

Abstract
In central nervous system drug discovery programs, early development of new chemical entities (NCEs) requires a multidisciplinary strategy and a translational approach to obtain proof of distribution, proof of occupancy, and proof of function in specific brain circuits. Positron emission tomography (PET) provides a way to assess in vivo the brain distribution of NCEs and their binding to the target of interest, provided that radiolabeling of the NCE is possible or that a suitable radioligand is available. PET is therefore a key tool for early phases of drug discovery programs. This review will summarize the main applications of PET in early drug development and discuss the usefulness of PET microdosing studies performed with direct labelling of the NCE and PET occupancy studies. The purpose of this review is also to propose an alignment of the nomenclatures used by drug metabolism and pharmacokinetic scientists and PET imaging scientists to indicate key pharmacokinetic parameters and to provide guidance in the performance and interpretation of PET studies.
Positron emission tomography (PET) is a molecular imaging technique widely used to measure the distribution of radiolabeled compounds, as well as functional parameters (e.g., blood flow and glucose metabolism), and the availability of different biological targets (e.g., receptors and enzymes). PET is a key experimental tool used in neuroscience drug discovery and development for assessment of exposure of new chemical entities (NCEs) in the central nervous system (CNS) and for quantitative assessment of target occupancy. The quantitative properties of PET and the ability to assess biological functions are also applied in other therapeutic areas. For instance, whole‐body hybrid PET imaging combined with computed tomography and/or magnetic resonance is used for the diagnosis and staging of solid tumors, and the development of new radioligands for specific targets (e.g., [68Ge]DOTATOC or [68Ge]DOTATATE for neuroendocrine tumors or [68Ge]PSMA for prostate cancer) has enabled the evaluation of tumors that would be otherwise difficult to access. 1 , 2 , 3
The importance of PET in CNS drug development was highlighted by Morgan et al., 4 where the term “three pillars of survival” was coined based on a systematic evaluation of phase II failures, uncovering that in almost 50% of the drug development programs investigated it was not possible to conclude whether the drug mechanism had been tested adequately due to unknown target site, drug exposure or target occupancy. 4 The three pillars have undoubtedly been implemented in many pharma companies. In this context, it will be called (i) proof of distribution; (ii) proof of occupancy, and (iii) proof of function. The last term can be further subdivided into proof of mechanism, proof of principle, etc. However, for our purpose proof of function indicates exposure of the drug at the site of action as well as target occupancy, which leads to engagement of the target and downstream functional effect.
PET radioligands developed for binding to specific targets associated with CNS disorders are developed as biomarkers for disease diagnosis, patient stratification, and assessment of disease progression (Figure 1 ). One example is the development of amyloid and tau PET radioligands for in vivo identification of Alzheimer pathology, Braak staging (tau), and assessment of a biological effect of a compound (reduction of amyloid plaques or tau accumulation). 5
Figure 1.
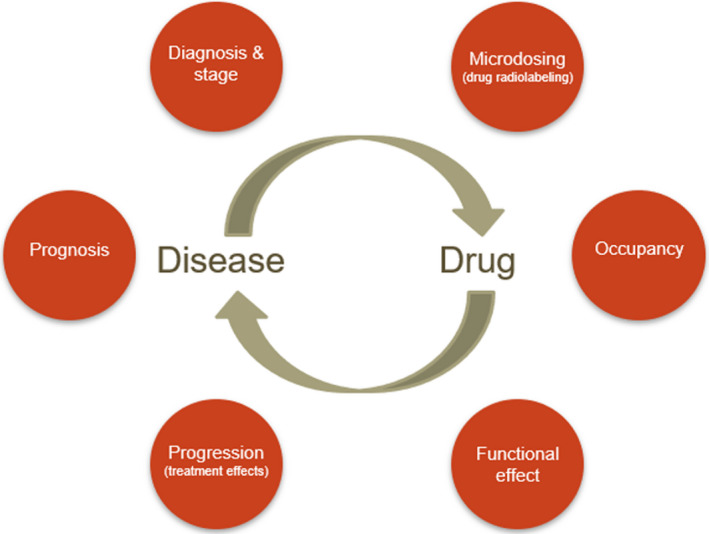
Positron emission tomography (PET) can be applied for different purposes, in relation to the characterization of the drug or to the study of different aspects of the disease. This review will focus on the methods/endpoints highlighted in the right‐hand side of the circle.
A comprehensive evaluation of the different applications of PET in drug development is beyond the scope of this review. Therefore, this review will focus on the application of PET in relation to the translatability of NCEs from experimental animals to humans and not on the application of PET for disease diagnosis, progression, etc. (Figure 1 ). For CNS drugs, brain exposure and target occupancy are key parameters that influence decision making in early drug development programs and are considered two important pillars for survival 4 (Table 1 ). PET studies intended to measure brain exposure are part of the development plan and characterization of NCEs, which requires continuous interaction between PET and drug metabolism and pharmacokinetic (DMPK) experts. Key pharmacokinetic (PK) parameters of NCEs are often described differently in publications that present PET or DMPK studies. In addition, PET occupancy studies are keys not only to define target engagement but also to enable decisions on therapeutic doses applied in early drug development studies. Overall, this review aims to provide an update on the application of PET in early drug development neuroscience programs with a specific focus on the assessment of drug brain exposure and target occupancy, as well as to propose an alignment of the nomenclatures used in PET and DMPK.
Table 1.
Three pillars of survival of new chemical entities, according to (Morgan, P. et al., Drug Discovery Today 2012, 17, 419–424)
| Animal | Human | |
|---|---|---|
|
Exposure at site of action (brain exposure) |
Bioanalysis vs. PET labeling of drug candidate | PET labeling of drug candidate |
|
Binding to target (occupancy) |
PET study using target specific PET ligand In vivo / ex vivo binding using [3H]labeled tracers |
PET study using target specific PET ligand |
| Pharmacodynamic effect a | Not discussed here | Partially discussed here |
PET, positron emission tomography.
In the original publication, this pillar was referred to as “Expression of pharmacology.” However, it has been changed to “Pharmacodynamic effect” for alignment with the content of the Review.
CONCEPT OF MICRODOSING STUDIES BY RADIOLABELING OF DRUG MOLECULES
Brain exposure and distribution of an NCE can be evaluated with direct radiolabeling of the compound with a short‐lived radioactive isotope, such as 11C (half‐life ~ 20 minutes) or 18F (half‐life ~ 2 hours). The possibility to perform such studies is dependent on whether such isotopes can be installed late in the synthesis of the target molecule by radiosynthesis and in practice often as the last step. This process is not always possible for many drug molecules and needs to be evaluated for each individual drug candidate. For instance, only some molecules can be labeled with 18F because fluorine is not part of all drug molecules. In other cases, the radiochemistry may not be feasible for either 11C or 18F.
If successful radiolabeling of the drug candidate can be achieved, this can be used for PK and distribution studies in animals and humans. This type of approach is referred to as microdosing, because it consists of the administration of a negligible amount of the radiolabeled compound, without any pharmacological effect. According to the European Medicines Agency (EMA) guidelines, a dose is considered a microdose when it is < 100 micrograms. 6 , 7 However, in PET studies, the amount of radiolabeled compound in most cases is < 10 micrograms. This method is typically used in early drug development when a PET radioligand for the target of interest is not available for direct assessment of target occupancy. In such cases, the assessment of brain distribution is an important parameter that can be measured in vivo (proof of distribution). Under the assumption that the in vitro and in vivo affinity (K i) are the same, the microdosing approach can also be used to estimate the occupancy of the drug to the target. 8 , 9
The microdosing PET studies are typically validated preclinically, by examining the kinetic and distributional properties of the radiolabeled drug in experimental animals, such as rats, pigs, and non‐human primates (NHPs), using both invasive methods and PET to build confidence in extrapolating the findings into the clinic. This translational phase is key to advance the program to phase 0/first‐in‐human (FIH) studies. The confirmation of sufficient brain exposure in humans is an important stage gate for the decision to advance the program into the next phases of development.
These microdosing studies can also be performed in combination with the administration of a pharmacological dose of the compound. The purpose with this approach is to examine whether the uptake of the radiolabeled compound is influenced by, for example, the activity of transporters located on the blood‐brain barrier (BBB) and/or to make sure that only the nondisplaceable signal of the radiolabeled compound is used for measuring the ratio of brain to plasma. The evaluation of the brain distribution of the muscarinic M1 positive allosteric modulator [11C]GSK1034702 in the living human brain is a good example of using PET microdosing to demonstrate brain uptake and blood‐brain barrier (BBB) passage consistent with passive diffusion or active influx, providing information to progress the molecule into the next stage of clinical development. 10 Another example comes from the observation that [11C]osimertinib, a tyrosine kinase inhibitor of the mutated epidermal growth factor receptor, that has shown clinical efficacy in the treatment of brain metastasis from non‐small cell lung cancer, displayed a favorable PK profile in healthy volunteers, with rapid brain distribution and higher uptake in grey than in white matter. 11 PET microdosing studies can also be performed with peripheral drug molecules to demonstrate limited brain availability, avoiding potential side effects on the CNS, that can be important for progressing molecules for peripheral indications. Such a study was done in NHPs with the cannabinoid receptor agonist AZD1940 that was confirmed to have limited brain availability. 12
CONCEPT OF TARGET OCCUPANCY STUDIES
Target occupancy is the other key parameter that influences the chance of clinical success. The proof that the compound interacts with the target of interest following drug administration (proof of occupancy) is needed in order to establish a link between target occupancy and pharmacological effect in vivo. Preclinically, these pharmacodynamic (PD) studies are often run in parallel with the occupancy studies that can be performed in experimental animals using both invasive ex vivo binding methods and noninvasive PET. The translational value of this approach with initial occupancy studies in experimental animals (rodents, pigs, or NHPs) followed by confirmatory studies in human subjects is the ideal de‐risking strategy to ensure that the drug has been tested at an adequate dose/exposure level in relation to the project hypothesis. This approach is described later in this article for the multimodal antidepressant vortioxetine. The process favors the discovery of compounds that are more likely to show efficacy in later phase Ib or phase II studies. PET occupancy studies are possible in all cases when a PET radioligand is available for imaging the target of interest. Well‐established PET tracers are available for several pharmacological targets in the CNS (e.g., dopamine D1 or D2/D3 receptors, dopamine or serotonin transporters, serotonin 5‐HT1A, 5‐HT1B, 5‐HT2A, 5‐HT4, 5‐HT6 receptors, GABA‐A receptors, phosphodiesterase 4 and 10, etc.), but, in many cases, for new drug targets, the discovery of PET radioligands is an integrated part of the drug development program.
DMPK PARAMETERS, PET OUTCOME MEASURES, AND METHODOLOGICAL CONSIDERATIONS
The nomenclature used by DMPK scientists to refer to parameters that characterize CNS compounds is different from the nomenclature used by PET scientists. Because the terminology is different depending on the context, it is important to align the DMPK and PET nomenclature (Table 2 ).
Table 2.
Definition of the key PK parameters of CNS candidate drugs according to the DMPK and PET nomenclatures
| Parameter | Nomenclature | Calculation | ||
|---|---|---|---|---|
| DMPK | PET | DMPK | PET | |
| Total concentration of drug in the brain | Cbrain | CT | ||
| Free fraction of drug in the brain | f u,brain a or f u,b | f ND | ||
| Free concentration of drug in the brain | Cu,brain | CFT | Cbrain ● f u,brain | CT ● f ND |
| Concentration of drug in the plasma | Cplasma | CP | ||
| Free fraction of drug in plasma | f plasma | f P | ||
| Free concentration of drug in plasma | Cu, plasma | CFP | Cplasma ● f plasma | CP ● f P |
| Brain to plasma ratio of the drug | Kp | V ND | Cbrain/ Cplasma | CT/ CP |
| Ratio of free concentration of drug in brain to free concentration of drug in plasma | Kp,uu | CFT/CFP | fu,brain/ fplasma ● Kp | f ND/f P ● V ND |
CNS, central nervous system; DMPK, drug metabolism and pharmacokinetics; PET, positron emission tomography; PK, pharmacokinetic.
f u,brain can be measured using brain homogenates (f u(brain)) or on brain slices (Vu(brain)).
In DMPK nomenclature, the brain‐to‐plasma ratio of the total drug concentrations is referred to as KP (Figure 2 ). The total concentration in the brain is the sum of the concentration specifically bound to a target (CS), the free or unbound (CFT), and nonspecifically bound (CND) concentrations of the compound, where the abbreviations in the parenthesis refer to the PET nomenclature. 13 , 14 KP is equivalent to V T in PET nomenclature (Figure 2 ), which denotes the total distribution volume of a compound and is equal to CT/CP. CT is the total concentration of the compound in the brain tissue and CP is the total concentration of the parent compound in plasma. Both concentrations are assumed to be measured at the equilibrium between the brain and plasma compartments. Because steady‐state conditions are typically not achieved during a PET experiment (unless the radiolabeled compound is administered as a bolus followed by constant infusion), the conventional way to derive V T is by using kinetic analysis with compartmental modeling.
Figure 2.
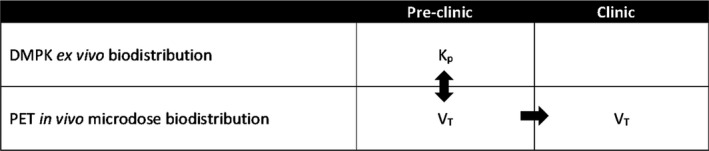
Rigorous preclinical characterization and alignment of biodistribution technologies before moving into the clinic with confidence using positron emission tomography (PET) microdosing.
The brain radioactivity is measured with dynamic PET acquisition and the plasma radioactivity is obtained through the measurement of the arterial input function. 15 For tracers that equilibrate very rapidly between the brain and plasma compartments (ideal tracers), the brain uptake and washout can be described by a one‐tissue compartment model. In this case, V T = K 1 / k 2, where K 1 is the rate of transfer of the tracer from plasma to brain (mL ● min−1 ● g−1) and k 2 is the rate of transfer from brain to plasma (min−1).
In most of the cases, though, the tracer does not equilibrate rapidly, and the brain uptake and washout can then often be described by a two‐tissue compartment model. In this case, V T = K 1 / k 2 (1 + k 3 / k 4). In this model, two additional rate constants describing the transfer of the molecule from a fast equilibrating to a slow equilibrating compartment (k 3, min−1) and vice versa (k 4, min−1) are required to be able to fit the data. It is important to clarify that k 3 and k 4 are conventionally used in PET quantification to describe the transfer of a radioligand from the nondisplaceable to the specific compartment (k 3) and vice versa (k 4). According to the PET nomenclature, the concentrations in the two compartments are referred to as CND and CS. 13 , 14
Therefore, the nondisplaceable distribution volume of the tracer is V ND = CND/CP and the specific distribution volume is V S = CS/CP. 13 , 14 V ND can be estimated from the measurement of the brain radioactivity of the tracer in a region of the brain devoid of receptors (e.g., cerebellum for the D2/D3 ligand [11C]raclopride), which is referred to as the reference region.
Because V ND represents the ratio at equilibrium between the free and nonspecific concentration in the brain and the concentration of the compound in plasma, V ND is the distribution volume that most appropriately represent Kp. If specific binding is negligible under the microdosing conditions and no regional differences in brain distribution of the radiolabeled compound are observed from the analysis of the brain PET data, V T can be considered as V ND and equivalent to Kp (Figure 3 ). If specific binding can be detected in the brain, a possible approach to estimate V ND is to block the binding of the radiolabeled compound with a pharmacological dose of the cold compound (Figure 3 ). In case of drugs, such as selegiline (MAO‐B inhibitor) and doxepin (H1 receptor antagonist), the experiments performed with pharmacological doses of the cold compounds showed that the binding of the radiolabeled drug was blocked by the concomitant administration of the cold drug, indicating the presence of specific binding. 16 Because both selegiline and doxepin have been developed as MAO‐B and H1 receptor radioligands, the results obtained after co‐administration of the cold drug are not surprising, but are relevant to indicate that the co‐administration of the cold drug can be important to demonstrate the presence of specific binding and for obtaining V ND that can be used to estimate KP 17 (Figure 3 ).
Figure 3.
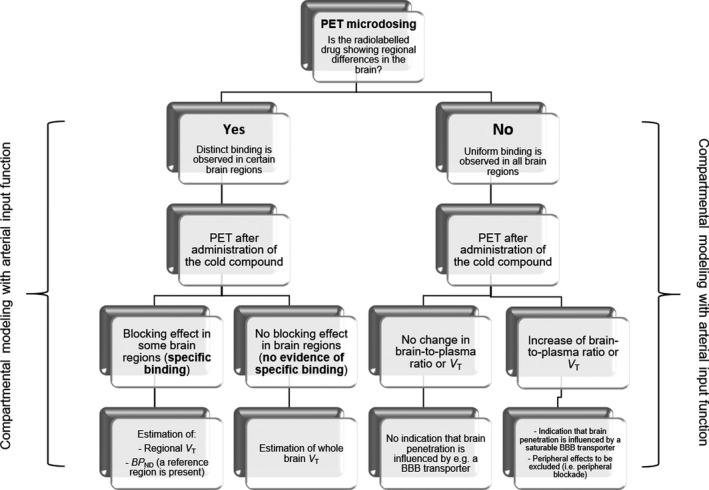
Suggested flowchart for the assessment of the pharmacokinetic properties of radiolabeled central nervous system (CNS) drugs using the microdosing approach. PET, positron emission tomography.
It is not always possible to estimate the distribution volume in the brain using standard compartmental modeling. In such cases, an alternative approach is to use the ratio of the area under the curve of the brain (AUCbrain) to the metabolite corrected plasma (AUCplasma). Although the ratio AUCbrain/AUCplasma can be used as a proxy of V T(V ND), it is important to clarify that the ratio consistently underestimates V T(V ND). 16
Radioactive metabolites
One important aspect to consider for the quantification of data from PET studies is whether there are brain penetrant metabolites formed during the experimental investigations that will complicate the quantification of V T. In the early phase of drug development, a comprehensive understanding of metabolites across species is seldom available, but in vivo preclinical studies together with in vitro metabolite profiling in liver microsomes and/or hepatocytes across species will give some valuable information about the potential risk. Combining such in vitro and in vivo studies with metabolic soft‐spot analysis using high‐performance liquid chromatography (HPLC) tandem mass spectrometry (MS/MS) can provide early indications of likely major metabolites. In general, radiometabolites that are more lipophilic than the parent compound and that are formed during the time window of the PET study are more likely to enter the brain and affect PET quantification. Less lipophilic radiometabolites may have less chance to cross the BBB, although this cannot be excluded unless metabolite analysis is conducted on plasma and brain homogenates after the administration of the NCE (e.g., in rodents).
In case of radioligands, such as [11C]PE2I (DAT), [11C]PBB3 (tau), and [11C]SMW139 (P2X7R), radiometabolite peaks that were less lipophilic than the parent compounds were observed from the analysis of the plasma samples. The same radiometabolite peaks were also observed from the analysis ex vivo of brain extracts. 18 , 19 , 20 In PET radioligand development and microdosing studies, the extent to which brain penetrant metabolites affect the estimation of the distribution volume of the parent will depend on the molecular structure of the metabolite that can help predicting the relative abundance of the metabolite in the brain. The identification of the structure of the radiometabolites requires the use of HPLC‐MS/MS. The synthesis of the metabolite might be needed to confirm the identity of the metabolite from the analysis. A recent review has summarized the main findings of studies on the radiometabolism of well‐known PET ligands. 21 Ex vivo experiments in rodents using carrier‐added PET radioligands (i.e., the unlabeled ligand in concentrations above microdoses is added to the radioligand formulation) might be helpful to elucidate which radioactive metabolites are present in the brain. 20
In case of potential brain penetrant metabolites of an NCE, the generation of radioactive metabolites will depend on the route of metabolism and the position of the radioisotope in the NCE. The two different positions of labeling the 5‐HT1A receptor antagonist PET ligand WAY‐100,635 illustrates the principle that the labeling of the molecule in the position that generates the more hydrophilic and non‐brain penetrant radioactive metabolite leads to the cleanest brain signal of the parent. 22
In case potentially brain‐penetrating radiolabeled metabolites are generated, an indirect way to assess the presence of potential radioactive metabolites is to examine the time stability of V T. 23 In general, the contribution of brain penetrant radioactive metabolites is more relevant toward the later part of the dynamic PET measurement, in which the contribution of the parent radioligand to the total PET signal is lower. If the estimate of V T progressively decreases by reducing the duration of image analysis, this pattern might be indicative of potential brain penetrant radioactive metabolite(s). Methods for estimation of distribution volume of the parent in presence of a brain penetrant radioactive metabolite are not used conventionally and have been described for radioligands such as [123I]epidepride 24 and [11C]PBB3, 25 based on the experimental evidence of the presence of lipophilic metabolites in the brain.
ASSESSMENT OF BRAIN EXPOSURE BY MICRODOSING
The main interest from a neuroscience drug development perspective is to assess the extent of brain penetration in quantitative terms according to the free drug hypothesis. 26 In DMPK, the relevant distribution parameter is referred to as Kp,uu, which is the ratio between the free drug concentration in the brain to the free drug concentration in the plasma. 27
In preclinical species, the unbound concentrations are typically derived from the measurement of total concentrations in vivo multiplied by the free fraction (f u) obtained in vitro in the brain and plasma. The accuracy of the measurement of f u in the brain and plasma is critical for the calculation of Kp,uu and this can be challenging for compounds that are highly bound.
Generally, the free fraction of the compound in the brain, f u,brain (DMPK) or f ND (PET) is measured through in vitro experiments in brain homogenates (f u(brain)) or brain slices (Vu(brain)) from rodents and is assumed to be similar across species. 8 , 28 The free fraction of the compound in plasma is measured through the ultrafiltration method for the radiolabeled compound and with the equilibrium dialysis method for the non‐radiolabeled compound. This latter measurement is considered most accurate, because the ultrafiltration method is sensitive to the physicochemical properties of the compound and the recovery from the filter, which might be low for lipophilic compounds.
The free concentration in the brain (CFT) and the free concentration in the plasma (CFP) are assumed to be equal if a compound only distributes in the brain by passive diffusion. 13 , 17 In this case, CFT/CFP = 1, which is the general assumption for PET radioligand quantification (similar to Kp,uu of 1 in DMPK nomenclature). In case of compounds developed as drugs, CFT/CFP can be < 1 in case the molecule is substrate for BBB efflux transporters, such as P‐glycoprotein (P‐gP) or breast cancer resistance protein (BCRP), or > 1 in case the passage of the molecule to the brain is mediated by active transport. 17
According to the standard PET nomenclature:
| (1) |
In the general case, because V ND = CND/CP
| (2) |
Therefore, Kp,uu can be calculated as:
| (3) |
if f ND and f P can be measured reliably and V ND can be estimated by compartmental modeling. 17
In case of passive diffusion:
| (4) |
Therefore,
| (5) |
For the equivalence of the parameters used in DMPK and PET, see Table 2 .
The estimation of Kp,uu is key to make decisions on advancing the early development plan. There is no established threshold of Kp,uu and the decision to progress a potential drug candidate is based on project specific parameters, linked to the overall feasibility of the target, drug, and disease for a given program.
In a study assessing the Kp,uu measured in rats, only 7 out of 57 marketed CNS drugs had Kp,uu < 0.3. 29 In a microdosing PET study in NHPs, the Kp,uu of 10 reference CNS drugs, calculated using the free fraction measured on brain slices (Vu(brain)), ranged from 0.42 (caffeine) to 4.8 (clomipramine). 16
The measurement of f u,brain (f ND) is a critical element for the calculation of Cu,brain. The Cu,brain has been shown to be the parameter that best predicts the D2 receptor occupancy by established antipsychotics and is superior to the concentration of the drug in the cerebral spinal fluid or in the blood. 30 The ability of Cu,brain to predict target occupancy has also been observed in studies evaluating the relationship between occupancy and PK parameters of inhibitors of dopamine and serotonin transporters. 31 , 32 In those studies, the animals were given drug and 3H‐labelled radioligands in vivo, but the target occupancy by the drugs was measured after the animals were killed by decapitation. The brains were quickly removed, and the drug occupancy was either determined using counts from target specific homogenates or by using brain sections and autoradiography. The Cbrain was measured from brain homogenates and f u,brain was measured with the dialysis method. The relationship between Cu,brain and target occupancy was evaluated by normalizing Cu,brain by the in vitro K i. The in vitro K i was measured with classical competition experiments with 3H‐labelled tracers using brain homogenates.
To our knowledge, a similar approach using in vivo measurement of receptor occupancy with PET instead of ex vivo using, for example, autoradiography has not been reported. From a theoretical standpoint, if Cu,brain and K i are available, receptor occupancy can be estimated as: RO% = Cu,brain 100 / (K i + Cu,brain). 8 , 9 This calculation can be used to make predictions on the potential activity of the drug in early development phase. However, in vivo measurement of receptor occupancy with PET is still the preferred method, if a PET radioligand is available, to advance early candidates to FIH studies (see section on receptor occupancy).
ASSESSMENT OF TARGET OCCUPANCY
The possibility to perform occupancy studies relies on the availability of a suitable PET radioligand. For some brain targets, such radioligands exist but for most new drug targets this is not the case. Therefore, a key aspect to consider in new drug project is whether an imaging ligand exists for target occupancy studies and whether there is need for further qualification of a radioligand to be used for both preclinical as well as clinical studies. If an imaging ligand does not exist, it is important to start the search as early as possible and optimally in parallel with the discovery of the drug candidate. In practice, this means that the medicinal chemists will initiate both hit‐to lead and hit‐to imaging ligand programs in parallel. However, as drug molecules and successful PET ligands often display different molecular properties, this is two parallel tracks in the medicinal chemistry program.
A comprehensive discussion of the optimal properties of a PET radioligand is beyond the scope of this review, but the process includes the measurement of in vitro target affinity across species, selectivity, and the establishment of the quantification and test‐retest properties of the radioligand in vivo in experimental animals and in human subjects. Overall, the identification of a successful clinical PET ligand for a new target is a major endeavor.
The outcome measure used to calculate target occupancy, in case there is a reference region in the brain, is BP ND, which is calculated from the distribution volumes or estimated using reference region models (Table 3 ). In the general case, when no reference region is present, V T is the outcome measure of choice. In this case, the revised Lassen plot 33 is the method used to derive an occupancy measure.
Table 3.
PET outcome measures for calculation or estimation of target occupancy
| Outcome measure | Estimation | |||
|---|---|---|---|---|
| Definition | Rate constants | Distribution volumes | Occupancy calculation | |
| BP ND (specific to nondisplaceable binding potential) | B max/K D ● f ND | k 3/k 4 a | V T/V ND − 1 b | BP ND baseline − BP ND drug / BP ND baseline |
| V T | V S+V ND | K 1/k 2 (1+k 3/k 4) | ‐ | V T drug − V T baseline = OCC ● (V T − V ND) |
| λk3 c | λ = K 1/k 2 | K1/k2 ● k 3 | ‐ | λk3 baseline − λk3 drug / λk3 baseline |
B max, max number of available receptors(targets); K D, dissociation rate constant; PET, positron emission tomography; V ND, nondisplaceable distribution volume (usually measured in a reference region); V S, distribution volume of the radioligand bound to the target.
k 3/k 4 can be estimated with 2‐tissue compartment model (TCM) or with simplified reference tissue model (SRTM).
V T and V ND can be estimated with 1‐TCM or 2‐TCM or with linear or multilinear graphical analysis (Logan plot or multilinear analysis) or with reference Logan or MRTM in case a reference region in the brain is present.
λk3 is the outcome measure conventionally used in case of an irreversible radioligand.
The revised Lassen plot uses a linear regression analysis approach, in which the difference between estimates of V T post drug (V T drug) and V T at baseline (V T baseline) obtained across several brain regions is plotted against V T baseline. The method is applied under the assumption that V ND is the same across regions, as well as the fractional occupancy (%) at the given target. The slope of the regression line provides an estimate of the occupancy and the intercept on the x axis provides an estimate of V ND. The revised Lassen plot can also be used to validate the use of cerebellum as reference region, in case V ND cannot be estimated from full blocking studies. The method shows a tendency to overestimate fractional occupancy at low occupancy levels and becomes more accurate when high occupancy is measured. To overcome potential inaccuracies connected with the linear graphical approach, an alternative method based on maximum likelihood estimation has been proposed. 34
Examples of successful applications of target occupancy studies in translational PK/PD assessments: Serotonin transporter occupancy by multimodal serotonergic drug vortioxetine
Vortioxetine that is available worldwide as an antidepressant drug was brought into phase II studies based on clinical PET occupancy data. The project was paused due to challenges in setting the dose for the phase II study. The dose was initially based on efficacious plasma exposure in experimental animals, but this could not be directly translated into the clinical situation in this case. This stipulates that important species‐dependent parameters need to be accounted for when translating plasma exposure levels between species for a given drug. Firstly, target affinity may differ between species, requiring information on in vitro Ki in both humans and in the relevant species used in the preclinical pharmacology studies. Second, it is widely accepted that the unbound concentrations are responsible for engaging the target and are therefore the relevant exposure metric to consider. 8 , 26 Thus, species differences in plasma protein binding also needs to be accounted for. Third, species differences in drug uptake and/or efflux transporters at the BBB level may lead to species differences in the extent of CNS penetration. 35
As vortioxetine is an inhibitor of serotonin (5‐HT) reuptake at the serotonin transporter (SERT), a SERT occupancy PET study was undertaken using the selective PET ligand [11C]3‐amino‐4‐((2‐((dimethylamino)methyl)phenyl)thio)benzonitrile ([11C]DASB). The PET study was suggested based on an extensive preclinical data package linking the SERT occupancy to serotonin release, as measured using microdialysis. Acutely, vortioxetine dose‐dependently occupied the SERT in rats, thus verifying its brain penetration. 36 , 37 Following 3 days of continuous dosing in rats, a significant increase in brain extracellular 5‐HT was observed, whereas only 41% SERT occupancy was observed. 36 This indicated that relevant functional effects at the neurotransmitter level could be obtained at low SERT occupancy compared to classical selective serotonin reuptake inhibitors and serotonin norepinephrine reuptake inhibitors, which typically require 80% SERT occupancy for a functional and therapeutic effect. 38 , 39 In a PET study of vortioxetine in healthy volunteers performed at different oral doses, the relationship between plasma concentrations and SERT occupancy showed that doses of 5–10 mg/day resulted in occupancies around 40–55%. 40 Later, a wide range of efficacy studies in depressed patients confirmed that these doses were therapeutically effective and well‐tolerated. 41 , 42 , 43 However, vortioxetine is also an agonist at 5‐HT1A receptors, a partial agonist at 5‐HT1B receptors, and an antagonist at 5‐HT3, 5‐HT1D, and 5‐HT7 receptors. 44 SERT and 5‐HT3 receptors are primarily occupied at 5 mg, whereas at 20 mg, all targets are likely occupied at functionally relevant levels. 45 The multimodal profile of vortioxetine has been demonstrated in vitro and in vivo in rats using ex vivo binding studies but because no PET ligands exist for most of these targets, clinical PET studies have only been performed on 5‐HT1A besides from SERT. 46 Thus, the clinical SERT occupancy study was prioritized as PET ligands for SERT were made available at the time but the translation using PET could in principle have been done using one of the other targets as well.
For vortioxetine, retrospective PK/PD evaluation showed that ~ 10‐fold higher total plasma concentrations were required in rats compared with humans to achieve the same SERT occupancy. 36 , 40 Detailed evaluation of the species‐dependent parameters showed that vortioxetine displayed approximately a six‐fold weaker SERT Ki in rats compared with humans, thus increasing the predicted required equivalent exposure in rats correspondingly. As to plasma protein binding, vortioxetine is highly bound to plasma proteins in both rats and humans (around 99% bound), making it difficult to quantitatively assess whether this parameter would have implications for translating effective drug exposure between these species.
Regarding extent of brain penetration, low involvement of active efflux at the BBB from P‐gp was suggested from in vitro permeability assessment and in vivo brain distribution studies in P‐gp knock‐out mice. 47 Overall, it seems likely that the species difference in SERT affinity is the main contributor to the observed difference between rats and humans in the SERT occupancy PK/PD relationship for vortioxetine. Hence, whereas the applicability of CNS target occupancy as quantitative translational biomarker between animal and humans have been widely implemented in the industry, a rigorous evaluation of potential interspecies differences is important in the prospective prediction of CNS occupancy in humans. 48
D2/D3 receptor occupancy by antipsychotic drugs
Several studies in healthy volunteers and patients with schizophrenia have been conducted to measure the occupancy of first‐ and second‐generation antipsychotics to D2/D3 receptors using PET and single‐photon computed emission tomography (SPECT). 49 , 50 The classical antipsychotics (e.g., haloperidol) are known to produce extrapyramidal side effects and these effects have been related to the high (> 80%) receptor occupancy to striatal D2/D3 receptors. 49 On the other hand, therapeutic efficacy is achieved with striatal receptor occupancy between 70% and 80%, making receptor occupancy studies extremely important to provide guidance on dose setting. 51 , 52
The pharmacological profile of atypical or second‐generation antipsychotics has been found to be different, because these drugs provide clinical efficacy at a lower striatal D2/D3 receptor occupancy and at higher D2/D3 occupancy in extrastriatal regions, such as the temporal cortex. In a meta‐analysis, PET and SPECT studies in patients with schizophrenia (total n = 139) have been reviewed and articles were selected based on the reported occupancy in striatum and temporal cortex after chronic dosing (steady‐state). 50 SPECT studies were performed with [123I]epidepride, a high‐affinity radioligand used to image striatal and extrastriatal D2/D3 receptor. PET studies were performed with [11C]raclopride (striatal D2/D3 receptors) and [18F]fallypride (striatal and extrastriatal D2/D3 receptors), [76Br]FLB‐457, or [11C]FLB‐457 (extrastriatal D2/D3 receptors). The meta‐analysis also included studies that examined 5‐HT2A receptor occupancy with [18F]setoperone or [11C]N‐methylspiperone. The main findings of the meta‐analysis were that first‐generation antipsychotics provided statistically higher D2/D3 receptor occupancy than second generation antipsychotics. The difference of receptor occupancy between first‐ and second‐generation antipsychotics in the striatum (79% vs. 49%) was larger than the difference observed in the temporal cortex (77% vs. 67%). For second‐generation antipsychotics, the correlation between the dose achieving maximal receptor occupancy and the clinically effective dose tended to be higher in the temporal cortex (r = 0.95, P < 0.001) than in the striatum (r = 0.76, P = 0.046). Overall, the results of the meta‐analysis corroborated the knowledge that clinical efficacy of first‐ and second‐generation antipsychotics is related to their striatal and cortical occupancy, and that the onset of extrapyramidal side effects of first‐generation antipsychotics is related to their higher striatal receptor occupancy. In the same meta‐analysis, the relationship between 5‐HT2A receptor occupancy and clinically effective dose was also evaluated. The available data were more limited than those available for D2/D3 receptor occupancy, however, the analysis did not show a significant correlation between 5‐HT2A receptor occupancy and clinically relevant doses, suggesting that 5‐HT2A antagonism by second‐generation antipsychotics is less likely related to clinical efficacy.
Another meta‐analysis of 12 PET studies (11 with [11C]raclopride and 1 using both [11C]raclopride and [11C]FLB457 for striatal and extrastriatal D2/D3 receptors) including 82 subjects, specifically examined the relationship between D2/D3 receptor occupancy and severity of extrapyramidal side effects (65 subjects) as well as treatment response (70 subjects) assessed through clinical scales (25% or greater or 50% or greater reduction in the Positive and Negative Syndrome Scale (PANSS) or the Brief Psychiatric Rating Scale (BPRS)). 53 The mean dopamine D2/D3 receptor occupancy was significantly higher in subjects who presented extrapyramidal side effects (n = 12; 77 ± 9%) than those who did not (n = 53; vs. 63 ± 17%, P = 0.011). Higher D2/D3 receptor occupancy (66 ± 14%) was observed in patients who presented a 25% reduction in the PANSS or BPRS (n = 43) as compared with the occupancy observed in patients who did not show symptom improvement (n = 27, 58 ± 19%). The difference did not reach statistical significance (P = 0.054), but the greatest accuracy in predicting a 25% and 50% of reduction in PANSS or BPRS corresponded to occupancy measures of 60% and 78%, respectively, confirming that this is the range of occupancy associated with a clinical effect.
On the contrary, third‐generation antipsychotics like aripiprazole, cariprazine, and brexpiprazole exhibit a higher D2/D3 receptor occupancy at clinically relevant doses (closer to 90% or 95%), without increasing the risk of extrapyramidal side effects. 54 , 55 , 56 This has been rationalized based on the partial D2 receptor agonism of those compounds that are different from the antagonism of both first‐ and second‐generation antipsychotics. 55 , 57 Finally, a special consideration should be given to clozapine, a highly efficacious antipsychotic used for the treatment of schizophrenia. The efficacy of clozapine is achieved at much lower D2/D3 receptor occupancy than first‐ and second‐generation antipsychotics. 49 This “atypical” property has been linked to the higher in vitro affinity of clozapine for D4 receptors as compared with D2/D3 receptors 58 and to the evidence from human PET studies of equivalent occupancy of clozapine for the D1 and D2/D3 receptors. 59 , 60
ASSESSMENT OF PHARMACODYNAMIC EFFECT
The PD effect of a drug can be studied in vivo either by assessing the direct effect on the modulation of neurotransmitter release or by evaluating the downstream effect of the drug on neuronal activity or specific brain circuits. The first approach benefits from the capability of PET to measure changes in endogenous neurotransmitter levels, whereas the second approach uses a multimodal evaluation of PET receptor occupancy and pharmacological‐induced changes in specific brain circuits measured with functional magnetic resonance imaging (fMRI).
The downstream effect related to target occupancy or to neurotransmitter release can be close in time with drug administration or occur more distant in time. The latter situation is especially relevant for disease‐modifying drugs, which recently have attracted much attention by the approval of aducanumab for Alzheimer’s disease. In this case, amyloid‐β (Aβ) PET ligands that bind to Aβ plaques 61 were developed in parallel with ongoing drug discovery programs targeting Aβ and used to show reduction in Aβ plaques. A recent paper exemplifies, for example, that aducanumab shows a dose‐ and time‐dependent reductions in the amyloid PET standard uptake value ratio as measured by 18F‐labeled florbetapir. 62 Thus, PET as a technology has had major impact on the approval of aducanumab by the US Food and Drug Administration (FDA). Neurofibrillary tangles that are aggregates of hyperphosphorylated tau is another important target for ongoing drug development programs with disease modifying potential. For these aggregates, PET ligands have also been successfully developed and will most likely have impact on future approvals of drug for AD and other tauopathies. 61 , 63
With regard to measuring endogenous neurotransmitters, a classical occupancy model has been described to explain the effects of changes in endogenous dopamine on the binding of a PET radioligand (e.g., [11C]raclopride) to the D2/D3 receptors. 64 , 65 According to the model, the radioligand, at tracer doses, occupies a fraction of the receptors on the postsynaptic site. The increase in the levels of endogenous dopamine, induced pharmacologically by the administration of a dopamine releasing drug, such as amphetamine, 66 will lead to an increased occupancy of dopamine to the D2/D3 receptors and a displacement of the PET radioligand from the D2/D3 receptor, resulting in a decrease of BP ND. The opposite effect (i.e., an increase) on BP ND is observed in cases of a decrease in dopamine levels, induced for instance by administration of a dopamine depleting drug, such as alpha‐methyl‐para‐tyrosine. 67 , 68
Studies in NHPs and in human subjects using agonist ([11C]MNPA or [11C]PHNO) and antagonist ([11C]raclopride) PET radioligands have shown that changes in striatal BP ND of [11C]MNPA or [11C]PHNO induced by amphetamine were larger than the changes of BP ND observed with [11C]raclopride, 69 , 70 indicating a higher sensitivity of D2 agonist PET radioligands to measure endogenous dopamine levels. PET imaging with [11C]MNPA or [11C]PHNO before and after the administration of a drug modulating dopamine release will enable the assessment of the pharmacological effect of the drug on synaptic neurotransmitter release. An example of such an approach is the examination of the effect of the orphan Gprotein‐coupled receptor GPR139 agonist TAK‐041 on the attenuation of the amphetamine‐induced release of endogenous dopamine in the human brain using [11C]PHNO PET. 71 A dose of 20 or 40 mg TAK‐041 administered 2 hours prior to the oral administration of 0.5 mg/kg d‐amphetamine, significantly attenuated the decrease of BP ND of [11C]PHNO in the putamen, ventral striatum, and substantia nigra, with 40 mg being associated with less reduction of BP ND that the 20 mg dose. 71 A mechanism postulated to explain these findings has been that TAK‐041 modulates D2 autoreceptors, reducing dopamine synthesis and hence synaptic dopamine release. The pharmacological modulation of neurotransmitter release has been applied also to other monoaminergic and non‐monoaminergic receptor and enzyme systems, as reviewed by Finnema et al. 65 It should be noted that the same technology can be used to investigate disease‐related changes in endogenous neurotransmitter release. For instance, studies conducted in patients with schizophrenia examining the changes of D2 receptor availability after dopamine depletion or amphetamine‐related dopamine release have suggested that patients with schizophrenia have increased synaptic dopamine concentration and increased dopamine release as compared with controls. 72 , 73 , 74
The second approach has been used to evaluate the relationship between receptor occupancy and functional hemodynamic changes measured with fMRI, by measuring the changes in cerebral blood volume (CBV; neurovascular response) or the changes in blood oxygen level‐dependent (BOLD) response. With the advent of the hybrid PET/MRI systems, studies have been conducted to address the relationship between pharmacological modulation of neurotransmitter systems and functional brain changes. In an early study performed in anesthetized NHPs, the relationship between D2/D3 receptor occupancy and changes of CBV was examined. 75 Receptor occupancy was measured using [11C]raclopride as PET radioligand and pharmacological doses of raclopride. Increasing D2/D3 receptor occupancy by pharmacological doses of raclopride (4.5 to 40 µg/kg) was associated with increased CBV changes. The relationship between D2/D3 receptor occupancy and CBV changes was monotonically increasing, with the putamen exhibiting approximately twice the CBV magnitude compared with caudate. The increase of CBV observed with increased receptor occupancy was explained with the fact that the fractional occupancy of dopamine to D2/D3 receptors is the parameter that drives the fMRI signal. The occupancy of D2/D3 receptors by raclopride reduces the fractional occupancy of dopamine to D2/D3 receptors, producing an increase in CBV. This interplay between D2/D3 receptor occupancy and changes in CBV suggest a link between fractional occupancy of dopamine and neuronal activity in the striatum.
On the contrary, in a study in which the D2/D3 receptor agonist ropinirole was used, the occupancy of D2/D3 receptors measured by the degree of displacement of [11C]raclopride, was associated with a decrease of CBV. 76 A similar inverse relationship has been found between displacement of [11C]carfentanil (µ opioid receptor agonist PET radioligand) by naltrexone (µ opioid receptor antagonist) and a decrease in CBV. 76
Effects of naltrexone and other opioid antagonists on the dopamine system have been linked to their ability to attenuate reward properties. In a combined PET and fMRI study, receptor occupancy of the µ opioid receptor antagonist GSK1521498 and its effects on brain function were assessed and compared with naltrexone. 77 Mu‐opioid receptor occupancy of GSK1521498 and naltrexone was measured with [11C]carfentanil PET. An fMRI paradigm comparing BOLD‐response associated with a palatile stimulus vs. purified water was used to measure food‐related activation in limbic brain regions (amygdala and nucleus accumbens). The 50% effective dose for GSK1521498 and naltrexone were 1.5 and 5.6 mg. The relationship between plasma concentration of GSK1521498 and receptor occupancy was time‐independent. On the other hand, for naltrexone and its metabolite 6‐b‐naltrexol, at the same plasma concentration, the receptor occupancy was time‐dependent, suggesting that probably other metabolites or a longer residence time at the µ opioid receptor could contribute to the time‐dependent occupancy relationship. The oral administration of GSK1521498 was associated with a significant decrease of BOLD‐response in the amygdala, whereas no differences in any of the brain regions were observed for naltrexone. Overall, the higher selectivity for µ opioid receptors, in association with the direct plasma occupancy relationship and the functional effect observed (attenuation of food‐related activation in the amygdala), indicated improved pharmacological properties of GSK1521498 compared with naltrexone.
CONCLUSIONS
The strategy needed to advance NCEs from research to early stages of development is multidisciplinary and takes advantage of several in vitro, in vivo, and ex vivo methods. Molecular imaging with PET is a methodology that supports the development strategy by providing in vivo evidence of NCE brain exposure and proof of occupancy of the NCE to the target of interest. Standard methods of quantification used for PET radioligands can be applied to describe the PKs of radiolabeled NCEs and obtain key parameters of brain exposure as Kp and Kp,uu. The availability of suitable PET radioligands is a prerequisite to be able to demonstrate proof of occupancy, but if receptor occupancy studies are not possible, microdosing studies can be used to make predictions of brain exposure to support early decisions. Multimodal imaging approaches combining PET with fMRI can be used to link target occupancy of NCEs with PD effects on specific brain regions or circuits.
CONFLICT OF INTEREST
A.V., C.B., and B.B.‐A. are employed at H. Lundbeck A/S.
- 1. Sanli, Y. et al. Neuroendocrine tumor diagnosis and management: (68)Ga‐DOTATATE PET/CT. Am. J. Roentgenol. 211, 267–277 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Ling, S.W. et al. Comparison of (68)Ga‐labeled Prostate‐specific Membrane Antigen Ligand Positron Emission Tomography/Magnetic Resonance Imaging and Positron Emission Tomography/Computed Tomography for Primary Staging of Prostate Cancer: a systematic review and meta‐analysis. Eur. Urol. Open Sci. 33, 61–71 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chavoshi, M. , Mirshahvalad, S.A. , Metser, U. & Veit‐Haibach, P. (68)Ga‐PSMA PET in prostate cancer: a systematic review and meta‐analysis of the observer agreement. Eur. J. Nucl. Med. Mol. Imaging 49, 1021–1029 (2022). [DOI] [PubMed] [Google Scholar]
- 4. Morgan, P. et al. Can the flow of medicines be improved? Fundamental pharmacokinetic and pharmacological principles toward improving Phase II survival. Drug Discov. Today 17, 419–424 (2012). [DOI] [PubMed] [Google Scholar]
- 5. Schwarz, A.J. The use, standardization, and interpretation of brain imaging data in clinical trials of neurodegenerative disorders. Neurotherapeutics 18, 686–708 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verbruggen, A. et al. Guideline to regulations for radiopharmaceuticals in early phase clinical trials in the EU. Eur. J. Nucl. Med. Mol. Imaging 35, 2144–2151 (2008). [DOI] [PubMed] [Google Scholar]
- 7. EMA . ICH guideline M3(R2) on non clinical safety studies for the conduct of human clinical trials and marketing authorisation for pharmaceuticals. (ed. European Medicine Agency, S.M.H.) (2009).
- 8. Read, K.D. & Braggio, S. Assessing brain free fraction in early drug discovery. Expert Opin. Drug Metab. Toxicol. 6, 337–344 (2010). [DOI] [PubMed] [Google Scholar]
- 9. Gunn, R.N. & Rabiner, E.A. Imaging in central nervous system drug discovery. Semin. Nucl. Med. 47, 89–98 (2017). [DOI] [PubMed] [Google Scholar]
- 10. Ridler, K. et al. An evaluation of the brain distribution of [(11)C]GSK1034702, a muscarinic‐1 (M 1) positive allosteric modulator in the living human brain using positron emission tomography. EJNMMI Res. 4, 66 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Varrone, A. et al. A PET study in healthy subjects of brain exposure of (11)C‐labelled osimertinib ‐ A drug intended for treatment of brain metastases in non‐small cell lung cancer. J. Cereb. Blood Flow Metab. 40, 799–807 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schou, M. , Varnas, K. , Jucaite, A. , Gulyas, B. , Halldin, C. & Farde, L. Radiolabeling of the cannabinoid receptor agonist AZD1940 with carbon‐11 and PET microdosing in non‐human primate. Nucl. Med. Biol. 40, 410–414 (2013). [DOI] [PubMed] [Google Scholar]
- 13. Innis, R.B. et al. Consensus nomenclature for in vivo imaging of reversibly binding radioligands. J. Cereb. Blood Flow Metab. 27, 1533–1539 (2007). [DOI] [PubMed] [Google Scholar]
- 14. Innis, R.B. & Carson, R. Consensus nomenclature: its time has come. Eur. J. Nucl. Med. Mol. Imaging 34, 1239 (2007). [DOI] [PubMed] [Google Scholar]
- 15. Varnäs, K. , Varrone, A. & Farde, L. Modeling of PET data in CNS drug discovery and development. J. Pharmacokinet. Pharmacodyn. 40, 267–279 (2013). [DOI] [PubMed] [Google Scholar]
- 16. Schou, M. et al. Large variation in brain exposure of reference CNS drugs: a PET study in nonhuman primates. Int. J. Neuropsychopharmacol. 18, pyv036 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gunn, R.N. et al. Combining PET biodistribution and equilibrium dialysis assays to assess the free brain concentration and BBB transport of CNS drugs. J. Cereb. Blood Flow Metab. 32, 874–883 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hashimoto, H. et al. Radiosynthesis, photoisomerization, biodistribution, and metabolite analysis of 11C‐PBB3 as a Clinically Useful PET Probe for Imaging of Tau Pathology. J. Nucl. Med. 55, 1532 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Janssen, B. et al. Identification of the allosteric P2X7 receptor antagonist [11C]SMW139 as a PET tracer of microglial activation. Sci. Rep. 8, 6580 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shetty, H.U. et al. Identification and regional distribution in rat brain of radiometabolites of the dopamine transporter PET radioligand [11C]PE2I. Eur. J. Nucl. Med. Mol. Imaging 34, 667–678 (2007). [DOI] [PubMed] [Google Scholar]
- 21. Ghosh, K.K. , Padmanabhan, P. , Yang, C.T. , Mishra, S. , Halldin, C. & Gulyas, B. Dealing with PET radiometabolites. EJNMMI Res. 10, 109 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Osman, S. et al. Characterisation of the appearance of radioactive metabolites in monkey and human plasma from the 5‐HT1A receptor radioligand, [carbonyl‐11C]WAY‐100635–explanation of high signal contrast in PET and an aid to biomathematical modelling. Nucl. Med. Biol. 25, 215–223 (1998). [DOI] [PubMed] [Google Scholar]
- 23. Kim, M.J. et al. First‐in‐human evaluation of [(11)C]PS13, a novel PET radioligand, to quantify cyclooxygenase‐1 in the brain. Eur. J. Nucl. Med. Mol. Imaging 47, 3143–3151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fujita, M. et al. Kinetic and equilibrium analyses of [(123)I]epidepride binding to striatal and extrastriatal dopamine D(2) receptors. Synapse 34, 290–304 (1999). [DOI] [PubMed] [Google Scholar]
- 25. Kimura, Y. et al. PET quantification of tau pathology in human brain with 11C‐PBB3. J. Nucl. Med. 56, 1359–1365 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Smith, D.A. , Di, L. & Kerns, E.H. The effect of plasma protein binding on in vivo efficacy: misconceptions in drug discovery. Nat. Rev. Drug Discov. 9, 929–939 (2010). [DOI] [PubMed] [Google Scholar]
- 27. Hammarlund‐Udenaes, M. , Fridén, M. , Syvänen, S. & Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 25, 1737–1750 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ryu, S. et al. Evaluation of fraction unbound across 7 tissues of 5 species. J. Pharm. Sci. 109, 1178–1190 (2020). [DOI] [PubMed] [Google Scholar]
- 29. Zhang, Y.Y. , Liu, H. , Summerfield, S.G. , Luscombe, C.N. & Sahi, J. Integrating in silico and in vitro approaches to predict drug accessibility to the central nervous system. Mol. Pharm. 13, 1540–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 30. Watson, J. et al. Receptor occupancy and brain free fraction. Drug Metab. Dispos. 37, 753 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Bundgaard, C. , Sveigaard, C. , Brennum, L.T. & Stensbøl, T.B. Associating in vitro target binding and in vivo CNS occupancy of serotonin reuptake inhibitors in rats: the role of free drug concentrations. Xenobiotica 42, 256–265 (2012). [DOI] [PubMed] [Google Scholar]
- 32. Liu, X. , Vilenski, O. , Kwan, J. , Apparsundaram, S. & Weikert, R. Unbound brain concentration determines receptor occupancy: a correlation of drug concentration and brain serotonin and dopamine reuptake transporter occupancy for eighteen compounds in rats. Drug Metab. Dispos. 37, 1548–1556 (2009). [DOI] [PubMed] [Google Scholar]
- 33. Cunningham, V.J. , Rabiner, E.A. , Slifstein, M. , Laruelle, M. & Gunn, R.N. Measuring drug occupancy in the absence of a reference region: the Lassen plot re‐visited. J. Cereb. Blood Flow Metab. 30, 46–50 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schain, M. , Zanderigo, F. & Todd Ogden, R. Likelihood estimation of drug occupancy for brain PET studies. NeuroImage 178, 255–265 (2018). [DOI] [PubMed] [Google Scholar]
- 35. Toth, M. et al. ABC transporter‐dependent brain uptake of the 5‐HT1B receptor radioligand [(11)C]AZ10419369: a comparative PET study in mouse, rat, and guinea pig. EJNMMI Res. 4, 64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mørk, A. et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J. Pharmacol. Exp. Ther. 340, 666–675 (2012). [DOI] [PubMed] [Google Scholar]
- 37. Pehrson, A.L. et al. Lu AA21004, a novel multimodal antidepressant, produces regionally selective increases of multiple neurotransmitters–a rat microdialysis and electrophysiology study. Eur. Neuropsychopharmacol. 23, 133–145 (2013). [DOI] [PubMed] [Google Scholar]
- 38. Kreilgaard, M. , Smith, D.G. , Brennum, L.T. & Sánchez, C. Prediction of clinical response based on pharmacokinetic/pharmacodynamic models of 5‐hydroxytryptamine reuptake inhibitors in mice. Br. J. Pharmacol. 155, 276–284 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Meyer, J.H. Imaging the serotonin transporter during major depressive disorder and antidepressant treatment. J. Psychiatry Neurosci. 32, 86–102 (2007). [PMC free article] [PubMed] [Google Scholar]
- 40. Areberg, J. , Luntang‐Jensen, M. , Søgaard, B. & Nilausen, D.Ø. Occupancy of the serotonin transporter after administration of Lu AA21004 and its relation to plasma concentration in healthy subjects. Basic Clin. Pharmacol. Toxicol. 110, 401–404 (2012). [DOI] [PubMed] [Google Scholar]
- 41. Alvarez, E. , Perez, V. , Dragheim, M. , Loft, H. & Artigas, F. A double‐blind, randomized, placebo‐controlled, active reference study of Lu AA21004 in patients with major depressive disorder. Int. J. Neuropsychopharmacol. 15, 589–600 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Henigsberg, N. , Mahableshwarkar, A.R. , Jacobsen, P. , Chen, Y. & Thase, M.E. A randomized, double‐blind, placebo‐controlled 8‐week trial of the efficacy and tolerability of multiple doses of Lu AA21004 in adults with major depressive disorder. J. Clin. Psychiatry 73, 953–959 (2012). [DOI] [PubMed] [Google Scholar]
- 43. Katona, C. , Hansen, T. & Olsen, C.K. A randomized, double‐blind, placebo‐controlled, duloxetine‐referenced, fixed‐dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int. Clin. Psychopharmacol. 27, 215–223 (2012). [DOI] [PubMed] [Google Scholar]
- 44. Bang‐Andersen, B. et al. Discovery of 1‐[2‐(2,4‐dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J. Med. Chem. 54, 3206–3221 (2011). [DOI] [PubMed] [Google Scholar]
- 45. Sanchez, C. , Asin, K.E. & Artigas, F. Vortioxetine, a novel antidepressant with multimodal activity: review of preclinical and clinical data. Pharmacol. Ther. 145, 43–57 (2015). [DOI] [PubMed] [Google Scholar]
- 46. Stenkrona, P. , Halldin, C. & Lundberg, J. 5‐HTT and 5‐HT(1A) receptor occupancy of the novel substance vortioxetine (Lu AA21004). A PET study in control subjects. Eur. Neuropsychopharmacol. 23, 1190–1198 (2013). [DOI] [PubMed] [Google Scholar]
- 47. Bundgaard, C. , Eneberg, E. & Sanchez, C. P‐glycoprotein differentially affects escitalopram, levomilnacipran, vilazodone and vortioxetine transport at the mouse blood‐brain barrier in vivo. Neuropharmacology 103, 104–111 (2016). [DOI] [PubMed] [Google Scholar]
- 48. Bundgaard, C. , Pehrson, A.L. , Sánchez, C. & Bang‐Andersen, B. Case Study 2. The discovery and development of the multimodal acting antidepressant vortioxetine. In Blood‐Brain Barrier in Drug Discovery: Optimizing Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs (eds. Di, L. and Kerns, E.H. ) 505–520 (John Wiley & Sons Inc., Hoboken, NJ, 2015). [Google Scholar]
- 49. Lako, I.M. , van den Heuvel, E.R. , Knegtering, H. , Bruggeman, R. & Taxis, K. Estimating dopamine D₂ receptor occupancy for doses of 8 antipsychotics: a meta‐analysis. J. Clin. Psychopharmacol. 33, 675–681 (2013). [DOI] [PubMed] [Google Scholar]
- 50. Stone, J.M. , Davis, J.M. , Leucht, S. & Pilowsky, L.S. Cortical dopamine D2/D3 receptors are a common site of action for antipsychotic drugs–an original patient data meta‐analysis of the SPECT and PET in vivo receptor imaging literature. Schizophr. Bull. 35, 789–797 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nord, M. & Farde, L. Antipsychotic occupancy of dopamine receptors in schizophrenia. CNS Neurosci. Ther. 17, 97–103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tauscher, J. & Kapur, S. Choosing the right dose of antipsychotics in schizophrenia: lessons from neuroimaging studies. CNS Drugs 15, 671–678 (2001). [DOI] [PubMed] [Google Scholar]
- 53. Uchida, H. , Takeuchi, H. , Graff‐Guerrero, A. , Suzuki, T. , Watanabe, K. & Mamo, D.C. Dopamine D2 receptor occupancy and clinical effects: a systematic review and pooled analysis. J. Clin. Psychopharmacol. 31, 497–502 (2011). [DOI] [PubMed] [Google Scholar]
- 54. Yokoi, F. et al. Dopamine D2 and D3 receptor occupancy in normal humans treated with the antipsychotic drug aripiprazole (OPC 14597): a study using positron emission tomography and [11C]raclopride. Neuropsychopharmacology 27, 248–259 (2002). [DOI] [PubMed] [Google Scholar]
- 55. Wong, D.F. et al. An open‐label, positron emission tomography study of the striatal D2/D3 receptor occupancy and pharmacokinetics of single‐dose oral brexpiprazole in healthy participants. Eur. J. Clin. Pharmacol. 77, 717–725 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Girgis, R.R. et al. Preferential binding to dopamine D3 over D2 receptors by cariprazine in patients with schizophrenia using PET with the D3/D2 receptor ligand [(11)C]‐(+)‐PHNO. Psychopharmacology 233, 3503–3512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gründer, G. , Carlsson, A. & Wong, D.F. Mechanism of new antipsychotic medications: occupancy is not just antagonism. Arch. Gen. Psychiatry 60, 974–977 (2003). [DOI] [PubMed] [Google Scholar]
- 58. Van Tol, H.H.M. et al. Cloning of the gene for a human dopamine D4 receptor with high affinity for the antipsychotic clozapine. Nature 350, 610–614 (1991). [DOI] [PubMed] [Google Scholar]
- 59. Farde, L. , Nordström, A.L. , Wiesel, F.A. , Pauli, S. , Halldin, C. & Sedvall, G. Positron emission tomographic analysis of central D1 and D2 dopamine receptor occupancy in patients treated with classical neuroleptics and clozapine. Relation to extrapyramidal side effects. Arch. Gen. Psychiatry 49, 538–544 (1992). [DOI] [PubMed] [Google Scholar]
- 60. Tauscher, J. et al. Equivalent occupancy of dopamine D1 and D2 receptors with clozapine: differentiation from other atypical antipsychotics. Am. J. Psychiatry 161, 1620–1625 (2004). [DOI] [PubMed] [Google Scholar]
- 61. van Waarde, A. , Marcolini, S. , de Deyn, P.P. & Dierckx, R. PET Agents in dementia: an overview. Semin. Nucl. Med. 51, 196–229 (2021). [DOI] [PubMed] [Google Scholar]
- 62. Chiao, P. et al. Impact of reference and target region selection on amyloid PET SUV ratios in the phase 1b PRIME study of aducanumab. J. Nucl. Med. 60, 100–106 (2019). [DOI] [PubMed] [Google Scholar]
- 63. Van Wambeke, E. , Gerard, T. , Lhommel, R. & Hanseeuw, B. Disclosing tau tangles using PET imaging: a pharmacological review of the radiotracers available in 2021. Acta Neurol. Belg. https://doi.org/ 10.1007/s13760-021-01797-w. [e‐pub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 64. Laruelle, M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J. Cereb. Blood Flow Metab. 20, 423–451 (2000). [DOI] [PubMed] [Google Scholar]
- 65. Finnema, S.J. et al. Application of cross‐species PET imaging to assess neurotransmitter release in brain. Psychopharmacology 232, 4129–4157 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Narendran, R. et al. In vivo vulnerability to competition by endogenous dopamine: comparison of the D2 receptor agonist radiotracer (‐)‐N‐[11C]propyl‐norapomorphine ([11C]NPA) with the D2 receptor antagonist radiotracer [11C]‐raclopride. Synapse 52, 188–208 (2004). [DOI] [PubMed] [Google Scholar]
- 67. Fujita, M. et al. Imaging extrastriatal dopamine D(2) receptor occupancy by endogenous dopamine in healthy humans. Eur. J. Pharmacol. 387, 179–188 (2000). [DOI] [PubMed] [Google Scholar]
- 68. Laruelle, M. et al. Imaging D2 receptor occupancy by endogenous dopamine in humans. Neuropsychopharmacology 17, 162–174 (1997). [DOI] [PubMed] [Google Scholar]
- 69. Finnema, S.J. et al. Dopamine D(2/3) receptor occupancy of apomorphine in the nonhuman primate brain–a comparative PET study with [11C]raclopride and [11C]MNPA. Synapse 63, 378–389 (2009). [DOI] [PubMed] [Google Scholar]
- 70. Shotbolt, P. et al. Within‐subject comparison of [(11)C]‐(+)‐PHNO and [(11)C]raclopride sensitivity to acute amphetamine challenge in healthy humans. J. Cereb. Blood Flow Metab. 32, 127–136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Rabiner, E.A. et al. Endogenous dopamine release in the human brain as a pharmacodynamic biomarker: evaluation of the new GPR139 agonist TAK‐041 with [(11)C]PHNO PET. Neuropsychopharmacology. https://doi.org/ 10.1038/s41386-021-01204-1. [e‐pub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Kegeles, L.S. et al. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch. Gen. Psychiatry 67, 231–239 (2010). [DOI] [PubMed] [Google Scholar]
- 73. Abi‐Dargham, A. et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc. Natl. Acad. Sci. USA 97, 8104–8109 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abi‐Dargham, A. , van de Giessen, E. , Slifstein, M. , Kegeles, L.S. & Laruelle, M. Baseline and amphetamine‐stimulated dopamine activity are related in drug‐naïve schizophrenic subjects. Biol. Psychiatry 65, 1091–1093 (2009). [DOI] [PubMed] [Google Scholar]
- 75. Sander, C.Y. et al. Neurovascular coupling to D2/D3 dopamine receptor occupancy using simultaneous PET/functional MRI. Proc. Natl. Acad. Sci. USA 110, 11169–11174 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sander, C.Y. , Hansen, H.D. & Wey, H.Y. Advances in simultaneous PET/MR for imaging neuroreceptor function. J. Cereb. Blood Flow Metab. 40, 1148–1166 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Rabiner, E.A. et al. Pharmacological differentiation of opioid receptor antagonists by molecular and functional imaging of target occupancy and food reward‐related brain activation in humans. Mol. Psychiatry 16, 826–835, 785 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]