

Abstract
Dysregulated chronic inflammation plays a crucial role in the pathophysiology of atherosclerosis and may be a result of impaired resolution. Thus, restoring levels of specialized pro‐resolving mediators (SPMs) to promote the resolution of inflammation has been proposed as a therapeutic strategy for patients with atherosclerosis, in addition to standard clinical care. Herein, we evaluated the effects of the SPM lipids, lipoxin A4 (LXA4) and lipoxin B4 (LXB4), on neutrophils isolated from patients with atherosclerosis compared with healthy controls. Patients displayed altered endogenous SPM production, and we demonstrated that lipoxin treatment in whole blood from atherosclerosis patients attenuates neutrophil oxidative burst, a key contributor to atherosclerotic development. We found the opposite effect in neutrophils from healthy controls, indicating a potential mechanism whereby lipoxins aid the endogenous neutrophil function in health but reduce its excessive activation in disease. We also demonstrated that lipoxins attenuated upregulation of the high‐affinity conformation of the CD11b/CD18 integrin, which plays a central role in clot activation and atherosclerosis. Finally, LXB4 enhanced lymphatic transmigration of human neutrophils isolated from patients with atherosclerosis. This finding is noteworthy, as impaired lymphatic function is now recognized as an important contributor to atherosclerosis. Although both lipoxins modulated neutrophil function, LXB4 displayed more potent effects than LXA4 in humans. This study highlights the therapeutic potential of lipoxins in atherosclerotic disease and demonstrates that the effect of these SPMs may be specifically tailored to the need of the individual.
Keywords: atherosclerosis, cardiovascular disease, CD11b/CD18 integrin, lipoxins, lymphatics transmigration, neutrophils, reactive oxygen species, resolution of inflammation, specialized pro‐resolving mediators
Abbreviations
- DHR
dihydrorhodamine 123
- E. coli
Escherichia coli
- fMLP
N‐formyl‐Met‐Leu‐Phe
- IL‐8
interleukin‐8
- LEC
lymphatic endothelial cell
- LXA4
lipoxin A4 [5(S),6(R),15(S)‐trihydroxy‐7E,9E,11Z,13E‐eicosatetraenoic acid]
- LXB4
lipoxin B4 [5(S),14(R),15(S)‐trihydroxy‐6E,8Z,10E,12E‐eicosatetraenoic acid]
- MFI
mean fluorescence intensity
- PBS
phosphate‐buffered saline
- PMA
phorbol 12‐myristate 13‐acetate
- ROS
reactive oxygen species
- SPMs
specialized pro‐resolving mediators
- TNF‐α
tumor necrosis factor‐α
1. INTRODUCTION
Dysregulated chronic inflammation is widely recognized to play a crucial role in the pathophysiology of atherosclerosis and cardiovascular disease. 1 , 2 Evidence suggests that the chronic inflammation observed within atherosclerotic lesions may result from a failure to induce the resolution of inflammation. 3 Thus, restoring levels of specialized pro‐resolving mediators (SPMs) to promote the resolution of inflammation is emerging as a potential anti‐atherogenic therapeutic strategy, in addition to standard clinical care. 4
In healthy individuals, inflammation consists of two phases; the initial acute phase, followed by the resolving phase. 5 The latter phase is orchestrated by SPMs derived from either omega‐3 fatty acids (eicosapentaenoic acid or docosahexaenoic acid) or omega‐6 fatty acids (arachidonic acid). 5 , 6 , 7 , 8 SPMs exert their pro‐resolving effects through various mechanisms, including regulating transcription factors, cytokine release, receptor expression, immune cell migration, and modulating cell phenotype, among others. 9 , 10 , 11 , 12 , 13 , 14 , 15 , 16 , 17 An ample body of evidence demonstrates that promoting the resolution of inflammation in atherosclerosis through the therapeutic application of SPMs has beneficial effects. Atherosclerotic animal models demonstrate that treatment with SPMs or peptides binding to their receptors attenuates atherosclerotic development, decreases intimal hyperplasia, and increases plaque stability. 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 For instance, the omega‐6‐derived lipid mediator, lipoxin A4 (LXA4), significantly reduces atherosclerotic lesions in the aortic arch of mice. 28 Interestingly, in humans, an imbalance of the ratio between pro‐resolving and pro‐inflammatory lipid mediators indicates more severe plaque phenotypes 18 and increased intima media thickness. 29 Recent evidence also suggests that the anti‐atherogenic therapeutic potential of SPMs may translate from experimental models to humans. For example, we have previously demonstrated that LXA4 treatment attenuates human blood clot formation by manipulating neutrophil integrin activation. 10 In addition, ex vivo treatment of human carotid plaques with LXA4 attenuates inflammation through inhibition of pro‐inflammatory cytokine release. 28 Together, these findings provide a solid foundation to continue to explore the role of lipoxins in resolving chronic inflammation in atherosclerosis.
Here, we continued to investigate the therapeutic effect of lipoxins in human atherosclerosis. We studied cellular responses in patients with atherosclerosis and healthy controls, as SPM treatment may elicit varied responses in health versus disease. We used an ex vivo treatment model of neutrophils from atherosclerosis patients and healthy volunteers, as these cells play an important role in creating a pro‐atherogenic environment. 30 , 31 , 32 , 33 , 34 , 35 We found that lipoxins modify reactive oxygen species (ROS) production, integrin receptor expression, and lymphatic migration of neutrophils isolated from patients with atherosclerosis. Additionally, some of these mechanisms are differentially regulated by the lipoxins in health versus disease.
2. MATERIALS AND METHODS
2.1. Patient recruitment
We included patients with peripheral artery disease and concomitant advanced atherosclerosis (n = 10) scheduled to undergo vascular surgery for revascularization, as stated in Table 1. We also included a control group consisting of reportedly healthy volunteers without diagnosed or treated diabetes, hypertension, or hyperlipidemia (n = 10). Given the nature of the atherosclerotic disease, the patient group consisted of older individuals (75.1 ± 1.5 years). Therefore, the inclusion criteria for the control group stated that healthy volunteers must be more than 50 years old to minimize the confounding effects of “inflammaging”. 36 Study participants underwent a detailed medical history and a health exam measuring blood pressure, height, weight, sagittal height, and body temperature before donating blood. Patients in drug trials or who were deemed unable to adequately comprehend the informed consent were excluded from the recruitment process. All study participants were enrolled in accordance with the Helsinki Declaration and provided written informed consent. The study was approved by the Swedish Ethical Review Authority #Dnr 660‐18 (ClinicalTrials.gov NCT03732612).
TABLE 1.
Clinical characteristics of control and patient cohorts
| Variables | Controls | Patients with atherosclerosis | p‐Value | Significance |
|---|---|---|---|---|
| Cohort characteristics | ||||
| n (%) | 10 | 10 | N/A | N/A |
| Sex | 1 ♂/9 ♀ | 5 ♂/5 ♀ | .1409 | ns |
| Age (years) | 59.6 ± 4.88 | 75.1 ± 4.84 | .0002 | *** |
| Body temperature (°C) | 36.5 ± 0.25 | 36.45 ± 0.35 | >.999 | ns |
| Anthropometrics | ||||
| Weight (kg) | 63.87 ± 5.35 | 77.7 ± 17.93 | .0342 | * |
| Body mass index (BMI, kg/m2) | 22.34 ± 1.64 | 25.8 ± 4.90 | .0686 | ns |
| Height (cm) | 169.3 ± 6.20 | 173.1 ± 7.72 | .3840 | ns |
| Waist circumference (cm) | 79.9 ± 5.77 | 101.8 ± 15.62 | .0028 | ** |
| Waist‐to‐height ratio (%) | 0.47 ± 0.04 | 0.59 ± 0.09 | .0032 | ** |
| Sagittal height (cm) | 16.67 ± 2.07 | 23.35 ± 4.29 | .0013 | ** |
| Surgical indication | ||||
| Aneurism indication with concomitant atherosclerosis (n, %) | N/A | 0/10 (0%) | N/A | N/A |
| Aortoiliac disease (n, %) | N/A | 1/10 (10%) | N/A | N/A |
| Critical limb ischemia (n, %) | N/A | 3/10 (30%) | N/A | N/A |
| Claudication (n, %) | N/A | 6/10 (60%) | N/A | N/A |
| Risk factors cardiovascular disease | ||||
| Diagnosed hypertension (n, %) | 0/10 (0%) | 8/10 (80%) | .0007 | *** |
| Diagnosed diabetes (n, %) | 0/10 (0%) | 1/10 (10%) | >.999 | ns |
| High low‐density lipoprotein (LDL)‐Cholesterol (n, %) | 1/10 (10%) | 0/10 (0%) | >.999 | ns |
| Low high‐density lipoprotein (HDL)‐Cholesterol (n, %) | 0/10 (0%) | 5/10 (50%) | .0325 | * |
| Hyperlipidemia (n, %) | 1/10 (10%) | 9/10 (90%) | .0011 | ** |
| Aorta iliac atherosclerosis (n, %) | N/A | 6/10 (60%) | N/A | N/A |
| Peripheral atherosclerosis (n, %) | N/A | 10/10 (100%) | N/A | N/A |
| Additional diagnosed medical conditions | ||||
| Gastrointestinal disease (n, %) | 0/9 a (0%) | 3/10 (30%) | .2105 | ns |
| Skin conditions (n, %) | 1/9 a (11.11%) | 2/10 (20%) | >.999 | ns |
| Lifestyle factors | ||||
| Alcohol use (n, %) | 8/9 a (88.89%) | 5/10 (50%) | .1409 | ns |
| Diet including red meat (n, %) | 9/9 a (100%) | 10/10 (100%) | >.999 | ns |
| Tobacco use present (n, %) | 1/9 a (11.11%) | 1/10 (10%) | >.999 | ns |
| Tobacco use past (n, %) | 1/9 a (11.11%) | 10/10 (100%) | .0001 | *** |
| Clinical measurements | ||||
| Blood pressure | ||||
| Diastolic blood pressure (mmHg) | 75.4 ± 10.13 | 77.4 ± 11.63 | .7910 | ns |
| Systolic blood pressure (mmHg) | 126.9 ± 19.30 | 151 ± 12.63 | .0040 | ** |
| Pulse (beats/min) | 65.4 ± 7.56 | 70.4 ± 14.01 | .2720 | ns |
| Inflammation | ||||
| C‐reactive protein (mg/L) | 1.13 ± 1.14 | 3.42 ± 3.05 | .0445 | * |
| White blood cell count (10⁹/L) | 4.79 ± 0.97 | 7.06 ± 2.04 | .0188 | * |
| Liver function enzymes | ||||
| AST (µkat/L) | 0.4 ± 0.06 | 0.49 ± 0.25 | .9090 | ns |
| ALT (µkat/L) | 0.34 ± 0.11 | 0.43 ± 0.27 | .8200 | ns |
| ALP (µkat/L) | 1.18 ± 0.27 | 1.84 ± 0.92 | .0615 | ns |
| GGT (µkat/L) | 0.34 ± 0.22 | 1.29 ± 1.35 | .0586 | ns |
| Metabolic analytes | ||||
| Fasting glucose, mmol/L | 5.38 ± 0.44 | 6.09 ± 1.20 | .2270 | ns |
| HbA1C (mmol/mol) | 38 ± 2.31 | 40.7 ± 6.22 | .4030 | ns |
| Insulin (mIE/L) | 4.59 ± 1.52 | 12.88 ± 8.20 | .0017 | ** |
| HOMA %S (insulin sensitivity) | 188.89 ± 43.99 | 100.54 ± 56.95 | .0093 | ** |
| HOMA %B (pancreatic function) | 54.84 ± 12.37 | 74.72 ± 19.46 | .0673 | ns |
| HOMA‐IR (insulin resistance) | 0.56 ± 0.16 | 1.36 ± 0.81 | .0081 | ** |
| Blood lipids | ||||
| Cholesterol (mmol/L) | 6.15 ± 0.74 | 3.52 ± 0.69 | .0002 | *** |
| LDL cholesterol (mmol/L) | 4.22 ± 0.85 | 1.87 ± 0.61 | .0003 | *** |
| HDL cholesterol (mmol/L) | 1.82 ± 0.51 | 1.33 ± 0.39 | .0401 | * |
| Triglycerides (mmol/L) | 1.01 ± 0.26 | 1.27 ± 0.59 | .4950 | ns |
| Platelet number and coagulation | ||||
| Platelet count (10⁹/L) | 270.5 ± 38.39 | 246.5 ± 73.71 | .3070 | ns |
| APTT (sec) | 26.1 ± 1.37 | 28.4 ± 3.53 | .1030 | ns |
| PCC (INR) | 1.02 ± 0.11 | 1.05 ± 0.10 | .3830 | ns |
| Blood proteins | ||||
| Albumin (g/L) | 39.4 ± 2.17 | 36.78 ± 0.97 | .0034 | ** |
| Medications | ||||
| Anti‐coagulant (n, %) | 0/10 (0%) | 3/10 (30%) | .2105 | ns |
| Anti‐hypertensive (n, %) | 0/10 (0%) | 9/10 (90%) | .0001 | *** |
| Anti‐inflammatory (n, %) | 0/10 (0%) | 3/10 (30%) | .2105 | ns |
| Diabetes (n, %) | 0/10 (0%) | 2/10 (20%) | .4737 | ns |
| Immunosuppressive (n, %) | 0/10 (0%) | 0/10 (0%) | >.999 | ns |
| Lipid lowering (n, %) | 0/10 (0%) | 8/10 (80%) | .0007 | *** |
| Platelet inhibitor (n, %) | 0/10 (0%) | 10/10 (100%) | <.0001 | **** |
| Other (n, %) | 3/10 (30%) | 9/10 (90%) | .0198 | * |
| Specialized pro‐resolving mediator‐related medications | ||||
| Low‐dose aspirin (n, %) | 0/10 (0%) | 9/10 (90%) | .0001 | *** |
| Statins (n, %) | 0/10 (0%) | 9/10 (90%) | .0001 | *** |
Data are presented as mean ± SD. p‐Values for continuous variables were calculated using Mann–Whitney U. p‐Values for categorical variables were calculated using Fisher's exact test.
Abbreviations: ALT, alanine transaminase; ALP, alkaline phosphatase; APTT, activated partial thromboplastin time; AST, aspartate transaminase; HOMA, homeostatic model assessment; GGT, gamma‐glutamyltransferase; PCC, prothrombin complex concentrate.
One participant declined to answer lifestyle questions and diagnosed medical conditions.
2.2. Blood collection
Venous blood samples were collected after an overnight fast in tubes spray‐coated with K2EDTA (Greiner Bio One, Kremsmünster, Upper Austria, Austria), citrate (Greiner Bio One), sodium‐heparin (Becton Dickinson and Company, Franklin Lakes, New Jersey, United States), or lithium‐heparin (Becton Dickinson and Company) as described. 37 Routine clinical tests were performed using lithium‐heparin blood (liver status, blood lipids, C‐reactive protein, insulin), citrate blood (coagulation status), and EDTA blood (HbA1c, pan‐leukocyte, and platelet counts), as detailed in Table 1. For experimental procedures, sodium‐heparin blood was used for whole blood stimulation experiments, as described below, and EDTA blood was used for neutrophil transmigration experiments.
2.3. Lipoxin treatment and inflammatory stimulation of whole blood samples
Whole blood collected in sodium‐heparin tubes was stimulated with vehicle (0.16% ethanol), LXA4 (500 nM, #437725, Calbiochem, San Diego, United States) or LXB4 (500 nM, #90420, Cayman Chemical, Ann Arbor, Michigan, United States) for 10 min at room temperature, after which it was cooled for 10 min on ice before stimulation. Briefly, 100 μl whole blood was stimulated for 20 min at 37°C with either 0.4 μM chemotactic peptide N‐formyl‐Met‐Leu‐Phe (fMLP), 1.5 × 107 opsonized Escherichia coli, or 1 μM phorbol 12‐myristate 13‐acetate (PMA), all part of the PhagoBurst kit (#341058, Becton Dickinson and Company, Franklin Lakes, New Jersey, United States). Sodium chloride was added to the unstimulated samples as a vehicle treatment to ensure equal final volumes of all tests. Subsequently, neutrophil oxidative burst/ROS production and cellular receptor expression were quantified using flow cytometry as described below.
2.4. Measuring neutrophil reactive oxygen species production
Oxidative burst/ROS production was measured using the PhagoBurst assay, whereby ROS is quantified when the fluorogenic substrate, dihydrorhodamine 123 (DHR), is taken up by the cell and oxidized by ROS into the fluorescent rhodamine 123. Briefly, 20 μl of DHR was prepared as per the manufacturer's instructions (PhagoBurst, #341058, Becton Dickinson and Company, Franklin Lakes, New Jersey, United States) and added to the whole blood while resting on ice, immediately before the 37°C stimulation with inflammatory stimuli (described above). After stimulation, the tubes were placed on ice for 5 min to halt the ROS production, and cells were stained with CD16 (BV510, clone 3G8 #302048, BioLegend, San Diego, California, United States) in Brilliant Stain Buffer (Becton Dickinson and Company, Franklin Lakes, New Jersey, United States) in the dark for 10 min on ice and subsequently 20 min at room temperature. To lyse red blood cells, FACS lysing solution (#349202, Becton Dickinson and Company, Franklin Lakes, New Jersey, United States) was added and incubated for 15 min at room temperature. The stained blood samples were centrifuged (460g, 5 min, 4°C), and stained cells were washed in FACS buffer (phosphate‐buffered saline [PBS] with 2% fetal bovine serum) twice (460g, 5 min, 4°C) and analyzed immediately with a Beckman Coulter Gallios flow cytometer. A total of 25000 events in the granulocyte gate (forward scatter [FSC]high side scatter [SSC]high) were acquired. The data were analyzed with the FlowJo software (version 10.7, Tree Star, Ashland, Oregon, United States). Neutrophil gates were made based on SSChighCD16bright events and single‐cell gating before analysis of DHR positivity (Figure S1A,B).
2.5. PhagoTest assay
Samples from healthy controls were prepared as described in Section 2.3. The phagocytosis rate of E. coli in neutrophils was measured as per manufacturer's instructions (PhagoTest, #341060, Becton Dickinson and Company, Franklin Lakes, New Jersey, United States), using 1.5 × 107 opsonized FITC‐labelled E. coli per sample. Neutrophil gates were made based on live leukocytes determined by DNA staining, followed by characteristic FSChigh/SSChigh neutrophils and single‐cell gating, before analysis of FITC positive neutrophils (Figure S3). A sample kept on ice was used as a gating control, as the cold prevents the cellular uptake of FITC‐E. coli.
2.6. Measuring neutrophil receptor expression
Neutrophil expression of SPM receptors and the CD11b integrin were quantified by flow cytometry. Blood was placed on ice for 5 min and stained in Brilliant Stain Buffer with CD11b‐FITC (high‐affinity conformation, clone CBRM1/5, #11‐0113‐41, eBioscience, San Diego, California, United States) and FPRL1/FPR2‐PE (aka FPR2/ALX, clone 304405, #FAB3479P, R&D Systems Inc., Minneapolis, Minnesota, United States). To facilitate gating of neutrophils, the cells were also stained with CD45‐PerCP (Clone 2D1, #345809), CD16‐APC‐H7 (Clone 3G8, #560248), and CD14‐BV786, (Clone M5E2, #563699), which were obtained from Becton Dickinson and Company, Franklin Lakes, New Jersey, United States. Following the addition of antibodies, the samples were vortexed and incubated in the dark for 15 min at room temperature. To lyse the red blood cells, FACS lysing solution was added to the samples, which were then vortexed and incubated in the dark for 15 min at room temperature. The samples were centrifuged (320g, 5 min, room temperature), and stained cells were washed with PBS with human serum albumin twice (320g, 5 min, room temperature) before acquiring using BD FACSLyric flow cytometer. One hundred thousand events were acquired in the leukocyte gate using BD FACS Suite (version 1.4, Becton Dickinson and Company, Franklin Lakes, New Jersey, United States). The data were analyzed in Kaluza (version 2.1, Beckman Coulter, Brea, California, United States) and FlowJo software (version 10.7, Tree Star, Ashland, Oregon, United States), and gates were made as described in Figure S1C,D.
2.7. Neutrophil lymphatic transmigration assay
2.7.1. Cell culture
Human dermal lymphatic microvascular endothelial cells (LECs) were purchased from Lonza (#CC‐2810, Basel city, Basel, Switzerland). LECs were cultured in the culture media recommended by the company, that is, EGM‐2MV media (#CC‐3202, Lonza, Basel city, Basel, Switzerland) containing the included SingleQuots Bullet kit with fetal bovine serum, hydrocortisone, hFGF‐B, VEGF, R3‐IGF‐1, ascorbic acid, hEGF, and GA‐100 in 75 cm2 culture flasks coated with 0.1% gelatin in a horizontal position. Cells were cultured in a 5% CO2 atmosphere at 37°C. Once cells reached 70% to 85% confluency, they were released with trypsin‐EDTA (4–7 min) and split at a concentration of 5000 cells/cm2 into new flasks. LECs were used for transmigration experiments between passages 5–11.
2.7.2. Seeding lymphatic endothelial cells onto transwells
LECs were seeded on a 24‐well transwell plate insert with 5 μm pore size (#3421, Corning International, New York, New York, United States), as previously described. 38 Briefly, transwells were flipped upside‐down into a 12‐well plate, where four empty wells contained PBS for a humidified atmosphere. The transwells were coated with 100 μl of 0.1% gelatin for a minimum of 30 min at 37°C. The remaining gelatin liquid was aspirated off, and LECs (100,000 cells in 100 μl complete EGM‐2MV media) were seeded on the upside‐down flipped transwells. The plate lid of the 12‐well plate was gently placed on top of the plate, thus allowing the media to touch the lid, whereby the liquid surface tension with the humidified atmosphere ensured that the media did not evaporate. The media was refreshed after 48 hours. On the third day post‐seeding, the transwells were washed with migration media, defined as Iscove's Modified Dulbecco's Medium (#12440‐046, Thermo Fisher Scientific Life Sciences, Waltham, Massachusetts, United States), insulin‐transferrin 1:100 (#41400‐045, Thermo Fisher Scientific Life Sciences) and 1% bovine serum albumin (#700‐107P, GeminiBio, West Sacramento, California, United States). The transwells were immediately flipped (180°) into 600 μl of migration media, containing either vehicle or 5 ng/ml interleukin (IL)‐8 (R&D Systems Inc., Minneapolis, Minnesota, United States).
2.7.3. Neutrophil isolation and staining
Human neutrophils were isolated using negative bead isolation with the EasySep Direct Human Neutrophil Isolation kit (#19666, StemCell, Vancouver, British Columbia, Canada) with minor modifications. In order to obtain a purer population of neutrophils (98%–99% of isolated cells were characterized as CD45+CD16+CD66b+ neutrophils), we added an additional amount (50% of recommended concentration) of the EasySep Pan‐Granulocyte Isolation Cocktail (#19659C, StemCell, Vancouver, British Columbia, Canada) to the original volume of Neutrophil Isolation Cocktail. Isolated neutrophils were then centrifuged (400g, 7 min, 4°C) and washed in PBS without calcium or magnesium (400g, 7 min, 4°C) before being resuspended in migration media (defined above) containing 5 µM CellTracker Green (Thermo Fisher Scientific Life Sciences, Waltham, MA, United States). Cells were stained on a shaker at 300 rpm, 15 min at 37°C. Following a wash (400g, 7 min, 4°C), cells were resuspended in migration media and recovered for 45–60 min at 37°C in a 5% CO2 atmosphere.
2.7.4. Lipoxin treatment of isolated neutrophils
As opposed to whole blood, where a higher lipoxin concentration is needed due to the presence of serum albumin, 10 isolated neutrophils were treated with vehicle (0.01% ethanol), LXA4 (1 nM), or LXB4 (1 nM) for 15 min at 37°C. Subsequently, 5 × 105 neutrophils in 100 µl of migration media were added to the upper chamber of the transwell, which had just been flipped into migration media containing vehicle or IL‐8 (5 ng/ml). Additional vehicle (0.01% ethanol), LXA4 (1 nM), or LXB4 (1 nM) was added to the lower chamber of the transwells.
2.7.5. Neutrophil transmigration assay
After the neutrophils migrated for 3 hours toward either vehicle or the chemoattractant, IL‐8, cells were collected from the lower chamber of the transwell. The cells were counted using an Accuri C6 flow cytometer (Becton Dickinson and Company, Franklin Lakes, New Jersey, United States) by acquiring 50 μl of cell suspension and back calculating the cell concentration based on expected volume (600 μl) in the lower chamber. CellTracker Green labelled neutrophils were easily distinguishable from contaminating LECs, which may have loosened from the transwell. The percentage of transmigrated neutrophils was calculated based on the lower chamber's cell count divided by the total sum of cells added multiplied by 100.
2.8. Lipid mediator profiling
Lipid mediators in human serum samples were extracted, identified, and quantified as described. 39 In brief, after the addition of four volumes of ice‐cold methanol containing d5‐RvD2, d5‐RvE1, d5‐RvD3, d5‐17R‐RvD1, d5‐LXA4, d5‐MaR2, d5‐MaR1, d4‐PGE2, d5‐LTC4, d5‐LTD4, d5‐LTE4, and d5‐LTB4, samples were placed at −20°C for 45 min to allow for protein precipitation. The lipid mediators were extracted using an ExtraHera liquid handling system (Biotage, Uppsala, Sweden) and solid phase extraction techniques with Isolute C18 500mg columns (Biotage). Methyl formate and methanol fractions were collected, brought to dryness and resuspended in phase (methanol/water, 1:1, vol/vol) for injection on a Shimadzu LC‐20AD HPLC and a Shimadzu SIL‐20AC autoinjector, paired with a QTrap 6500+ or QTrap 5500 (Sciex, Framingham, Massachusetts, United States). For the identification and quantitation of mediators eluted in methyl formate fractions the QTRAP was operated in negative ion mode using a multiple reaction monitoring method coupled with an information dependent acquisition to an Enhanced Product Ion experiment. An Agilent Poroshell 120 EC‐C18 column (100 mm × 4.6 mm × 2.7 μm) was kept at 50°C and mediators eluted using a mobile phase consisting of methanol/water/acetic acid of 20:80:0.01 (vol/vol/vol) that was ramped to 50:50:0.01 (vol/vol/vol) over 0.5 min and then to 80:20:0.01 (vol/vol/vol) from 2 min to 11 min, maintained till 14.5 min and then rapidly ramped to 98:2:0.01 (vol/vol/vol) for the next 0.1 min. This was subsequently maintained at 98:2:0.01 (vol/vol/vol) for 5.4 min, and the flow rate was maintained at 0.5 ml/min. In the analysis of mediators eluted in the methanol fraction, the QTrap 6500+ was operated in positive ion mode using a multiple reaction monitoring method. An Agilent Poroshell 120 EC‐C18 column (100 mm × 4.6 mm × 2.7 μm) was kept at 50°C and mediators eluted using a mobile phase consisting of methanol/water/acetic acid 55:45:0.5 (vol/vol/vol) over 5 min, that was ramped to 80:20:0.5 (vol/vol/vol) for 2 min, maintained at 80:20:0.5 (vol/vol/vol) for the successive 3 min and ramped to 98:2:0.5 (vol/vol/vol) over 3 min. This condition was maintained for 3 min. Each lipid mediator was identified using established criteria, 39 , 40 including the following: (1) matching retention time to synthetic or authentic standards, (2) ≥8 data points, and (3) matching of MS/MS spectrum to that of reference standard in representative samples. For quantitation the following criteria were used: (1) matching retention time to synthetic or authentic standards, (2) ≥8 data points, (3) signal to noise ratio ≥4. Calibration curves were obtained for each mediator using synthetic compound mixtures at 0.78, 1.56, 3.12, 6.25, 12.5, 25, 50, 100, and 200 pg that gave linear calibration curves with R 2 values of 0.98–0.99.
2.9. Statistical analyses
Data were analyzed using GraphPad (version 7.04, San Diego, California, United States). Assuming non‐Gaussian distribution of the human samples, statistical significance (p < .05) for continuous variables was determined using Mann–Whitney U test when comparing two groups, and Kruskal‐Wallis test with Dunn's post hoc comparisons when comparing over two groups. Statistical significance (p < .05) for categorical variables was calculated using Fisher's exact test. All statistical significance for lipoxin treatments were calculated based on the log2 of the fold change relative to vehicle treatment. Data are presented as mean ± standard error of the mean (SEM) or mean ± standard deviation (SD), as stated in respective figure or table legend.
3. RESULTS
3.1. Cohort characteristics
We compared the clinical status of the patients diagnosed with atherosclerosis and the healthy controls. Briefly, their clinical characteristics, anthropometrics, cardiovascular risk factors, medical history, and drug regimen were recorded (Table 1). The patients with atherosclerosis were older than the control group (59.6 ± 4.88 vs. 75.1 ± 4.84 years: p = .0002). However, as the healthy controls were over 50 years old, the potential impact of “inflammaging” should be minimized. 36 One patient with atherosclerosis was diagnosed with type 2 diabetes mellitus. Both the patient and control groups had well‐controlled blood sugar, as evidenced by normal levels of whole blood glycosylated hemoglobin (HbA1c), which provides the average blood sugar levels for the 6–12 weeks prior to the blood draw. There was no difference in body mass index (BMI) between the groups, although the patients with atherosclerosis had signs of elevated central adiposity (i.e., higher sagittal height, waist circumference, and waist‐to‐height ratio), compared with the control group. We unexpectedly found that compared with healthy controls, the atherosclerosis cohort had lower cholesterol (6.15 ± 0.74 vs. 3.52 ± 0.69 mmol/L, respectively: p = .0002) and low‐density lipoprotein (LDL) cholesterol (4.22 ± 0.85 vs. 1.87 ± 0.61, respectively: p = .0003). This is likely due to lipid‐lowering drugs (statins) that were used by 90% of the patients (Table 1). Patients with atherosclerosis displayed elevated markers of systemic inflammation compared with controls, that is, plasma levels of C‐reactive protein (1.13 ± 1.14 vs. 3.42 ± 3.05 mg/ml, respectively: p = .0445) and circulating leukocytes (4.79 ± 0.97 vs. 7.06 ± 2.04 × 109 cells/L, respectively: p = .0188). However, there were no differences between the groups regarding liver function tests, that is, alanine transaminase, aspartate transaminase, alkaline phosphatase, or gamma‐glutamyltransferase levels. All patients with atherosclerosis had a history of smoking, whereas only one healthy control was a former smoker (p = .0001). It may be important to note that the patients with atherosclerosis were taking medications that may alter endogenous SPM production, such as low‐dose aspirin and statins, 20 , 41 , 42 , 43 which is, therefore, highlighted in Table 1.
3.2. Neutrophils reactive oxygen species production is elevated in patients with atherosclerosis compared with healthy controls and is inversely modulated by lipoxins
The neutrophil oxidative burst process is critical to the host's defense against certain pathogens. 44 However, exacerbated ROS production amplifies the pro‐atherogenic inflammatory environment. 35 The PhagoBurst assay was used to determine differences in neutrophil ROS production between patients with atherosclerosis and healthy controls (Figure 1A). We analyzed both the percentages of cells having produced intracellular ROS (i.e., DHR+ cells) and the mean fluorescence intensity (MFI) of DHR in the neutrophil population. ROS production was measured both at baseline (unstimulated cells) and in response to inflammatory stimuli: chemotactic peptide (fMLP) and whole bacteria (opsonized E. coli). Maximal ROS production was defined as the cellular response to PMA stimulation.
FIGURE 1.
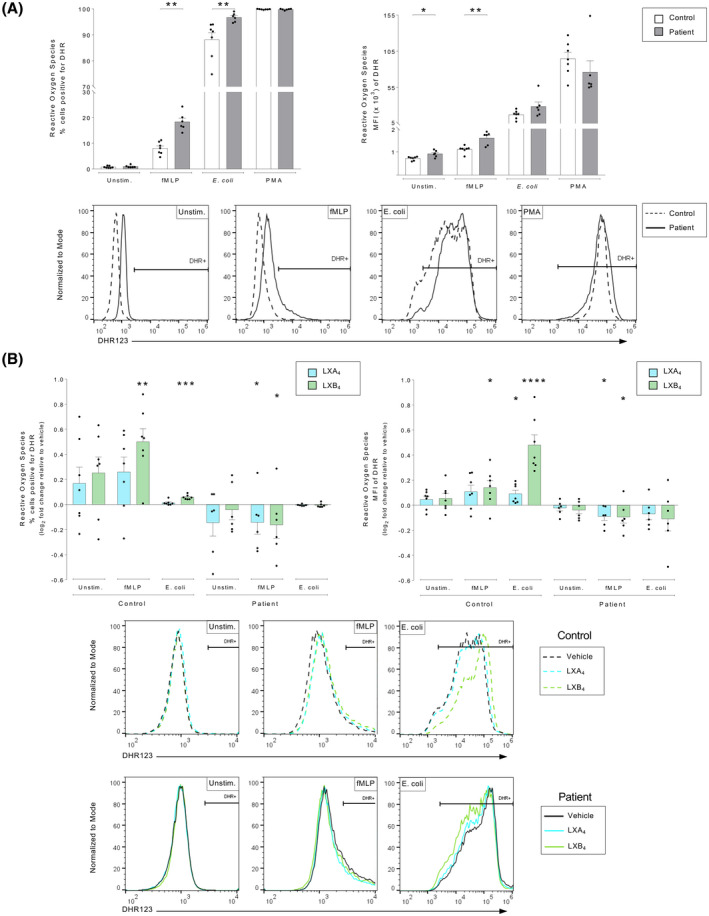
Lipoxins modulate oxidative burst in human neutrophils from patients with atherosclerosis and healthy controls. Whole blood from healthy controls (n = 7) or patients with atherosclerosis (n = 6) was exposed to an inflammatory stimulus, either in the absence or presence of lipoxin A4 (LXA4: 500 nM) or lipoxin B4 (LXB4: 500 nM). Neutrophil reactive oxygen species (ROS) production was measured by flow cytometry and reported as DHR positivity. The percentage ROS‐positive neutrophils (DHR+) was determined in relation to a 1% baseline, which was set using an unstimulated control sample and kept constant throughout the analysis. (A) Neutrophil ROS production was measured as % positive cells (left panel) and cellular mean fluorescence intensity (MFI) (right panel), in controls (white bars) and patients (gray bars). The cells were untreated (Unstim.) or stimulated with chemotactic peptide N‐formyl‐Met‐Leu‐Phe (fMLP, 0.4 μM) opsonized Escherichia coli (1.5 × 107), or phorbol 12‐myristate 13‐acetate (PMA, 1 μM). Representative histograms of the MFI DHR‐signal in respective conditions are shown for controls (dashed line) and patients (solid line). (B) LXA4 (blue bars) and LXB4 (green bars)‐induced changes to ROS production was calculated as the log2 fold change relative to respective vehicle‐treated condition. The cells were stimulated as indicated. The bar graphs show ROS levels as % positive cells (left panel) and cellular mean fluorescence intensity (MFI) (right panel). Representative histograms of the MFI DHR‐signal in respective conditions are shown for vehicle (black line), LXA4 (blue line), and LXB4 (green line). Assuming non‐Gaussian distribution of the human samples, statistical analysis was determined using Mann–Whitney U test when comparing two groups, and Kruskal–Wallis test with Dunn's post hoc comparisons when comparing more than two groups. Data are presented as mean ± standard error of the mean (SEM). Statistical significance is indicated as *p < .05, **p < .01, ***p < .001, ****p < .0001
At baseline, the percentages of DHR+ neutrophils were similar in control and patient cohorts. However, neutrophils from patients with atherosclerosis produced more ROS per cell (i.e., elevated MFI values) than healthy controls (p = .0221). In response to fMLP, the percentage of DHR+ neutrophils was approximately doubled in patients with atherosclerosis compared with controls (p = .001). In addition, ROS production per cell after fMLP stimulation was also higher in neutrophils from patients (p = .005). In response to E. coli stimulation, a significantly higher percentage of neutrophils from atherosclerosis patients produced ROS compared with the healthy controls (p = .001), although the ROS production per cell did not differ between groups. PMA stimulation represents the maximum ROS production, and it did not differ between patients and controls.
To assess the effects of lipoxins on ROS production, whole blood was treated with vehicle (0.16% ethanol) or lipoxins (500 nM), prior to exposure to inflammatory stimuli (Figure 1B). To highlight drug‐mediated effects, the fluorescence scale on the x‐axes of the representative histograms were adjusted for respective condition, and raw data may be found in Figure S2. The lipoxins manipulated neutrophil ROS production differently in the two cohorts. In healthy controls, LXB4 amplified fMLP‐ and E. coli–induced ROS production, both regarding percentage of DHR+ neutrophils (p = .002, p < .001, respectively) and the MFI (p = .035, p ≤ .001, respectively). In addition, in healthy individuals, LXA4 increased E. coli–induced ROS production based on MFI (p = .0316). The lipoxin‐mediated increase in E. coli–induced ROS in the healthy controls was not related to an increased phagocytosis rate (Figure S3). In contrast, the lipoxins reduced the upregulated ROS production observed in the patient cohort. Specifically, both LXA4 and LXB4 treatment reduced the fMLP‐induced increase in ROS production, both with respect to percentage of DHR+ neutrophils (p = .047, p = .047, respectively), and the MFI (p = .013, p = .013, respectively).
3.3. Expression of neutrophil specialized pro‐resolving mediator receptors in atherosclerosis patients versus healthy controls
To determine the individual's capacity to induce the resolution of inflammation, we quantified the expression of the LXA4 receptor, FPR2/ALX, on neutrophils from healthy controls and patients with atherosclerosis (Figure 2A, Figure S1). Patients and healthy controls expressed similar FPR2/ALX receptor levels at baseline. In contrast, fMLP stimulation caused a significant decrease in the percentage of cells expressing FPR2/ALX in the patient cohort (p = .032), and a similar decreasing trend was observed in the healthy controls (p = .056). No significant difference in the MFI of FPR2/ALX was observed in either group in response to fMLP stimulation (p > .999). We further investigated if lipoxin treatment could alter the SPM receptor expression. Thus, whole blood from patients and healthy controls was incubated with vehicle (0.16% ethanol) or lipoxins (500 nM) for 15 min prior to fMLP stimulation. Under these conditions, we found no difference in FRP2/ALX receptor expression in response to lipoxins in either patients or controls (Figure 2B). As an additional measurement of endogenous resolution, we quantified the levels of pro‐resolving lipids in serum of healthy controls and patients (Table 2). Factors that drive the separation between the cohorts were identified by a supervised partial least squares‐discriminant analysis (PLS‐DA) (Figure 3A). To identify those lipid mediators that drive the group separation, we calculated the variable importance in projection (VIP) score. This analysis identified 10 mediators with VIP scores >1, of which Maresin conjugates in tissue regeneration 3 (MCTR3) and 15‐epi LXA4 concentrations were observed to display the highest differences between the two groups (Figure 3B). Differences in the concentrations of these mediators were further corroborated following additional statistical evaluation. Of note, this analysis also demonstrated that 17R‐PD1, the aspirin triggered epimer of PD1, was also upregulated in patients with cardiovascular disease, likely reflecting aspirin intake in these patients.
FIGURE 2.
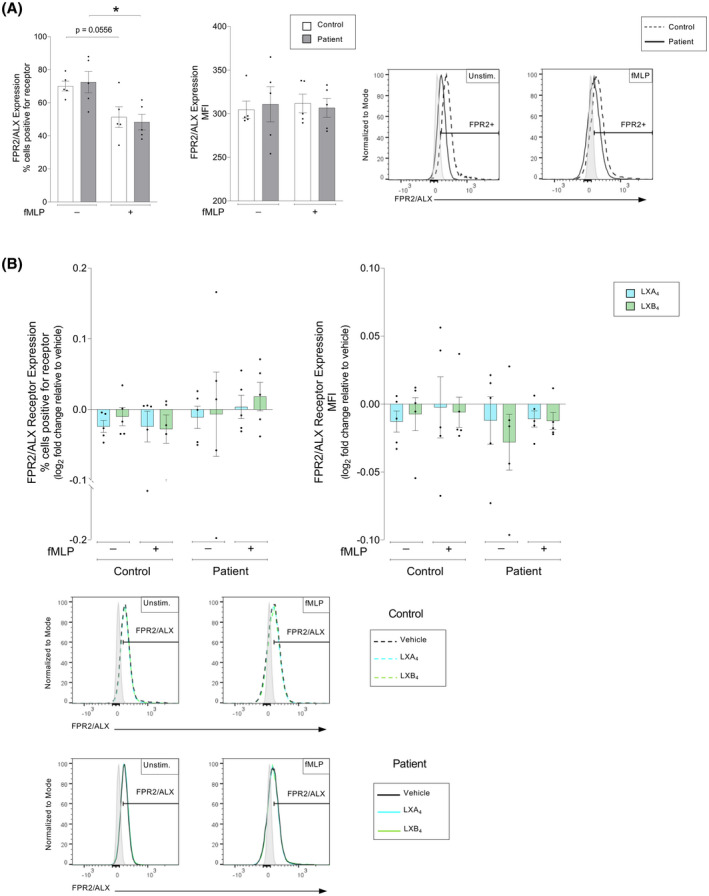
Lipoxin A4 receptor, FPR2/ALX, expression on neutrophils from patients with atherosclerosis versus healthy controls. Whole blood from healthy controls (n = 5) or patients with atherosclerosis (n = 5) was exposed to inflammatory stimulus, either in the absence or presence of lipoxin A4 (LXA4: 500 nM) or lipoxin B4 (LXB4: 500 nM). The neutrophil surface expression of FPR2/ALX was measured by flow cytometry. (A) Neutrophil FPR2/ALX expressions were measured as % positive cells (left panel) and cellular mean fluorescence intensity (MFI) (right panel). The expression was measured in controls (white bars) and patients (gray bars). The cells were untreated (Unstim.) or stimulated with chemotactic peptide N‐formyl‐Met‐Leu‐Phe (fMLP, 0.4 μM). Representative histograms for the MFI of respective receptor expression and indicated conditions are shown for controls (dashed line) and patients (solid line), where the gates were determined using a negative population (gray shaded peaks). (B) LXA4 (blue bars) and LXB4 (green bars)‐induced changes to FPR2/ALX expression was calculated as the log2 fold change relative to respective vehicle‐treated condition. The samples were stimulated as indicated. The bar graphs show levels as % marker positive cells (left panel) and MFI of indicated markers (right panel). Representative MFI histograms for each receptor expression under respective treatment conditions are shown for vehicle (black line), LXA4 (blue line) and LXB4 (green line), where the gates were determined using a negative population (gray shaded peaks). Assuming non‐Gaussian distribution of the human samples, statistical analysis was determined using Mann–Whitney U test when comparing two groups, and Kruskal–Wallis test with Dunn's post hoc comparisons when comparing more than two groups. Data are presented as mean ± standard error of the mean (SEM). Statistical significance is indicated as *p < .05, **p < .01, ***p < .001
TABLE 2.
Serum lipid mediator profiles in controls and patient cohorts
| Q1 | Q3 | Patients with atherosclerosis (pg/ml) | Controls (pg/ml) | p‐Value | Significance | |
|---|---|---|---|---|---|---|
| DHA bioactive metabolome | ||||||
| RvD1 | 375 | 215 | 0.08 ± 0.15 | 0.05 ± 0.04 | .0371 | * |
| 17R‐RvD1 | 375 | 215 | 0.01 ± 0.03 | 0.09 ± 0.08 | .1662 | |
| RvD2 | 375 | 215 | 0.23 ± 0.75 | 0.19 ± 0.21 | .2053 | |
| RvD3 | 375 | 147 | – | – | – | |
| 17R‐RvD3 | 375 | 137 | – | – | – | |
| RvD4 | 375 | 225 | 0.23 ± 0.86 | 0.65 ± 0.35 | .3936 | |
| RvD5 | 359 | 141 | – | – | – | |
| RvD6 | 359 | 101 | 0.13 ± 0.39 | – | – | |
| PD1 | 359 | 153 | – | – | – | |
| 10S,17S‐diHDHA | 359 | 153 | – | – | – | |
| 22‐OH‐PD1 | 359 | 153 | – | – | – | |
| 17R‐PD1 | 359 | 153 | 0.09 ± 0.11 | 0.07 ± 0.06 | .0491 | * |
| PCTR1 | 650 | 231 | – | 0.34 ± 0.38 | – | |
| PCTR2 | 521 | 231 | 0.19 ± 0.25 | 0.30 ± 0.11 | .2133 | |
| PCTR3 | 464 | 231 | 0.52 ± 2.22 | 0.39 ± 0.27 | .2240 | |
| MCTR1 | 650 | 191 | – | 1.39 ± 1.29 | – | |
| MCTR2 | 521 | 191 | 0.03 ± 0.09 | 0.11 ± 0.07 | .2858 | |
| MCTR3 | 464 | 191 | 0.15 ± 0.54 | 1.75 ± 0.16 | .0003 | *** |
| MaR1 | 359 | 221 | 0.18 ± 0.64 | 1.08 ± 0.87 | .2276 | |
| 7S,14S‐diHDHA | 359 | 221 | – | – | – | |
| MaR2 | 359 | 221 | – | – | – | |
| 4S,14S‐diHDHA | 359 | 101 | – | – | – | |
| n−3 DPA bioactive metabolome | ||||||
| RvT1 | 377 | 193 | – | – | – | |
| RvT2 | 377 | 197 | – | – | – | |
| RvT3 | 377 | 197 | 0.02 ± 0.05 | – | – | |
| RvT4 | 361 | 193 | 0.12 ± 0.34 | 0.17 ± 0.09 | .2844 | |
| RvD1n−3 DPA | 377 | 215 | 0.05 ± 0.10 | 0.06 ± 0.03 | .1697 | |
| RvD2n−3 DPA | 377 | 261 | – | – | – | |
| RvD5n−3 DPA | 361 | 143 | 0.03 ± 0.11 | 0.04 ± 0.03 | .3143 | |
| PD1n−3 DPA | 361 | 183 | 0.03 ± 0.14 | 0.51 ± 0.56 | .2173 | |
| PD2n−3 DPA | 361 | 233 | – | – | – | |
| 10S, 17S‐diHDPA | 361 | 183 | – | – | – | |
| MaR1n−3 DPA | 361 | 223 | – | – | – | |
| MaR2n−3 DPA | 361 | 193 | – | – | – | |
| 7S,14S‐diHDPA | 361 | 223 | – | – | – | |
| EPA bioactive metabolome | ||||||
| RvE1 | 161 | – | – | – | ||
| RvE2 | 333 | 199 | – | – | – | |
| RvE3 | 333 | 201 | 0.29 ± 0.53 | 0.23 ± 0.19 | .0794 | |
| RvE4 | 333 | 115 | 0.79 ± 4.32 | 5.20 ± 3.04 | .1627 | |
| AA bioactive metabolome | ||||||
| LXA4 | 351 | 115 | 0.02 ± 0.05 | 0.06 ± 0.04 | .2502 | |
| LXB4 | 351 | 115 | 0.22 ± 0.58 | 0.34 ± 0.17 | .2974 | |
| 5S,15S‐diHETE | 335 | 235 | 0.52 ± 1.81 | 1.83 ± 1.01 | .2941 | |
| 15‐epi‐LXA4 | 351 | 115 | 0.06 ± 0.03 | 0.02 ± 0.02 | .0003 | *** |
| 15‐epi‐LXB4 | 351 | 115 | 0.29 ± 1.12 | 0.97 ± 0.56 | .3264 | |
| LTB4 | 335 | 195 | 4.80 ± 5.91 | 10.83 ± 1.69 | .4867 | |
| 5S,12S‐diHETE | 335 | 195 | – | – | – | |
| LTC4 | 626 | 189 | 0.47 ± 1.21 | 1.59 ± 0.40 | .1950 | |
| LTD4 | 497 | 189 | 0.22 ± 0.18 | 0.42 ± 0.07 | .2705 | |
| LTE4 | 440 | 189 | 0.72 ± 0.75 | 1.48 ± 0.12 | .3629 | |
| PGE2 | 351 | 189 | 0.02 ± 0.07 | 0.26 ± 0.08 | .0199 | * |
| PGD2 | 351 | 189 | 0.03 ± 0.05 | 0.24 ± 0.09 | .0376 | * |
| PGF2a | 353 | 193 | 0.06 ± 0.08 | 0.21 ± 0.06 | .1537 | |
| TxB2 | 369 | 169 | 0.22 ± 0.67 | 8.68 ± 2.88 | .0131 | * |
Serum was collected from healthy volunteers (n = 6) and patients with cardiovascular disease (n = 6), lipid mediators were extracted, identified and quantified using LC‐MS/MS‐based lipid mediator profiling (See methods for details). – = Below limits of quantification. Assuming non‐Gaussian distribution of the human samples, statistical analysis was determined using Mann–Whitney U test when comparing two groups. Data are presented as mean ± standard error of the mean. Statistical significance is indicated as *p < .05, ***p < .001.
FIGURE 3.
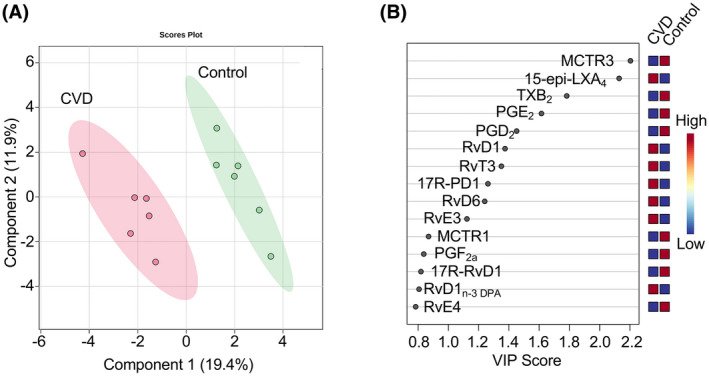
Serum lipid mediator concentrations are altered in patients with atherosclerosis versus healthy controls. Serum was collected from healthy volunteers (n = 6) and patients with cardiovascular disease (n = 6). Lipid mediators were extracted, identified and quantified using LC‐MS/MS‐based lipid mediator profiling (See methods for details). (A) Differences in lipid mediator concentrations were evaluated using PLS‐DA two‐dimensional scores plot, and (B) corresponding VIP plot of serum lipid mediators. Shaded areas represent 95% confidence interval
3.4. Lipoxin treatment significantly downregulates neutrophil CD11b expression in patients with atherosclerosis
The expression of the high‐affinity state of the CD11b integrin is closely linked to the pathophysiology of atherosclerosis as it regulates chemotaxis of neutrophils and neutrophil‐platelet aggregation. 10 , 45 We measured the expression of the CD11b high‐affinity conformation on neutrophils and whether this could be altered by lipoxin treatment. As expected, all neutrophils were 100% positive for the CD11b high‐affinity conformation integrin receptor, and the MFI increased in response to fMLP stimulation on the neutrophils from both healthy controls and patients with atherosclerosis (p = .008 for all) (Figure 4A). Lipoxin treatment did not alter the receptor expression at baseline in either group. The fMLP‐induced increase of CD11b expression significantly decreased in atherosclerosis patient neutrophils in response to both LXA4 and LXB4 (p = .045 and p < .001, respectively) (Figure 4B).
FIGURE 4.
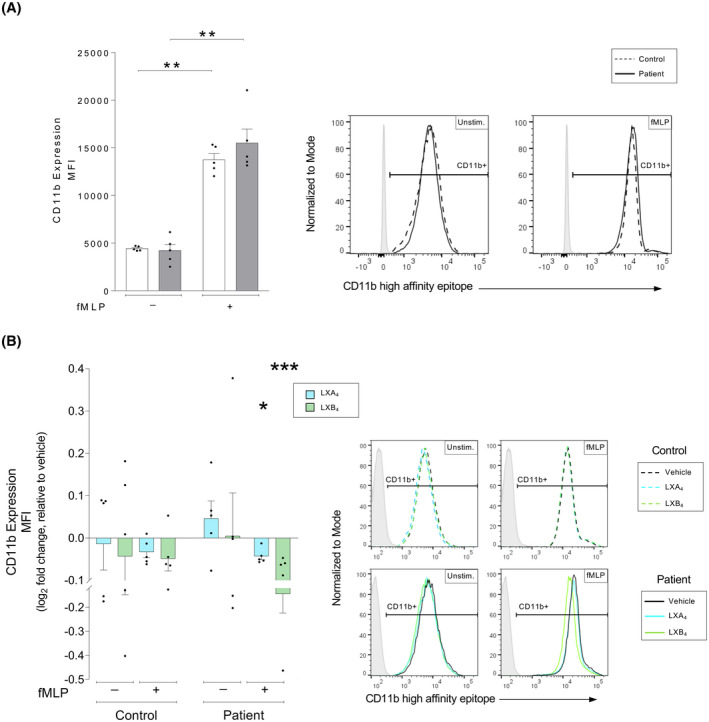
Lipoxin‐mediated changes to the high‐affinity conformation of the CD11b receptor in human neutrophils from patients with atherosclerosis versus healthy controls. Whole blood from healthy controls (n = 5) or patients with atherosclerosis (n = 5) was exposed to inflammatory stimulus as indicated, either in the absence or presence of lipoxin A4 (LXA4: 500 nM) or lipoxin B4 (LXB4: 500 nM). Neutrophil expression of the CD11b high‐affinity conformation was measured by flow cytometry. (A) Neutrophil expression of the CD11b high‐affinity conformation was measured as the cellular mean fluorescence intensity (MFI). The expression was measured in controls (white bars) and patients (gray bars). The cells were untreated (Unstim.) or stimulated with chemotactic peptide N‐formyl‐Met‐Leu‐Phe (fMLP, 0.4 μM). Representative MFI histograms for CD11b expression and respective conditions are shown for controls (dashed line) and patients (solid line), where the gates were determined using a negative population (gray shaded peaks). (B) LXA4 (blue bars) and LXB4 (green greens)‐induced changes to the neutrophil expression of the CD11b high‐affinity conformation was calculated as the log2 fold change relative to respective vehicle‐treated condition. The samples were stimulated as indicated. The bar graphs show levels of cellular CD11b MFI. Representative histograms for the expression of CD11b and respective conditions are shown for vehicle (black line), LXA4 (blue line), and LXB4 (green line), where the gates were determined using a negative population (gray shaded peaks). Assuming non‐Gaussian distribution of the human samples, statistical analysis was determined using Mann–Whitney U test when comparing two groups, and Kruskal–Wallis test with Dunn's post hoc comparisons when comparing more than two groups. Data are presented as mean ± standard error of the mean (SEM). Statistical significance is indicated as *p < .05, **p < .01, ***p < .001
3.5. Lipoxin B4 treatment enhances lymphatic transmigration of neutrophils from patients with atherosclerosis
Leukocyte egression from the plaques plays a critical role in atherosclerosis. 46 , 47 Thus, we quantified the ability of neutrophils, isolated from either healthy controls or patients, to migrate across a monolayer of LECs in the presence or absence of a chemoattractant (IL‐8: 5 ng/ml) (schematic illustration, Figure 5A).
FIGURE 5.
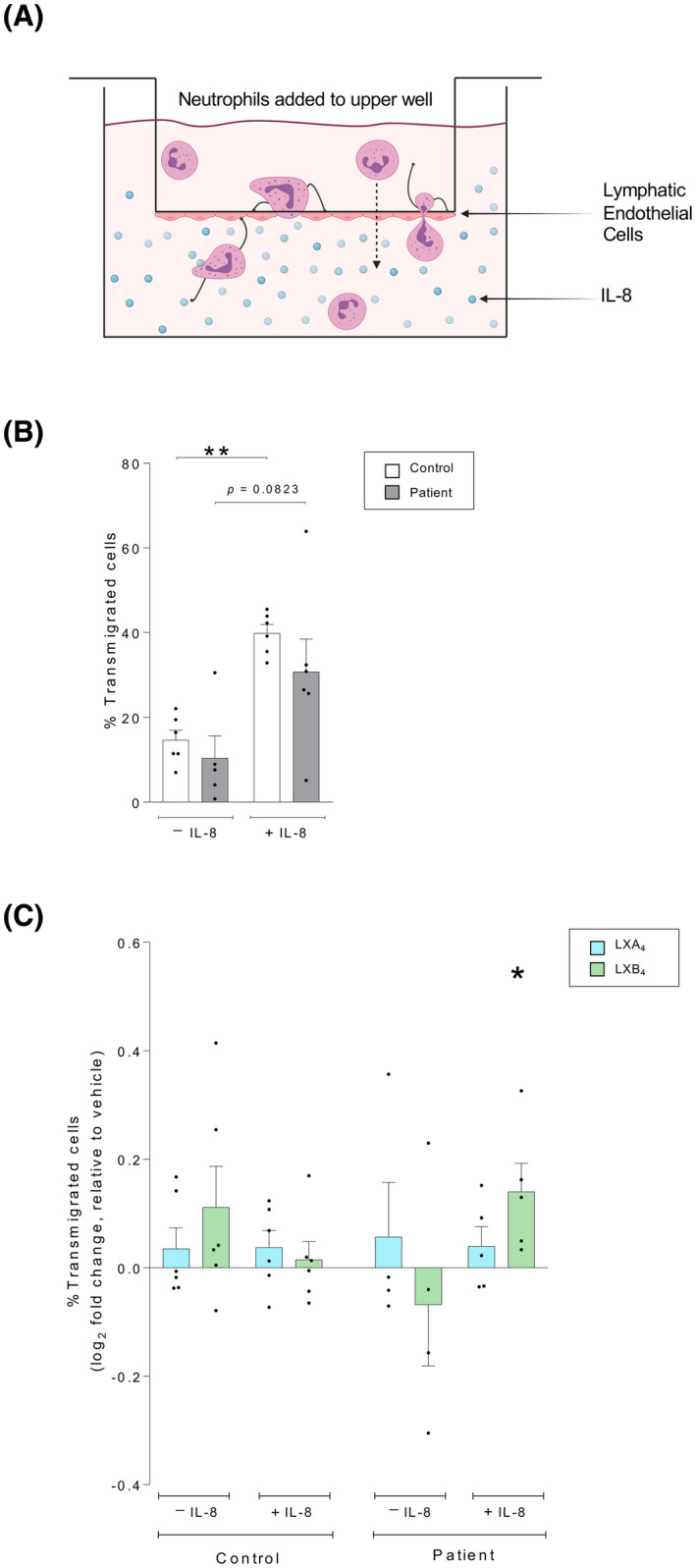
Lipoxin B4 enhances the lymphatic transmigration of neutrophils from patients with atherosclerosis but does not affect neutrophils from healthy controls. Neutrophils were isolated from healthy controls or patients with atherosclerosis and added to the upper well of a transwell with a confluent lymphatic endothelial cell monolayer. The neutrophils transmigrated for three hours, after which the cells in the lower well were counted to determine the migration rate. The migration experiment was conducted using either vehicle or the chemoattractant interleukin (IL)‐8 (5 ng/ml) in the lower well. (A) A schematic illustration of the transmigration assay is shown. (B) The percentage of neutrophils that migrated across lymphatic endothelial cell monolayer were quantified, either spontaneously or in response to the chemoattractant IL‐8. The migration pattern was investigated in neutrophils isolated from healthy controls (n = 6, white bars) and patients with atherosclerosis (n = 6, gray bars). (C) Neutrophils were treated with vehicle (0.01% ethanol), lipoxin A4 (LXA4:1 nM, blue bars), or lipoxin B4 (LXB4: 1 nM, green bars) for 15 minutes prior to the transmigration assay. Lipoxin‐induced changes to the neutrophil migration rate were calculated as the log2 fold change relative to respective vehicle‐treated condition. Assuming non‐Gaussian distribution of the human samples, statistical analysis was determined using Mann–Whitney U test when comparing two groups, and Kruskal–Wallis test with Dunn's post hoc comparisons when comparing over two groups. Data are presented as mean ± standard error of the mean (SEM). Statistical significance is indicated as *p < .05, **p < .01
There was no difference in spontaneous transmigration of neutrophils from controls or patients (Figure 5B). In healthy controls, IL‐8 significantly enhanced the rate of neutrophil migration across the LEC monolayer (p = .002). Neutrophils isolated from patients with atherosclerosis tended to migrate to a lesser extent than healthy controls, although there was not a significant difference between the groups (Figure 5B).
To investigate if SPMs could modulate the lymphatic transmigration, isolated neutrophils were treated with vehicle (0.01% ethanol) or lipoxins (1 nM) for 15 min prior to the migration assay. The lipoxins did not affect the migration of neutrophils isolated from healthy controls (Figure 5C). However, neutrophils isolated from patients with atherosclerosis responded to LXB4 treatment with a significantly increased transmigration across the lymphatic monolayer (p = .0235) (Figure 5C).
4. DISCUSSION
Mortality related to cardiovascular disease accounted for a third of deaths globally (18.5 million lives) in 2019, indicating the pressing need for effective therapeutics. 48 It is well established that inflammation fuels the development of atherosclerosis. 2 , 3 , 4 , 49 , 50 , 51 Thus, attenuating inflammation has emerged as a promising anti‐atherogenic therapeutic strategy, 3 in addition to standard clinical care with lipid‐lowering medication. For instance, the Canakinumab Anti‐inflammatory Thrombosis Outcome Study, CANTOS, presented evidence that attenuating the levels of the pro‐inflammatory cytokine IL‐1β reduced nonfatal myocardial infarction, nonfatal stroke, and cardiovascular death; an effect independent of the patients’ LDL/HDL levels. 52 , 53 However, the Canakinumab‐mediated effect varied significantly between subgroups, and furthermore, these anti‐inflammatory drugs may increase susceptibility to infections. 52 , 53 An alternative approach to attenuate chronic pro‐atherogenic inflammation is to enhance endogenous pathways that promote the resolution of inflammation. 3 , 4 , 54 , 55 The strategy of boosting resolution through administration of exogenous SPMs has been shown to reduce the risk of cardiovascular disease in experimental models, 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 and some evidence suggests that these findings may translate to humans. 10 , 28 Herein, we demonstrate the potent anti‐atherogenic effects of lipoxins on human neutrophils from patients with atherosclerosis, which differs from the effects in healthy controls.
Neutrophil ROS production plays a critical role in the antibacterial host defense, 35 , 44 although an uncontrolled continuous oxidative burst response can be detrimental. We demonstrate that neutrophils isolated from patients with atherosclerosis produce significantly more ROS than cells from healthy controls, both at baseline and on stimulation with fMLP or E. coli. This is likely due to these patients’ elevated C‐reactive protein levels, which may prime the respiratory burst response and sustain inflammation. 35 In patients with atherosclerosis, we also demonstrated that treating whole blood with lipoxins reduced the excessive ROS production elicited by the inflammatory triggers. In support of these findings, we have previously demonstrated that LXA4 treatment reduces Porphyromonas gingivalis–induced leukocyte ROS production in human whole blood. 10 Reduced ROS production after LXA4 treatment has also been observed in other cell types following various inflammatory challenges in vitro, for example, in endothelial, 56 , 57 pancreatic, 58 and microglia 59 cell lines. As elevated ROS production may promote leukocyte infiltration, plaque development, and plaque rupture, 33 , 60 an SPM‐mediated attenuation may benefit patients with atherosclerosis. Our data concomitantly show that lipoxin treatment enhanced fMLP‐ and E. coli–induced ROS production from neutrophils of healthy controls, with LXB4 having a more significant effect. The lipoxin‐induced manipulation of E. coli–induced ROS in healthy controls was not related to an increased phagocytosis rate. Published data on the effect of SPMs in healthy controls are sparse. However, one study demonstrated that LXA4 treatment of healthy Wistar rat aorta led to an increase in ROS production ex vivo. 61 Thus, it is plausible that lipoxins can upregulate ROS production in healthy individuals, which may be beneficial given that an efficient, but controlled, oxidative burst response is required by the immune system. In conclusion, we found that neutrophils from patients with atherosclerosis displayed an excessive oxidative burst response when activated, compared with neutrophils from healthy controls. Our findings demonstrate that the lipoxin‐mediated effect on ROS production differs in health versus disease. Lipoxins only attenuate the oxidative burst response if cells are derived from an inflammatory environment, for instance, in the patients with atherosclerosis who have elevated systemic inflammation. This is important to consider when pursuing the therapeutic application of SPMs, as their effect may differ depending on the inflammatory status of the individual patient.
SPMs exert their biological effects through the action of G‐coupled protein receptors, for example, ALX/FPR2, ERV1/ChemR23, DRV1/GPR32, and DRV2/GPR18, 5 , 13 , 15 , 62 although the LXB4 receptor remains to be identified. Characterizing the expression of these receptors in health and disease may provide important mechanistic clues of inflammatory resolution, and these receptors may also represent therapeutic targets. However, it is noteworthy that pro‐inflammatory ligands also activate some of these receptors, and conflicting findings regarding the regulation of these receptors in human atherosclerosis have been reported. 55 We focused our investigations on the expression of the main LXA4 receptor FPR2/ALX. 9 FPR2/ALX is a pluripotent receptor that recognizes both pro‐resolving (e.g., LXA4 and annexin A1) and pro‐inflammatory (e.g., fMLP) signals and elicits different responses depending on the ligand and the subsequent conformational change of the receptor. 63 , 64 These effects include altered leukocyte chemotaxis, cytokine production, and efferocytosis. 65 , 66 , 67 In mouse models, FPR2/ALX receptor knockouts are characterized by delayed atherosclerosis development. 19 In humans, FPR2/ALX expression is increased in atherosclerotic plaques. 19 We observed no difference in the neutrophil FPR2/ALX expression in either group at baseline. Neutrophil FPR2/ALX expression was reduced by fMLP in both groups. As a measurement of endogenous resolution we also quantified serum levels of SPMs in healthy controls and patients with atherosclerosis. MCTR3 and 15‐epi LXA4 were identified as the main mediators that drive the separation observed between healthy controls and patients with atherosclerosis. MCTR3 is a peptide–lipid‐conjugated mediator previously shown to have potent pro‐resolving properties, for instance upregulating human neutrophil and macrophage phagocytic responses. 68 Thus, the fact that the patient cohort has reduced levels of the pro‐resolving factor MCTR3 may indicate reduced resolution capability of this group. The elevated levels of 15‐epi LXA4 observed in the patient cohort may be explained by the fact that these individuals were treated with low‐dose aspirin and statins, as these drugs induce the formation of epi‐lipoxins. 69 , 70 , 71 We would like to point out that the concentrations of this mediator are in line with concentrations observed for many other lipid mediators in these samples. Lipid mediator concentrations in samples are dependent on a number of factors that include substrate availability, activation status of the cells carrying the biosynthetic enzymes as well as their expression levels and activities, and the expression and activity of enzymes involved in their further metabolism. Our analysis only offers a snapshot of this very dynamic process. Nonetheless, concentrations of both 15‐epi LXA4 and those of another aspirin‐triggered SPM, 17R‐PD1, were both increased in samples from patients with atherosclerosis, in line with the known role of aspirin to upregulate these autacoids. In summary, we did not find any evidence indicating that neutrophil surface expression of the lipoxin receptor FPR2/ALX was altered in the atherosclerotic cohort, but lipid mediator profiling analyses revealed that the endogenous resolution capacity may be altered in atherosclerosis.
The integrin complex CD11b/CD18 is expressed by many leukocyte subsets and is important in several biological processes related to atherosclerosis. 10 In addition to regulating leukocyte transendothelial migration, CD11b/CD18 mediates platelet–leukocyte aggregation and associated ROS production by binding to the platelet GPIIb/IIIa receptor via fibrinogen. 10 Previously, we have demonstrated that lipoxins can reduce P. gingivalis–induced thrombotic events in human blood by manipulating the neutrophil expression of both the total CD11b/CD18 integrin complex and the upregulation of its high‐affinity conformation. 10 In this study, we found no differences in the CD11b high‐affinity conformation expression comparing controls with patients in response to inflammatory stimuli. However, LXA4 and LXB4 treatment reduced the fMLP‐induced upregulation of the CD11b high‐affinity conformation in patients with atherosclerosis, although not in healthy controls. This again highlights the possibility that lipoxins exert their actions in a manner tailored to the needs of the individual.
Lymphatic dysfunction precedes atherosclerosis, and stimulation of lymphangiogenesis limits plaque formation and improves inflammatory cell migration through the lymphatics. 72 The lymphatics play an important role in reverse cholesterol transport, macromolecule clearance, and immune cell egression in atherosclerosis. 73 , 74 Thus, we investigated the migratory capacity of neutrophils from patients and healthy controls in an ex vivo transmigration assay with human LECs. We demonstrated that neutrophils from patients with atherosclerosis moved at a slower rate across lymphatic endothelium than controls. LXB4 treatment significantly increased the lymphatic neutrophil transmigration from patients with atherosclerosis, but lipoxins did not affect the migration of neutrophils from controls. Future studies are needed to determine the mechanism of how LXB4 restoration of lymphatic transmigration in neutrophils from patients is mediated. We hypothesize that the role of LXB4 on neutrophil migration may be of high relevance in atherosclerosis.
In conclusion, we have evaluated the effects of lipoxins on the function of neutrophils isolated from patients with atherosclerosis compared with neutrophils from healthy controls. Patients demonstrated altered endogenous SPM production and lipoxins attenuated neutrophil oxidative burst, a key driver of the atherosclerotic process, in the patient cohort. Importantly, this effect was restricted to neutrophils from patients with atherosclerosis. Conversely, in neutrophils from healthy controls, lipoxins increased the oxidative burst, indicating that the lipoxin effect depends on the inflammatory status of the individual. In neutrophils from patients with atherosclerosis, lipoxins also reduced upregulation of the high‐affinity conformation of the CD11b integrin, which has a central role in clot activation. Finally, LXB4 enhanced the lymphatic transmigration of human neutrophils isolated from atherosclerosis patients across LECs ex vivo, which may translate to a pro‐resolving effect of enhanced lymphatic egression. Although both lipoxins modulated neutrophil function in most assays investigated, LXB4 displayed more potent effects than LXA4. This study highlights the therapeutic potential of lipoxins in atherosclerosis and demonstrates that the pro‐resolving effect of lipoxins is related to the individual's inflammatory status.
DISCLOSURES
All the authors declare that they have no competing interests.
AUTHOR CONTRIBUTIONS
Emma Börgeson, Jamie D. Kraft, Robert Blomgran, Ida Bergström, Sofia Nyström, and Per Skoog were involved in conception and study design. Jamie D. Kraft, Robert Blomgran, Ida Bergström, Matúš Soták, Madison Clark, and Jesmond Dalli performed the experiments and data analysis. Per Skoog was medically responsible for the clinical recruitment of patients, and the patient samples were collected and processed by Jamie D. Kraft, Alankrita Rani, Meenu Rohini Rajan, Matúš Soták, and Catherine Åhlund. All authors contributed to the writing and editing of the manuscript and the interpretation of data.
Supporting information
Fig S1
Fig S2
Fig S3
Text S1
ACKNOWLEDGMENTS
We gratefully acknowledge the contribution of Catherine Åhlund, who was responsible for organizing the collection of clinical material. The research was funded by the Wallenberg Centre for Molecular & Translational Medicine at University of Gothenburg and Knut & Alice Wallenberg Foundation, the Swedish Research Council (#2016/82), SSMF (#S150086), and an ERC StG (#804418) and grant support from NIH (#R37AI062765#).
Kraft JD, Blomgran R, Bergström I, et al. Lipoxins modulate neutrophil oxidative burst, integrin expression and lymphatic transmigration differentially in human health and atherosclerosis. FASEB J. 2022;36:e22173. doi: 10.1096/fj.202101219RR
Data Availability Statement
All raw data (lipdomics, flow cytometry, transmigration and clinical characteristics data) that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1. Bäck M, Yurdagul A, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat Rev Cardiol. 2019;16:389‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Libby P, Ridker PM, Maseri A. Inflammation and atherosclerosis. Circulation. 2002;105:1135‐1143. [DOI] [PubMed] [Google Scholar]
- 3. Fredman G, Tabas I. Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol. 2017;187:1211‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kasikara C, Doran AC, Cai B, Tabas I. The role of non‐resolving inflammation in atherosclerosis. J Clin Invest. 2018;128:2713‐2723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Serhan CN. Pro‐resolving lipid mediators are leads for resolution physiology. Nature. 2014;510:92‐101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Titos E, Claria J. Omega‐3‐derived mediators counteract obesity‐induced adipose tissue inflammation. Prostaglandins Other Lipid Mediat. 2013;107:77‐84. [DOI] [PubMed] [Google Scholar]
- 7. Vasconcelos DP, Costa M, Amaral IF, Barbosa MA, Aguas AP, Barbosa JN. Modulation of the inflammatory response to chitosan through M2 macrophage polarization using pro‐resolution mediators. Biomaterials. 2015;37:116‐123. [DOI] [PubMed] [Google Scholar]
- 8. Tabas I, Glass CK. Anti‐inflammatory therapy in chronic disease: challenges and opportunities. Science. 2013;339:166‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chandrasekharan JA, Sharma‐Walia N. Lipoxins: nature's way to resolve inflammation. J Inflamm Res. 2015;8:181‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Borgeson E, Lonn J, Bergstrom I, et al. Lipoxin A(4) inhibits porphyromonas gingivalis‐induced aggregation and reactive oxygen species production by modulating neutrophil‐platelet interaction and CD11b expression. Infect Immun. 2011;79:1489‐1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Borgeson E, Johnson AM, Lee YS, et al. Lipoxin A4 attenuates obesity‐induced adipose inflammation and associated liver and kidney disease. Cell Metab. 2015;22:125‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Serhan CN. Discovery of specialized pro‐resolving mediators marks the dawn of resolution physiology and pharmacology. Mol Aspects Med. 2017;58:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Serhan CN, Chiang N, Dalli J, Levy BD. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol. 2014;7:a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Headland SE, Norling LV. The resolution of inflammation: Principles and challenges. Semin Immunol. 2015;27:149‐160. [DOI] [PubMed] [Google Scholar]
- 15. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. 2017;31:1273‐1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Börgeson E, McGillicuddy FC, Harford KA, et al. Lipoxin A4 attenuates adipose inflammation. FASEB J. 2012;26:4287‐4294. [DOI] [PubMed] [Google Scholar]
- 17. Borgeson E, Docherty NG, Murphy M, et al. Lipoxin A(4) and benzo‐lipoxin A(4) attenuate experimental renal fibrosis. FASEB J. 2011;25:2967‐2979. [DOI] [PubMed] [Google Scholar]
- 18. Fredman G, Hellmann J, Proto JD, et al. An imbalance between specialized pro‐resolving lipid mediators and pro‐inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. 2016;7:12859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Petri MH, Laguna‐Fernandez A, Gonzalez‐Diez M, Paulsson‐Berne G, Hansson GK, Back M. The role of the FPR2/ALX receptor in atherosclerosis development and plaque stability. Cardiovasc Res. 2015;105:65‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Petri MH, Laguna‐Fernandez A, Arnardottir H, et al. Aspirin‐triggered lipoxin A4 inhibits atherosclerosis progression in apolipoprotein E−/− mice. Br J Pharmacol. 2017;174:4043‐4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petri MH, Laguna‐Fernandez A, Tseng C‐N, Hedin U, Perretti M, Bäck M. Aspirin‐triggered 15‐epi‐lipoxin A₄ signals through FPR2/ALX in vascular smooth muscle cells and protects against intimal hyperplasia after carotid ligation. Int J Cardiol. 2015;179:370‐372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fredman G, Kamaly N, Spolitu S, et al. Targeted nanoparticles containing the proresolving peptide Ac2‐26 protect against advanced atherosclerosis in hypercholesterolemic mice. Sci Transl Med. 2015;7:220‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Drechsler M, Jong R, Rossaint J, et al. Annexin A1 counteracts chemokine‐induced arterial myeloid cell recruitment. Circ Res. 2015;116:827‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Viola JR, Lemnitzer P, Jansen Y, et al. Resolving lipid mediators Maresin 1 and Resolvin D2 prevent atheroprogression in mice. Circ Res. 2016;119:1030‐1038. [DOI] [PubMed] [Google Scholar]
- 25. Hasturk H, Abdallah R, Kantarci A, et al. Resolvin E1 (RvE1) attenuates atherosclerotic plaque formation in diet and inflammation‐induced atherogenesis. Arterioscler Thromb Vasc Biol. 2015;35:1123‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salic K, Morrison MC, Verschuren L, et al. Resolvin E1 attenuates atherosclerosis in absence of cholesterol‐lowering effects and on top of atorvastatin. Atherosclerosis. 2016;250:158‐165. [DOI] [PubMed] [Google Scholar]
- 27. Liu G, Gong Y, Zhang R, et al. Resolvin E1 attenuates injury‐induced vascular neointimal formation by inhibition of inflammatory responses and vascular smooth muscle cell migration. FASEB J. 2018;32:5413‐5425. [DOI] [PubMed] [Google Scholar]
- 28. Brennan EP, Mohan M, McClelland A, et al. Lipoxins protect against inflammation in diabetes‐associated atherosclerosis. Diabetes. 2018;67:2657‐2667. [DOI] [PubMed] [Google Scholar]
- 29. Thul S, Labat C, Temmar M, Benetos A, Bäck M. Low salivary resolvin D1 to leukotriene B4 ratio predicts carotid intima media thickness: A novel biomarker of non‐resolving vascular inflammation. Eur J Prev Cardiol. 2020;24:903‐906. [DOI] [PubMed] [Google Scholar]
- 30. Ionita MG, van den Borne P, Catanzariti LM, et al. High neutrophil numbers in human carotid atherosclerotic plaques are associated with characteristics of rupture‐prone lesions. Arterioscler Thromb Vasc Biol. 2010;30:1842‐1848. [DOI] [PubMed] [Google Scholar]
- 31. Drechsler M, Döring Y, Megens RT, Soehnlein O. Neutrophilic granulocytes ‐ promiscuous accelerators of atherosclerosis. Thromb Haemost. 2011;106:839‐848. [DOI] [PubMed] [Google Scholar]
- 32. Silvestre‐Roig C, Braster Q, Ortega‐Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. 2020;17:327‐340. [DOI] [PubMed] [Google Scholar]
- 33. Döring Y, Drechsler M, Soehnlein O, Weber C. Neutrophils in atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:288‐295. [DOI] [PubMed] [Google Scholar]
- 34. Cochain C, Ait‐Oufella H, Zernecke A. Neutrophils promote atherosclerotic plaque destabilization in a mouse model of endotoxinaemia. Cardiovasc Res. 2018;114:1573‐1574. [DOI] [PubMed] [Google Scholar]
- 35. Chistiakov DA, Bobryshev YV, Orekhov AN. Neutrophil's weapons in atherosclerosis. Exp Mol Pathol. 2015;99:663‐671. [DOI] [PubMed] [Google Scholar]
- 36. Fülöp T, Larbi A, Witkowski JM. Human inflammaging. Gerontology. 2019;65:495‐504. [DOI] [PubMed] [Google Scholar]
- 37. Rajan MR, Sotak M, Barrenas F, et al. Comparative analysis of obesity‐related cardiometabolic and renal biomarkers in human plasma and serum. Sci Rep. 2019;9:15385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Xiong Y, Brinkman CC, Famulski KS, et al. A robust in vitro model for trans‐lymphatic endothelial migration. Sci Rep. 2017;7:1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gomez EA, Colas RA, Souza PR, et al. Blood pro‐resolving mediators are linked with synovial pathology and are predictive of DMARD responsiveness in rheumatoid arthritis. Nat Commun. 2020;11:5420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Koenis DS, Beegun I, Jouvene CC, et al. Disrupted resolution mechanisms favor altered phagocyte responses in COVID‐19. Circ Res. 2021;129:e54‐e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gilroy DW. The role of aspirin‐triggered lipoxins in the mechanism of action of aspirin. Prostaglandins Leukot Essent Fatty Acids. 2005;73:203‐210. [DOI] [PubMed] [Google Scholar]
- 42. Elajami TK, Colas RA, Dalli J, Chiang N, Serhan CN, Welty FK. Specialized proresolving lipid mediators in patients with coronary artery disease and their potential for clot remodeling. FASEB J. 2016;30:2792‐2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Birnbaum Y, Ye Y, Lin Y, et al. Augmentation of myocardial production of 15‐epi‐lipoxin‐A4 by pioglitazone and atorvastatin in the rat. Circulation. 2006;114:929‐935. [DOI] [PubMed] [Google Scholar]
- 44. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect Microbiol. 2017;7:373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Särndahl E, Bergström I, Brodin VP, Nijm J, Setterud HL, Jonasson L. Neutrophil activation status in stable coronary artery disease. PLoS One. 2007;2:e1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rademakers T, van der Vorst EPC, Daissormont ITMN, et al. Adventitial lymphatic capillary expansion impacts on plaque T cell accumulation in atherosclerosis. Sci Rep. 2017;7:45263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yeo KP, Lim HY, Angeli V. Leukocyte trafficking via lymphatic vessels in atherosclerosis. Cells. 2021;10:1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kauffman D. Cardiovascular disease burden, deaths are rising around the world. American College of Cardiology; 2020. [Google Scholar]
- 49. Geovanini GR, Libby P. Atherosclerosis and inflammation: overview and updates. Clin Sci (Lond). 2018;132:1243‐1252. [DOI] [PubMed] [Google Scholar]
- 50. Fava C, Montagnana M. Atherosclerosis is an inflammatory disease which lacks a common anti‐inflammatory therapy: How human genetics can help to this issue. A narrative review. Front Pharmacol. 2018;9:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruparelia N, Chai JT, Fisher EA, Choudhury RP. Inflammatory processes in cardiovascular disease: a route to targeted therapies. Nat Rev Cardiol. 2017;14:133‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ridker PM, Everett BM, Thuren T, et al. Antiinflammatory therapy with Canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119‐1131. [DOI] [PubMed] [Google Scholar]
- 53. Husten L. Experts caution on CANTOS and Canakinumab's future. MedPage Today; 2017. [Google Scholar]
- 54. Conte MS, Desai TA, Wu B, Schaller M, Werlin E. Pro‐resolving lipid mediators in vascular disease. J Clin Invest. 2018;128:3727‐3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Pirault J, Bäck M. Lipoxin and resolvin receptors transducing the resolution of inflammation in cardiovascular disease. Front Pharmacol. 2018;9:1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nascimento‐Silva V, Arruda MA, Barja‐Fidalgo C, Fierro IM. Aspirin‐triggered lipoxin A4 blocks reactive oxygen species generation in endothelial cells: a novel antioxidative mechanism. Thromb Haemost. 2007;97:88‐98. [PubMed] [Google Scholar]
- 57. Zhou Y, You H, Zhang A, et al. Lipoxin A4 attenuates uric acid‐activated, NADPH oxidase‐dependent oxidative stress by interfering with translocation of p47phox in human umbilical vein endothelial cells. Exp. Ther. Med. 2020;20:1682‐1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Zong L, Li J, Chen X, et al. Lipoxin A4 attenuates cell invasion by inhibiting ROS/ERK/MMP pathway in pancreatic cancer. Oxid Med Cell Longev. 2016;2016:6815727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wu Y, Zhai H, Wang Y, et al. Aspirin‐triggered lipoxin A4 attenuates lipopolysaccharide‐induced intracellular ROS in BV2 microglia cells by inhibiting the function of NADPH oxidase. Neurochem Res. 2012;37:1690‐1696. [DOI] [PubMed] [Google Scholar]
- 60. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875‐888. [DOI] [PubMed] [Google Scholar]
- 61. Wenceslau CF, McCarthy CG, Szasz T, Webb RC. Lipoxin A4 mediates aortic contraction via RHOA/RHO kinase, endothelial dysfunction and reactive oxygen species. J Vasc Res. 2014;51:407‐417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Basil MC, Levy BD. Specialized pro‐resolving mediators: endogenous regulators of infection and inflammation. Nat Rev Immunol. 2016;16:51‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gavins FNE. Are formyl peptide receptors novel targets for therapeutic intervention in ischaemia–reperfusion injury? Trends Pharmacol Sci. 2010;31:266‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cooray SN, Gobbetti T, Montero‐Melendez T, et al. Ligand‐specific conformational change of the G‐protein‐coupled receptor ALX/FPR2 determines proresolving functional responses. Proc Natl Acad Sci U S A. 2013;110:18232‐18237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Petri MH, Thul S, Andonova T, et al. Resolution of Inflammation through the lipoxin and ALX/FPR2 receptor pathway protects against abdominal aortic aneurysms. JACC Basic Transl Sci. 2018;3:719‐727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Maderna P, Cottell DC, Toivonen T, et al. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin‐derived peptide‐stimulated phagocytosis. FASEB J. 2010;24:4240‐4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Perretti M, Godson C. Formyl peptide receptor type 2 agonists to kick‐start resolution pharmacology. Br J Pharmacol. 2020;177:4595‐4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dalli J, Sanger JM, Rodriguez AR, Chiang N, Spur BW, Serhan CN. Identification and actions of a novel third maresin conjugate in tissue regeneration: MCTR3. PLoS One. 2016;11:e0149319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Levy BD. Myocardial 15‐epi‐lipoxin A4 generation provides a new mechanism for the immunomodulatory effects of statins and thiazolidinediones. Circulation. 2006;114:873‐875. [DOI] [PubMed] [Google Scholar]
- 70. Zhou G, Ge S, Liu D, et al. Atorvastatin reduces plaque vulnerability in an atherosclerotic rabbit model by altering the 5‐lipoxygenase pathway. Cardiology. 2010;115:221‐228. [DOI] [PubMed] [Google Scholar]
- 71. Serhan CN. Lipoxins and aspirin‐triggered 15‐epi‐lipoxins are the first lipid mediators of endogenous anti‐inflammation and resolution. Prostaglandins Leukot Essent Fatty Acids. 2005;73:141‐162. [DOI] [PubMed] [Google Scholar]
- 72. Milasan A, Smaani A, Martel C. Early rescue of lymphatic function limits atherosclerosis progression in Ldlr(‐/‐) mice. Atherosclerosis. 2019;283:106‐119. [DOI] [PubMed] [Google Scholar]
- 73. Llodrá J, Angeli V, Liu J, Trogan E, Fisher EA, Randolph GJ. Emigration of monocyte‐derived cells from atherosclerotic lesions characterizes regressive, but not progressive, plaques. Proc Natl Acad Sci U S A. 2004;101:11779‐11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kraft JD, Blomgran R, Lundgaard I, Quiding‐Järbrink M, Bromberg JS, Börgeson E. Specialized pro‐resolving mediators and the lymphatic system. Int J Mol Sci. 2021;22:2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Text S1
Data Availability Statement
All raw data (lipdomics, flow cytometry, transmigration and clinical characteristics data) that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.