

Author Roles
Research project – design: C.S.S., V.B.; execution: C.F., F.F., G.J.B., C.S.S.; analysis: C.F., F.F., G.J.B., C.S.S.; manuscript – writing of the first draft: C.F., V.B.; review and critique: F.F., G.J.B., C.S.S.; final approval: all authors.
Financial Disclosures
Vincenzo Bonifati receives research grants from the Stichting Parkinson Fonds and from Alzheimer Nederland (the Netherlands); honoraria from the International Parkinson and Movement Disorder Society, as chair of the Congress Scientific Program Committee 2019–2021; and from Elsevier Ltd, as Co‐Editor‐in‐Chief of Parkinsonism & Related Disorders. All other authors (C.F., F.F., G.J.B., C.S.S., V.B.) report no disclosures relevant to the manuscript.
Biallelic loss‐of‐function (LOF) variants in the AOPEP gene were recently identified in five European patients with generalized or multifocal dystonia.1, 2 In this letter, we present an Indian case with early‐onset, generalized dystonia carrying a novel homozygous LOF AOPEP variant.
A 31‐year‐old man born to consanguineous parents of Indian ancestry (Fig. 1A, II‐1) presented with complaints of involuntary posturing of the neck, trunk, and both upper limbs since 20 years of age. His symptoms started after an injury resulting in the fracture of his right thumb. Within a month after the injury, he developed abnormal posturing of the right hand. After a few months, abnormal posturing involved both upper limbs, and subsequently the neck and the trunk. He later experienced changes in his voice and difficulty in uttering words and swallowing. His family history was negative for neurological disorders. Perinatal history and medical history were unremarkable.
FIG 1.
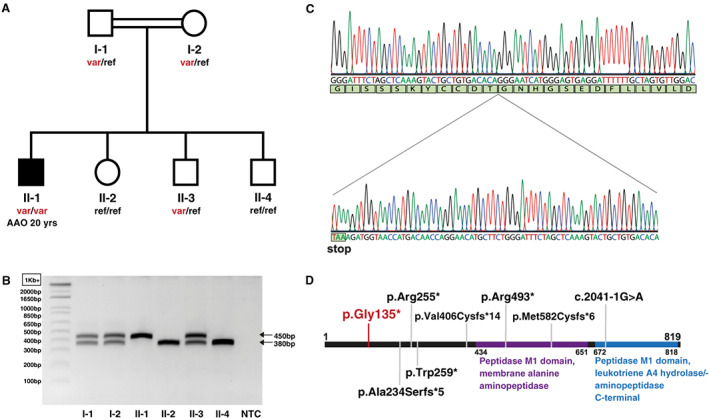
Pedigree and genetic results. (A) The AOPEP variant is present in homozygous state in the subject affected by dystonia (black symbol) but in none of his unaffected relatives (white symbols). (B) Agarose gel electrophoresis of PCR fragments containing the AOPEP exon 2; wild‐type allele: 380 bp; variant allele: 450 bp. (C) Electropherogram showing the 70‐nucleotide duplication in AOPEP exon 2 in the DNA amplified from the affected subject. (D) Schematic representation of the AOPEP protein with known functional domains 4 and variants reported in patients with dystonia in recent studies (gray anchors) and in this study (in red). AAO, age at onset; NTC, negative control; ref, reference (wild‐type) allele; var, AOPEP c.333_402dup (p.Gly135*) variant allele. [Color figure can be viewed at wileyonlinelibrary.com]
Upon examination, we found marked dysarthria, laryngeal dysphonia, kyphoscoliosis, retrocollis, and truncal and bilateral hand dystonia (Supporting Information Supplementary Video S1). Hypertonia was evident in both upper and lower limbs. Assessment of deep tendon reflexes and limb muscle strength as well as cerebellar examination, eye movements, sensory examination, and cognitive functions were normal. The patient was able to walk without support but with extensor trunk posture. Brain and cervical spine magnetic resonance imaging was normal.
To identify the disease‐causing gene, we carried out high‐density single‐nucleotide polymorphism genome‐wide genotyping in all DNA samples available from the family and whole‐exome sequencing (WES) in the patient. We ran linkage analysis assuming an autosomal recessive mode of inheritance and parental consanguinity, which yielded a list of candidate genomic regions (Supporting Information Appendix S1). By inspecting the WES data of the patient for rare homozygous variants with predicted coding or splicing effect (Supporting Information Appendix S1), we identified a 70‐nucleotide duplication in exon 2 of AOPEP (NM_001193329.1), leading to premature termination in the encoded protein: c.333_402dup (p.Gly135*). This variant is absent in gnomAD. 3 Agarose gel electrophoresis (Fig. 1B), as well as Sanger sequencing (Fig. 1C), confirmed the variant and showed its presence in homozygous state in the affected subject but in none of his unaffected relatives. Furthermore, WES analysis demonstrated no definitive disease‐causing variants in other known dystonia genes, nor compelling variants in other genes in the candidate genomic regions (Supporting Information Appendix S1). We therefore consider the novel LOF AOPEP variant (Fig. 1D) as disease causing in this patient.
The recently described cases presented with progressive dystonia, predominantly involving upper and lower limbs, with variable involvement of craniocervical and truncal districts. 2 The age at onset ranged from childhood to early adulthood. In three of the four families reported, dystonia was isolated. Our patient also manifested dystonia in the upper limbs in early adulthood, which progressed to the craniocervical and truncal segments.
This work provides further, independent evidence for the involvement of AOPEP in early‐onset dystonia. Future clinical studies will contribute to better delineating the phenotypic spectrum of AOPEP‐related dystonia, while functional work is warranted to provide insights into the mechanisms by which AOPEP LOF leads to dystonia.
Supporting information
Appendix S1. Supporting Information
Video S1. The video documents dystonia in the neck, the trunk and both upper limbs.
Acknowledgments
We are indebted to all the participating subjects. V.B. acknowledges financial support from the Stichting Parkinson Fonds (the Netherlands) to his research on Genetics of Movement Disorders (grant SPF‐1870).
Christina Fevga and Federico Ferraro contributed equally to this work.
Relevant conflicts of interest/financial disclosures: Nothing to report.
Full financial disclosures and author roles may be found in the online version of this article.
Data Availability Statement
The data that support the findings of this study are available from the authors upon reasonable request.
References
- 1. Zech M, Jech R, Boesch S, et al. Monogenic variants in dystonia: an exome‐wide sequencing study. Lancet Neurol 2020;19(11):908–918. 10.1016/S1474-4422(20)30312-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zech M, Kumar KR, Reining S, et al. Biallelic AOPEP loss‐of‐function variants cause progressive dystonia with prominent limb involvement. Mov Disord 2022;37(1):137–147. 10.1002/mds.28804 [DOI] [PubMed] [Google Scholar]
- 3. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blum M, Chang HY, Chuguransky S, et al. The InterPro protein families and domains database: 20 years on. Nucleic Acids Res 2021;49(D1):D344–D354. 10.1093/nar/gkaa977 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information
Video S1. The video documents dystonia in the neck, the trunk and both upper limbs.
Data Availability Statement
The data that support the findings of this study are available from the authors upon reasonable request.