

Abstract
Introduction/Aims
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative illness with great unmet patient need. We aimed to evaluate whether mesenchymal stem cells induced to secrete high levels of neurotrophic factors (MSC‐NTF), a novel autologous cell‐therapy capable of targeting multiple pathways, could safely slow ALS disease progression.
Methods
This randomized, double‐blind, placebo‐controlled study enrolled ALS participants meeting revised El Escorial criteria, revised ALS Functional Rating Scale (ALSFRS‐R) ≥25 (screening) and ≥3 ALSFRS‐R points decline prior to randomization. Participants received three treatments of MSC‐NTF or placebo intrathecally. The primary endpoint evaluated efficacy of MSC‐NTF through a responder analysis and safety. A change in disease progression post‐treatment of ≥1.25 points/mo defines a clinical response. A pre‐specified analysis leveraged baseline ALSFRS‐R of 35 as a subgroup threshold.
Results
Overall, MSC‐NTF treatment was well tolerated; there were no safety concerns. Thirty‐three percent of MSC‐NTF and 28% of placebo participants met clinical response criteria at 28 wk (odds ratio [OR] = 1.33, P = .45); thus, the primary endpoint was not met. A pre‐specified analysis of participants with baseline ALSFRS‐R ≥ 35 (n = 58) showed a clinical response rate at 28 wk of 35% MSC‐NTF and 16% placebo (OR = 2.6, P = .29). Significant improvements in cerebrospinal biomarkers of neuroinflammation, neurodegeneration, and neurotrophic factor support were observed with MSC‐NTF, with placebo unchanged.
Discussion
The study did not reach statistical significance on the primary endpoint. However, a pre‐specified subgroup suggests that MSC‐NTF participants with less severe disease may have retained more function compared to placebo. Given the unmet patient need, the results of this trial warrant further investigation.
Keywords: ALSFRS‐R, amyotrophic lateral sclerosis, biomarker, clinical trial, stem cells
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is an adult neurodegenerative disease, characterized by progressive degeneration and loss of cortical and spinal motor neurons. Neuroinflammation is believed to play a prominent role. 1 , 2 , 3
There is currently no cure for ALS, and no treatment halts or reverses disease progression. A cell‐based therapy that targets multiple disease pathways has the potential to tackle the multifaceted pathogenesis of ALS by supporting survival of diseased neuronal cells, both by secreting neuroprotective factors and by modulating neuroinflammatory and neurodegenerative pathways. 4
Mesenchymal stem cells (MSC) derived from adult bone marrow were induced under proprietary ex vivo conditions to secrete high levels of neurotrophic factors (MSC‐NTF). MSC‐NTF has been shown to favorably modify neuroprotective and neuroinflammatory cerebrospinal fluid (CSF) biomarkers following single intrathecal administration 5 with safety and preliminary efficacy demonstrated in prior studies 5 , 6 in ALS participants. Here, we report results of the BCT‐002 phase 3 clinical trial conducted to evaluate whether repeat treatment with MSC‐NTF would safely slow the rate of ALS disease progression.
2. METHODS
2.1. Study design and participants
The BCT‐002 study (NCT03280056), a parallel‐group, randomized, double‐blind, placebo‐controlled study, was conducted at six US academic centers. Informed consent was obtained and documented from all participants, and the study was approved by the Institutional Review Boards of all centers. After an 18‐wk pre‐treatment period, including an outpatient bone marrow aspiration to harvest cells for autologous product manufacturing, participants received study treatment intrathecally at weeks 0, 8, and 16, followed by a 12‐wk observation period (Supporting Information Figure S1, which is available online).
Eligible participants were 18–60 y old with possible, laboratory‐supported probable, probable, or definite ALS per the revised‐El Escorial criteria, 7 symptom onset ≤24 mo prior to screening, upright slow vital capacity (SVC) ≥65% of predicted value and revised ALS Functional Rating Scale 8 (ALSFRS‐R) total score ≥ 25 at screening. Participants on a stable dose of riluzole were permitted. Prior stem cell therapy and active participation in an ALS interventional study were prohibited.
Following the first 12 wk in the pre‐treatment period, one final inclusion criteria was required prior to randomization, ie, a decline of ≥3 ALSFRS‐R points. Once randomized, participants received intrathecal MSC‐NTF (NurOwn®, manufactured by Dana‐Farber Cancer Institute, Boston, MA or City of Hope, Duarte, CA; see Supporting Information Methods) or placebo by standard lumbar puncture.
In March 2020, the protocol was amended to allow remote visits due to coronavirus disease 2019 (COVID‐19), with a major impact on the collection of SVC assessments.
2.2. Randomization and masking
Eligible participants were randomized 1:1 to MSC‐NTF: placebo. Investigators, participants, and all personnel involved in the study conduct were blinded to treatment assignments except for the cell manufacturing and site teams that administered treatment. These unblinded personnel did not otherwise interact with participants.
2.3. Primary and secondary objectives
The primary efficacy endpoint, a responder analysis based on change in disease progression, was determined by the rate of change in ALSFRS‐R per month using linear regression, with a separate line fit to pre‐treatment and post‐treatment data through week 28. A participant was defined as a responder if their change in disease progression post‐treatment compared to pre‐treatment was ≥1.25 points/mo, reflecting slowing of decline or improvement of function. Participants who died due to disease progression were considered non‐responders.
Key secondary endpoints were the percentage of participants with post‐treatment rate of decline improved by ≥100% as measured by ALSFRS‐R slope, change from baseline to week 28 in ALSFRS‐R, the combined analysis of function and survival (CAFS), 9 and SVC.
The safety objective was assessed through outcomes of adverse events (AEs), the Columbia‐Suicide Severity Rating Scale (CSSRS), and laboratory, physical examination, vital signs, and electrocardiogram (ECG) assessments. Secondary outcomes for survival included time to death due to disease progression and for any cause through week 32.
2.4. Statistical analysis
To calculate the sample size for the study, the PRO‐ACT database and phase 2 study BCT‐001 (NCT02017912) were analyzed. It was estimated that the true percentage of responders expected to improve by ≥1.25 points/mo was 35% MSC‐NTF and 15% placebo. Using a chi‐square test with α = .05 (two‐sided) and 90% power, 97 participants/treatment were required.
The primary efficacy population was the modified intention‐to‐treat population (mITT), which included participants who received at least one treatment and had at least one pre‐treatment, baseline and at least one post‐treatment ALSFRS‐R assessment. The ITT population included all participants who were randomized. The baseline ALSFRS‐R assessment is defined as the last ALSFRS‐R prior to the first treatment. Safety analyses were performed on the safety population, which included all participants who received at least one treatment. Deaths that occurred during the trial are also reported on the ITT population, to capture all deaths following randomization.
Pre‐specified analyses by subgroups were conducted including a baseline ALSFRS‐R threshold of 35 (the anticipated baseline average). The value of 35 was identified and pre‐defined during the planning portion of the trial based upon the experience of the clinical investigators, data from phase 2 trial of MSC‐NTF and analysis of a database of prior clinical trial participants (PRO‐ACT).
The primary efficacy endpoint and secondary efficacy endpoints were tested sequentially to account for multiplicity and preserve overall type I error. No adjustments were made for multiple comparisons in testing exploratory efficacy endpoints.
Given the higher than anticipated number of participants with advanced ALS disease, post‐hoc sensitivity analyses focused on participants with baseline ALSFRS‐R > 25 (77% of the trial participants). To evaluate robustness of results additional baseline thresholds are presented considering thresholds between 25 and 34 (ie, up to the pre‐specified threshold of 35).
Handling of missing ALSFRS‐R data due to deaths or discontinuations were pre‐specified in the analysis plan. These analyses used multiple imputation methodology under assumptions of missing at random (MI‐MAR) and missing not at random (MI‐MNAR). An additional post‐hoc evaluation of deaths and missing data was undertaken to better understand the impact of both on estimates of treatment effects with the secondary endpoint, change from baseline to 28 wk in ALSFRS‐R. 10
Safety was assessed based on the incidence of treatment‐emergent adverse events (TEAEs) and clinically relevant changes in vital signs, clinical laboratory assessments, physical and neurological examinations, and ECG tests.
2.5. Biomarkers
CSF samples were collected seven times for each participant (Supporting Information Figure S1) and were immediately centrifuged at 1750g for 10 min and stored at −80 °C. A pre‐specified selection of biomarkers important to ALS disease were specified in the statistical analysis plan, including vascular endothelial growth factor (VEGF), monocyte chemoattractant protein‐1 (MCP‐1), and neurofilament light chains (NfL). VEGF is a neurotrophic factor that has demonstrated motor neurons protection. 12 Additionally, VEGF gene expression appears to be deficient in ALS patient fibroblasts, 13 and lower CSF VEGF levels are associated with disease progression. 11 MCP‐1 is a β‐chemokine involved in neuroinflammatory pathways that correlates with ALS progression and reduced survival. 14 NfL is a biomarker known to be increased several‐fold in ALS, reflecting the extensive damage of motor neurons and axons, 15 and correlate with ALS disease progression. 16
Since biomarker data are highly skewed, data were log transformed for analysis using a MMRM model with covariates from the primary endpoint. A pre‐specified stepwise regression model was used to select biomarkers that were predictive of the primary endpoint, from a set of all biomarkers. Biomarkers with term P‐values <.05 were required to enter the model and to be retained in the final model.
3. RESULTS
3.1. Baseline and disease characteristics
Between August 2017 to September 2020, 263 persons with ALS were screened for eligibility; 196 were randomly assigned to treatment group and 189 received at least one treatment (Figure 1). Seven participants did not receive treatment. In the mITT population, more than 75% of participants completed the treatment regimen. A total of 45 participants discontinued the study prematurely.
FIGURE 1.
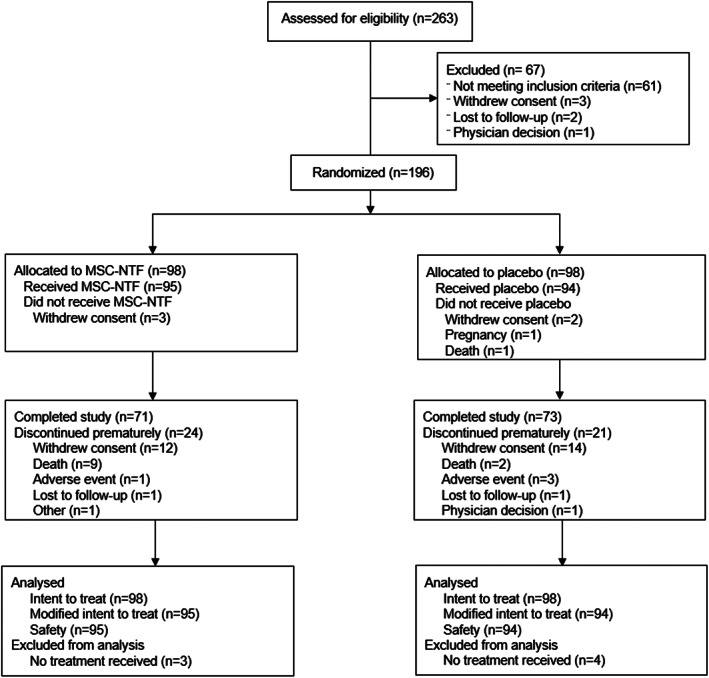
CONSORT diagram. Phase 3 randomized, placebo‐controlled study to evaluate MSC‐NTF effects on safety, tolerability, biomarkers, and clinical outcomes in amyotrophic lateral sclerosis. CONSORT methodology is used for consistent documentation of the flow of participants through a trial. The categories within each box are reported as they were captured on the electronic Case Report Form. There are participants who discontinued the trial reasons other than death and then subsequently died. For a complete list of deaths for all participants randomized in the trial, see Table 2
The mean age of study participants was 49 y, and 67% were male (Table 1). Baseline characteristics between treatment groups were generally well‐balanced, indicating that randomization was effective. The trial enrolled participants with advanced disease (ALSFRS‐R ≤ 25) resulting in an average baseline ALSFRS‐R value of 31 and inclusion of some participants starting the trial with a value of zero on ALSFRS‐R individual items, therefore unable to show disease progression on these items. In participants with baseline ALSFRS‐R ≤ 25, Fine and Gross Motor ALSFRS‐R subscales had more individual items with baseline scores of zero than other subscales, with 43%, 43%, and 39% of participants with zero values on each Fine Motor item and 23%, 14%, and 75% on each Gross Motor item. For average number of participants with zero values on items across subscales in this subgroup, see Table 1.
TABLE 1.
Demography and baseline characteristics
| Characteristic | MSC‐NTF | Placebo |
|---|---|---|
| (N = 95) | (N = 94) | |
| Age (y), mean (SD) | 48.1 (9.71) | 49.1 (8.38) |
| Sex | ||
| Female, nn (%) | 27 (28.4) | 35 (37.2) |
| Male, n (%) | 68 (71.6) | 59 (62.8) |
| Baseline ALSFRS‐R, mean (SD) | 30.3 (6.5) | 31.4 (6.1) |
| ≤25, n (%) | 23 (24.2) | 21 (22.3) |
| > 25, n (%) | 72 (75.8) | 73 (77.7) |
| Baseline SVC (% predicted), mean (SD) | 76.2 (20.9) | 75.0 (19.8) |
| Time from first symptom to first treatment (mo), mean (SD) | 19.6 (5.17) | 19.1 (4.90) |
| Time from diagnosis (mo), mean (SD) | 6.8 (4.35) | 6.1 (4.80) |
| Site of disease onset | ||
| Limb, n (%) | 80 (84.2) | 73 (77.7) |
| Limb and bulbar, n (%) | 15 (15.8) | 21 (22.3) |
| El Escorial criteria | ||
| Possible, n (%) | 6 (6.3) | 6 (6.4) |
| Lab‐supported probable, n (%) | 15 (15.8) | 23 (24.5) |
| Probable, n (%) | 24 (25.3) | 31 (33.0) |
| Definite, n (%) | 50 (52.6) | 34 (36.2) |
| Use of riluzole at baseline | ||
| Yes, n (%) | 65 (68.4) | 56 (59.6) |
| No, n (%) | 30 (31.6) | 38 (40.4) |
| ALSFRS‐R | MSC‐NTF | Placebo |
|---|---|---|
| (N = 23) | (N = 21) | |
| Trial participants with ALSFRS‐R total score ≤ 25 at baseline | ||
| Average bulbar items with value of 0 at baseline, % | 7 | 6 |
| Average fine motor items with value of 0 at baseline, % | 49 | 33 |
| Average gross motor items with value of 0 at baseline, % | 42 | 32 |
| Average respiratory items with value of 0 at baseline, % | 1 | 0 |
3.2. Safety
Almost all participants experienced mild‐to‐moderate TEAEs (Table 2). The most commonly reported TEAEs were predominantly associated with pain, were transient, and many were related to the treatment administration lumbar puncture procedure (Supporting Information Table S1). Post‐procedural AEs associated with study procedures (bone marrow aspiration and/or lumbar puncture) were commonly seen in active and placebo treated participants and were mostly mild to moderate in severity. These AEs were reported more frequently in subjects who had received MSC‐NTF. Only one participant in each treatment group had a related SAE, and more withdrawals due to TEAEs were reported in the placebo group. A small percentage (2.1%) of participants withdrew from the study due to AEs (Figure 1). There were no instances of tracheostomy and permanent ventilation in the study.
TABLE 2.
Safety results
| MSC‐NTF | Placebo | |
|---|---|---|
| (N = 95) | (N = 94) | |
| n (%) | n (%) | |
| Safety overview, safety population | ||
| TEAEs | 94 (98.9) | 92 (97.9) |
| TEAE related to the study medication | 76 (80.0) | 66 (70.2) |
| Severe TEAEs | 29 (30.5) | 19 (20.2) |
| Severe‐related TEAEs | 7 (7.4) | 3 (3.2) |
| Serious TEAEs | 23 (24.2) | 17 (18.1) |
| Serious‐related TEAEs | 1 (1.1) | 1 (1.1) |
| Procedure‐related TEAEs | 89 (93.7) | 82 (87.2) |
| TEAEs leading to treatment withdrawal | 1 (1.1) | 3 (3.2) |
| Deaths, ITT population | MSC‐NTF | Placebo |
|---|---|---|
| (N = 98) | (N = 98) | |
| AEs leading to death | 10 (10.2) | 6 (6.1) |
| Deaths related to study treatment | 0 | 0 |
| Cause of death | ||
| Disease progression | 8 (8.2) | 4 (4.1) |
| Other | 2 (2.0) | 2 (2.0) |
| Deaths by baseline ALSFRS‐R score | ||
| Baseline ALSFRS‐R ≤ 25 | 6 (6.1) | 4 (4.1) |
| Baseline ALSFRS‐R > 25 | 4 (4.1) | 2 (2.0) |
There were no clinically significant differences between treatment groups at any timepoint nor in change from baseline in clinical laboratory values, vital signs, physical examination findings, and ECG results over the course of the study. Assessments of suicidal ideation and behavior using the CSSRS were similar between treatment groups.
During the study, 16 participants died after being randomized to treatment (Table 2). These participants had more advanced disease than the broader study population. Fourteen participants died after receiving treatment, two before. Most deaths occurred in participants with baseline ALSFRS‐R scores ≤25. All except one were directly attributed to disease progression. No deaths were reported as related to study treatment by the investigator or the sponsor. Event free probability for death due to disease progression and death due to any cause for both treatments was >88% and were not statistically different between treatments (Table 3). Overall, MSC‐NTF was well tolerated. There were no safety concerns.
TABLE 3.
Efficacy results: primary and secondary endpoints at 28 wk, mITT population
| All participants | MSC‐NTF | Placebo |
|---|---|---|
| (N = 95) | (N = 94) | |
| Primary endpoint, through week 28 | ||
| ≥1.25 points improvement in ALSFRS‐R slope, n (%) | 31 (32.6) | 26 (27.7) |
| OR (95% CI), P‐value | 1.33 (0.63, 2.80), P = .45 | |
| Secondary endpoints, through week 28 | ||
| ≥100% improvement in ALSFRS‐R slope, n (%) | 13 (13.7) | 13 (13.8) |
| OR (95% CI), P‐value | 0.998 (0.42, 2.40), P = 0.997 | |
| ALSFRS‐R Total score, LS mean change from baseline (SE) | −5.52 (0.67) | −5.88 (0.67) |
| LS mean difference (95% CI), P‐value | 0.37 (−1.47, 2.20), P = .69 | |
| CAFS, LS mean at week 28 (SE) | 73.74 (5.21) | 72.21 (4.89) |
| LS mean difference (95% CI), P‐value | 1.53 (−10.65, 13.72), P = .80 | |
| Slow vital capacity % predicted, LS mean change from baseline (SE) | −12.94 (1.80) | −11.55 (1.81) |
| LS mean difference (95% CI), P‐value | −1.39 (−6.15, 3.38), P = .56 | |
| Secondary endpoints, through week 32 | ||
| Tracheostomy‐free survival (no tracheostomies during the study) | n/a | n/a |
| Hazard ratio (95% CI) | n/a | |
| Event free probability (K‐M) for deaths due to disease progression | 90.43 | 92.24 |
| (95% CI), | (81.71, 95.12) | (70.98, 98.12) |
| P‐Value versus placebo | .21 | |
| Event free probability (K‐M) for death due to any cause | ||
| (95% CI) |
88.32 (79.32, 93.56) |
89.17 (69.08, 96.51) |
| P‐Value versus placebo | .11 | |
| ALSFRS‐R ≥ 35 | ALSFRS‐R < 35 | |||
|---|---|---|---|---|
| Pre‐specified subgroup | MSC‐NTF | Placebo | MSC‐NTF | Placebo |
| ALSFRS‐R Total baseline | (N = 26) | (N = 32) | (N = 69) | (N = 62) |
| Primary endpoint through week 28 | ||||
| ≥1.25 ALSFRS‐R slope improvement, n (%) | 9 (34.6) | 5 (15.6) | 22 (31.9) | 21 (33.9) |
| OR (95% CI), P‐value | 2.6 (0.45, 14.36), P = .29 | 0.87 (0.36, 2.07), P = .74 | ||
| Secondary endpoints through week 28 | ||||
| ≥100% improvement in ALSFRS‐R slope, n (%) | 7 (26.9) | 5 (15.6) | 6 (8.7) | 8 (12.9) |
| OR (95% CI), P‐value | 1.80 (0.40, 8.15), P = .45 | 0.65 (0.21, 2.09), P = .47 | ||
| ALSFRS‐R Total score, LS mean change (SE) | −1.77 (1.20) | −3.78 (1.07) | −6.87 (0.76) | −6.96 (0.79) |
| LS mean difference (95% CI), P‐value | 2.01 (−1.06, 5.08), P = .20 | 0.09 (−2.00, 2.18), P = .93 | ||
| CAFS, LS mean at week 28 (SE) | 93.27 (10.24) | 74.84 (9.11) | 66.25 (6.34) | 71.96 (6.29) |
| LS mean difference (95% CI), P‐value | 18.42 (−3.59, 40.44), P = .10 | −5.71 (−20.12, 8.71), P = .44 | ||
Abbreviations: CI, confidence interval; K‐M, Kaplan Meier.
Note: Results from secondary endpoints through week 32 do not include two deaths that occurred in participants randomized to placebo which occurred prior to treatment.
3.3. Primary endpoint
The study did not meet the primary endpoint. The percentages of participants in the mITT population who met the criteria for clinical response at 28 wk on the primary endpoint were 33% MSC‐NTF and 28% placebo (odds ratio [OR] = 1.33, P = .45) (Table 3). As a result of the planned hierarchical testing approach to control type I error, all remaining P‐values in the manuscript are nominal and not controlled at the .05 level. A pre‐specified sensitivity analysis accounting for all deaths as non‐responders had identical results. In a prespecified subgroup of participants with less severe disease at baseline (ALSFRS‐R ≥ 35), MSC‐NTF treated participants achieved 35% response versus 16% placebo, which was not statistically significant (Table 3). In participants with more severe disease at baseline (ALSFRS‐R < 35), response rates were similar. When the observed baseline study mean (ALSFRS‐R ≥ 31) was used instead of the anticipated baseline study mean (ALSFRS‐R ≥ 35), MSC‐NTF treated participants achieved 35.4% response versus 15.4% with placebo, which was nominally statistically significant (P < .05, Table 4).
TABLE 4.
Efficacy analyses across ALSFRS‐R baseline thresholds
| Percent response across ALSFRS‐R baseline thresholds, primary endpoint | ALSFRS‐R MMRM change from baseline to week 28 secondary endpoint | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| ALSFRS‐R baseline score | n (%) total participants | % MSC‐NTF | % placebo | P‐Value | n (%) total participants | MSC‐NTF LS mean (SE) | Placebo LS mean (SE) | Delta | P‐Value |
| ≥34 | 68 (36) | 37.9 | 15.4 | .163 | 57 (30) | −1.58 (1.13) | −4.32 (0.96) | 2.74 | 0.057 |
| ≥33 | 83 (44) | 35.1 | 15.2 | .088 | 69 (37) | −2.36 (1.01) | −4.58 (0.91) | 2.23 | 0.092 |
| ≥32 | 96 (51) | 37.0 | 16.0 | .056 | 78 (41) | −2.22 (0.90) | −4.32 (0.87) | 2.10 | 0.081 |
| ≥31 | 100 (53) | 35.4 | 15.4 | .043 | 80 (42) | −2.44 (0.89) | −4.83 (0.86) | 2.39 | 0.045 |
| ≥30 | 115 (61) | 35.7 | 16.9 | .103 | 92 (49) | −3.14 (0.82) | −5.53 (0.81) | 2.39 | 0.032 |
| ≥29 | 123 (65) | 34.5 | 18.5 | .056 | 97 (51) | −3.66 (0.82) | −5.77 (0.78) | 2.11 | 0.054 |
| ≥28 | 131 (69) | 36.5 | 19.1 | .022 | 105 (56) | −3.49 (0.78) | −5.58 (0.75) | 2.09 | 0.048 |
| ≥27 | 138 (73) | 35.4 | 20.5 | .026 | 110 (58) | −3.95 (0.76) | −5.89 (0.72) | 1.94 | 0.06 |
| ≥26 | 145 (77) | 34.7 | 20.5 | .053 | 116 (61) | −4.61 (0.75) | −5.81 (0.75) | 1.20 | 0.247 |
Note: Hypothesis testing performed using logistic regression adjusted for baseline ALSFRS‐R total score, duration from onset of symptoms to first treatment, site of onset (Limb versus Limb & Bulbar). Riluzole use at baseline and ALSFRS‐R slope pre‐treatment were used to test the hypothesis of an OR of 1 between the two treatment groups.
Pre‐specified analyses accounting for missing data through MI‐MAR and MI‐MNAR resulted in increased confidence in the MSC‐NTF treatment effect from this trial in the presence of missing data and deaths. When leveraging different approaches for handling missing data, treatment differences favoring MSC‐NTF increased by more than 45% and the P‐values substantially decreased (Supporting Information Table S2).
3.4. Secondary endpoints
There were no statistically significant differences between treatment groups on the secondary endpoints (Table 3). The analysis of participants who achieved 100% improvement was 14% across both treatment groups. The least squares (LS) mean change from baseline to 28 wk treatment difference, and the average rank score on the CAFS both favored MSC‐NTF, but the difference was relatively small. The change from baseline in % predicted SVC was smaller for placebo.
In the pre‐specified subgroup analysis, the treatment difference observed with MSC‐NTF participants with less severe baseline disease (≥35, Table 3) was more pronounced compared to the analysis of all participants. For example, MSC‐NTF participants progressed on average two points less on the ALSFRS‐R compared to placebo, while for participants with more advanced disease the change from baseline to week 28 was similar between treatment groups. In the participants with more severe baseline disease (<35) the treatment groups were generally more similar across endpoints, with placebo participants having a higher CAFS rank.
Analysis of the ALSFRS‐R subscales showed changes in each subscale favored MSC‐NTF, except for the Respiratory subscale, which favored placebo (Supporting Information Figure S2A). The largest differences between MSC‐NTF and placebo were seen in the Fine Motor subscale and Gross & Fine Motor subscales combined. Similar to the analysis of the ALSFRS‐R total score, treatment differences across the subscales were larger in participants with less severe disease at baseline (Figure S2B and S2C).
Pre‐specified analyses for the LS mean change from baseline to 28 wk accounting for missing data through MI‐MAR and MI‐MNAR, in addition to a post hoc model estimating the joint impact of survival and missing data treatment, confirmed the MSC‐NTF treatment effect from this trial in the presence of missing data and deaths (Supporting Information Tables S3 and S4).
3.5. Post hoc efficacy analyses
Given the higher‐than‐expected number of participants randomized with more advanced ALS, in order to better understand the difference between the pre‐specified subgroups and to include a higher percentage of participants in the subgroup analysis of participants with less severe disease (ALSFRS‐R > 25, 77% participants versus ALSFRS‐R ≥ 35, 31% participants) we conducted a post‐hoc analysis in participants with baseline ALSFRS‐R score > 25 for all endpoints (Supporting Information Figure S3). The percentage of these participants who met the primary endpoint response criteria at 28 wk were 35% MSC‐NTF and 21% placebo (OR = 2.53, Table 4). In this same subgroup, the secondary endpoint of >100% response showed a higher response rate with MSC‐NTF than placebo, a larger treatment difference from baseline to 28 wk on the ALFSRS‐R and the average rank score on the CAFS of 12 points higher.
Further sensitivity analysis aimed at exploring the consistency of treatment effect across subgroup thresholds progressing from baseline scores of >25 (≥26) up to ≥34, were conducted (Table 4). On the primary endpoint, a consistent response rate was observed in the MSC‐NTF treatment group, with an average MSC‐NTF response rate above 34.5% across all thresholds. In contrast, the placebo response was inconsistent across baseline thresholds with the highest response rate observed with the threshold containing participants with the most advanced disease at baseline (20.5%, baseline >25) and decreasing by 5% at the threshold of 34 (15.4%, baseline ≥34). The difference in response rates between treatments across baseline thresholds all favored MSC‐NTF, exceeding placebo response by 14% to 23% (Supporting Information Figure S4A, Table 4).
On the secondary endpoint, change from baseline to week 28 in the ALSFRS‐R, the LS mean for MSC‐NTF participants across baseline thresholds varied by baseline severity with participants with less advanced disease at baseline losing less function compared to those with more advanced disease. LS means for placebo exhibited far less spread in the LS mean (range) change compared to MSC‐NTF across baseline thresholds: MSC‐NTF (−1.58, −4.61), placebo (−4.32, −5.81), meaning placebo‐treated participants lost 4–5 points on average in 28 wk across baseline thresholds while MSC‐NTF‐treated participants lost less function at higher compared to lower baseline levels. Comparison of the treatment groups across baseline thresholds highlighted that MSC‐NTF‐treated‐participants lost on average, 2 points of function less compared to placebo‐treated‐participants, which was maintained or became larger through the prespecified threshold of 35 (Supporting Information Figure S4B, Table 4, Table 3). Changes in ALSFRS‐R over time showed how quickly the MSC‐NTF diverged from placebo, across baseline thresholds, using the pre‐specified MMRM model and the post‐hoc sensitivity analysis that accounted for death and missing data (Supporting Information Figure S5).
These post hoc analyses suggest that MSC‐NTF participants with less advanced disease may have retained more function compared to placebo.
3.6. Biomarkers
CSF collected from all participants revealed large and statistically significant CSF biomarker improvements from baseline with MSC‐NTF treatment, particularly in biomarkers related to neuroinflammation, neurodegeneration, and NTF, while placebo remained unchanged. At week 20, CSF VEGF increased two‐fold from baseline in the MSC‐NTF group and was significantly higher than placebo at all visits (P < .05, Figure 2A), with MSC‐NTF values 258% of placebo at week 20, an overall treatment effect P‐value of P < .0001. At all timepoints, CSF MCP‐1 was significantly reduced with MSC‐NTF relative to placebo (P < .05, overall treatment effect P < .0001), with week 20 MSC‐NTF values 74% of placebo (Figure 2B). At week 20, NfL levels with MSC‐NTF were 82% relative to placebo (Figure 2C). Statistical models demonstrated that these biomarkers, in addition to MSR1, Fetuin‐A, change in MCP‐1, change in Fetuin‐A and the ENCALS model term, were important predictors of the treatment response in this trial (Table 5). The estimates and P‐values associated with each term retained in the final model and the receiver operating characteristic curve (ROC) both highlight strong performance (82.5% accuracy) of the biomarker data in the model predicting clinical response (Supporting Information Figure S6). This model supports the hypothesis that MSC‐NTF treatment impacted ALS disease progression and provides additional evidence linking the mechanism of action to MSC‐NTF's impact on ALS disease progression. A thorough analysis of biomarker data will be published separately.
FIGURE 2.
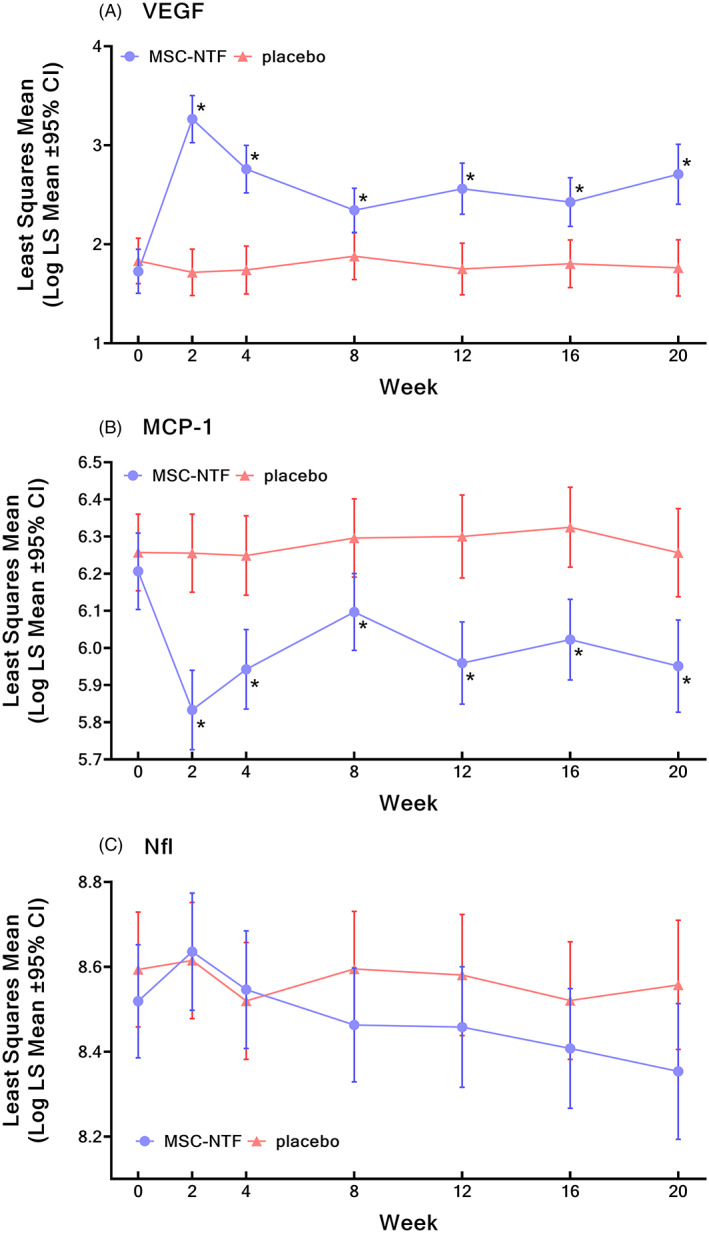
Longitudinal changes in biomarkers (log transformed). CI, confidence interval. Longitudinal changes in biomarkers (log transformed) over the course of the study in least square mean cerebrospinal fluid levels of VEGF (A), MCP‐1 (B), and NfL (C). *P < .05
TABLE 5.
Biomarker terms with significant explanatory value for the primary endpoint
| Biomarker (post baseline) | Estimate | P‐Value |
|---|---|---|
| BMKCHG‐Fetuin‐A | 1.31 | <.0001 |
| BMKCHG‐MCP‐1 | 1.33 | .0002 |
| Fetuin‐A | −0.50 | .0089 |
| ENCALS | 0.71 | <.0001 |
| MCP‐1 | −1.03 | <.0001 |
| MCP‐1*treatment | −0.53 | .0074 |
| MSR1 | −0.65 | <.0001 |
| NfL | −0.59 | .0006 |
| Treatment | 3.72 | .0027 |
| VEGF‐A | −0.25 | .0248 |
Note: A stepwise forward regression model using the primary endpoint as dependent variable and all the biomarkers as independent variables or predictor variables was performed, in the attempt to build a potential composite index that can predict the outcomes more efficiently than being performed one by one. Along with CSF biomarkers, the European Network to Cure ALS (ENCALS) risk profile 18 was also included as a possible model term. The ENCALS risk profile is a criterion to evaluate the risk of a patient dying (a measure of severity).
Abbreviations: BMKCHG, change in biomarker across the treatment period.
4. DISCUSSION
The primary and secondary efficacy endpoints of this phase 3, placebo‐controlled, randomized trial were not statistically significant; however, the study provided significant information about the study design and potential biomarkers of treatment response for use in future clinical trials, in addition to important information about MSC‐NTF. This trial confirmed the safety of repeat intrathecal administration of autologous mesenchymal stem cell therapy in patients with ALS and demonstrated statistically significant improvements across multiple biomarkers of neuroinflammation, neurodegeneration, and NTF support with MSC‐NTF.
Overall, participants enrolled in this study had more advanced disease than in other late phase ALS trials, which may have had a dilution effect, reducing the ability to show a treatment effect in the overall study population. In a pre‐specified subgroup with baseline ALSFRS‐R ≥ 35, there was a higher response rate on the primary endpoint with MSC‐NTF compared to placebo along with a larger treatment effect across all endpoints with MSC‐NTF compared to placebo, both of which did not reach statistical significance. This subgroup threshold was selected as the anticipated baseline mean; however, because the participants enrolled had more advanced ALS disease, only 31% of participants had baseline ALSFRS‐R ≥ 35, reducing power associated with the statistical test. Using the observed baseline study mean (ALSFRS‐R ≥ 31), the difference in the percentage of MSC‐NTF versus placebo responders increased, and the increase in sample size produced a nominally significant P ‐value (P < .05, Table 4). A 2.39‐point treatment difference was observed at this threshold in the change from baseline to week 28 on the ALSFRS‐R, also with a nominally significant P ‐value (P = .045).
There were a few limitations for this study providing learning for future trials. We recently highlighted the difficulty in ALS trials because of disease heterogeneity. 17 This phase 3 trial is one of a few that included a sizeable group of participants with advanced ALS at baseline and is more representative of the broader patient population. While a strength in some ways, one lesson learned is that it perhaps makes this trial more subject to the impact of the floor effect in the ALSFRS‐R scale. A key question is whether the insensitivity of the scale, particularly in participants with the lowest ALSFRS‐R scores who were close to this floor, could result in an apparent improvement that was, in fact, a misclassification of response given the primary endpoint was defined by slowing of progression.
Had the inclusion criteria of baseline ALSFRS‐R > 25 been used at baseline instead of screening, post‐hoc power calculations reveal that the same sized trial (n = 196 randomized) in this set of participants would have likely been underpowered, given conventional levels for a phase 3 trials. (ie, leveraging the response rate observed in the trial from the subgroup of participants with ALSFRS‐R > 25 [34.7% response MSC‐NTF, 20.5% response placebo], the estimated power would have been 60%).
Another observation is the primary endpoint, a threshold‐based analysis, appears to be more greatly influenced by participants at the lower end of the scale. The likelihood of meeting the threshold of 1.25 points/mo is influenced by the rate of decline (associated with baseline level), eg, a patient losing 3 points/mo is more likely to achieve a 1.25 point/mo improvement than someone losing 1 point/mo. A more homogeneous set of participants would have likely resulted in more consistent properties with this endpoint. The threshold‐based sensitivity analysis for the primary endpoint shows that placebo participants with lower values had higher rates of clinical response while MSC‐NTF had more stable rates of clinical response across these same thresholds. However, a similar sensitivity applied to average changes on the ALSFRS‐R highlights that placebo participants were consistently losing far more function over time than MSC‐NTF, with the range of LS means on placebo being much tighter (−4.32, −5.81) across 28 wk compared to MSC‐NTF (−1.58, −4.61). This analysis reveals patterns more typical of a placebo arm in a clinical trial and suggests that participants earlier in their disease course had a more evident treatment response on MSC‐NTF than those that were further progressed, a pattern one would expect from an effective treatment.
Biomarker data from this study provide important information about disease mechanism of action, which targets multiple pathways including neuroprotection, neuroinflammation, and neurodegeneration. These significant, robust, and sustained changes in all MSC‐NTF‐treated participants in many neuroinflammatory and neurodegenerative biomarkers, including MCP‐1 and NfL, were consistent with earlier trials 5 and provide support for a treatment associated with disease progression. Statistical modeling of CSF biomarker data and the ENCALS model term provides evidence linking a targeted therapeutic mechanism of action and prediction of personalized prognosis 18 to the preservation of ALS function. 19 The authors acknowledge that additional data, adding to the data in this trial, will increase the acceptance of these biomarkers and their utility in predicting clinical response.
In summary, we present a thorough analysis of MSC‐NTF phase 3 data, demonstrating a potential treatment effect on ALS disease progression linked to target biomarker changes and confirming treatment safety. While the primary and secondary endpoints failed to reach statistical significance in the overall study population, a pre‐specified subgroup suggests across endpoints that MSC‐NTF participants with less severe disease may have retained more function compared to placebo. Pre‐specified and post‐hoc analyses that accounted for the ALSFRS‐R floor effects, missing data, and deaths suggest a potential treatment effect with MSC‐NTF across the primary and secondary efficacy (change from baseline to 28 wk) outcomes. Pre‐specified biomarker analyses provide evidence for target engagement that is compatible with a proposed mechanism of action of MSC‐NTF therapy relevant to multiple known ALS disease pathways. These data will contribute to advancing the role of MSC‐NTF cell therapy in ALS, the relationship between biomarkers and disease progression and to further benefit the broader ALS community. Given the unmet patient need, the results of this trial warrant further investigation.
CONFLICT OF INTEREST
D.A.B. reports personal fees from Brainstorm Cell Therapeutics. J.D.B. reports personal fees from Biogen, Clene Nanomedicine, MT Pharma of America, MT Pharma Holdings of America, Janssen; grants from Alexion, Biogen, Amylyx Therapeutics, MT Pharma of America, Anelixis Therapeutics, Genentech, Rapa Therapeutics, MT Pharma Holdings of America, nQ Medical, NINDS, Muscular Dystrophy Association, ALS One, ALS Association, ALS Finding A Cure, Rapa Therapeutics; and has equity in ReactNeuro. J.K. reports consulting fees from MT Pharma, Denali Therapeutics, Biogen, Genetech, Amylyx, Cytokinetics, Wave, and Calico. K.N. reports personal fees from Alector LLC, AI Therapeutics, Biogen, Biohaven, MT Pharma, Wave Therapeutics, Enclear, and Regeneron; grants from ALS Association, Muscular Dystrophy Association, ALS Finding a Cure, and Target ALS. M.C. reports personal fees from Lilly Avexis, Pontifax, Orion, MT Pharma, Denali, Biogen, Pharmnext, Treeway, Revalasio, Takeda, Aclipse, Biohaven, Sunovian, Anelixis, Disarm, ALS Biopharma, Cytokinetics, RRD, Immunity Pharma, Helixsmith, Wave, Transposon, Quralis, Faze, Regeneron, and AB Sciences. N.A.G. received research support from Alexion, Brainstorm Cell Therapeutics, Cytokinetics, Fulcrum, Healey Platform, Kezar, Medicinova, Octapharma, Orion, Orphazyme; served on Advisory Boards for Acceleron, Alexion, Argenx, CSL Behring, MT Pharma, Sanofi Genzyme, Sarepta, UCB; received travel reimbursement and honoraria; and served on the speaker's bureau for CSL. N.P.S. reports research support from the National Institutes of Health (R01 CA21887), Regenerative Medicine Minnesota, Target ALS, and ALS Association. R.H.B. Jr. reports personal fees from Wave Lifesciences and serves on advisory boards of ALS Finding a Cure, Project ALS, and NEALS. R.L. received research support from Argenx, Annexon, Biotest, CSL Behring, Grifols, Pharnext, Sanofi‐Genzyme, Seattle Genetics, UCB, and Pfizer; honoraria from Medscape, Alnylam, Akcea; and served on advisory boards for GBS‐CIDP Foundation International, Myasthenia Gravis Foundation of America, MGF of California, and Board of Directors for Peripheral Nerve Society. S.R.L., R.K., Y.G, Y.S.L., and R.A are employees of and hold stock in Brainstorm Cell Therapeutics. A.J.W., L.J., M.A.O., M.B., R.M., and T.M. have no conflicts of interest to report.
Abbreviations
- AE
adverse event
- ALS
amyotrophic lateral sclerosis
- ALSFRS‐R
Revised ALS Functional Rating Scale
- CAFS
Combined Analysis of Function and Survival
- CSF
cerebrospinal fluid
- CSSRS
Columbia‐Suicide Severity Rating Scale
- ECG
electrocardiogram
- LS
least squares
- MCP‐1
monocyte chemoattractant protein‐1
- MI‐MAR
multiple imputation methodology under assumptions of missing at random
- MI‐MNAR
multiple imputation methodology under assumptions of missing not at random
- mITT
modified intention‐to‐treat population
- MMRM
mixed effect repeated measures
- MSC
mesenchymal stem cells
- NfL
neurofilament light chain
- NTF
neurotrophic factors
- OR
odds ratio
- ROC
receiver operating characteristic curve
- SVC
slow vital capacity
- TEAE
treatment‐emergent adverse event
- VEGF
vascular endothelial growth factor
PRESENTATIONS MADE ON THIS TRIAL
(1) Merit Cudkowicz, Ralph Kern; 31st International Symposium ALS/MND, 12/9/2020 (fully virtual meeting from a COVID‐secure studio).
(2) Ralph Kern, 11th Annual California ALS Research Summit, January 21, 2021 (fully virtual meeting due to COVID‐19).
(3) James Berry; Northeast Amyotrophic Lateral Sclerosis Consortium® (NEALS), 10/6/2021 (fully virtual meeting due to COVID‐19).
ETHICAL PUBLICATION STATEMENT
We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this report is consistent with those guidelines.
Supporting information
Appendix Supplementary Information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
This study was funded by Brainstorm Cell Therapeutics. Except for Brainstorm, no funding organization was involved in the data collection, analysis, interpretation and writing of the manuscript. The study was also supported by two grants: the California Institute for Regenerate Medicine (CIRM, CLIN2‐09894) and a grant from the ALS Association and I AM ALS. With the exception of Brainstorm, no funding organization was involved in the data collection, analysis, interpretation and writing of the manuscript. The authors thank the study participants and their families without whom this study would not have been accomplished. We also wish to thank the Data Safety Monitoring Board: Carlayne Jackson MD, Richard Bedlack MD, and David Schoenfeld, PhD. Brainstorm Cell Therapeutics wishes to thank: the team of statisticians and programmers at Tigermed led by Munish Mehra, PhD; and at Pharmalex led by Bruno Boulanger, PhD.; Brainstorm R&D; Brainstorm Manufacturing, Quality Control and Quality Assurance. We also thank Dana‐Farber Cancer Institute (DFCI) in Boston, MA for work carried out in the Connell and O’Reilly Families Cell Manipulation Core Facility (CMCF) led by Dr. Jerome Ritz, which manufactured autologous MSC‐NTF for patients enrolled in this trial. We also thank the Center for Biomedicine & Genetics (CBG) at City of Hope (COH) lead by Dr. Joseph Gold, which manufactured autologous MSC‐NTF for patients enrolled in this trial. We thank our principal investigators and their dedicated study teams who are individually acknowledged in the Supporting Information text. Sean M. Healey and AMG Center for ALS at Massachusetts General Hospital wishes to thank their Investigators, Coordinators, TCRC Leads, Hematologists and CTTL. University of California at Irvine wishes to thank the Sub‐Investigators (Sarita Said MD, Jonathan Cauchi MD), Coordinators, CTU manager, Hematologist, and the Alpha Stem Cell Clinic. California Pacific Medical Center wishes to thank Dr. Brian Bronzo, Dr. Olufoladare Olorunsola, Dr. Ricky Tong, Marguerite Engel, Marian Leon, Chow Saephanh. Cedars‐Sinai Medical Center wishes to thank Ashraf Elsayegh, Philip Ng, Michele Tagliati, Michael Yang, Peggy Allred, Jeena Cha, Carolyn Prina, Jillian Doherty, and the rest of our invaluable coordinator and pharmacy staff. Mayo Clinic College of Medicine wishes to thank the Neurologist Sub‐Investigators, Carol Denny and the other Coordinators/Support staff, Hematologist and IMPACT Cell Therapy Laboratory. University of Massachusetts Medical School wishes to thank Diane McKenna‐Yasek, Catherine Douthwright PhD, Jeff Gao.
Cudkowicz ME, Lindborg SR, Goyal NA, et al. A randomized placebo‐controlled phase 3 study of mesenchymal stem cells induced to secrete high levels of neurotrophic factors in amyotrophic lateral sclerosis. Muscle & Nerve. 2022;65(3):291-302. doi: 10.1002/mus.27472
Funding information Amyotrophic Lateral Sclerosis Association; California Institute for Regenerate Medicine, Grant/Award Numbers: CIRM, CLIN2‐09894; I AM ALS
DATA AVAILABILITY STATEMENT
The anonymized patient data are not being publicly shared as they are the focus of regulatory interactions for future regulatory approval.
REFERENCES
- 1. Brown RH, Al‐Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377(2):162‐172. doi: 10.1056/NEJMra1603471 [DOI] [PubMed] [Google Scholar]
- 2. Chen JJ. Overview of current and emerging therapies for amytrophic lateral sclerosis. Am J Manag Care. 2020;26(suppl 9):S191‐S197. doi: 10.37765/ajmc.2020.88483 [DOI] [PubMed] [Google Scholar]
- 3. Thonhoff JR, Simpson EP, Appel SH. Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr Opin Neurol. 2018;31(5):635‐639. doi: 10.1097/WCO.0000000000000599 [DOI] [PubMed] [Google Scholar]
- 4. Abati E, Bresolin N, Comi G, Corti S. Advances, challenges, and perspectives in translational stem cell therapy for amyotrophic lateral sclerosis. Mol Neurobiol. 2019;56(10):6703‐6715. doi: 10.1007/s12035-019-1554-x [DOI] [PubMed] [Google Scholar]
- 5. Berry JD, Cudkowicz ME, Windebank AJ, et al. NurOwn, phase 2, randomized, clinical trial in patients with ALS: safety, clinical, and biomarker results. Neurology. 2019;93(24):e2294‐e2305. doi: 10.1212/WNL.0000000000008620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of Mesenchymal stem cells secreting Neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis: results of phase 1/2 and 2a clinical trials. JAMA Neurol. 2016;73(3):337‐344. doi: 10.1001/jamaneurol.2015.4321 [DOI] [PubMed] [Google Scholar]
- 7. Brooks BR, Miller RG, Swash M, Munsat TL. World Federation of Neurology Research Group on motor neuron diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293‐299. doi: 10.1080/146608200300079536 [DOI] [PubMed] [Google Scholar]
- 8. Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS‐R: a revised ALS functional rating scale that incorporates assessments of respiratory function. BDNF ALS study group (phase III). J Neurol Sci. 1999;169:13‐21. doi: 10.1016/s0022-510x(99)00210-5 [DOI] [PubMed] [Google Scholar]
- 9. Berry JD, Miller R, Moore DH, et al. The combined assessment of function and survival (CAFS): a new endpoint for ALS clinical trials. Amyotroph Lateral Scler Frontotemporal Degener. 2013;14(3):162‐168. doi: 10.3109/21678421.2012.762930 [DOI] [PubMed] [Google Scholar]
- 10. Guo X, Carlin BP. Separate and joint modeling of longitudinal and event time data using standard computer packages. Am Stat. 2004;58(1):16‐24. doi: 10.1198/0003130042854 [DOI] [Google Scholar]
- 11.Guo J, Yang X, Gao L, Zang D. Evaluating the levels of CSF and serum factors in ALS. Brain Behav. 2017;7:1‐8. doi: 10.1002/brb3.637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruiz de Almodovar C, Lambrechts D, Mazzone M, Carmeliet P. Role and therapeutic potential of VEGF in the nervous system. Physiol Rev. 2009;89(2):607‐648. doi: 10.1152/physrev.00031.2008 [DOI] [PubMed] [Google Scholar]
- 13. Raman R, Allen SP, Goodall EF, et al. Gene expression signatures in motor neurone disease fibroblasts reveal dysregulation of metabolism, hypoxia‐response and RNA processing functions. Neuropathol Appl Neurobiol. 2015;41(2):201‐226. doi: 10.1111/nan.12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gille B, De Schaepdryver M, Dedeene L, et al. Inflammatory markers in cerebrospinal fluid: independent prognostic biomarkers in amyotrophic lateral sclerosis? J Neurol Neurosurg Psychiatry. 2019;90(12):1338‐1346. doi: 10.1136/jnnp-2018-319586 [DOI] [PubMed] [Google Scholar]
- 15. Bridel C, van Wieringen WN, Zetterberg H, et al. Diagnostic value of cerebrospinal fluid Neurofilament light protein in neurology: a systematic review and meta‐analysis. JAMA Neurol. 2019;76(9):1035‐1048. doi: 10.1001/jamaneurol.2019.1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Poesen K, Van Damme P. Diagnostic and prognostic performance of neurofilaments in ALS. Front Neurol. 2019;9:1167. doi: 10.3389/fneur.2018.01167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Goyal NA, Berry JD, Windebank A, et al. Addressing heterogeneity in amyotrophic lateral sclerosis CLINICAL TRIALS. Muscle Nerve. 2020;62(2):156‐166. doi: 10.1002/mus.26801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westeneng H‐J, Debray TPA, Visser AE, et al. Prognosis for patients with amyotrophic lateral sclerosis: development and validation of a personalised prediction model. Lancet Neurol. 2018;17(5):423433. doi: 10.1016/S1474-4422(18)30089-9 [DOI] [PubMed] [Google Scholar]
- 19. Cudkowicz ME, Brown RH, Aricha R, et al. CSF biomarker correlations with primary outcome in NurOwn phase 3 clinical trial. Presented by Berry JD at Northeast Amyotrophic Lateral Sclerosis Consortium® (NEALS). 2021. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix Supplementary Information
Appendix S1: Supplementary Information
Data Availability Statement
The anonymized patient data are not being publicly shared as they are the focus of regulatory interactions for future regulatory approval.