant associations (α = 5 × 10−8) were investigated in a replication cohort (α = 0.05) of functional assay-confirmed HIT cases (n = 177), antibody-positive (function assay-negative) controls (n = 258), and antibody-negative controls (n = 351). We observed a strong association for positive functional assay with increasing PF4/heparin immunoglobulin-G (IgG) level (odds ratio [OR], 16.53; 95% confidence interval [CI], 13.83-19.74; P = 1.51 × 10−209) and female sex (OR, 1.15; 95% CI, 1.01-1.32; P = .034). The rs8176719 C insertion variant in ABO was significantly associated with positive functional assay status in the discovery cohort (frequency = 0.41; OR, 0.751; 95% CI, 0.682-0.828; P = 7.80 × 10−9) and in the replication cohort (OR, 0.467; 95% CI, 0.228-0.954; P = .0367). The rs8176719 C insertion, which encodes all non-O blood group alleles, had a protective effect, indicating that the rs8176719 C deletion and the O blood group were risk factors for HIT (O blood group OR, 1.42; 95% CI, 1.26-1.61; P = 3.09 × 10−8). Meta-analyses indicated that the ABO association was independent of PF4/heparin IgG levels and was stronger when functional assay-positive cases were compared with antibody-positive (functional assay-negative) controls than with antibody-negative controls. Sequencing and fine-mapping of ABO demonstrated that rs8176719 was the causal single nucleotide polymorphism (SNP). Our results clarify the biology underlying HIT pathogenesis with ramifications for prediction and may have important implications for related conditions, such as vaccine-induced thrombotic thrombocytopenia.
Introduction
Heparin-induced thrombocytopenia (HIT) is an antibody-mediated adverse drug reaction that results from abnormal platelet activation in patients receiving unfractionated heparin (UFH) and low molecular weight heparin (LMWH).1,2 HIT develops in up to 2.4% of patients treated with UFH, has a significant mortality rate, and results in potentially catastrophic thromboembolic complications, including life- and limb-threatening thrombosis.3,4 Prevention of HIT-related thrombosis is currently possible only after manifestations of HIT are evident and the disease process has already begun.2,5 The inability to predict HIT thus represents a liability with heparin administration. Emerging evidence also implicates platelet-activating antiplatelet factor 4 (PF4) antibodies in adenoviral vector severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) vaccine-induced thrombotic thrombocytopenia (VITT), which is characterized by high levels of HIT-mimicking antibodies to PF4-polyanion complexes, unusual thrombotic sites, and high mortality, similar to autoimmune HIT.6-8 Identification of genetic risk factors for HIT has the potential to both inform our knowledge of HIT and related conditions, such as VITT, and to elucidate clinically implementable biomarkers for prevention of HIT.9
Despite decades of research into the immunopathology of HIT, fundamental knowledge gaps remain.10,11 Although multiple risk factors are known, triggers for antibody formation remain poorly defined. The clinical significance of nonpathogenic PF4/heparin antibodies and the molecular basis that distinguishes them from pathogenic PF4/heparin antibodies remain unclear. Although informative, prior targeted molecular approaches have not fully identified the mechanisms of HIT, likely due to the complicated and unusual nature of the HIT immune response as well as the narrow focus of these targeted approaches. Discovery-oriented approaches, such as genome-wide association studies (GWAS), have the potential to identify genetic risk factors without reliance on prior biological knowledge. While prior GWAS have identified potential genetic risk factors associated with the development of HIT, these studies have been limited by a small number of HIT cases. The largest HIT GWAS to date included 96 cases in the discovery cohort and 86 cases in the replication cohort.12,13 These studies are also limited by the lack of functional assay confirmation of these cases and the lack of a PF4/heparin antibody-positive (functional assay-negative) control group, which enables differentiation between genetic predictors of HIT and PF4/heparin antibody production.
The objective of this study was to identify and validate genetic predictors of platelet reactivity and HIT using a GWAS approach. We conducted this study in a large observational population of patients who underwent laboratory testing for PF4/heparin antibodies and the heparin-induced platelet activation (HIPA) assay using the platelet laboratory at Greifswald University Hospital (Greifswald, Germany; discovery cohort). Significant associations with platelet reactivity from the discovery cohort were tested for association in an independent, prospectively collected replication cohort from the University Hospital of Tours (Tours, France) (replication cohort).14,15
Methods
Study populations
Discovery cohort
An observational cohort of patients who underwent laboratory testing for PF4/heparin antibodies and HIPA at the platelet laboratory at Greifswald University Hospital constituted the discovery cohort. Greifswald University Medicine is used as a central testing facility for HIT diagnostic assays across multiple institutions and uses standardized polyclonal enzyme-linked immunosorbent assay (ELISA) for PF4/heparin antibodies, including immunoglobulin-G (IgG), IgA, and IgM quantification, and HIPA for functional assay confirmation of HIT. HIPA is a washed platelet assay with very similar characteristics to the serotonin release assay (SRA) used in North America.16 The HIPA assay has been extensively validated in comparison with the SRA and by analysis of prospective clinical study data.16-19 Samples for this GWAS were referred due to clinical suspicion of HIT, were tested between 2002 and 2015, and were completely deidentified before GWAS genotyping. The discovery cohort was divided into 3 groups: (1) functional assay-positive cases with both positive HIPA and positive PF4/heparin antibodies; (2) antibody-positive (functional assay-negative) controls with negative HIPA and positive PF4/heparin antibodies; and (3) PF4/heparin antibody-negative controls with both negative functional assay (HIPA) and PF4/heparin antibody-negative results. This research received ethical approval from the institutional ethics committee at Greifswald University.
Replication cohort
The replication cohort was made up of 2 previously described cohorts recruited prospectively at the University Hospital of Tours (Tours, France).14,15 The replication cohort was similarly divided into 3 groups: (1) SRA-confirmed HIT cases; (2) antibody-positive (functional assay [SRA]-negative) controls with positive PF4/heparin antibodies; and (3) PF4/heparin antibody-negative controls with both negative functional assay (SRA) and PF4/heparin antibody results. Both PF4-specific ELISA and SRA were positive in every HIT case. More detailed clinical data were available on these prospectively collected patients, including age, sex, UFH vs LMWH treatment, platelet counts before and following heparin treatment, and whether patients underwent cardiopulmonary bypass surgery. This research received ethical approval from both the University Hospital of Tours ethics committee and the Ministry of Research. Additional details regarding the discovery and replication populations are described in the supplemental Materials.
Genotyping, imputation, and quality control
GWAS genotyping was performed at the Laboratory for Genotyping Development, RIKEN Center for Integrative Medical Sciences (IMS). All samples in the discovery and replication cohorts were genotyped using the lllumina Infinium HumanOmniExpressExome BeadChip, which contains 958 497 markers, including 273 000 functional exonic markers. Quality control was accomplished using sample and marker call rate ≥98%, sex mismatch, duplicate concordance, and removal of related samples through identity by descent. Genomic imputation was performed for samples using Minimac4 and the phase3 v5 reference panel data from the 1000 genomes project.20 To ensure consistency of our results with alternative imputation platforms, genomic imputation was also performed using the Trans-Omics for Precision Medicine (TOPMed) Imputation Server.21 During statistical analyses, SNPs were removed for Hardy-Weinberg equilibrium (HWE) P < .001 and minor allele frequency (MAF) <0.01.
Analysis and adjustment for ancestry
To minimize confounding by population stratification, GWAS analyses were adjusted for the first 3 principal components (PCs). Since the overwhelming majority of patients in both cohorts were of European ancestry, the primary GWAS analysis was restricted to these individuals (supplemental Figures 1 and 2). Estimated proportions of global ancestry were determined using ancestry-informative markers input into STRUCTURE with HapMap reference populations.22 In order to incorporate samples with non-European ancestry, we also performed a mixed-model analysis using Genome-wide complex trait analysis (GCTA),23 which included successfully genotyped individuals with any ancestry and included related samples.
Sequencing and fine-mapping of GWAS association
In order to investigate functional markers in genome-wide associated regions, multiplex polymerase chain reaction-based target sequencing was performed at RIKEN IMS, as previously described.24 Sequencing was performed in all discovery and replication cohort samples to acquire full coverage of the ABO gene ±5000 base pairs (kbps). Fine-mapping of causal variants from the association with functional assay-positive status summary statistics was carried out using variants derived from ABO sequencing in Probabilistic Annotation INtegraTOR (PAINTOR) V3.0, a Bayesian probabilistic framework algorithm that integrates association strength with functional genomic annotation data to improve accuracy in selecting functional SNPs.25 Genomic regions for significant markers from genome-wide meta-analysis were plotted and visually inspected alongside PAINTOR results. Functional Mapping and Annotation of Genome-Wide Association Studies (FUMA) was also used to facilitate functional annotation of GWAS results, gene prioritization, and interactive visualization.26
Statistical analysis
Differences in demographics, clinical data, and laboratory characteristics between case groups were tested in both cohorts using Fisher’s Exact test and Kruskal-Wallis analysis of variance. Demographic variables were also tested in logistic regressions for univariate association with functional assay-confirmed case status in both cohorts independently. The primary GWAS analysis used multivariable logistic regression assuming an additive genetic model with functional assay-confirmed case status as the outcome. Both PF4/heparin antibody-negative and antibody-positive controls were considered controls in the primary analysis. Logistic regressions were used to generate odds ratios (ORs) and 95% confidence intervals (CIs) after adjustment for age, sex, and the first 3 PCs. Results were considered significant with a Bonferroni-corrected, 2-sided α = 5 × 10−8 in the discovery cohort. In order to determine genomic associations independent of the most strongly associated loci, additional covariate adjustment for genome-wide associated SNP(s) was performed in the discovery cohort. X chromosome variation was also tested for association with positive functional assay status using similar statistical models stratified by females and males. A secondary GWAS analysis was performed with positive PF4/heparin antibody status as the outcome. In this analysis, all PF4/heparin antibody-positive individuals, including both functional assay-positive and antibody-positive (functional assay-negative) individuals, were considered cases, and all PF4/heparin antibody-negative individuals were considered controls.
Significantly associated genomic loci from the discovery cohort were then interrogated in the replication cohort after adjustment for clinical covariates, including age, sex, optical density level, platelet count before heparin administration, and PCs 1-3 with 2-sided α = 0.05. We then performed a genome-wide meta-analysis of the discovery and replication cohorts using a random-effects model. The meta-analysis included GWAS results adjusted for age, sex, and the first 3 PCs in an additive genetic model for both cohorts. Meta-analysis was weighted using standard errors of ORs. Sensitivity analyses were performed to determine the robustness of significant associations. In meta-analyses, functional assay-positive cases were compared with antibody-positive (functional assay-negative) controls alone (excluding antibody-negative controls) and to antibody-negative controls alone (excluding antibody-positive controls). The association of significant variants was then adjusted for PF4/heparin antibody IgG levels.
Because both HIT and ABO SNPs are associated with thromboembolism,27 we performed analyses to exclude the possibility that the ABO association was due to SNP association with thrombosis rather than functional assay-positive case status. First, we generated polygenic risk scores (PRSs) using a set of SNPs significantly associated (P < 5 × 10−8) with venous thromboembolism from genome-wide data for each sample as previously reported.28 In order to exclude variants from the ABO gene in PRS, we excluded all SNPs located on chromosome 9. The validity of PRSs was also assessed in BioVU, Vanderbilt University’s DNA biorepository coupled to deidentified electronic medical records resource using a phenome-wide association study approach (supplemental Materials; supplemental Figure 3).29,30 A meta-analysis was performed with adjustment for PRS in addition to age, sex, and PCAs 1-3. We then tested for the association of known thrombosis risk SNPs with functional assay-positive case status. ABO blood groups (A, B, AB, and O) were then derived based on haplotypes of rs8176719 and rs8176746, as previously described, and tested for association with functional assay-positive status in both cohorts combined.31,32 All statistical analyses were performed using R v4.04 (R Core Team, Vienna, Austria), PLINK v1.90,33 and SAS v9.4 (SAS, Cary, NC). Additional details related to statistical analysis, genotyping, imputation, quality control, sequencing, fine-mapping analysis, and PRS are described in the supplemental Materials. Genome-wide data and phenotype data used in this analysis have been provided on the database of Genotypes and Phenotypes, at accession #phs002863.
Results
After quality control, a total of 1269 functional assay-positive cases, 1131 antibody-positive (functional assay-negative) controls, and 1766 antibody-negative controls were included in the discovery cohort for GWAS analysis. Functional assay-positive cases were more likely to be female and have higher PF4/heparin IgG levels compared with both PF4/heparin antibody-positive and PF4/heparin antibody-negative controls (Table 1). In univariate logistic regressions, odds of functional assay-positive status increased with PF4/heparin IgG levels (OR, 16.53; 95% CI, 13.83-19.74; P = 1.51 × 10−209) and female sex (OR, 1.15; 95% CI, 1.01-1.32; P = .034). A total of 177 HIT cases, 258 PF4/heparin antibody-positive controls, and 351 antibody-negative controls were included in the replication cohort. Similarly, HIT cases were more likely to be female and have higher PF4/heparin IgG levels (Table 1). Platelet count before heparin administration was also significantly increased in HIT cases. In univariate logistic regressions, odds of HIT were increased with PF4/heparin IgG levels (OR, 18.53; 95% CI, 12.02-28.57; P = 7.25 × 10−40), female sex (OR, 3.02; 95% CI, 2.14-4.26; P = 3.38 × 10−10), and platelet count prior to heparin (OR, 1.03; 95% CI, 1.00-1.06; P = .033 per increase of 10 × 103 per µL). In both discovery and replication cohorts, age was not significantly associated with functional assay-positive status.
Table 1.
Demographic and laboratory characteristics of discovery and replication cohorts included in genome-wide association study
| Characteristic | Functional assay-positive* | Antibody-positive (functional assay-negative) controls*,† | Antibody-negative controls*,† | P value‡ |
|---|---|---|---|---|
| Discovery cohort | ||||
| Total (n) | 1269 | 1131 | 1766 | — |
| Female, n (%) | 535 (42.2) | 412 (36.4) | 697 (39.5) | .016 |
| Age (yr) | 66.5 (13.6) | 64.7 (15.6) | 67.3 (13.5) | .0004 |
| IgG (OD)† | 1.79 (0.71) | 0.90 (0.40) | 0.17 (0.12) | <.0001 |
| IgA (OD)† | 0.74 (0.60) | 0.50 (0.46) | 0.21 (0.24) | <.0001 |
| IgM (OD)† | 0.70 (0.43) | 0.66 (0.42) | 0.35 (0.25) | <.0001 |
| Ancestry, n (%)§ | ||||
| European | 1252 (98.7) | 1108 (98.0) | 1735 (98.2) | .408 |
| African | 2 (0.2) | 2 (0.2) | 3 (0.2) | 1.00 |
| Asian | 1 (0.1) | 1 (0.1) | 2 (0.1) | 1.00 |
| Admixed | 14 (1.1) | 20 (1.8) | 26 (1.5) | .395 |
| Ancestral proportion (%)§ | ||||
| European | 0.988 | 0.984 | 0.986 | .233 |
| African | 0.006 | 0.008 | 0.007 | .236 |
| Asian | 0.006 | 0.008 | 0.007 | .348 |
| Replication cohort | ||||
| Total (n) | 177 | 258 | 351 | — |
| Female, n (%) | 98 (55.7) | 90 (34.9) | 89 (25.4) | <.0001 |
| Age (yr) | 67.2 (14.0) | 67.1 (13.0) | 69.8 (12.1) | .014 |
| Platelet count prior to heparin (×103/µL) | 227.3 (63.3) | 221.8 (63.3) | 207.5 (51.0) | .004 |
| PF4/heparin antibodies (OD)† | 2.47 (0.72) | 1.17 (0.56) | 0.21 (0.07) | <.001 |
| Ancestry, n (%)§ | ||||
| European | 170 (96.1) | 250 (96.9) | 341 (97.2) | .782 |
| African | 3 (1.7) | 2 (0.8) | 1 (0.3) | .173 |
| Asian | 1 (0.6) | 0 (0) | 1 (0.3) | .707 |
| Admixed | 3 (1.7) | 6 (2.3) | 8 (2.3) | .904 |
| Ancestral proportion (%)§ | ||||
| European | 0.962 | 0.977 | 0.980 | .625 |
| African | 0.026 | 0.018 | 0.013 | .629 |
| Asian | 0.012 | 0.005 | 0.007 | .866 |
OD, optical density.
Values represent mean (standard deviation) unless otherwise specified.
Functional assay-positive status was determined using the HIPA test in the discovery cohort and the serotonin release assay in the replication cohort.
PF4/heparin antibody levels were determined using ELISA in both discovery and replication cohorts.
P values for comparison across 3 groups were generated using Fisher’s exact test for dichotomous variables and Kruskal-Wallis test for continuous variables.
Ancestral proportions were generated using ancestry-informative markers from genome-wide arrays input into STRUCTURE with HapMap reference panels. Individuals were considered to have European, African, or Asian ancestry if relevant ancestral proportions were ≥80 percent. Otherwise, individuals were considered admixed since patient-reported race/ethnicity data were not available.
GWAS analysis in the discovery cohort included 9 065 510 assayed and imputed genomic markers after quality control filters (supplemental Table 1). A total of 47 SNPs were significantly associated with functional assay-positive case status at a genome-wide significance level with no evidence of genomic inflation (λ = 0.998) (Figure 1; supplemental Figure 4). All of these SNPs were within a 6.4 kb region of the ABO gene locus on chromosome 9. The most strongly associated allele was the rs36058710 T insertion allele (MAF = 0.36; OR, 0.730; 95% CI, 0.661-0.807; P = 7.96 × 10−10), which was imputed and in linkage disequilibrium (LD) with the rs8176719 C insertion polymorphism (r2 = 0.79; D’ = 0.98). The rs8176719 C insertion/deletion allele was considered to be the functional polymorphism responsible for this association based on the biological plausibility of ABO blood groups in platelet reactivity and HIT risk. The rs8176719 C insertion allele was significantly protective for functional assay-positive case status (MAF = 0.41; OR, 0.751; 95% CI, 0.682-0.828; P = 7.80 × 10−9). In the replication cohort, the rs8176719 C insertion allele was also significantly protective (α = 0.05) for functional assay-confirmed HIT with the same direction of effect and similar frequency (MAF = 0.38; OR, 0.467; 95% CI, 0.228-0.954; P = .0367). Rs8176719 genotypes were in HWE in both the discovery cohort (P = .749) and the replication cohort (P = .489). The protective effect of the rs8176719 C insertion indicated that the rs8176719 C deletion was a risk factor for HIT. Deletion of the ABO rs8176719 C allele produces O blood group individuals, whereas insertion of the rs8176719 C allele produces A, B, or AB blood group individuals (supplemental Figure 5).34
Figure 1.

Genome-wide association with functional assay-positive status in the discovery cohort. P values were generated using logistic regression adjusted for age, sex, and principal components 1-3 in an additive model. P values on the −log10 scale are plotted on the left vertical axis, and the chromosomal position is plotted along the horizontal axis. The significance threshold of 5 × 10−8 is indicated by the solid red horizontal line, and the suggestive threshold of 1 × 10−6 is indicated by the red dashed horizontal line. ORs are indicated by dot color as described in the legend on the right.
Several suggestive associations were also observed with P values <1 × 10−6 in the discovery cohort. On chromosome 19, the rs59537957 T insertion allele was associated with functional assay-positive case status (MAF = 0.32; OR, 1.296; 95% CI, 1.175-1.43; P = 2.37 × 10−7). This SNP is downstream of both the solute carrier family 1 member 5 gene (SLC1A5) and the fukutin-related protein gene (FKRP). On chromosome 12, the rs12825115 T allele was protective for functional assay-positive case status (MAF = 0.12; OR, 0.691; 95% CI, 0.598-0.790; P = 5.77 × 10−7). This intronic SNP is located in the adhesion G protein-coupled receptor D1 gene (ADGRD1). After adjustment for the rs8176719 locus, no SNPs reached the genome-wide significance threshold, but the suggestive signals on chromosomes 19 and 12 remained (supplemental Figure 6). Similar results were also observed when utilizing TOPMed for genomic imputation (supplemental Figure 7). No X chromosome variation was associated with functional assay-positive case status (supplemental Figure 8). In the secondary GWAS analysis of positive PF4/heparin antibody status, no genome-wide significant associations were observed (supplemental Figure 9). In the random-effects meta-analysis, the rs8176719 C insertion allele was strongly protective with an OR of 0.758 and P = 2.88 × 10−9 (Figure 2; supplemental Figure 10). For this SNP, the P value for Cochrane's Q statistic was 0.592, and the I2 heterogeneity index was 0, indicating a lack of heterogeneity. The rs36058710 T insertion allele was again the most significant association observed (OR, 0.739; P = 4.12 × 10−10; Q = 0.466; I2 = 0). Multiethnic mixed-model association analysis produced consistent results compared with the primary meta-analysis (supplemental Figure 11). The rs8176719 C insertion allele was strongly protective for functional assay-positive status (OR, 0.879; P = 8.42 × 10−9).
Figure 2.

SNPs in ABO and the surrounding region that were most strongly associated with positive functional assay status from (A) meta-analysis of association with functional assay-positive status and (B) fine-mapping of association in PAINTOR. (A) P values were generated using a random-effects meta-analysis of logistic regressions adjusted for age, sex, and principal components 1-3 in an additive model for combined discovery and replication cohorts. P values on the −log10 scale are plotted on the left vertical axis, and the chromosomal position is plotted along the horizontal axis along with the gene names and size of flanking region. The significance threshold of 5 × 10−8 is indicated by the black horizontal line. Pairwise LD (r2) with this SNP is indicated by dot color as described in the legend in the upper left corner. The right vertical axis indicates the regional recombination rate (cM/Mb), which is overlaid in blue. (B) PAINTOR posterior probabilities based on the association of sequenced ABO SNPs in combined discovery and replication cohorts with functional annotations from FANTOM5, GENCODE, and Transcription Factor Binding Site datasets. Posterior probabilities values on the log10 scale are plotted on the left vertical axis, and the chromosomal position is plotted along the horizontal axis along with ABO exonic/intronic regions. Z Scores for association with functional assay-positive status are indicated by dot color as described in the legend in the upper right corner.
Sensitivity analyses for genomic association
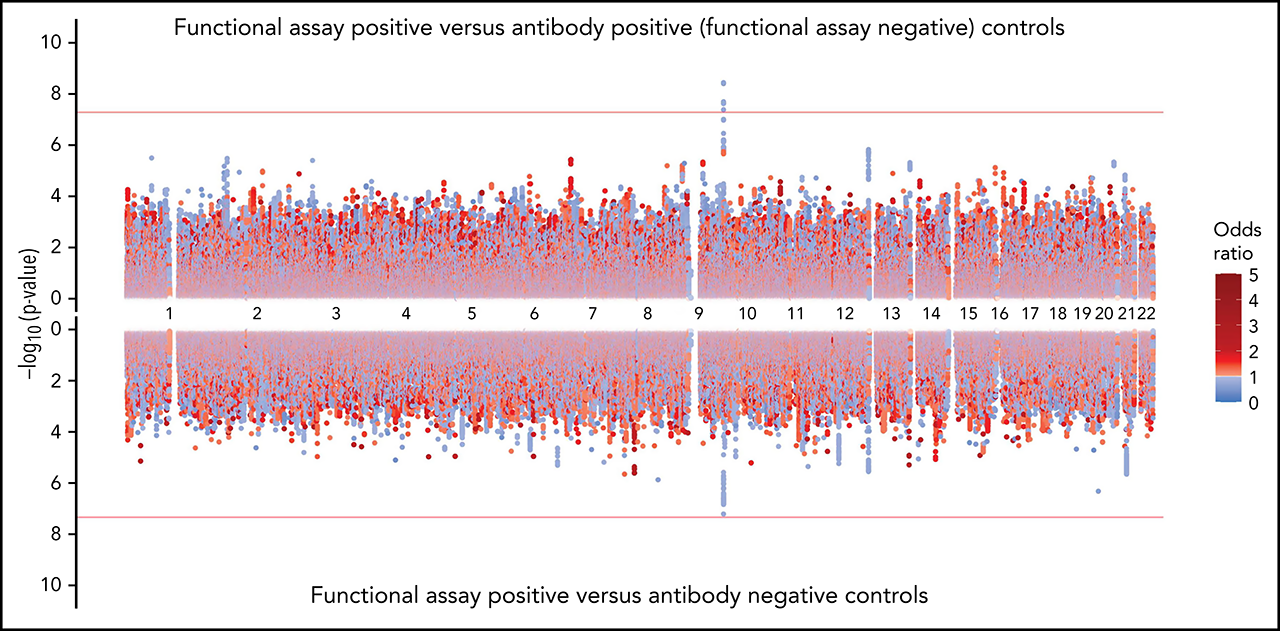
The rs8176719 C insertion allele was significantly protective for functional assay-positive status when compared with antibody-positive controls but not when compared with antibody-negative controls (Figure 3). Meta-analysis after adjustment for PF4/heparin antibody levels indicated that the strength of the ABO association was reduced but was still present at a genome-wide level (supplemental Figure 12). Adjustment for a thromboembolism PRS had little effect on the association of ABO with functional assay-positive status (supplemental Figure 13). Furthermore, no other SNPs from known thrombosis-associated genes showed a significant association (α = 0.05) in meta-analysis results (supplemental Table 2).
Figure 3.

Genome-wide association results for positive functional assay status vs patients with positive antibodies (above) and vs patients with negative antibodies (below) from the meta-analysis of discovery and replication cohorts. P values were generated using random-effects meta-analysis logistic regression adjusted for age, sex, and principal components 1-3 in an additive model. P values on the −log10 scale are plotted on the left vertical axis, and the chromosomal position is plotted along the horizontal axis. The significance threshold of 5 × 10−8 is indicated by the red horizontal lines. ORs are indicated by dot color as described in the legend on the right.
Fine-mapping of ABO association
Sequencing was performed for the ABO gene ±5 kbp, constituting a 30 068 bp region. At least 20× coverage was achieved in 99% of the target region. A total of 956 polymorphic sites were identified, including 335 variants with MAF >0.01. PAINTOR identified rs8176719 as the SNP with the highest posterior probability (0.994) in the ABO sequence dataset of being the functional polymorphism responsible for the association (Figure 2; supplemental Table 3). The aggregated results of FUMA also indicated that rs8176719 was the most likely causal SNP, based on functional annotations and deleterious score (supplemental Table 4). FUMA-based Annotate Variation (ANNOVAR)35 results ranked rs8176719 as the most likely causal variant based on LD (r2) and functional consequences, while FUMA-based Combined Annotation-Dependent Depletion (CADD)36 identified rs8176719 as the top SNP based on a deleterious score with a CADD score of 15.84, twice as large as the next closest variant.
Association of ABO blood groups
Following imputation of blood groups, 35.2% of patients in both cohorts were assigned the O group, 47.3% were assigned the A group, 11.5% were assigned the B group, and 6.0% were assigned the AB group. The observed protective effect from our GWAS of the rs8176719 C insertion, which encodes all non-O blood group alleles, indicated that the rs8176719 C deletion, which determines the O blood group allele, was a risk factor for HIT. The O blood group, corresponding to the deletion of the rs8176719 C allele, was a strong risk factor for functional assay-positive status compared with all other blood groups (OR, 1.42; 95% CI, 1.26-1.61; P = 3.09 × 10−8) (Figure 4). We also did not observe an increased OR in individuals with the A2 blood group allele, defined based on 3 SNP ABO haplotypes as previously reported.37 The effect of the O blood group was also more pronounced when functional assay-positive cases were compared with antibody-positive controls than when compared with antibody-negative patients. The effect of the O blood group was also robust to adjustment for PF4/heparin IgG levels.
Figure 4.

Forest plot of ABO blood group associations with positive functional assay status in discovery and replication cohorts combined. ORs, 95% CIs, and P values were generated using logistic regression adjusted for age, sex, principal components 1-3, and cohort (discovery vs replication) in an additive model. Logistic regressions for the association of blood groups compare the relevant blood group (eg, O, A, B, or AB) to all other blood groups combined. Sensitivity analyses included functional assay-positive cases compared with PF4/heparin antibody-positive controls alone (excluding antibody-negative controls) and functional assay-positive cases compared with PF4/heparin antibody-negative controls alone (excluding antibody-positive controls) and after adjustment for PF4/heparin antibody IgG levels.
Discussion
Here we present results from the largest available study of genetic determinants of platelet activation capacity of anti-PF4/heparin IgG antibodies in HIT, strengthened by functional assay confirmation of cases and by the inclusion of both antibody-positive (functional assay-negative) and antibody-negative control groups. Our study adds to prior evidence of female sex as a risk factor for HIT and the strong influence of PF4/heparin antibodies on HIT risk. Our independently replicated GWAS results describe a novel ABO association with HIT, implicating ABO blood groups in HIT pathogenesis and the O blood group as a risk factor for HIT. Our results suggest that the observed association between the O blood group and HIT is largely independent of PF4/heparin antibody levels. Furthermore, this association was observed to be stronger when functional assay-positive cases were compared with antibody-positive controls relative to antibody-negative controls. Our results may have important implications for the understanding of HIT pathogenesis, for HIT risk prediction, and potential prevention of HIT, but likely also for other Fc-γ receptor IIa (FcγRIIa)-mediated disease,38,39 of which VITT is the most recent highly relevant example.
Two previous reports have used GWAS to study HIT, and neither implicated ABO variation.12,13 However, these studies included a small number of suspected HIT cases and did not confirm cases using a functional assay, relying instead on PF4/heparin antibody test results and 4Ts scores. The lack of a functional assay test is known to result in misclassification, and a confirmatory functional assay is clinically recommended for antibody-positive patients since at least half of ELISA-positive patients will test negative in the functional assay.2,40,41 SNPs may exert their effects only at distinct stages in the progression of HIT, and different SNPs may be predictors of PF4/heparin antibody formation, thrombocytopenia, or thrombosis. To differentiate genetic determinants of HIT vs PF4/heparin antibody production, the current study divided discovery and replication cohorts into 3 groups. Prior GWAS did not use this approach, potentially explaining the difference in results in addition to small sample sizes and the lack of functional assay testing.9 Indeed, the results of the present study indicate that functional assay characterization and differentiation of risk between antibody-positive and antibody-negative groups were necessary to observe the O blood group association.
Our results strongly suggest HIT is influenced by variation in ABO, which codes for the glycosyltransferase enzyme that is responsible for the histo-blood group ABO, the major human alloantigen system that determines blood groups. Our observed ABO association may not be due to a direct effect of the O blood group but may be mediated through other ABO-associated traits. The ABO locus has been repeatedly associated with multiple phenotypes through GWAS, including thromboembolism and p-selectin, a marker of platelet activation.27,42 P-selectin is elevated in HIT cases as well as O blood group individuals, and functional assay-positive samples induce platelet p-selectin expression when PF4 is present.43-45 Alternatively, von Willebrand Factor (VWF) and factor VIII (FVIII) levels are lower in O blood group patients, and VWF has been shown to strongly bind PF4.46,47 Lower VWF levels might leave more PF4 available for the formation of PF4/heparin complexes on the platelet surface, increasing HIT risk in O blood group individuals. However, many O blood type individuals have low VWF levels, and this does not explain the increased capacity of anti-PF4/heparin IgG antibodies to activate washed platelets in vitro. Our observed association, which was independent of antibody titers, may indicate the capacity of PF4/heparin antibodies to interact with Fc-receptors, especially when the FcγRIIa is modulated by the ABO blood group. Mendelian randomization studies would help to establish causal relationships between HIT, ABO variation, and ABO-associated biomarkers, such as p-selectin, FVIII, and VWF.
PF4/heparin antibody tests are not able to differentiate patients who will and will not develop HIT, and functional assay confirmation is required to confirm HIT. Because the SRA and HIPA tests are technically demanding, often restricted to specialized laboratories, and may result in significant delays in HIT diagnosis, alternative approaches to HIT risk stratification have a potential role in HIT diagnosis and treatment. The ABO association for the present study was evident when comparing functional assay-positive cases to antibody-positive (functional assay-negative) controls, indicating an interaction between O blood type and PF4/heparin antibody levels. The production of PF4/heparin antibodies in the presence of type O blood might predispose a patient to HIT, suggesting that O blood type could be used to better differentiate patients that will develop HIT vs patients with nonpathogenic PF4/heparin antibodies. ABO blood group might conceivably be used alongside other clinical characteristics to better predict functional assay-positive status before the acquisition of a laboratory result of a functional HIT test, which is not always widely available. However, the translational potential of the ABO blood group is limited by the modest OR for HIT risk and the high prevalence of the O blood group. Furthermore, the potential for positive antibody and functional assay results likely evolves with a patients’ changing clinical circumstances. Additional studies will be necessary to determine whether ABO blood groups or other ABO variation has clinical utility in predicting HIT.
Genetic predictors of platelet reactivity in HIT identified in our study may inform the pathogenesis of rare adverse drug reactions related to COVID-19 vaccination. Two adenoviral vector vaccines containing the SARS-CoV-2 spike protein have been reported to cause the rare immune, thrombotic disorder VITT. VITT is characterized by high levels of HIT-mimicking antibodies to PF4-polyanion complexes and unusual thrombotic sites, similar to autoimmune HIT.6-8,48,49 However, anti-PF4 antibodies occurring after vaccination bind to different sites on PF4 and also cause different reactivity patterns.50 Furthermore, Fc receptor-dependent platelet activation has been observed in COVID-19 patients, and prior studies have also observed inconsistent associations between the ABO locus and COVID-19 infection and severity.51-53
Several limitations are worthy of mention in this study. Although our study is one of the largest studies of HIT to date, the sample size is small relative to emerging GWAS literature. We are underpowered to detect variants with low effect sizes and frequencies. A definitive diagnosis of HIT was not possible in the discovery cohort due to the lack of clinical data, although all samples were referred due to clinical suspicion of HIT. Thus platelet activation assay reactivity was used as a model for HIT in the discovery cohort, confirming these associations in a well-characterized and prospectively-collected replication cohort. Our study also included only patients that underwent functional assay and antibody testing due to clinically suspected HIT in the context of heparin/LMWH exposure. We acknowledge that our control populations may be enriched for conditions leading to thrombocytopenia, which may confound our genetic association. We attempted to adjust for a confounding effect due to genetic predisposition to thromboembolism, and our analyses suggest that enrichment of SNPs associated with thromboembolism was not responsible for our observed ABO association. Furthermore, our association between the O blood group and HIT is in the opposite direction of what would be expected with an overabundance of thrombosis in HIT cases since thromboembolism is most strongly associated with the A blood group.27 Due to the lack of ABO serotyping data in our 2 cohorts, we were also unable to confirm the existence of rare or novel ABO variation that resulted in the O blood group. Finally, the overwhelming majority of our cohorts were of European ancestry, and our results may not be generalizable to more diverse populations, especially considering the differential frequencies of ABO blood groups across different racial/ethnic groups.54,55
We present results from the largest available study of genetic determinants of platelet reactivity in HIT, strengthened by functional assay confirmation of cases and by the inclusion of both a PF4/heparin antibody-positive (functional assay-negative) control group and a PF4/heparin antibody-negative control group. Our independently replicated GWAS results describe a novel ABO association with platelet reactivity in HIT, strongly implicating ABO variation in HIT pathogenesis and the O blood group as a risk factor for HIT. Our results indicate that this association is stronger when functional assay-positive cases were compared with antibody-positive (functional assay-negative) controls than to antibody-negative controls. These findings have important implications for the understanding of HIT pathogenesis, HIT risk prediction, and possibly related conditions, such as VITT, and indicate that the ABO blood group might be a modifier of FcγRIIa-mediated disease.
Supplementary Material
The online version of this article contains a data supplement.
{kind=link}
Acknowledgments
The authors thank the staff of the Laboratory for Genotyping Development, RIKEN Center for the Integrative Medical Sciences. They are grateful to Ulrike Strobel, Carmen Freyer, Katrin Stein, Ines Warnig, Ricarda Raschke, and Jessica Fuhrmann for excellent technical support in performing all HIT assays in the discovery cohort.
Genome-wide data and ABO sequence data were provided by the Pharmacogenomics Research Network (PGRN)-RIKEN Collaborative. This research is funded by the National Institutes of Health’s (NIH) National Heart, Lung, and Blood Institute (NHLBI) under awards K01HL143137 (J.H.K.), R01HL156993 (J.H.K.), R01HL158686 (J.H.K.), and U19 HL065962 (D.M.R.), the National Institute of General Medical Sciences (NIGMS) under award P50GM115305 (D.M.R.), and the National Institute of Environmental Health Sciences (NIEHS) under award T32 ES007091 (J.B.G.). Acquisition of the replication cohort from the University of Tours with the independent replication population was supported by the IRTH (Institut pour la Recherche sur la Thrombose et l’Hémostase) and by a PHRC grant (PHRN09-YG/FRIGTIH). The study was supported by the Deutsche Forschungsgemeinschaft (DFG; German Research Foundation) Project Number 374031971–TRR240 (A.G.). Dataset(s) obtained from Vanderbilt University Medical Center’s BioVU are supported by numerous sources: institutional funding, private agencies, and federal grants. These include the NIH-funded Shared Instrumentation Grant S10RR025141 and CTSA grants UL1TR002243, UL1TR000445, and UL1RR024975. Genomic data are also supported by investigator-led projects that include U01HG004798, R01NS032830, RC2GM092618, P50GM115305, U01HG006378, U19HL065962, and R01HD074711. Additional funding sources are listed at https://victr.vumc.org/biovu-funding.
Footnotes
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.H.K., J.R., C.P., N.M.H., E.J.P., T.E.W., Y.G., A.G., and D.M.R. designed the research; J.H.K., J.R., Y.M., C.I., K.S., T.T., T.M., M.K., C.P., Y.G., A.G., and D.M.R. performed the research; J.H.K., J.B.G., K.L.M., H.E.S., C.M.S., A.B., M.S., J.D.M., and I.S. analyzed the data; and J.H.K., J.R., Y.G., A.G., and D.M.R. wrote the manuscript.
Conflict-of-interest disclosure: T.E.W. received lecture honoraria from Alexion and Instrumentation Laboratory; received royalties from Informa (Taylor & Francis); has provided consulting services to Aspen Canada, Aspen Global, Bayer, CSL Behring, Ergomed, and Octapharma; has received research funding from Instrumentation Laboratory; and has provided expert witness testimony relating to HIT and non‐HIT thrombocytopenic and coagulopathic disorders. A.G. reports grants and nonfinancial support from Aspen, Boehringer Ingelheim, MSD, Bristol Myers Squibb (BMS), Paringenix, Bayer Healthcare, Gore Inc., Rovi, Sagent, Biomarin/Prosensa, Portola, Ergomed, and GTH e.V.; and personal fees from Aspen, Boehringer Ingelheim, MSD, Macopharma, BMS, Chromatec, and Instrumentation Laboratory. K.S. reports research funding from Immucor; personal fees from Aspen and Viatris; and nonfinancial support from SOBI outside the submitted work. All remaining authors declare no competing financial interests.
Correspondence: Jason H. Karnes, University of Arizona College of Pharmacy, 1295 N Martin Ave, Tucson, AZ 85721; e-mail: karnes@pharmacy.arizona.edu.
REFERENCES
- 1.Greinacher A. CLINICAL PRACTICE. Heparin-induced thrombocytopenia. N Engl J Med. 2015;373(3):252-261. [DOI] [PubMed] [Google Scholar]
- 2.Cuker A, Arepally GM, Chong BH, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia. Blood Adv. 2018;2(22):3360-3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Girolami B, Prandoni P, Stefani PM, et al. The incidence of heparin-induced thrombocytopenia in hospitalized medical patients treated with subcutaneous unfractionated heparin: a prospective cohort study. Blood. 2003;101(8):2955-2959. [DOI] [PubMed] [Google Scholar]
- 4.Greinacher A, Farner B, Kroll H, Kohlmann T, Warkentin TE, Eichler P. Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis. A retrospective analysis of 408 patients. Thromb Haemost. 2005;94(1):132-135. [DOI] [PubMed] [Google Scholar]
- 5.Linkins LA, Dans AL, Moores LK, et al. Treatment and prevention of heparin-induced thrombocytopenia: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e495S-e530S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greinacher A, Thiele T, Warkentin TE, Weisser K, Kyrle PA, Eichinger S. Thrombotic thrombocytopenia after ChAdOx1 nCov-19 vaccination. N Engl J Med. 2021;384(22):2092-2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vayne C, Rollin J, Gruel Y, et al. PF4 Immunoassays in vaccine-induced thrombotic thrombocytopenia. N Engl J Med. 2021;385(4):376-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scully M, Singh D, Lown R, et al. Pathologic antibodies to platelet factor 4 after ChAdOx1 nCoV-19 vaccination. N Engl J Med. 2021;384(23):2202-2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karnes JH. Pharmacogenetics to prevent heparin-induced thrombocytopenia: what do we know? Pharmacogenomics. 2018;19(18):1413-1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arepally GM. Heparin-induced thrombocytopenia. Blood. 2017;129(21):2864-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Khandelwal S, Arepally GM. Immune pathogenesis of heparin-induced thrombocytopenia. Thromb Haemost. 2016;116(5):792-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Witten A, Bolbrinker J, Barysenka A, et al. Targeted resequencing of a locus for heparin-induced thrombocytopenia on chromosome 5 identified in a genome-wide association study. J Mol Med (Berl). 2018;96(8):765-775. [DOI] [PubMed] [Google Scholar]
- 13.Karnes JH, Cronin RM, Rollin J, et al. A genome-wide association study of heparin-induced thrombocytopenia using an electronic medical record. Thromb Haemost. 2015;113(4):772-781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gruel Y, Vayne C, Rollin J, et al. Comparative analysis of a French prospective series of 144 patients with heparin-induced thrombocytopenia (FRIGTIH) and the literature. Thromb Haemost. 2020;120(7):1096-1107. [DOI] [PubMed] [Google Scholar]
- 15.Rollin J, Pouplard C, Sung HC, et al. Increased risk of thrombosis in FcγRIIA 131RR patients with HIT due to defective control of platelet activation by plasma IgG2. Blood. 2015;125(15):2397-2404. [DOI] [PubMed] [Google Scholar]
- 16.Eekels JJM, Althaus K, Bakchoul T, et al. An international external quality assessment for laboratory diagnosis of heparin-induced thrombocytopenia. J Thromb Haemost. 2019;17(3):525-531. [DOI] [PubMed] [Google Scholar]
- 17.Greinacher A, Amiral J, Dummel V, Vissac A, Kiefel V, Mueller-Eckhardt C. Laboratory diagnosis of heparin-associated thrombocytopenia and comparison of platelet aggregation test, heparin-induced platelet activation test, and platelet factor 4/heparin enzyme-linked immunosorbent assay. Transfusion. 1994;34(5):381-385. [DOI] [PubMed] [Google Scholar]
- 18.Lo GK, Juhl D, Warkentin TE, Sigouin CS, Eichler P, Greinacher A. Evaluation of pretest clinical score (4 T’s) for the diagnosis of heparin-induced thrombocytopenia in two clinical settings. J Thromb Haemost. 2006;4(4):759-765. [DOI] [PubMed] [Google Scholar]
- 19.Greinacher A, Ittermann T, Bagemühl J, et al. Heparin-induced thrombocytopenia: towards standardization of platelet factor 4/heparin antigen tests. J Thromb Haemost. 2010;8(9):2025-2031. [DOI] [PubMed] [Google Scholar]
- 20.Abecasis GR, Altshuler D, Auton A, et al. ; 1000 Genomes Project Consortium . A map of human genome variation from population-scale sequencing [published correction appears in Nature. 2011;473(7348):544]. Nature. 2010;467(7319):1061-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taliun D, Harris DN, Kessler MD, et al. ; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium . Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature. 2021;590(7845):290-299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155(2):945-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J, Lee SH, Goddard ME, Visscher PM. Genome-wide complex trait analysis (GCTA): methods, data analyses, and interpretations. Methods Mol Biol. 2013;1019:215-236. [DOI] [PubMed] [Google Scholar]
- 24.Momozawa Y, Akiyama M, Kamatani Y, et al. Low-frequency coding variants in CETP and CFB are associated with susceptibility of exudative age-related macular degeneration in the Japanese population. Hum Mol Genet. 2016;25(22):5027-5034. [DOI] [PubMed] [Google Scholar]
- 25.Kichaev G, Yang W-Y, Lindstrom S, et al. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10(10):e1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8(1):1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vasan SK, Rostgaard K, Majeed A, et al. ABO blood group and risk of thromboembolic and arterial disease: a study of 1.5 million blood donors. Circulation. 2016;133(15):1449-1457, discussion 1457. [DOI] [PubMed] [Google Scholar]
- 28.Klarin D, Busenkell E, Judy R, et al. Genome-wide association analysis of venous thromboembolism identifies new risk loci and genetic overlap with arterial vascular disease. 2019;51(11):1574-1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26(9):1205-1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Karnes JH, Bastarache L, Shaffer CM, et al. Phenome-wide scanning identifies multiple diseases and disease severity phenotypes associated with HLA variants. Sci Transl Med. 2017;9(389):eaai8708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Groot HE, Villegas Sierra LE, Said MA, Lipsic E, Karper JC, van der Harst P. Genetically determined ABO blood group and its associations with health and disease. Arterioscler Thromb Vasc Biol. 2020;40(3):830-838. [DOI] [PubMed] [Google Scholar]
- 32.Kolin DA, Kulm S, Christos PJ, Elemento O. Clinical, regional, and genetic characteristics of Covid-19 patients from UK Biobank. PLoS One. 2020;15(11):e0241264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559-575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto F, Clausen H, White T, Marken J, Hakomori S. Molecular genetic basis of the histo-blood group ABO system. Nature. 1990;345(6272):229-233. [DOI] [PubMed] [Google Scholar]
- 35.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310-315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goumidi L, Thibord F, Wiggins KL, et al. Association between ABO haplotypes and the risk of venous thrombosis: impact on disease risk estimation. Blood. 2021;137(17):2394-2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hubbard JJ, Pyzik M, Rath T, et al. FcRn is a CD32a coreceptor that determines susceptibility to IgG immune complex-driven autoimmunity. J Exp Med. 2020;217(10):e20200359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lande R, Lee EY, Palazzo R, et al. CXCL4 assembles DNA into liquid crystalline complexes to amplify TLR9-mediated interferon-α production in systemic sclerosis. Nat Commun. 2019;10(1):1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Greinacher A, Juhl D, Strobel U, et al. Heparin-induced thrombocytopenia: a prospective study on the incidence, platelet-activating capacity and clinical significance of antiplatelet factor 4/heparin antibodies of the IgG, IgM, and IgA classes. J Thromb Haemost. 2007;5(8):1666-1673. [DOI] [PubMed] [Google Scholar]
- 41.Warkentin TE, Sheppard JI, Moore JC, Sigouin CS, Kelton JG. Quantitative interpretation of optical density measurements using PF4-dependent enzyme-immunoassays. J Thromb Haemost. 2008;6(8):1304-1312. [DOI] [PubMed] [Google Scholar]
- 42.Kiechl S, Paré G, Barbalic M, et al. Association of variation at the ABO locus with circulating levels of soluble intercellular adhesion molecule-1, soluble P-selectin, and soluble E-selectin: a meta-analysis. Circ Cardiovasc Genet. 2011;4(6):681-686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Larson NB, Bell EJ, Decker PA, et al. ABO blood group associations with markers of endothelial dysfunction in the multi-ethnic study of atherosclerosis. Atherosclerosis. 2016;251:422-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fareed J, Walenga JM, Hoppensteadt DA, et al. Selectins in the HIT syndrome: pathophysiologic role and therapeutic modulation. Semin Thromb Hemost. 1999;25(Suppl 1):37-42. [PubMed] [Google Scholar]
- 45.Campello E, Radu CM, Duner E, et al. Activated platelet-derived and leukocyte-derived circulating microparticles and the risk of thrombosis in heparin-induced thrombocytopenia: a role for PF4-bearing microparticles? Cytometry B Clin Cytom. 2018;94(2):334-341. [DOI] [PubMed] [Google Scholar]
- 46.Albánez S, Ogiwara K, Michels A, et al. Aging and ABO blood type influence von Willebrand factor and factor VIII levels through interrelated mechanisms. J Thromb Haemost. 2016;14(5):953-963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Johnston I, Sarkar A, Hayes V, et al. Recognition of PF4-VWF complexes by heparin-induced thrombocytopenia antibodies contributes to thrombus propagation. Blood. 2020;135(15):1270-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Simpson CR, Shi T, Vasileiou E, et al. First-dose ChAdOx1 and BNT162b2 COVID-19 vaccines and thrombocytopenic, thromboembolic and hemorrhagic events in Scotland. Nat Med. 2021;27(7):1290-1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brodard J, Kremer Hovinga JA, Fontana P, Studt JD, Gruel Y, Greinacher A. COVID-19 patients often show high-titer non-platelet-activating anti-PF4/heparin IgG antibodies. J Thromb Haemost. 2021;19(5):1294-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huynh A, Kelton JG, Arnold DM, Daka M, Nazy I. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature. 2021;596(7873):565-569. [DOI] [PubMed] [Google Scholar]
- 51.COVID-19 Host Genetics Initiative . Mapping the human genetic architecture of COVID-19. Nature. 2021;600(7889):472-477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellinghaus D, Degenhardt F, Bujanda L, et al. ; Severe Covid-19 GWAS Group . Genomewide association study of severe COVID-19 with respiratory failure. N Engl J Med. 2020;383(16):1522-1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Daviet F, Guervilly C, Baldesi O, et al. Heparin-induced thrombocytopenia in severe COVID-19. Circulation. 2020;142(19):1875-1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hodonsky CJ, Baldassari AR, Bien SA, et al. Ancestry-specific associations identified in genome-wide combined-phenotype study of red blood cell traits emphasize benefits of diversity in genomics. BMC Genomics. 2020;21(1):228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Garratty G, Glynn SA, McEntire R; Retrovirus Epidemiology Donor Study . ABO and Rh(D) phenotype frequencies of different racial/ethnic groups in the United States. Transfusion. 2004;44(5):703-706. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.