

Abstract
A key pathological process in Parkinson's disease (PD) is the transneuronal spreading of α‐synuclein. Alpha‐synuclein (α‐syn) is a presynaptic protein that, in PD, forms pathological inclusions. Other hallmarks of PD include neurodegeneration and microgliosis in susceptible brain regions. Whether it is primarily transneuronal spreading of α‐syn particles, inclusion formation, or other mechanisms, such as inflammation, that cause neurodegeneration in PD is unclear. We used a model of spreading of α‐syn induced by striatal injection of α‐syn preformed fibrils into the mouse striatum to address this question. We performed quantitative analysis for α‐syn inclusions, neurodegeneration, and microgliosis in different brain regions, and generated gene expression profiles of the ventral midbrain, at two different timepoints after disease induction. We observed significant neurodegeneration and microgliosis in brain regions not only with, but also without α‐syn inclusions. We also observed prominent microgliosis in injured brain regions that did not correlate with neurodegeneration nor with inclusion load. Using longitudinal gene expression profiling, we observed early gene expression changes, linked to neuroinflammation, that preceded neurodegeneration, indicating an active role of microglia in this process. Altered gene pathways overlapped with those typical of PD. Our observations indicate that α‐syn inclusion formation is not the major driver in the early phases of PD‐like neurodegeneration, but that microglia, activated by diffusible, oligomeric α‐syn, may play a key role in this process. Our findings uncover new features of α‐syn induced pathologies, in particular microgliosis, and point to the necessity for a broader view of the process of α‐syn spreading.
Keywords: alpha‐synuclein spreading, disease pathways, microgliosis, neurodegeneration, neuroinflammation, Parkinson's disease, transcriptional profiling, translational
Main Points
Striatal injection of synuclein fibrils in mice to model PD.
No correlation between inclusions, neurodegeneration, microgliosis.
Transcriptomics reveals microglia response preceding neurodegeneration.
Molecular pathways overlap with those of PD.
Abbreviations
- 13 dpi
13 days post‐injection
- 6‐OHDA
6‐hydroxydopamine
- 90 dpi
90 days post‐injection
- AD
Alzheimer's disease
- Aβ
amyloid beta peptide
- BP
biological process
- CD68
cluster of differentiation 68
- CNS
central nervous system
- Contra
contralateral
- COX2
cyclooxygenase 2
- DA
dopamine
- DAM
disease‐associated microglia
- DAT
dopamine transporter
- DEG
differentially expressed gene
- ES
enrichment score
- FDR
false discovery rate
- GO
gene ontology
- GSEA
gene set enrichment analysis
- Iba1
ionized calcium binding adaptor molecule
- Ipsi
ipsilateral
- PBS
phosphate‐buffered saline
- PET
paraffin‐embedded tissue blot
- PFF
pre‐formed fibril
- Pfp
percent false positives
- PLA
proximity ligation assay
- p‐SER129‐α‐syn
alpha‐synuclein phosphorylated at serine position 129
- SN
substantia nigra
- TDP43
TAR DNA‐binding protein 43
- TH
tyrosine‐hydroxylase
- TLR2
toll‐like receptor 2
- WB
Western blot
- α‐syn
alpha‐synuclein
1. INTRODUCTION
Protein misfolding and aggregation are central pathological processes in neurodegenerative diseases, where they are believed to play a key role in driving the pathology (Chiti & Dobson, 2017; Selkoe, 2003). Proteins such as the amyloid beta peptide (Aβ) and tau in Alzheimer's disease (AD), TAR DNA‐binding protein 43 (TDP43) in motor neuron disease, prion in Creutzfeldt‐Jakob disease, and finally alpha‐synuclein (α‐syn) in Parkinson's disease (PD), are all examples of physiologically occurring proteins that, upon pathological misfolding, form oligomers, fibrils, and extracellular (Aβ, prion) or intracellular (TDP43, tau, α‐syn) deposits, and injure neurons in the process (Goedert, 2015; Ross & Poirier, 2004; Scheckel & Aguzzi, 2018).
An important property of these disease‐associated proteins is their ability to self‐propagate, a process first described in prion diseases, in which disease‐associated misfolding proteins induce the disease when transposed into a susceptible recipient host (Scheckel & Aguzzi, 2018; Walker & Jucker, 2015). They do so by acting as a seed and corrupting the endogenous form of the protein, leading it to aggregate and form, over time, inclusions along interconnected neuronal pathways (Jucker & Walker, 2018; Mezias et al., 2020). The “spreading hypothesis” posits that misfolded/aggregated particles of a disease protein move trans‐synaptically from neuron to neuron, causing dysfunction and damage along the way (Goedert, 2015; Mezias et al., 2020). Major support for this hypothesis comes from two observations. First, the neuropathological studies by Braak and colleagues, staging tau inclusions in AD (Braak & Braak, 1995), and Lewy inclusions in PD (Braak et al., 2003), suggest a progression, starting first in a population of susceptible neurons, of proteinaceous intraneuronal inclusions, a process that takes place over decades. Second, postmortem studies of PD patients that had received striatal fetal neuron transplants to combat dopamine loss, revealed Lewy bodies in a subset of the grafted neurons, indicating a spreading of abnormal α‐syn from the diseased neurons of the recipient to those of the donor (Brundin et al., 2016; Chu & Kordower, 2010).
Alpha‐syn is a presynaptic protein that normally is involved in the regulation of the synaptic vesicle cycle (Bendor et al., 2013; Burre et al., 2018). Its involvement in PD was discovered when it was identified as an essential component of a PD pathological hallmark, the Lewy body (Spillantini et al., 1998), and when mutations in its gene, as well as dupli‐ or triplication thereof, were shown to lead to hereditary forms of the disease (Lin & Farrer, 2014; Singleton & Hardy, 2019). Its prion‐like spreading properties have been demonstrated in in vitro and in vivo model systems, using intracranial injection of Lewy‐body containing brain extracts, of viral‐construct mediated α‐syn overexpression, or administration of pre‐formed fibrils (PFFs) from recombinant α‐syn as seeds, to induce spreading and progressive aggregation (Chu et al., 2019) (Luk et al., 2012; Luna & Luk, 2015; Rey et al., 2016; Ulusoy et al., 2013).
The role of α‐syn spreading and inclusion formation in PD pathogenesis is still unclear, since no correlation between PD symptoms and α‐syn inclusion load was consistently found (Dijkstra et al., 2014; Espay & Marras, 2019; Jellinger, 2009a, 2009b). Different possibilities that could explain what ultimately causes neuronal dysfunction and injury and, hence, neurological symptoms, have surfaced (Poewe et al., 2017; Surmeier et al., 2017b). Oligomers, rather than deposited forms of α‐syn, may be more neurotoxic that deposited forms (Bengoa‐Vergniory et al., 2017; Walsh & Selkoe, 2004). While some studies report the toxicity of α‐syn oligomers, notably through activation of microglia (Bengoa‐Vergniory et al., 2017; Helwig et al., 2016), others contend only inclusions cause neuronal dysfunction and injury (Abdelmotilib et al., 2017; Osterberg et al., 2015).
In this study, we addressed this issue by inducing α‐syn seeding/spreading in wildtype mice (Luk et al., 2012). We used intracranial administration of recombinant murine α‐syn PFFs to induce α‐syn spreading and inclusion formation in the brain, and examined neurodegeneration and microgliosis in regions with α‐syn inclusions and, importantly, in those without. We observed neurodegeneration in both cases, indicating that neuronal injury can occur independently of the formation of α‐syn inclusions. Because neuroinflammation has emerged as a key player in neurodegenerative disease (Hammond et al., 2019), and because microglia are the main cellular effectors of this process (Crotti & Ransohoff, 2016; Wolf et al., 2017), we measured microgliosis in our model. We noticed, in regions with or without inclusions, a surprisingly strong microgliosis (4–5× over baseline), which far surpassed that observed after administration of neurotoxins such as the dopaminergic lesioning agent 6‐hydroxydopamine (6‐OHDA). In contrast to mice injected with 6‐OHDA, neurodegeneration and microgliosis did not correlate with each other in the brains of α‐syn PFFs injected mice. Moreover, by measuring gene expression profiles after striatal α‐syn PFF injection, we observed numerous changes in inflammation‐related genes and pathways, and an unusual microglial molecular activation profile that preceded neurodegeneration and pointed toward an involvement of these cells in the process. We also saw that key PD pathways were recapitulated in this model.
These findings indicated that, in the α‐syn seeding/spreading mouse model, microgliosis does not occur primarily as a response to neuronal damage, but as part of a response that occurs independently of α‐syn inclusion formation. Our results demonstrate that PD‐like neurodegeneration can occur in the absence of α‐syn inclusions, and thus that PD‐like pathology is more than just the progressive formation of such inclusions. It may involve spreading of other, more soluble forms of toxic aggregates, such as oligomers, that could induce an excessive microglial response. We believe these results add an important aspect on how the pathogenic properties of “prion‐like” α‐syn should be viewed, highlight an important role of microglia in the process, and stress the translational relevance of the α‐syn seeding/spreading model.
2. MATERIALS AND METHODS
2.1. Expression and purification of recombinant murine α‐Syn, and generation of pre‐formed fibrils (PFFs), and of oligomers
Expression and purification of recombinant murine α‐syn and generation of PFFs were performed as described (Weihofen et al., 2019). PFFs were stored aliquoted at −80°C until use. For the preparation of oligomers, recombinant α‐syn was purchased from Analytik Jena (Jena, Germany). Oligomers were generated as described (Almandoz‐Gil et al., 2018; Malaplate‐Armand et al., 2006), by incubating soluble α‐syn in 10 mM Tris–HCl, 100 mM NaCl under continuous shaking in an Eppendorf Thermomixer at 650 rpm and 37°C for 24 h, then stored aliquoted at 2 mg/ml at −80°C until use.
2.2. Western blot of α‐syn PFFs and oligomers
The composition of α‐syn PFFs and oligomers was checked by non‐denaturing Western Blot. Three different concentrations of oligomers of PFFs (10, 100, and 500 ng), were loaded on 4%–10% Precast Gel Mini Protean TGX (BioRad) according to manufacturer's instructions. To reveal α‐syn bands, anti‐synuclein antibody clone 4D6 (Covance) was used at 1:2000 dilution (2 h at RT incubation), followed by IRDyeR 800 CW donkey anti‐mouse, diluted 1:10,000 (1 h at RT incubation). Image was captured with a LI‐COR Bioscience C‐Digit Chemoluminescence scanner.
2.3. Electron microscopy of a‐syn PFFs and oligomers
Samples were prepared for transmission electron microscopy by negative staining using a direct application method (Doane, 1987). The Formvar/carbon coated 100 mesh copper grids (EMS, FCF100H‐CU, Lot# 190405) were charged using a glow discharge apparatus (Cressington 208: 10s at HT10 0.1 bar). This renders the carbon surface negatively charged and allows the sample and stain to spread more effectively (Hayat & Miller, 1990). To load the samples (diluted to 0.2–0.5 μg/μl in PBS), a 2 μl drop was placed directly onto the carbon coated grid for 1 min without drying, and excess volume was blotted off the grid with a piece of filter paper, then the samples were let to air‐dry under a hood. Then the grids were washed three times, to remove salts, by applying a small (100 μl) droplet of sterile purified water (Millipore) to the sample side of the grid. Excess water was blotted off the grid with a piece of filter paper, and the samples were let to air‐dry. The samples were then stained with aqueous 2% uranyl acetate for 20 s. Excess liquid was blotted off the grid surface with filter paper, and the grid was let to dry. The samples were then examined and photographed in a scanning electron microscope (Zeiss GeminiSEM 300) using a scanning transmission electron detector at an accelerating voltage of 20 Kv.
2.4. Animals
All 3‐ to 6‐month‐old C57Bl/6J mice were purchased from Jackson via Charles River (Bois‐des‐Oncins, France), or Janvier Labs (Le‐Genet‐St.‐Isle, France). Mice were housed in individually‐ventilated cages (IVC) in a conventional animal facility of the University of Luxembourg, or in the facility of SynAging, in Vandeouvre‐les‐Nancy, France. All animal studies were in agreement with the requirements of the EU Directive 2010/63/EU and Commission recommendation 2007/526/EC. Male and female mice were housed, separated by sex, under a 12 h–12 h dark/light cycle with ad libitum access to water and food (#2016, Harlan, Horst, NL). For time point of 90 dpi PFF injections (see below), the youngest mice were used, for time point 13 dpi PFF injections, the oldest mice were used, so that, at euthanasia, all the mice were of comparable age (6–6.5 months). Animals were otherwise randomly and equitably assigned to groups. For quantitative histology (see below), ten‐eleven mice/group were injected and all were quantified, and for transcriptional profiling, six mice/group were injected and all were profiled. Such sample numbers are similar or higher than the ones used in the original study by Luk et al (Luk et al., 2012), and have proven sufficient in previous studies on different models of neurodegeneration, while also keeping in line with the rule of the “3Rs” (Buttini et al., 1999; Buttini et al., 2005; Cabeza‐Arvelaiz et al., 2011; Jaeger et al., 2015; Miller et al., 2007). Animals used for 6‐hydroxydopamine injection have been described elsewhere (Ashrafi et al., 2017). For injections of α‐syn oligomers, three mice/group were used, as this experiment was for qualitative purpose only (see results). Animal studies were approved by the institutional Animal Experimentation Ethics Committee of the University of Luxembourg and the responsible Luxembourg government authorities (Ministry of Health, Ministry of Agriculture). Alternatively, experiments done at the SynAging site were approved by ethics committee “Comité d'Ethique Lorrain en Matière d'Expérimentation Animale”, and by the governmental agency the “Direction Départementale de la Protection des Populations de Meurthe et Moselle‐ Domaine Expérimentation Animale”.
2.5. Striatal injections of α‐syn PFFs, α‐syn oligomers, and 6‐hydroxydopamine
Alpha‐syn PFFs were sonicated in a sonicating waterbath (Branson 2510, Danbury, CT) for 2 h at RT, keeping the temperature constant at 25°C by adding ice as needed, or using the Bioruptor UCD 300 (Diagenode, Seraing, Belgium) with 30 cycles of 15 s ON/15 s OFF at 4°C. Sonicated PFFs were kept on ice and used within 10 h. Mice were injected under isoflurane anesthesia (2%) on a heating pad. A 1 cm long mid‐line scalp incision was made into the desinfected surgical area and a 0.5 mm hole drilled unilaterally into the skull using stereotaxic coordinates for striatum according to the Mouse Brain Atlas of Franklin and Paxinos (Paxinos & Franklin, 2008). Ten μg of PFFs, or just PBS solution (control mice) were administered, in volumes of 2 μl, within the right dorsal striatum at the following relative‐to‐bregma coordinates: anterior +0.5 mm, lateral +2.1 mm; depth +3.2 mm. The 24‐gauge blunt tip needle of the Hamilton syringue (7105KH, Bonaduz, CH) was inserted down 3.3 mm for 10 s to form an injection pocket, and the needle remained in place for 2 min before and after the injection procedure. The hole was covered with bonewax (Lukens, Arlington, VA), and the wound closed using 7 mm Reflex wound clips (Fine Science Tools, Heidelberg, Germany). Two % xylocaine gel was applied to the wound, and mice were allowed to recover from anesthesia before being put back into their home cages. The day of injection of PFFs was named day 0. Same coordinates and a similar procedure were used for 6‐OHDA or α‐syn oligomers injections. Striatal injection of 6‐OHDA has been described elsewhere (Ashrafi et al., 2017). Striatal injections of α‐syn oligomers were done with 4 μg oligomers in 2 μl vehicle. Control mice received the same volume of vehicle (see above). Mice were euthanized in a deep anesthesia (i.p. injection of Medetomidin, 1 mg/kg and Ketamin, 100 mg/kg) by transcardial transfusion with PBS. PFF‐injected mice were euthanized either at day 13 (13 dpi) or at day 90 (90 dpi) after striatal injections (“day 0”: day of injection). Mice injected with oligomers or with 6‐OHDA were euthanized at 13 dpi.
2.6. Tissue extraction and preparation
For immunohistochemistry, extracted brains were fixed in in 4% buffered PFA for 48 h and kept in PBS with 0.1% NaN3 until they were cut with a vibratome (VT1000 S from Leica) into sagittal 50 μm free‐floating sections. Before the staining procedure, sections were kept at −20°C in a cryoprotectant medium (1:1 vol/vol PBS/ethylene glycol, 10 g/L polyvinyl pyrrolidone). Alternatively, for dopamine measurement or RNA extraction, after removal from the skull, brains were dissected on ice into regions. Isolated striatum and ventral midbrain were quickly weighted, then snap‐frozen on dry ice until further processing. Extraction and measurement of striatal dopamine (DA) has been described elsewhere (Jager et al., 2016). Briefly, after homogenization and derivatization, striatal metabolites were measured with a gas‐chromatography/mass‐spectrometry set‐up (Agilent 7890B GC – Agilent 5977A MSD, Santa Clara, CA). Absolute level of DA were determined using an internal standard, 2‐(3,4‐Dihydroxyphenyl)ethyl‐1,1,2,2‐d4‐amine HCl (D‐1540, C/D/N isotopes, Pointe‐Claire, Canada). For RNA extraction from the ventral midbrain, the RNEasy Universal Kit (Quiagen) was used. After homogenization of midbrain tissues in a Retsch MM 400 device (2 min at 22 Hz, Haan, Germany). RNA concentrations and integrity were determined using a Nanodrop 2000c (Thermo Scientific) and a BioAnalyzer 2100 (Agilent), respectively. Purified RNAs were considered of sufficient quality if their RNA Integrity Number (RIN) was above 8.5, their 260/230 absorbance ratio > 1, and their 260/280 absorbance ratio = 2.
2.7. Single and double‐label immunohistochemistry
Immunostaining procures followed standard protocols, as described (Buttini et al., 1999; Buttini et al., 2005). All stainings, except those for proteinase‐K resistant α‐syn inclusions (see below), were performed on free‐floating 50 μm‐thick sections. Table S1 lists all primary and secondary antibodies used in this study, as well as their dilutions. All other reagents were from Sigma unless indicated otherwise. All antibody incubations were at room temperature, except for the anti‐synaptophysin antibody, which was incubated at 4°C. Sections were washed 3× in PBS between each incubation step. To block endogenous peroxidases and for permeabilization, sections were incubated with 3% H2O2 vol/vol and 1.5% Triton ×100 vol/vol for 30 min. For immoperoxidase staining with anti‐synuclein antibody, this step was followed by an epitope unmasking step with 75% vol/vol formic acid for 5 min. To avoid unspecific antibody binding, sections were incubated with 5% serum (Vector Laboratories, Burlingame, CA) or 5% BSA wt/vol in PBS for 1 h before they were incubated with the respective primary antibody, or antibodies in case of double labeling. The following day, sections were incubated with a secondary antibody for 1–2 h (fluorophore‐coupled for immunofluorescence, or biotinylated for immunoperoxidase). Singly or doubly fluorescently‐stained sections were mounted on Superfrost plus slides (Thermoscientific, Walham, MA), air‐dried, and coverslipped using ProLong Gold antifade mounting medium (Life technologies, Darmstadt, Germany). For immunoperoxidase staining, antibody binding was visualized using an ABC Vectastain Kit (Vector Laboratories), followed by detection with diaminobenzidine (Merck) and H2O2 as peroxidase substrates. Sections were mounted, dried overnight and coverslipped with Neo‐mount (Merck) after soaking in Neo‐clear xylene substitute (Merck) for 10 min. Visualization of Proteinase‐K resistant α‐synuclein inclusions was done by Paraffin‐Embedded Tissue blot (PET blot) on 3 μm paraffin sections mounted on nitrocellulose membrane (0.45 μm, BioRad), as previously (Kramer & Schulz‐Schaeffer, 2007).
2.8. Proximity ligation assay
Protocol for proximity ligation assay (PLA) was adapted for free floating sections. All reactants were prepared according manufacturer's recommendations (Duolink, Sigma) and incubation times were as described (Trifilieff et al., 2011). Washes were performed in 24‐well plates at RT, and reactions volumes were 40 μl at 37°C. First, 20 μg of anti‐pSER129‐ α‐syn mouse monoclonal 11E5 antibody (Prothena Biosciences, see Table S1) were conjugated with either plus or minus oligonucleotide probes according manufacturer's recommendations, and stored 4°C until use. Free floating sections were washed in PBS and permeabilized as described above. Blocking was performed with DuoLink blocking solution for 2 h at RT. Sections were incubated overnight with both plus and minus probe‐linked antibody (1:1 1/750 in Duolink antibody diluent solution). For ligation of the probes, after washing of the probe‐linked antibodies (2 × 5 min in Duolink's Buffer A), the ligation‐ligase solution was added and incubated for 30 min at 37°C. For detection, after washing of the ligation‐ligase solution (2 × 5 min in Duolink's Buffer A), sections were incubated with the amplification‐polymerase solution for 2.5 h at 37°C. Sections were washed in Buffer B for 10 min, and in Buffer B 0.01× for a minute prior to mounting, then dried in the dark, and coverslipped. Z‐stacks of pictures were acquired at 40× with a Zeiss LabA1 microscope, a maximum intensity projection was created using the Zen Blue 2012 software (Zeiss).
2.9. Quantitative neuropathology on immunostained sections
Imaging of peroxidase‐ labeled sections for pSER129‐α‐syn, and of fluorescently labeled sections for tyrosine‐hydroxylase (TH), dopamine transporter (DAT), or ionized calcium binding adaptor molecule 1 (Iba1), was done using a Zeiss LabA1 microscope, coupled to a Zeiss Axiocam MRm3 digital camera, and to a PC running the Zeiss Zen Blue 2012 software.
Alpha‐syn inclusions were visualized by immunostaining for pSER129‐α‐syn. For the quantitation of α‐syn inclusions in the frontal cortex and the amygdala (basolateral nucleus), two immunoperoxidase labeled (see above) sections/animal were imaged, using the 10× objective (frontal cortex) or the 20× objective (amygdala). A total of four‐six images was collected for each region (10× objective, 2 × 1.52 mm2 each image), and digitized. After manually drawing regions of interests and thresholding, the percent image area occupied by immunopositive structures was determined using the ImageJ v. 1.45 (NIH, Bethesda, MD) public domain software. All values obtained from sections of the same animal were averaged. For the quantitation of α‐syn inclusions in the SN, double immunostainings for TH and pSER129‐α‐syn were performed using one section of each hemibrain for each animal. TH staining was used to locate the SN, and images were acquired at 10× magnification. Percent overlap of pSER129‐ α‐syn signal within the TH immunopositive area was calculated using ImageJ. For the quantitation of synaptophysin‐positive synaptic terminals, two fluorescently stained sections, selected randomly for each hemibrain, from each animal were viewed by a Zeiss LSM 710 laser‐scanning confocal microscope, using a 20× objective and a software magnification zoom factor was used to obtain images of 180 × 180 μm2 each. From each of the two hemibrains of all animals, a total of four‐six images were collected from the frontal cortex, and four from the hippocampal pyramidal region. Images were then transferred to a PC personal computer, and average intensity of positive presynaptic terminals was quantified for each image using the ImageJ software. Values from individual animals were averaged. This method to quantify synaptic integrity has been validated by electron microscopy quantitation of synaptic densities in a previous study (Buttini et al., 2005).
The quantitation of degeneration of TH positive neurons in the SN has been described, and results obtained with this approach have been shown to correlate with stereological cell counts (supplemental material in Ashrafi et al. (2017)). For the quantitation of striatal TH‐positive neuronal fibers and of DAT‐positive synaptic terminal, two doubly labeled sections with anti‐TH and anti‐DAT were used from each hemibrain for all animals. A total of six to nine 40× pictures (223.8 × 167.7 μm2 each) of the dorsal striatum, from two‐three sections per hemibrain, were acquired using the optical sectioning system Apotome.2 (Zeiss). The percent area occupied by TH and DAT was determined using Image J software and averaged for each mouse.
For the quantitation of microglial activation in the hippocampus and frontal cortex, two randomly selected sections/hemibrain for each animal were labeled for the microglial marker Iba1. For the frontal cortex, a total of six/hemibrain, and, for the hippocampus, a total of three‐four images/hemibrain were collected with a 40x objective (223.8 × 167.7 μm2 each image). Digitized images were transferred to a PC, and, with ImageJ v. 1.45, after thresholding, average area occupied by Iba1‐positive microglia was measured. All values obtained from sections of the same animal were averaged. For the quantitation of the microglial activation in the SN, TH, and Iba1‐ double‐labeled sections were imaged with a 10× objective. Average area covered by TH‐positive neurons in control mice was used to determine the region of interest, restricted to the SN, to measure microglial activation. Four subregions of the SN were imaged and quantified for each hemibrain of each mouse (Ashrafi et al., 2017). Iba1 immunopositive cells were quantified within each subregions, averaged for each of them, and converted in mm2. For each mouse, the sum of the four averaged subregions was used as a measure of microglial activation.
All quantitative neuropathological analyses were performed blinded on coded sections, and, for each of the measurements, codes were only broken when quantification for that measure in all animals was complete. For all measures, the ipsilateral and contralateral values of PBS‐injected control mice were similar (no statistical difference detected), thus these values were grouped. Statistics on quantitative histological data were done using the GraphPad Prism 8 software. Neurodegeneration and microgliosis measurements were analyzed by ANOVA followed by Dunnett's post hoc for all datasets, which were all parametric. Figures 2 and 3 are mixed graphs of bars and scattergrams. Bars represent means +/− SD, and points in the scatters represent the individual animal values. Pearson's test was used for linear correlations, except for correlations involving α‐syn inclusion load (non‐parametric dataset), for which Spearman's rank was used. Adjusted P values smaller than 5% were considered significant for all tests.
2.10. Microarray analysis and calculation of differentially expressed genes
GeneChip Mouse Gene 2.0ST Arrays (Affymetrix) were used for transcriptional profiling. Total RNAs (150 ng) were processed using the Affymetrix GeneChip® WT PLUS Reagent Kit according to the manufacturer's instructions (Manual Target Preparation for GeneChip® Whole Transcript [WT] Expression Arrays P/N 703174 Rev. 2). In this procedure, adapted from (Bougnaud et al., 2016), the purified, sense‐strand cDNA is fragmented by uracil‐DNA glycosylase (UDG) and apurinic/apyrimidinic endonuclease 1 (APE 1) at the unnatural dUTP residues and breaks the DNA strand. The fragmented cDNA was labeled by terminal deoxynucleotidyl transferase (TdT) using the Affymetrix proprietary DNA labeling reagent that is covalently linked to biotin; 5.5 μg of single‐stranded cDNA are required for fragmentation and labeling, then 3.5 μg of labeled DNA + hybridization controls were injected into an Affymetrix cartridge. Microarrays were then incubated in the Affymetrix Oven with rotation at 60 rpm for 16 h at 45°C, then the arrays were washed and scanned with the Affymetrix® GeneChip® Scanner 3000, based on the following protocol: UserGuide GeneChip® Expression Wash, Stain and Scan for Cartridge Arrays P/N 702731 Rev. 4, which generated the Affymetrix raw data CEL files containing hybridization raw signal intensities were imported into the Partek GS software. First, probe intensities were summarized to gene expression signals using Partek default options (GCcontent adjustment, RMA background correction, quantile normalization, log2 transformation and summarization by means).
For statistical analysis, the normalized and log2 transformed data was loaded into the R/Bioconductor statistical environment. The rank product (Package: RankProd) approach was chosen to determine the differentially expressed genes (DEGs) (Breitling et al., 2004; Del Carratore et al., 2017; Hong et al., 2006). Rank product statistics were computed, since they have been shown to enable a robust non‐parametric analysis of microarray datasets with limited number of samples (Breitling et al., 2004). Estimated p‐values and pfp (percentage of false prediction) values were determined and used as nominal and adjusted significance scores, respectively. Pfp scores estimate the significance of differential expression after adjusting for multiple hypothesis testing, and can have values larger than 1. The chosen significance cut‐offs were p‐value <.05 and pfp < 0.1. A cut‐off (pfp < 0.1 instead of <.05) was chosen to avoid loss of information for the subsequent enrichment analysis, which combines several genes below this threshold to enable detection of pathway alterations. No minimal fold change threshold was applied.
For visualization of differential gene expression, Venn diagrams and heatmaps were generated using the VennDiagram and gplots packages, respectively, in R. Data pre‐processing included removal of all transcripts missing gene IDs and duplicated entries (after ranking). Mouse “Gene Symbols” were used to calculate the overlapping DEGs between the different comparisons. Diagrams were generated for the following criteria and comparisons: (a) 13 and 90 dpi ‐ ipsiPFF versus ipsiPBS (p‐value <.05); (b) 13 and 90 dpi – ipsiPFF versus ipsiPBS (pfp < 0.1); (c) 13 and 90 dpi ‐ ipsiPFF versus contraPFF (p‐value <.05); (d) 13 and 90 dpi – ipsiPFF versus contraPFF (pfp < 0.1). In a second step, we were interested in investigating the expression direction of the overlapping transcripts between the early to late timepoint. Therefore, we extracted the probe IDs, matched the individual lists and grouped them into high and low expressed transcripts. Then, the above mentioned Venn diagrams were generated with these newly generated lists. Heatmaps were generated using the heatmap.2 function for ipsiPFF versus ipsiPBS and ipsiPFF versus contraPFF for p‐value <.05 and pfp < 0.1 at 13 and 90 dpi, respectively. The log2‐transformed data matrix was used to plot. Additionally, we applied hierarchical top down clustering (cor and hclust basic R functions) and the data matrix was scaled row‐by‐row generating Z‐scores.
2.11. Gene set enrichment analysis (GSEA)
The enrichment analysis for GO terms (biological processes [BP] only) was performed using the GUI (graphical user interface) version GSEA (version 3.0) published by the Broad Institute (download: http://software.broadinstitute.org/gsea/downloads.jsp) (Subramanian et al., 2005). All parameters were set to default in GSEA, except “Collapse dataset to gene symbols” was set to “false”, “Permutation type” was set to “gene_set” and “Max size: excluding larger sets” was set to “250”. One optimization step was introduced: a customized GMT/GMX file was generated in R/Bioconductor with mouse NCBI EntryzIDs as gene identifiers. This file was used as the “Gene sets database” in GSEA. The resulting enrichment scores (ES) were obtained applying the weighted Kolmogorov–Smirnov‐like statistics. ES reflect the level to which a gene set is overrepresented among the top up‐ or down‐regulated genes in a ranked gene list, then, the ES statistic was normalized (normalized enrichment scores, NES) as described (Subramanian et al., 2005). Finally, the p‐value significance scores were adjusted for multiple hypothesis testing (Benjamini, 1995) to provide final FDR scores. A network map of the enrichment analysis results was generated using Cytoscape (Shannon et al., 2003). The mapping parameters used in Cytoscape were: p‐value <.05, FDR Q‐value <0.1 (default setting is 1) and Overlap >0.5. The enrichment map was automatically launched from GSEA and created in Cytoscape. In the enrichment maps, nodes represent enriched gene sets associated with BPs, and edges the degree of similarity between them using the overlap coefficient (threshold >0.5). Further curation of gene sets was done manually. Since gene sets with similar gene compositions tend to group together, such gene set clusters were easily identifiable. Nodes grouped into more than one gene cluster according to this procedure were assigned to the most overlapping cluster, that is, the cluster they were associated with by a shorter sequence of connecting edges in the ontology graph. All softwares used are given in Table S2.
2.12. Identification of cellular source of DEGs
For the identification of the cellular source of specific DEGs, the public database GSE52564 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52564) was used. An easily accessible online resource of this database is found here: https://www.brainrnaseq.org/. Data were filtered based on FPKM values relative to the sum of FPKM per gene across different cell types. The filter criteria focused on isolating genes that were specifically expressed by individual cell types or cell type groups (mixed glial cells (e.g., for instance microglia and astrocytes), mixed oligodendrocytes (e.g., myelinated, precursors, and newly formed oligodendrocytes), and pan‐cellular (e.g., expressed in all cell types). The selection criteria were:
| Group description | Inclusion criteria | Exclusion criteria |
|---|---|---|
| Single cell type | ≥40% | >26.6% all other cell type |
| More than one cell type | ∑ %CellType > 50% + not in single cell type | >15% all other cell type |
| Pan cellular expression | All < 20% + not in single of more than one cell type | — |
Datasets were finally checked for their uniqueness to each specific group. No overlaps were found.
2.13. Characterization of microglial molecular profile
Overlaps were determined using the “calculate.overlap” function of the VennDiagram‐package in R, and, in select analyses, diagrams were generated with the “ggvenn” function of the package with the same name. Datasets from (Uriarte et al., 2021) were from Table S2 of that study. Datasets from (Holtman et al., 2015) were from Table S3 of that study. The datasets of that study were analyzed each separately (for each of the mouse models) and compared to our datasets. Then an overall overlap was generated from all the datasets in that study, to extract a “disease‐associated” microglial core gene signature, which was termed “disease‐associated microglia” (DAM) signature. This resulting dataset of 50 genes was compared to our datasets, to generate overlaps and Venn diagrams.
2.14. Translational relevance assessment: comparison of DEGs and of pathways between the α‐syn seeding/spreading model and different stages of PD
To generate overlaps and Venn diagrams of our data with PD human databases, datasets from two studies were used: (Dijkstra et al., 2015) (GSE49036), and (Glaab & Schneider, 2015). Gene symbols of these datasets were first changed to mouse gene symbols using the “convert_human_to_mouse” function of the NicheNet‐package (Browaeys et al., 2020). Then the same procedure to generate overlaps and Venn diagram was applied as described above.
To compare pathways between the mouse model used in this study and PD, the PD map (https://pdmap.uni.lu/minerva/) on the MINERVA platform was used (Gawron et al., 2016; Hoksza et al., 2019). All datasets were prepared appropriately before analysis. In short, a list of DEGs per dataset containing ‘Gene Symbols’ and the respective log‐fold changes was produced. First, Gene symbols had to be under the format proposed by the HUGO Gene Nomenclature Committee (HGNC). For mouse datasets, gene symbols were converted using the “convert_mouse_to_human” function from the NicheNet‐package in R. Next, log‐fold changes were altered as follows: arranged in decreasing order, all positive log‐fold‐changes were divided by the highest positive log‐fold‐change and analogous for the negative log‐fold‐changes. Finally, each DEG list in a tab delimited format was uploaded to https://pdmap.uni.lu/minerva/ and the integrated GSEA plugin was activated.
To visualize directionality of changes, the number and expression changes of DEGs in each pathway that mapped to the PD map was simply categorized as “Up” or “Down”, based on the original expression data. This was done by accessing the hits information in the PD map for the significantly altered gene sets, using the API calls of the MINERVA Platform that is hosting the map (https://minerva-web.lcsb.uni.lu).The extracted information was distilled to keep multi‐hit genes as unique hits only. The generated point plots allowed to differentiate between number or counts of DEGs and the expression directionality of those genes for pathways that were significantly altered in at least one of the datasets (human or mouse).
3. RESULTS
3.1. Western blot and electron microscopy characterization of α‐syn moieties
A non‐denaturing blot of the α‐syn moieties used in this study is shown in Figure S1 (upper panel). For striatal injections, α‐syn oligomers were used non‐sonicated, whereas α‐syn PFFs were sonicated. Based on their Western blot (WB) profile, the oligomer preparation was composed mainly of monomers, dimers, and trimers, as well as higher molecular weight species. The sonicated PFFs were composed mainly of monomers and dimers, and higher molecular weight species. When compared to their non‐sonicated counterparts, sonicated PFFs seemed to have less of all these components, consistent with a shearing effect of sonication that produces smaller α‐syn fragments, which may then act as seeds. Electron microscopy confirmed the presence of different sized moieties in the different preparations. At the ultrastructural level (Figure S1, lower panels), we observed that oligomers were mostly composed of small globular assemblies, unsonicated PFFs were formed of highly complex large agglomerates, and a number of smaller assemblies, and sonicated PFFs were mostly composed of small fibrillar fragments.
Because we wanted to follow the original protocol of Luk et al. (Luk et al., 2012), no effort was made to purify particular α‐syn moieties. In addition, defining the pathobiological properties of the different moieties, while also making sure these moieties stayed stable in their respective conformation over the whole course of an experiment, was beyond the scope of the present study. Other studies have looked at that issue (Grozdanov et al., 2019; Peelaerts et al., 2015; Rey et al., 2019). We were instead focused on determining whether the pathologies resulting from intracerebral injection of the moieties we used resulted in translationally relevant PD‐like phenotypes, and on understanding underlying mechanisms of neurodegeneration.
3.2. Striatal injection of PFFs causes bilateral α‐syn inclusions in multiple brain regions
Because we wanted to capture the early features of α‐syn spreading associated pathologies, we decided to focus our investigations on time points when these pathologies have not peaked yet (Luk et al., 2012). Since α‐syn inclusions have been suggested to be a major driver of PD‐like pathology (Abdelmotilib et al., 2017; Spillantini & Goedert, 2018), we first looked at the appearance of such inclusions in our model.
To determine if striatal injection of murine α‐syn PFF reliably induced propagation of fibrillar α‐syn in our mice, we performed immunohistochemistry against pSER129‐ α‐syn on sections of both brain hemispheres 13 and 90 days after they had been injected with PFFs (13 and 90 dpi). Immunostaining for pSER129‐ α‐syn is the most commonly used approach to detect α‐syn inclusions in rodent or human brain tissues (Vaikath et al., 2019).
At an early time point after α‐syn PFF administration (13 dpi), we only detected few pSER129‐α‐syn positive inclusions in frontal cortex, amygdala, and SN, and a few more in the ipsilateral striatum (Figure S2).
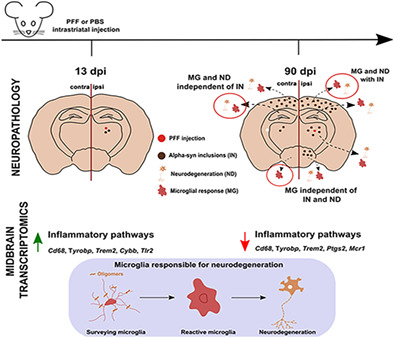
However, at 90 dpi, we observed robust appearance of pSER129‐ α‐syn positive cellular and neuritic inclusions ipsi‐ and contralaterally, in the same brain regions (Figure 1a). Quantitation of image area occupied revealed median coverage of 10% for the ipsilateral frontal cortex, 5% for the contralateral frontal cortex, 8% for the ipsilateral amygdala, 4.2% for the contralateral amygdala, and 12% for the ipsilateral SN. No or very few α‐syn inclusions were found in the contralateral SN and striatum, and no inclusions in either side of the hippocampus. Cells containing inclusions had neuronal morphology. In the ipsilateral SN, fluorescent double staining for pSER129‐α‐syn and TH, a marker for dopaminergic neurons in the SN, showed that 85% of inclusions co‐localized with TH‐positive neurons, indicating that most, if not all, inclusions were localized in neurons.
FIGURE 1.
Striatal injection of murine α‐syn PFFs induced α‐syn inclusions in various brain regions. Mice were euthanized 90 days after injection (90 dpi, n = 10–11/group). (a) PhosphoSER129 α‐syn immunostaining showed numerous α‐syn inclusions in neuritic and neuronal body structures in different brain regions. Widespread α‐syn inclusions were observed bilaterally in frontal cortex and the amygdala, ipsilaterally in the striatum and the substantia nigra (SN), and only minimally in the contralateral striatum and SN. None were observed in the hippocampus. No inclusions were observed in either side of the brains of PBS‐injected control mice. Pictures show the ipsilateral side of these mice. (b) Proteinase‐K digestion on thin sections generated from paraffin‐embedded tissue (PET) revealed the presence of digestion‐resistant α‐syn inclusions stained for PhosphoSER129 α‐syn. Shown here are ipsilateral striatum and amygdala for illustration. (c) Proximity‐ligation assay using a monoclonal PhosphoSER129 α‐syn antibody showed the presence of enhanced levels of oligomeric forms of α‐syn in the hippocampus of PFF‐injected mice, where no inclusions could be detected 90 dpi, compared to PBS‐injected controls. Scale bar = 250 μm (a), 250 μm (b), 25 μm (c)
To determine if pSER129‐α‐syn positive inclusions were Proteinase‐K resistant, we performed a Paraffin‐Embedded Tissue blot (Kramer & Schulz‐Schaeffer, 2007; Milber et al., 2012). We observed numerous pSER129‐α‐syn positive signals in these tissue sections (Figure 1b), indicating that most inclusions were proteinase‐K resistant.
Inclusions are not the only α‐syn species that have been suggested to be linked to neurodegeneration in PD. To determine if regions without detectable α‐syn inclusions, such as the hippocampus, were still affected by abnormal α‐syn after injection of PFFs, we performed a proximity ligation assay (Malaplate‐Armand et al., 2006). This assay has been used for detecting oligomeric forms of α‐syn in human (Roberts et al., 2015) and mouse models of PD (Ulusoy et al., 2015). We observed greatly enhanced signal intensity in the hippocampi of PFF‐injected mice than in those of control mice (Figure 1c), indicating the presence of abnormal levels of oligomeric α‐syn in that region.
Overall, the pattern of α‐syn inclusions we observed 90 dpi matched that described at a similar time point by Luk et al. (2012) (Luk et al., 2012). The robust appearance of intracellular α‐syn inclusions in this model, at just 90 dpi after injection of PFFs, opened up the possibility of analyzing how they are associated with other pathological hallmarks, such as neurodegeneration and –inflammation, and which of these events precedes the other.
3.3. Striatal injection of PFFs causes bilateral synaptic loss and unilateral dopaminergic neuron injury that was independent of α‐syn inclusions
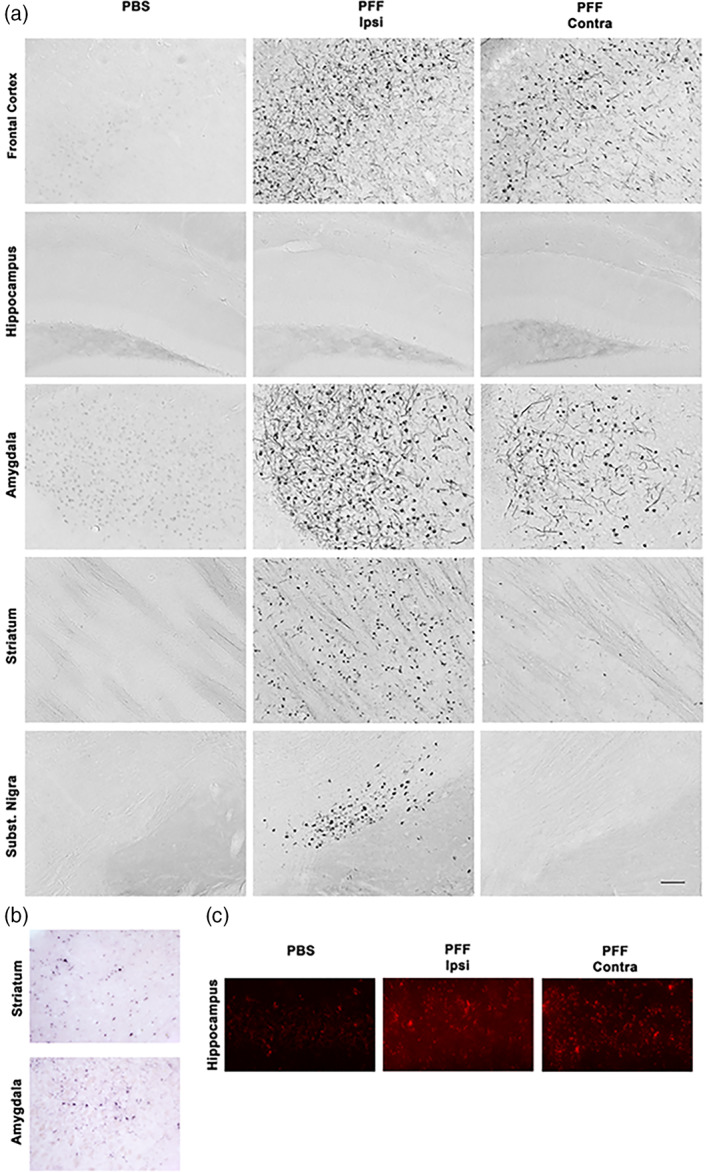
We set out to determine to what extent the presence of neuronal α‐syn deposition was linked to neurodegeneration, 90 dpi after striatal administration of PFFs. First, we analyzed synaptic degeneration in the hippocampus and frontal cortex. In these brain regions, we measured the level of the presynaptic protein synaptophysin. Synaptophysin is a good marker for synaptic integrity (Buttini et al., 2005; Calhoun et al., 1996; Zhan et al., 1993), and pathological synaptic alterations have been reported in PD post‐mortem tissues (Bellucci et al., 2016). Roughly 60% of PD patients suffer from cognitive impairments and dementia (Aarsland et al., 2017), indicating that their hippocampus and their higher cortical association areas are affected. Addition to PFFs to cultured primary hippocampal neurons was reported to affect these neurons' synaptic integrity and function (Wu et al., 2019). Thus, we measured synaptophysin ipsi‐ and contralaterally in these brain regions in mice 90 days after PFF administration (Figure 2). We found, in both regions, a highly significant, bilateral 20%–25% reduction of this protein in PFF‐injected mice. Interestingly, we noticed this decrease in the absence of α‐syn inclusions in the hippocampus. The α‐syn oligomers though (Figure 1) in that region colocalized with synaptophysin loss.
FIGURE 2.
Striatal injection of murine α‐syn PFFs induced neurodegeneration in various brain regions. Mice were euthanized 90 dpi. In the frontal cortex and the hippocampus, a significant bilateral loss of synaptophysin‐positive presynaptic terminals was observed (first two rows). In the striatum, a significant ispilateral loss of TH‐positive axonal fibers and DAT‐positive synaptic terminals was observed (3rd and 4th row). In the SN, a significant loss of TH‐positive neurons was observed only ipsilaterally. For group comparisons and graphing, ipsilateral PBS measures were combined contralateral PBS measures, since they were similar. Pictures show the ipsilateral side of PBS‐injected mice. ****p < .0001, **p < .01, compared to PBS controls by Dunnett's post hoc; n = 10–11/group; graphs are mixed scattergrams/bar diagrams, where points represent the individual values for each animal, and bars represent the means +/− SD; 95% confidence intervals of differences: Frontal cortex – PBS versus PFF ipsi: 11.6 to 28.7, PBS versus PFF contra: 11.42 to 28.52; hippocampus – PBS versus PFF ipsi: 16.2 to 30.5, PBS versus PFF contra: 13.9 to 28.3; striatum (TH) – PBS versus PFF ipsi: 3.56 to 16.3, PBS versus PFF contra: −9.8 to 2.9; striatum (DAT) – PBS versus PFF ipsi: 7.8 to 20.6; PBS versus PFF contra: −7.8 to 5.7; SN (TH) – PBS versus PFF ipsi: 0.002 to 0.152, PBS versus PFF contra: −0.017 to 0.112). Scale bars: 18 μm (for frontal cortical and hippocampal synaptophysin panels), 22.5 μm (for striatal TH and DAT panels), 80 μm (for Subst. Nigra panels)
Next, we examined the SN, because it contains dopaminergic neurons that are one of the most susceptible population in PD. We measured the area occupied by tyrosine hydroxylase (TH)‐positive neuronal profiles in the SN ipsilaterally, where α‐syn inclusions were present (see above), but also contralaterally, which was without such inclusions. We found a statistically significant 16% decrease of TH‐positive neurons in the ipsilateral SN, but not so in the contralateral SN (Figure 2). To determine if striatal axonal projections of dopaminergic neurons were affected in our model, we analyzed the morphological integrity of these projections and their synaptic terminals. We observed, 90 dpi, a significant decrease in TH‐positive axonal fibers as well as in dopamine transporter (DAT) positive synaptic terminals, in the ipsilateral, but not the contralateral striatum. We did not find any sign of degeneration in the striatum or SN at 13 dpi (Figure S3, 3 first rows).
To confirm ipsilateral striatal injury, we measured the neurotransmitter dopamine (DA) in dissected ipsi‐ and contralateral striata of PFF‐injected and PBS control mice (n = 8–12/group). We found a significant decrease in ipsilateral striatum of PFF mice compared their ipsilateral PBS controls (19.5+/−5.8 vs. 27.5+/−7.3 pmol/mg; p = .02 by ANOVA followed by Sidak's post hoc, results are means +/− SD), but no difference between contralateral striatum of PFF mice compared to their compared their ipsilateral PBS controls (27.3+/−3.7 pmol/mg vs. 28.5+/−5.7 pmol/mg). This substantiated our histological observations.
3.4. Striatal injection of α‐syn PFFs caused profound microgliosis in different brain regions that was independent of α‐syn inclusions
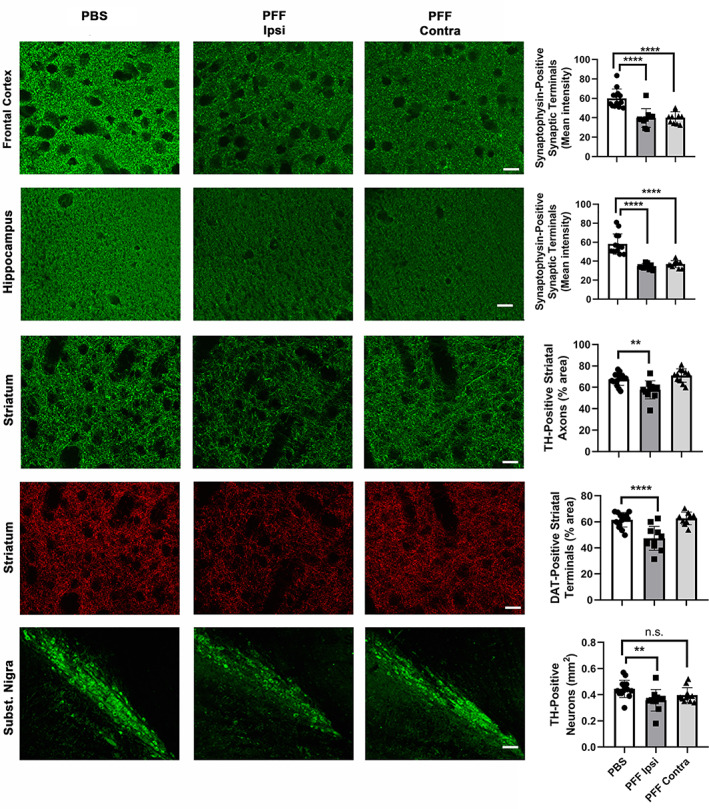
Microglia, the local CNS innate immune defense cells (Michelucci et al., 2018), react rapidly to CNS infection or injury. Functional imbalance of these cells can precipitate disease outcomes (Biber et al., 2014; Crotti & Ransohoff, 2016; Wolf et al., 2017). While strong microgliosis has been reported in PD and models thereof (Doorn et al., 2012; Joers et al., 2017; Tan et al., 2020), the role of these cells in disease initiation and progression is poorly understood.
To better understand the role of microglia in the context of α‐syn spreading, and more precisely to determine if these cells have a role in driving the neurodegeneration we observed, we first analyzed their response using a specific marker (Iba1), in mouse brains after injection of α‐syn PFFs. We observed a surprisingly strong (4–5 times over control) microgliosis in different brain regions (bilaterally in frontal cortex, amygdala, SN) at 90 dpi. The microgliosis was present in brain regions with inclusions, but also those without (hippocampus) or very little (contralateral SN) inclusions (Figure 3). While no significant Iba1 increase was seen at 90 dpi in the ipsilateral striatum in PFF injected mice, microglial Cluster‐of‐Differentiation 68 (CD68), a marker for phagocytic activity, was increased in that location, indicating that these cells, while having a different kind of response, were still activated. No significant microgliosis was observed in the SN at 13 dpi (Figure S2, lower row). Microglia in PFF‐injected mice had thickened, though still ramified, processes, and an intensely stained cell soma. In the cerebellum, which was devoid of α‐syn deposits in all mice, we could not detect any differences in Iba1 positive microglia between PFF‐injected and control PBS injected mice (not shown).
FIGURE 3.
Striatal injection of murine α‐syn PFFs induced widespread microgliosis in different brain regions. Mice were euthanized 90 dpi. Panels show microgliosis measured on Iba1‐stained sections of frontal cortex (upper row), hippocampus (second row), striatum (third row), and Subst. Nigra (last row), and on CD68‐stained sections of striatum (4th row). A very strong microgliosis (up to 4× over control) was observed bilaterally in frontal cortex, hippocampus, and SN. No increase in Iba1 signal, but a significant bilateral increase in CD68 signal was observed in the striatum of PFF‐injected mice. For group comparisons and graphing, ipsilateral PBS measures were combined contralateral PBS measures, since they were similar. Pictures show the ipsilateral side of PBS‐injected mice. ****p < .0001, *p < .05, compared to PBS controls by Dunnett's post hoc; n = 10–11/group; graphs are mixed scattergrams/bar diagrams, where points represent the individual values for each animal, and bars represent the means +/− SD; 95% confidence intervals of differences: Frontal cortex – PBS versus PFF ipsi: −23.9 to −14.5, PBS versus PFF contra: −19.5 to −10.1; hippocampus: – PBS versus PFF ipsi: −31.5 to −17.7, PBS versus PFF contra −31.0 to −17.2; striatum (Iba1) – PBS versus PFF ipsi: −2..9 to 1.1, PBS versus PFF contra: −1.9 to 2.1; striatum (CD68) – PBS versus PFF ipsi: −0.9 to −0.4, PBS versus PFF contra: −0.5 to −0.01; SN – PBS versus PFF ipsi:‐0.31 to −0.17, PBS versus PFF contra: −0.28 to −0.15. Scale bars: 22.5 μm (for all panels)
Our observations indicate that a robust, widespread microglial reaction is an important part of the α‐syn spreading process, and warranted further investigation into the pathological implications of that reaction.
3.5. Neurodegeneration and microgliosis correlated neither with α‐syn deposition, nor with each other
To gain insight into the pathological properties of α‐syn inclusions, we correlated the inclusion load with neurodegeneration and with microgliosis measured locally in frontal cortex and SN. We found that inclusion load correlated with neither of the two (Figure 4). Thus, neurodegeneration as well as microgliosis induced by α‐syn PFFs are probably independent of α‐syn deposition.
FIGURE 4.
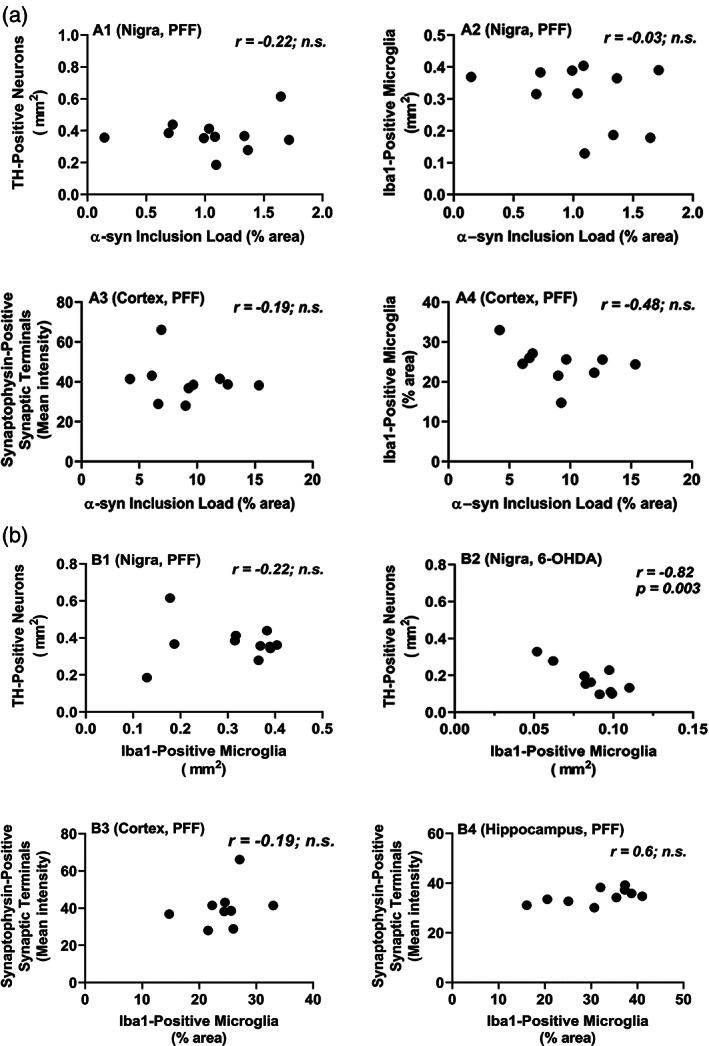
Different PD‐related pathologies in the brains of mice injected striatally with α‐syn PFFs do not correlate with each other. Mice were euthanized 90 dpi. (a) α‐syn inclusion load did no correlate with neurodegeneration (loss TH‐positive neurons, A1) or with microgliosis (A2) in the SN (Nigra), nor with neurodegeneration (loss of synaptophysin‐positive synaptic terminals, A3) or with microgliosis (A4) in the frontal cortex (cortex). (b) Microgliosis did not correlate with loss of TH‐positive neurons in the SN after intrastriatal PFF injection, but did so after intrastriatal injection of the toxin 6‐OHDA. The microgliosis, measured on Iba1‐stained section, was also much higher in the Subst. Nigra of PFF‐injected mice than in that of 6‐OHDA‐injected mice. All measures shown are from the ipsilateral brain sides; similar observations were made for the contralateral sides of PFF‐injected mice. Correlation analyses were done using Spearman rank test for data set including α‐syn inclusion load measures (non‐parametric), and with Pearson's test for data sets with the other measures (parametric)
The strong microgliosis in different brain regions after administration of PFFs prompted us to look into this observation further. In the brain, microglia react rapidly to tissue injury to control the damage and clear up cell debris (Fu et al., 2014; Wolf et al., 2017). Thus, microglial reaction is typically secondary to an underlying neurodegenerative process, and, as a consequence, increase of microglial reaction is directly associated with decrease of neuronal integrity. For instance, we have observed that microglial reaction (measured on Iba1 immunostained sections) correlated negatively with TH neuron loss in the ipsilateral SN after unilateral lesioning by 6‐OHDA (Figure 4b2), and with synapse or dendritic loss in the cortex after lesioning with the excitotoxin kainic acid (Jaeger et al., 2015). After intracerebral injection of α‐syn PFF though, we found that microglial reaction was not only much stronger than after injection of neurotoxins (4–5× vs. 2–3× over control), but also failed to correlate with measures of neurodegeneration (TH neuron loss in the SN, synaptophysin in the cortex and hippocampus) (Figure 4b). This observation indicates that the microglial reaction to α‐syn spreading may be a direct response to factors produced during that process, and not just a secondary response to neuronal degeneration.
3.6. Microglia across several brain regions reacted strongly to striatal injection of α‐syn oligomers
Several studies have indicated that microglia are activated in vitro by α‐syn oligomers (Hughes et al., 2019; Kim et al., 2013). As described above, we have observed the presence of α‐syn oligomers, notably in the hippocampus, after striatal injection of α‐syn PFFs. To test whether α‐syn oligomers could be the factor that led to a strong microglial reaction during the α‐syn spreading process, we injected such oligomers into the same location as the PFFs, the dorsal striatum. Just 13 dpi, we observed, on Iba1 stained sections, a strong microglial reaction in the ipsilateral striatum, frontal cortex, and hippocampus (Figure 5). Qualitatively, the reaction was even stronger than 90 dpi after PFF injection. We did not move forward in quantifying this reaction and its consequences, since our goal was just to show that microglia react to the injection α‐syn oligomers, even in regions distant to the injection site, and because the number of animals used to test this was only three per group. Further studies will investigate these issues. Thus, microglial cells in vivo respond strongly to α‐syn oligomers, and further analyzing the possible role of these cells was warranted.
FIGURE 5.
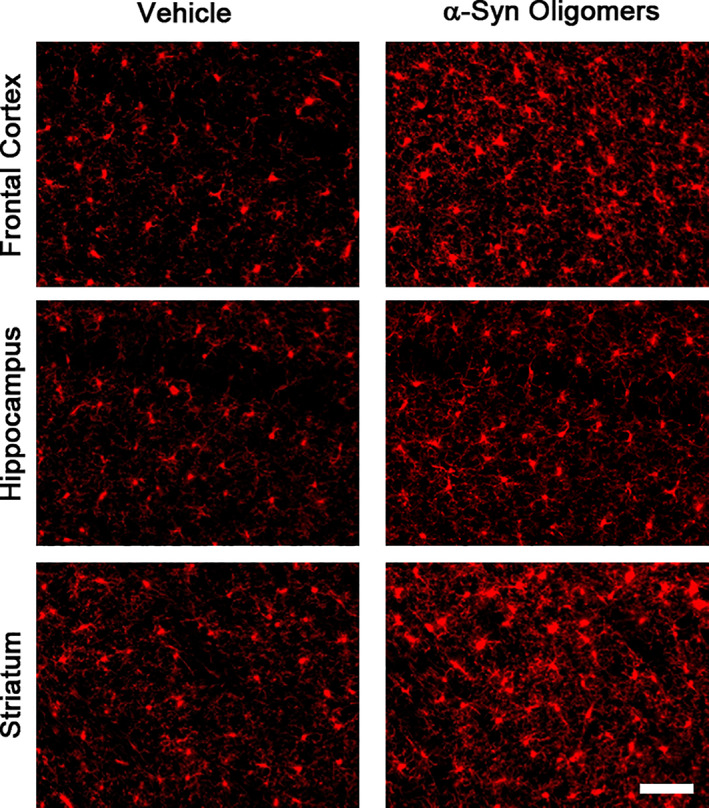
Strong microglial response after striatal injection of α‐syn oligomers. Oligomers were prepared and injections were performed as described in Materials and Methods, Section 2. A strong microgliosis was observed in different brain regions 13 dpi, confirming that these cells react strongly to oligomers. Scale bar = 40 μm
3.7. Transcriptional profiling of ventral midbrain revealed most gene expression changes occur 13 days after α‐syn PFF injection
To investigate the molecular underpinnings of the neurodegeneration and the microglial response accompanying α‐syn spreading, we generated a gene expression profile from ventral midbrain of PFF injected and control mice using the Affymetrix gene expression profiling platform. Because microglial response typically starts early after an insult (Michelucci et al., 2018; Tansey & Romero‐Ramos, 2019), we analyzed the midbrain gene expression profiles 13 dpi (no neurodegeneration) and 90 dpi (neurodegeneration in the ipsilateral striatum and midbrain) after striatal α‐syn PFF injection.
We focused on two comparisons of ventral midbrain gene expression profiles:
Ipsilateral midbrain of PFF‐injected mice (ipsi PFF, with degeneration of nigral TH neurons and their striatal projections) versus ipsilateral midbrain of control PBS‐injected mice (ipsi PBS);
Ipsilateral midbrain of PFF‐injected mice versus contralateral midbrain of the same, PFF‐injected, mice (contra PFF, without loss of nigral TH neurons and their striatal projections).
We figured that these two comparisons would be best suited to reveal relevant gene expression changes.
Venn diagrams for the number of DEGs that emerged in the comparisons between the two time (13 and 90 dpi) are shown in Figure S4. The heatmaps are shown in Figure S5. By comparing ipsi PFF to ipsi PBS, applying a cut‐off of p < .05, we found a total of 3.584 significant DEGs at 13 dpi, and significant 2.990 DEGs at 90 dpi, with 960 overlapping DEGs between the two time points. After correcting for multiple hypothesis testing at a cut‐off of pfp < 0.1, we found 308 DEGs at 13 dpi, and 94 DEGs at 90 dpi, with 37 DEGs overlapping between the two times points. The majority of overlapping DEGs showed enhanced expression at 13 dpi, but reduced expression at 90 dpi. By comparing ipsi PFF to contra PFF, we found 4.268 significant DEGs at 13 dpi, and 3.976 DEGs at 90 dpi, with 1312 overlapping DEGs. At pfp < 0.1, we found 674 DEGs at 13 dpi, and 688 DEGs at 90 dpi, with 210 overlapping DEGs. At 13 dpi, we found a similar number of DEGs with enhanced versus reduced expression, but at 90 dpi, we saw that most DEGs were, interestingly, had reduced expression. To capture all potentially relevant molecular changes, we used the all DEGs with p < .05 for the subsequent analyses.
Taken together, these two comparisons indicate that enhanced gene expression changes occurred in the ventral midbrains of both hemispheres at 13 dpi, possibly setting the stage for the subsequent pathological events. In contrast, at 90 dpi, in the ipsilateral midbrain, most DEGs dial their expression level back, indicating a reduction in gene transcription, while the major pathological events now appear to take place at the protein level, and are measurable with quantitative histology (see above).
3.8. Gene set enrichment revealed early involvement of inflammation in the α‐syn seeding/spreading process
To investigate which molecular pathways underlie the α‐syn spreading process and its associated pathologies, in particular microgliosis, we generated an enrichment map based on a Gene‐Set Enrichment Analysis (GSEA, see Materials and Methods, Section 2) for Biological Processes (BP, see Materials and Methods, Section 2). To obtain a global view of the BPs alterations during the evolution of α‐syn spreading induced pathologies, we used manual curation to group gene sets into biologically meaningful gene‐set clusters associated with high order pathological processes (Figure 6).
FIGURE 6.
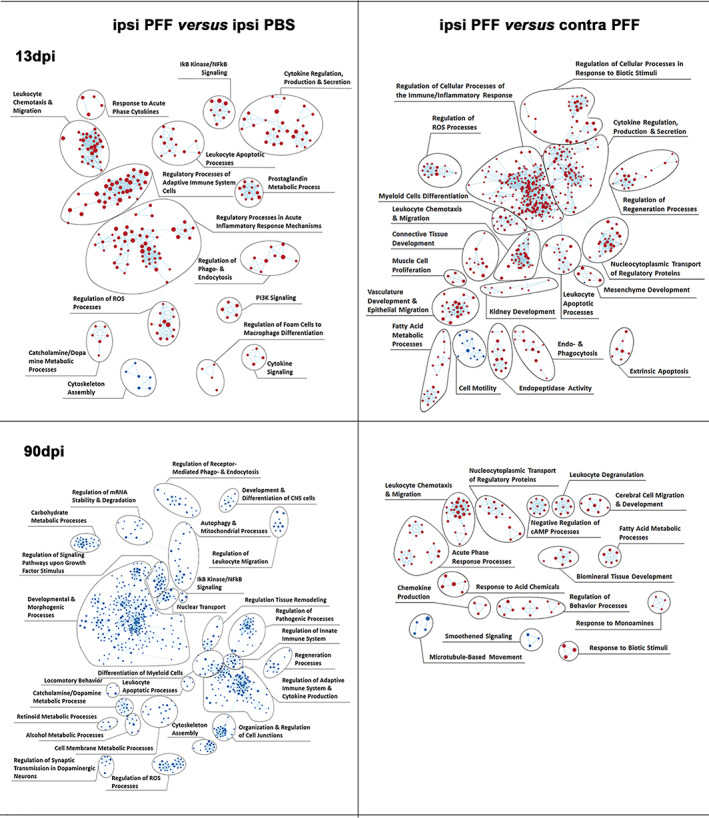
Enriched inflammatory pathways precede neurodegeneration in mouse ventral midbrains after intrastriatal α‐syn PFFs injection. Enrichment map of gene expression profiles were derived from GSEA. Statistics were done by weighted Kolmogorov–Smirnov, gene set size limits were set to min15 – max250. Details of curation procedure used to group BPs (represented as dots, either red if upregulated, or blue if downregulated) into high‐level functional gene set clusters of BPs of related biological function are described in Material & Methods. At 13 dpi, comparing ipsi PFF to either ipsi PBS or contra PFF, most BPs were upregulated and associated with gene sets related to immune and inflammation processes. This shows that, in the ipsilateral nigro‐striatum, neuroinflammation precedes neurodegeneration (measurable at 90 dpi), and might contribute to its development. At 90 dpi, comparing ipsi PFF to ipsi PBS, all BPs, including those associated with inflammation gene sets, were downregulated, possibly reflecting the neurodegenerative process itself. Comparing ipsi PFF to contra PFF at this time point, most BPs were upregulated
Our first observation was that, in ipsilateral midbrains of PFF‐injected mice compared to those of PBS‐injected ones, 261 BPs were enriched at 13 dpi, but, surprisingly none at 90 dpi. In contrast, we observed that, at 90 dpi, all BPs in ipsilateral midbrains of PFF‐injected mice versus those of PBS‐injected ones (total of 1067 BPs), showed reduced gene activity. This observation indicates a significant shift from enhanced to greatly reduced transcriptional activity in the time frame between 13 and 90 dpi, and confirms the observations on DEGs depicted in Figure S5.
We then observed that many gene set clusters with enhanced transcriptional activity at 13 dpi in ipsi PFF were associated with inflammation/immune processes (Figure 6, upper panels), while gene set clusters associated with similar profiles had reduced transcriptional activity at 90 dpi, in particular compared to ipsi PBS (Figure 6, lower left panel). This indicated that, after an initially enhanced activity of genes regulating inflammation/immune responses, that activity was strongly reduced at a stage when pathology was histologically detectable.
Another interesting observation we made was that some gene set clusters containing BPs associated with reduced gene activity at 90 dpi, were related to dopaminergic neuron function (e.g., catecholamine/dopamine metabolic processes, locomotor behavior, regulation of synaptic transmission regulation of signaling pathways upon growth factor stimulus). The reduced gene activity in midbrain dopaminergic neurons was likely a reflection of their pathological demise.
Taken together, these observations point to an important role for inflammatory/immune processes in the initiation and, possibly, progression of neurodegeneration in the context of α‐syn spreading.
3.9. Gene expression changes based on cell type confirmed early microglial response to α‐syn seeding/spreading
To identify the immune cell type(s) active in the inflammatory response to α‐syn seeding/spreading, we attributed all DEGs of ipsi PFF versus ipsi PBS and ipsi PFF versus contra PFF to cell types, using a widely used public database (see Materials and Methods, Section 2). We found that the majority of DEGs with increased expression at 13 dpi were microglial, whereas many of these DEGs had decreased expression at 90 dpi (Figure 7a).
FIGURE 7.

(a) Deconvolution of cell types for DEGs at 13 and 90 dpi after striatal injection of α‐syn PFFs. Many DEGs were expressed in multiple, but not all cell types. DEGs that were expressed in all cell types are named “pan‐cellular”. DEGs expressed in related cells (e.g., “multiple glia”, “multiple oligodendrocytes”), have been grouped for simplicity. The majority of DEGs, at 13dpi, expressed uniquely by one cell type were in microglia. (b) Top 20 DEGs in mouse ventral midbrain after striatal injection of α‐syn PFF indicate involvement of microglia in initial pathological events. At 13 dpi, comparing ipsi PFF with ipsi PBS or contra PFF, 45% and 40%, respectively, of the top 20 DEGs were microglial. At 90 dpi, comparing ipsi PFF with ipsi PBS, 50% of top 20 DEGs were neuronal, possibly a reflection of neurodegeneration. The bottom panel lists the gene products of the gene symbols, coded proteins, the associated cell type, the fold change (FC) and the pfp of all top 20 DEGs for each comparison
We also looked at the 20 top DEGs and their cellular source for each time points after PFF injection (Figure 7b). At 13 dpi, in both the ipsi PFF versus ipsi PBS as well as the ipsi PFF versus contra PFF comparison, we observed that the majority of these 20 DEGs with enhanced expression were microglial (ipsi PFF versus ipsi PBS: 9 out of 20, or 45%, ipsi PFF versus contra PFF: 8 out of 20, or 40%). This indicates a strong gene expression activity of these cells, well before pathological changes can be detected histologically. In contrast, at 90 dpi, we observed that only 1 out of 20 (5%) DEGs was microglial in both comparisons (ipsi PFF versus ipsi PBS, ipsi PFF versus contra PFF). Here, the majority (>50%) of DEGs in the ipsi PFF versus ipsi PBS comparison were neuronal.
The observation that the majority of DEGs at 13 dpi were microglial confirmed an early response of these cells to α‐syn spreading.
3.10. Unusual microglial molecular signature, induced by striatal injection of α‐syn PFFS, preceded neurodegeneration
Recent studies using transcriptomics technologies have shed light on microglial activation profiles at baseline and in disease models. In one study (Uriarte Huarte et al., 2021), single‐cell transcriptomics in mouse nigro‐striatal brain regions revealed an enrichment of a microglia subtype with an “immune‐alerted” expression signature of 68 genes, and 2 other subtypes (homeostatic, intermediate) with a much smaller number of signature genes. A list of the DEGs in our study that overlap with the genes for these microglial subtypes is given in Table S3. At 13 dpi, 20 microglial ipsi PFF versus ipsi PBS DEGs, and 21 microglial ipsi PFF versus contra PFF DEGs were of the “immune‐alerted” class. In contrast, at 90 dpi, the corresponding numbers were 9 microglial DEGs for ipsi PFF versus ipsi PBS, and 7 for ipsi PFF versus contra PFF. At both time points, the number of DEGs for other microglial subtypes (homeostatic, intermediate) was, for both comparisons, less than 5 (Table S3). This indicated a predominant participation of “immune‐alerted” microglia in the α‐syn seeding/spreading model.
In neurological disease and models thereof, the term “disease‐associated microglia” (DAM) defines molecular microglial states, as reflected by a gene expression signature, that are associated with these diseases and may modulate their pathologies (Hakim et al., 2021; Hammond et al., 2019; Keren‐Shaul et al., 2017; Mathys et al., 2017; Mrdjen et al., 2018; Tay et al., 2018; Uriarte Huarte et al., 2021). Some DAM genes show expression changes common to many diseases, while others may only change in a specific disease, or at specific time points across one or many diseases. There is little information on such microglial states for PD.
It is beyond the scope of our study to compare our data with all the available DAM gene expression profiles. One study though (Holtman et al., 2015) presents a meta‐analysis of aged mice and four different mouse neurological disease models. We compared our DEG datasets to each of these models, and the Venn Diagrams are shown in Figure S6. We then extracted the overlapping microglia genes from the five models in that study, and found a core DAM signature of 50 genes. We compared our datasets with those DAM core genes and found that, at 13 dpi, almost double as many genes overlap with these core genes than at 90 dpi. Venn diagrams illustrating these results are shown in Figure 8, and the list of overlapping genes are found in Table S4. Further studies, in particular on single microglial cells, will have to determine how that number changes over the course of the disease.
FIGURE 8.
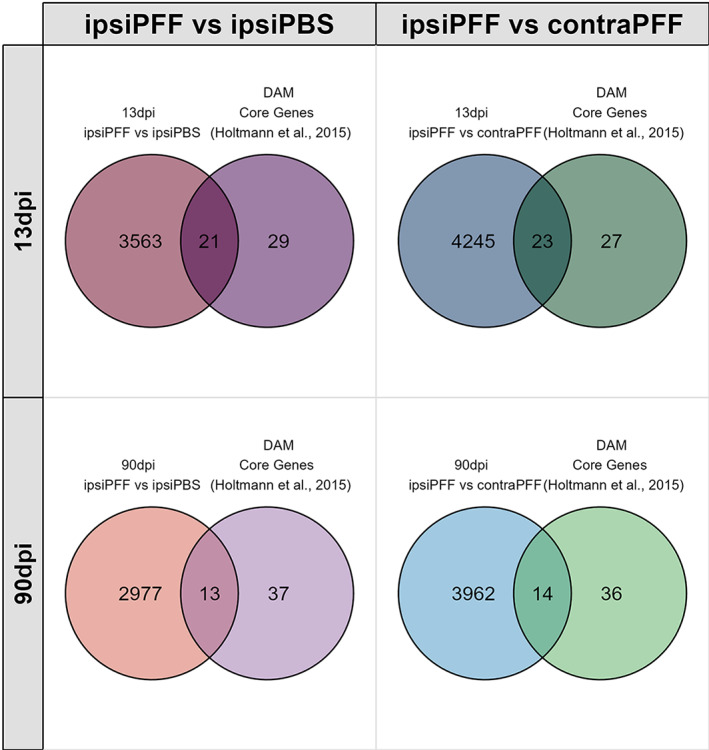
Venn diagrams of DEGs 13 and 90 dpi after striatal injections of α‐syn PFFs overlapping with a core gene signature of microglia associated with five different mouse neurological disease models (disease‐associated microglia, DAM). More DEGs at 13dpi overlap with the core DAM signature than at 90 dpi. See main text for details
To look at more unique characteristics of the microglial gene signature in the α‐syn seeding/spreading model, we looked at individual genes typically associated with inflammation during neurodegeneration (Table S5). Curiously, there were no changes, at 13 or 90 dpi, in the expressions of Il1b, Il6, Ccl2, Tnfa, or Nos2, genes that are often involved in inflammation. Gene expression was enhanced for Cybb, Ptgs2, and Cxcl10. NADPH oxidase 2 (Nox2), coded by Cybb, generates free oxygen radicals, which can harm neurons (Ma et al., 2017). Cyclooxygenase 2 (Cox2), coded by Ptgs2, generates arachidonic acid metabolites, some of which have been reported to be neurotoxic (Figueiredo‐Pereira et al., 2016) or form neurotoxic dopamine‐quinone adducts (Teismann et al., 2003). Thus, this may be the mediators of the neurodegeneration observed in ipsi PFF midbrains at 90 dpi. Other microglial activation markers whose products may be involved in promoting pathology were Mrc1, Cd68, Tyrobd, Trem2, Tlr2, P2ry6, and Aif1, which showed increased expression at 13 dpi and/or 90 dpi. Mrc1, Cd68, P2ry6, Aif1 gene products are all involved in phagocytic processes and/or signal transduction (Bhattacharya & Biber, 2016; Janda et al., 2018; Ransohoff & El Khoury, 2015). We had observed CD68 upregulation by immunostaining in the striatal projection area of dopaminergic neurons (Figure 3), indicating that the arrays' results reflect actual gene product changes. The Tlr2 gene product is a receptor for α‐syn, an interaction that elicits the production of microglial neurotoxins (Kim et al., 2016). Tyrobp and the gene for its receptor, Trem2, whose product is involved, among other processes in the regulation of microglial phagocytosis (Konishi & Kiyama, 2018), also showed enhanced expression.
Finally, to see if there was an astroglial and peripheral immune cell involvement in the α‐syn seeding/spreading model, we listed gene expression data for typical markers of these cells from our gene expression dataset (Table S6). Enhanced expression of a series of astroglial genes in ipsi PFF midbrain indicates a reaction of these cells. Enhanced expression of Ptprc, which codes for CD45, a marker that can be both expressed by microglia and invading macrophages, and of Cd4, which codes for the helper T cell antigen CD4, in the same region, indicated possible infiltration of peripheral immune cells that could contribute to neuronal injury (Brochard et al., 2009; Harms et al., 2017; Kannarkat et al., 2013).
Overall, we tentatively conclude that a unique molecular signature in the ipsilateral ventral midbrain at 13 dpi underlies the initial molecular events that lead to the neurodegeneration we observed at 90 dpi. Since neurodegeneration in the contralateral SN has been reported at later time points after PFF injection previously (Luk et al., 2012), in one study even in the absence of α‐syn inclusions (Paumier et al., 2015), one can speculate that, at a point past 13dpi, the same molecular signature appears there as well.
Our data indicate that inflammatory events, in particular those associated with microglia, but not α‐syn inclusion formation, could be initiators of neurodegeneration in the context of α‐syn spreading in the model used in our study.
3.11. Translational relevance assessment
Finally, we evaluated the translational value of the main pathological changes we observed in the α‐syn seeding/spreading model in this study. Pathologically, Braak staging indicates α‐syn spreading (Braak et al., 2004), and the presence of important microgliosis, detectable by medical imaging, are happening at early stages of PD (Terada et al., 2016). Hence, at that level of investigation, evidence suggests that the α‐syn seeding/spreading model holds translational value. There is however, to our knowledge, no studies systematically comparing the transcriptional profile of the ventral midbrain in the α‐syn seeding/spreading model to that of the SN in different stages of PD. There are numerous studies describing transcriptional profiles of the SN in PD, and comparing them all to our data is beyond the scope of this study. We therefore picked two studies that seemed particularly pertinent: one study lays out the transcriptional profiles of different Braak stages in PD (Dijkstra et al., 2015), and another one presents a meta‐analysis study of end‐stage PD studies (Glaab & Schneider, 2015).
We limited our analysis to comparing DEGs between the ipsi PFF versus ipsi PBS brain sides with human databases, since we did not expect the comparison of ipsi PFF versus contra PFF, (done to reveal molecular features of activated microglia in the presence (ipsi PFF) versus the absence (contra PFF) of neurodegeneration) with those databases to reveal translationally meaningful information. Venn diagrams of overlapping DEGs of the α‐syn model with different stages of PD were not very informative, as they revealed anywhere from around 300 to a bit less than 700 overlaps (Figure S6). Overlapping DEGs revealed no pattern and were seemingly random (list not shown).
In studies comparing mouse models of inflammatory diseases with their human counterparts, analysis of overlapping DEGs also show poor similarities (Seok et al., 2013). However, a follow‐up meta‐analysis, looking at pathways and biologically meaningful gene sets, revealed great similarities between the same mouse models and human inflammatory conditions (Takao & Miyakawa, 2015). Similar observations were made when comparing transcriptome profiles of Alzheimer's disease mouse models with those of Alzheimer brains (Wan et al., 2020). We thus proceeded to investigate overlapping pathways between the α‐syn seeding/spreading model and different stages of PD. To do this, we used the PD map online tool (Fujita et al., 2014) (see Materials and Methods, Section 2). By selecting pathways that were significantly represented in at least one of the human databases, as well as in at least one of the two mouse model datasets, we found 15 highly PD‐relevant pathways that were shared between the model and PD (Figure 9). Among those, “neuroinflammation” (highest score) and “microglial phagocytosis” were noteworthy, reinforcing the role of microglia in this PD mouse model and in human. While the pathways identified in the mouse model were, perhaps not unexpectedly (see Discussion, Section 4), not completely overlapping in extent and directionality of change, the similarities were surprisingly close. We thus tentatively conclude that the α‐syn seeding/spreading mouse model of PD provides a valuable preclinical tool to study disease mechanisms, as well as the efficacy of experimental therapeutics, in particular if they relate to microglial‐mediated neuroinflammation.
FIGURE 9.
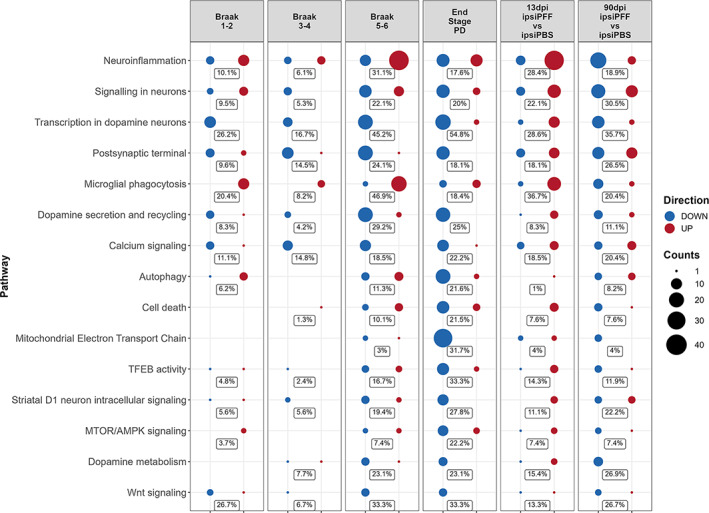
Translational relevance assessment of gene expression changes at 13 and 90 dpi after striatal injection of α‐syn PFFs. The PD map (https://pdmap.uni.lu/minerva/) was used to extract pathways from human databases (first 4 columns) and from the DEG datasets of this study (two last columns). The percentages in each row give the proportion of genes in each pathway that were found as DEGs in each dataset that were mapped. There were strong similarities between human PD gene expression changes and those found in the α‐syn seeding/spreading model. See main text for details
4. DISCUSSION
In this study, we have used a seeding/spreading model of α‐syn, based on striatal injection of α‐syn PFFs in the mouse brain, to investigate key questions on how two major pathological features of PD, α‐syn inclusion formation and neuroinflammation, contribute to neurodegeneration. Using a transcriptomics approach, we also shed light into the molecular underpinning the disease process in this model. We provide evidence that:
α‐syn inclusion formation did not correlate with neurodegeneration in areas with inclusions;
An exceptionally strong microglial response was seen across different brain regions, but this response did also not correlate with neurodegeneration;
The most likely driver of the microglial response were diffusible α‐syn oligomers;
Gene expression changes indicative of early neuroinflammatory events in the ventral midbrain, in particular in microglia, appeared before nigro‐striatal degeneration, and microglial factors could be the driver for downstream neurodegeneration;
A translational relevance analysis revealed that 15 key molecular pathways that change in the model overlap with those found altered in PD, with neuroinflammation scoring at the top. Our study provides novel insights into underlying pathological processes of α‐syn spreading mediated PD‐like neuronal injury, and stress the value of the α‐syn spreading model for investigation of disease mechanisms and preclinical testing of therapeutics.
We undertook this study because it is unclear how different pathological processes relate to each other in PD. In particular, it is debated whether α‐syn inclusion formation is the main driving force in disease initiation and progression, or whether other processes, such as neuroinflammation are (Brundin & Melki, 2017; Surmeier et al., 2017a; Walsh & Selkoe, 2016). The Braak hypothesis (Braak et al., 2003) posits that α‐syn inclusion pathology starts in lower motor nuclei of the brainstem (e.g., Dorsal Motor Nucleus of the Vagus), or even in the PNS, then gradually moves upwards and, in doing so, causes various PD symptoms, from early non‐motor to later motor and cognitive and psychiatric ones, to appear.
More direct evidence for the importance of α‐syn spreading in inducing PD‐like disease comes from experimental models. In rodent or primate models, direct injection, in different brain regions, of PD brain tissue, isolated Lewy bodies, PFF made out of recombinant α‐syn, or viral vector driven local overexpression of α‐syn, induces a variety of PD‐related pathologies, including α‐syn spreading and inclusion formation along connected neurons (Rey et al., 2016). Peripheral PFF injections, such as intramuscular, intestinal, or intravenous have been reported to also lead to PD‐like pathologies in the brain of rodents (Ayers et al., 2017; Holmqvist et al., 2014; Peelaerts et al., 2015; Sacino et al., 2014). These studies have cemented, experimentally, the process of “prion‐like” propagation and inclusion formation of α‐syn.
The mechanism of this process has been investigated in in vitro systems. Cultured neurons secrete as well as take up circulating α‐syn, and various underlying mechanisms have been proposed, such as unusual forms of endo‐ and exocytosis, or nanotubes (Rodriguez et al., 2018; Vasili et al., 2019). Ingested, presumably misfolded α‐syn, corrupts its endogenous counterpart and leads it to form pathological inclusions (Spillantini & Goedert, 2018). Glial cells have also been reported to take up α‐syn, and, in some cases, this can lead to pathological inclusions, as is the case for oligodendrocytes in Multiple System Atrophy (Filippini et al., 2019). Microglia, in particular, have recently been shown to uptake and degrade abnormal α‐syn, a process facilitated by through the formation of intercellular connections that distribute the “work‐load” through a microglial cellular network (Scheiblich et al., 2021).
The toxic potential of inclusions has also been investigated in vivo. In a mouse model of α‐syn spreading, where inclusion formation was followed in vivo by multi‐photon laser microscopy, the formation of intraneuronal inclusion was reported to coincide with neuronal dysfunction (Osterberg et al., 2015). Another study has shown a weak correlation between loss of TH neurons in the SN and a global score of inclusion load after striatal PFF injection in both mice and rats, but a strong correlation between the two measures after direct injection of PFFs into the SN of rats (Abdelmotilib et al., 2017). Thus, it is tempting to conclude that α‐syn inclusions are a major driver of PD pathology. But a closer look at other evidence reveals several unresolved questions in this otherwise elegant picture. In post‐mortem brain tissues of early or late PD, the correlation between α‐syn inclusion (Lewy body) load and nigral degeneration is unclear (Jellinger, 2009a, 2009b). Across different studies looking at various brain structures affected in PD, α‐syn inclusions have been reported in areas with high, moderate, or no neuronal loss (Jellinger, 2009b). Some PD patients, including some familial forms, have PD symptoms and loss of nigral neurons without detectable α‐syn inclusions (Surmeier et al., 2017b). Interestingly, one study, comparing Incipient Lewy Body Disease (ILBD) to PD autopsy material, reported that neuronal loss precedes α‐syn inclusion formation in the SN (Milber et al., 2012). In mice injected with α‐syn PFFs into the pedunculopontine nucleus, appearance of inclusions was not related to strength or extent of neuronal connection of that brain region (Henrich et al., 2020). In a rodent models where spreading is driven by viral overexpression of α‐syn in the Dorsal Motor Nucleus of the Vagus, while intact neuronal architecture was essential for the spreading process to happen, neurodegeneration and inclusion formation were also found to be independent processes (Ulusoy et al., 2015).
Non‐fibrillar forms of misfolded α‐syn, notably oligomers, diffusing for long distances have been suggested to drive neuronal dysfunction and degeneration (Bengoa‐Vergniory et al., 2017; Walsh & Selkoe, 2016). In our study, we indeed found evidence of neurodegeneration that was independent of inclusions, and, in the hippocampus, even appeared in the complete absence of them, but in the presence of oligomers. Published evidence suggests that the hippocampus remains devoid of α‐syn inclusions even 180 days after PFF injection into the striatum (Luk et al., 2012). Our data therefore does not support the notion of a direct relationship between the formation of α‐syn inclusions and neurodegeneration, but rather indicate that the α‐syn spreading process may lead to the formation of pathological oligomers, at least in the early stages of the disease.
Pathologically misfolded α‐syn can drive neuronal injury in PD by different means, including mitochondrial dysfunction, oxidative stress, endoplasmic reticulum stress and lysosomal dysfunction, disequilibrium in cytosolic Ca2+, neurotoxic oxidized dopamine, disruption of axonal transport, and, notably, neuroinflammation (Rocha et al., 2018). The relative contribution of these different processes to neuronal demise is unclear. Neuroinflammation has received particular attention because of its widespread involvement in various neurological diseases and the potential for therapeutic modulation (Hirsch & Standaert, 2020; Lema Tome et al., 2013; Tansey & Romero‐Ramos, 2019). The major cellular mediators of this process are microglia. Microglia are a particular kind of myeloid cells that originate from the yolk sack and populate the nervous system during early stages of development, where they act as the innate, resident immune cells (Michelucci et al., 2018; Ransohoff & El Khoury, 2015). During development and under normal conditions, they modulate nervous system homeostasis, prune synapses, and regulate their formation. Under pathological conditions, they act as the primary line of defense against infectious organisms, and clear endogenous tissue debris after injury (Michelucci et al., 2018; Ransohoff & El Khoury, 2015; Wolf et al., 2017). They undergo a substantial morphological and functional transition to activated, or reactive, microglia, which makes them functionally equivalent to macrophages (Michelucci et al., 2018). Evidence suggests though that, in many neurological conditions, they are not only reacting to disease, but also drive tissue injury (Ransohoff & El Khoury, 2015). This pathological process is, in particular in PD, incompletely understood. While microglial activation can be induced by neuronal injury and/or misfolded and aggregated protein, notably α‐syn oligomers or fibrils (Fellner et al., 2013), it is still unclear how and when microglial activation damages healthy tissue and exacerbates the neurological disease process. In PD, a strong microgliosis is observed post mortem in the SN (Croisier et al., 2005; Joers et al., 2017). Longitudinal imaging studies with PET ligands demonstrated an early microglial activation in various regions beyond the SN, such as cortex, hippocampus, basal ganglia, and pons, but no correlation with other pathological measures, including clinical scores, of PD emerged (Gerhard et al., 2006; Terada et al., 2016). Interestingly, in striatal fetal grafts implanted in PD patients (Chu & Kordower, 2010), microglia activation was observed years before the appearance of α‐syn inclusions (Olanow et al., 2019). In different toxin‐induced PD rodent models, microgliosis was reported to precede, coincide, or follow the appearance of neuronal demise (Joers et al., 2017), while in a transgenic human α‐syn model (Watson et al., 2012), and in rats injected with PFFs into the striatum (Duffy et al., 2018), microgliosis, measured histologically, was shown to precede neurodegeneration.
These studies are based mainly, if not exclusively, on the observation of morphological changes of microglial response using immunostaining techniques for generic cell markers. While informative, the detection of morphological changes indicating microglial activation does not yield enough information on the actual physiological or molecular profile of these cells. Microglia commonly have a spectrum of activation states that may change over the course of the disease (Ransohoff, 2016). Recent gene expression profiling approaches have revealed a bewildering complexity in microglial heterogeneity (Crotti & Ransohoff, 2016; Dubbelaar et al., 2018; Uriarte Huarte et al., 2021). Evidence suggests a “core” gene expression profile response that is associated with every neurodegeneration condition, while expression changes of a more restricted set of genes may be specific for each condition, leading to the concept of disease‐specific microglial signatures, or “disease‐associated microglia” (DAM) (Dubbelaar et al., 2018). Our study provides new insights into the molecular underpinnings of neuroinflammation preceding neuronal injury in the PD‐like context of α‐syn spreading, and highlights a microglial gene signature that may drive neurodegeneration. First, we show, at the level of gene expression, that neuroinflammation‐linked processes were activated, and that many microglial genes had increased expression levels early (13 dpi), which then were down‐regulated later (90 dpi) after PFF injection. Microglia genes that code for factors that could cause neurodegeneration showed increased expression 13 dpi only in the ipsilateral midbrain, where TH loss was observed later, at 90 dpi. Among these were Cybb, which codes for NAPDH oxidase 2, an enzyme that catalyzes the production of tissue harming free radicals (Ma et al., 2017), and Ptgs2, which codes for cyclooxygenase 2 (Cox2), an enzyme that forms prostanoids from arachidonic acid, some of which are neurotoxic (Figueiredo‐Pereira et al., 2016; Hsieh et al., 2011). Tlr2, Trem2, and Tyrobp RNAs showed increased levels in our model at 13 dpi. Many genes linked to microglial activation are regulated by Tyrobp, a tyrosine kinase binding protein that binds to Trem2. The Tyrobp/Trem2 pair triggers pathways that are involved in the inhibition of TLR‐mediated inflammation, and in the modulation of phagocytosis (Konishi & Kiyama, 2018). In prodromal PD, enhanced TLR2 immunoreactivity on microglia was observed, whereas in late stage PD, it wasn't (Doorn et al., 2014), indicating that, just like in our model, the microglial response happens in early phases of the disease and changes over time. Alpha‐syn, in particular in its oligomeric form, activates microglia in vitro through Toll‐like receptors (Fellner et al., 2013; Kim et al., 2013), and targeting TLR2 by immunotherapy was shown to be beneficial in α‐syn pathology models (Kim et al., 2018).
The absence of increased gene expression of common pro‐inflammatory mediators such as Il1b and Tnfa in our α‐syn spreading model is puzzling, since these are factors associated with most, if not all, inflammatory conditions. Of note though is that we also did not observe enhanced expression of these factors when primary microglia were exposed to our α‐syn PFFs, while they responded strongly to bacterial lipopolysaccharide (not shown). It is possible tough that the increased expression for these genes was missed and occurs at a time point after PFF injection that we have not looked at. Future studies will have to focus on isolated microglia and use the latest transcriptional technology in a longitudinal fashion in this model to better understand the specific microglial response, in particular the response of microglial subtypes.
It is crucial to demonstrate the translational relevance of a rodent model by drawing parallels between the disease manifestations and mechanisms observed in it with those found in human. Our comparative analysis of molecular pathways altered in the α‐syn seeding/spreading model revealed several overlaps of disease‐relevant pathways with the pathways that change in different stages of PD. There was no absolute 1–1 overlap of any of the two mouse datasets with any of the human datasets, but this may not be surprising. One of the limitations of rodent models is that studies are typically done in inbred strains (genetically identical subjects that are homozygotes in all loci), whereas humans are genetically quite heterogeneous. One can thus expect more variation in human than in the typical inbred strain mouse model. Another one is that, in the field of chronic neurological diseases, it is necessary to compress a disease that in human takes decades to develop into the comparatively short lifespan of a mouse. The similarities we saw though between the α‐syn seeding/spreading model and PD are encouraging.
Taken together, our data indicate that, at least in the initial period of PD‐like disease progression that is associated with α‐syn spreading, non‐deposited pathological forms of α‐syn, such as oligomers, may drive neurodegeneration in different brain regions via their action on microglia. Activated microglia respond early, before neurodegeneration is apparent, by producing potentially neurotoxic compounds. Our findings contribute toward answers to unresolved questions around neuroinflammation in PD (Hirsch & Standaert, 2020), and have important implications for the design of therapeutic interventions during the early stages of the disease.
CONFLICT OF INTEREST
Andreas Weihofen is affiliated with Biogen. The author has no financial interests to declare. Ahmad Allouche and Nicolas Fischer were affiliated with SynAging SAS, now with ETAP‐labs. These authors have no financial interests to declare. Annette Masuch is currently affiliated with Anklam Extrakt GmbH. The author has no financial interests to declare. Knut Biber is currently affiliated with AbbVie Pharmaceutical Research and Development. The author has no financial interests to declare.
AUTHORS CONTRIBUTION
Pierre Garcia, Wiebke Jürgens‐Wemheuer, Djalil Coowar, and Manuel Buttini, designed the study. Pierre Garcia, Wiebke Jürgens‐Wemheuer, Oihane Uriarte Huarte, Annette Masuch, Simone Brioschi, Eric Koncina, Tony Heurtaux, Andreas Weihofen, Carole Sousa, Alessandro Michelucci, Felix Kleine‐Borgmann, Tatjana Pfander, Ahmad Allouche, Nicolas Fischer, and Manuel Buttini did the experiments (stereotactic surgery, tissue processing, stainings, imaging, RNA extraction). Tony Kaoma, Nathalie Nicot generated the microarray data. Kristopher J Schmit, Enrico Glaab analyzed the microarray data. Pierre Garcia, Wiebke Jürgens‐Wemheuer, Oihane Uriarte Huarte, Kristopher J Schmit, Rudi Balling, Walter Schulz‐Schaeffer, Knut Biber, Michel Mittelbronn, Manuel Buttini analyzed and interpreted the data. Kristopher J. Schmit and Marek Ostaszewski did the transitional relevance analysis. Manuel Buttini wrote the paper. All authors read and approved the final manuscript. All authors have approved of the contents of this manuscript and provided consent for publication.
ETHICS STATEMENT
Animal studies performed at the Luxembourg Centre for Systems Biomedicine were approved by the institutional Animal Experimentation Ethics Committee of the University of Luxembourg, and the responsible Luxembourg government authorities (Ministry of Health, Ministry of Agriculture). Alternatively, experiments done at the SynAging site were approved by ethics committee “Comité d'Ethique Lorrain en Matière d'Expérimentation Animale”, and by the governmental agency the “Direction Départementale de la Protection des Populations de Meurthe et Moselle‐ Domaine Expérimentation Animale”. All followed EU directive 2010/63/EU.
Supporting information
Figure S1 Upper panel: Western blot characterization of α‐syn moities used for intrastriatal injections. Loading of the gel was as follows: lanes 1, 6, 14 – ladder; lanes 5, 10,11‐ blank; lanes 2,3,4 – α‐syn oligomer, 10, 100, and 500 ng respectively; lanes 7, 8, 9 – α‐syn PFFs, not sonicated, 10,100, 500 ng respectively; lanes 11, 12, 13 – α‐syn PFFs (sonicated), 10,100, 500 ng respectively. Bands from corresponding to the MW of α‐syn monomers, dimers, and trimmers are circled (red, blue, orange, respectively). Note the presence of high molecular weight moities (visible as a smear) after loading of non‐sonicated PFFs. The sonication process of PFFs appears to reduce all higher molecular weight species of α‐syn and the amount of dimers and monomers, since smears and monomer/dimer bands were visible after loading 100 ng of non‐sonicated PFFs, but not after loading the same amount of sonicated PFFs. Lower panels: Electron microscopy of α‐syn moities. A: Oligomers; B: unsonicated α‐syn PFFs; C: sonicated α‐syn PFFs. Oligomers were primarily composed of small globular moities. Unsonicated PFFs contained many large assemblies of α‐syn fibrils, whereas sonicated PFFs were composed mainly of small fibrillar fragments, resulting form the sonication‐induced disruption of the large assemblies. Scale bar = 152 nm.
Figure S2 Minimal presence of α‐syn inclusions in different brain regions 13 DPI after intrastriatal injection of α‐syn PFFs. Only few α‐syn inclusions were seen, ipsi‐ and contralaterally, in the frontal cortex, amygdala, amd ipsilaterally, but not contralaterally, in the striatum and SN. Scale bar = 130 μm.
Figure S3 No nigro‐striatal degeneration and microgliosis 13 dpi after intrastriatal injection of α‐syn PFFs. No loss of striatal TH‐positive axons (first row), striatal DAT‐positive synaptic terminals (second row), nigral TH‐positive neurons (third row) was observed 13 dpi after injection of PFFs. No increase of Iba1‐positive microglial reaction in the SN was observed (last row). Microphotographs show examples of PBS‐injected control brains (ipsilateral) and ipsilateral α‐syn PFF‐injected brains. Scale bars = 25 μm (striatal panels), 200 μm (Subst. Nigra panels).
Figure S4 Venn diagrams of DEGs induced in the ventral midbrain of mice at 13 dpi and 90 dpi after striatal injections of α‐syn PFFs. Comparisons for each time point were made between DEGs in ipsilateral ventral midbrain of PFF‐injected mice versus those in the ipsilateral ventral midbrain for PBS‐injected mice (ipsi PFF vs. ipsi PBS, n = 6/group; upper 4 panels), and between DEGs in ipsilateral ventral midbrain versus the contralateral ventral midbrains of PFF‐injected mice (ipsi PFF vs. contra PFF, n = 6/group; lower 4 panels). Diagrams on the left show Venn diagrams with numbers of DEGs with a significance level of p < .05 (n = 6 mice/group). Because some gene products have an effect within biological pathways while the expression of their genes may only change minimally, we show DEGs that emerge with this low level of statistical stringency. In ipsi PFF vs. ipsi PBS, the number of DEGs was 2631 at 13 DPI, and 2584 at 90 DPI, with 985 DEGs common to both time points. In ipsi PFF vs. 3584 at 13 DPI, and 2990 at 90 DPI, with 960 DEGs common to both time points. In ipsi PFF vs. contra PFF, the number of DEGs was 4268 at 13 DPI, and 3976 at 90 DPI, with 1312 DEGs common to both time points. Panels on the right show Venn diagrams with the number of DEGs after adjusting for multiple hypothesis testing at pfp < 0.1. In ipsi PFF vs. ipsi PBS, the number of DEGs was 308 at 13 DPI, and 94 at 90 DPI, with 37 DEGs common to both time points. In ipsi PFF vs. contra PFF, the number of DEGs was 674 at 13 DPI, and 688 at 90 DPI, with 210 DEGs common to both time points. The Venn diagrams below the main ones indicate the number of DEGs that show enhanced (“up”) versus decreased (“down”), as well as the overlaps, in the different comparisons.
Figure S5 Heatmaps illustrating the ventral midbrain gene expression patterns at 13 dpi and 90 dpi after intrastriatal injection of α‐syn PFFs. Heatmaps are shown for p‐values <0.05 or pfp < 0.1. Scaled row expression values (row Z‐score) in red indicate higher, in blue lower, and in white unchanged gene expression (see Materials and Methods for details). Comparisons of ipsi PFF versus ipsi PBS (upper panel row) and ipsi PBS versus contra PFF (lower panel row) reveal distinctive gene expression patterns at 13 dpi for both comparisons (whether higher, pfp < 0.1, or lower, p < 0.05, statistical stringency is used), whereas a distinctive pattern at 90 dpi only appears in the ipsi PFF versus contra PFF, but not in the ipsi PFF versus ipsi PBS, comparisons.
Figure S6 Venn diagrams of DEGs, induced in the ventral midbrain of mice at 13 dpi and 90 dpi after striatal injections of α‐syn PFFs, overlapping with DEGs of isolated microglia from different neurological disease models. APP stands for Alzheimer model mice, ERRCC1 mice are a model of premature aging, SOP1 mice are a model for amyothrophic lateral sclerosis, and LPS are mice injected with endotoxin. See main text for details and literature reference.
Figure S7 Venn diagrams of DEGs, induced in the ventral midbrain of mice at 13 dpi and 90 dpi after striatal injections of α‐syn PFFs, overlapping with DEGs from PD databases. No clear pattern was observed at this level of analysis. See man text for details and literature references.
Table S1 Antibodies used.
Table S2 Softwares used.
Table S3 Overlapping genes of the datasets of the α‐syn seeding/spreading model at 13dpi and 90 dpi with genes of microglial subtypes isolated from the nigro‐striatal circuit. See main text for details.
Table S4 Overlapping genes of the α‐syn seeding/preadig model at 13dpi and 90 dpi with genes of a disease‐associated microglia core signature (see main text for details).
Table S5 Unusual microglial gene expression signature in the α‐syn seeding/spreading model at 13dpi and 90 dpi. The bold higlighted genes may be involved in inducing neurodegeneration (see main text for details).
Table S6 Molecular astrocyte and peripheral immune cell genes after striatal PFF injection.
ACKNOWLEDGMENTS
Wiebke Jürgens‐Wemheuer and Kristopher J. Schmit were recipients, respectively, of a post‐doctoral (FNR AFR 5712281), and a pre‐doctoral (FNR AFR 12515776) fellowship from the Luxembourg National Research Fond. Michel Mittelbronn thanks the Luxembourg National Research Fond for support (FNR PEARL P16/BM/11192868). The authors thank Laurent Vallar (Luxembourg Institute of Health) for help with gene expression arrays, Christian Jaeger (Luxembourg Centre for Systems Biomedicine) for dopamine measurements, Eliezer Masliah (University of California, San Diego) for advice, Thierry Pillot and Violette Koziel (SynAging, France) for synuclein oligomers and advice, Yuting Liu (Biogen) for purifying recombinant murine α‐syn, Alessia Sciortino (University of Luxembourg) for help with Electron Microscopy sample preparation, Wagner Zago (Prothena Biosciences) for providing the 11A5 antibody.
Garcia, P. , Jürgens‐Wemheuer, W. , Uriarte Huarte, O. , Michelucci, A. , Masuch, A. , Brioschi, S. , Weihofen, A. , Koncina, E. , Coowar, D. , Heurtaux, T. , Glaab, E. , Balling, R. , Sousa, C. , Kaoma, T. , Nicot, N. , Pfander, T. , Schulz‐Schaeffer, W. , Allouche, A. , Fischer, N. , … Buttini, M. (2022). Neurodegeneration and neuroinflammation are linked, but independent of alpha‐synuclein inclusions, in a seeding/spreading mouse model of Parkinson's disease. Glia, 70(5), 935–960. 10.1002/glia.24149
Pierre Garcia, Wiebke Jürgens‐Wemheuer, Oihane Uriarte Huarte, and Kristopher J. Schmit contributed equally to this study.
Funding information Fonds National de la Recherche Luxembourg, Grant/Award Numbers: FNR AFR 5712281, FNR AFR 12515776, FNR PEARL P16/BM/11192868
DATA AVAILABILITY STATEMENT
Original datasets and list of genes from comparative analyses are available upon any reasonable request to the correspondence author (manuel.buttini@uni.lu). The GEO accession number of the microarray expression data is GSE15571. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155716).
REFERENCES
- Aarsland, D. , Creese, B. , Politis, M. , Chaudhuri, K. R. , Ffytche, D. H. , Weintraub, D. , & Ballard, C. (2017). Cognitive decline in Parkinson disease. Nature Reviews. Neurology, 13(4), 217–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmotilib, H. , Maltbie, T. , Delic, V. , Liu, Z. , Hu, X. , Fraser, K. B. , Moehle, M. S. , Stoyka, L. , Anabtawi, N. , Krendelchtchikova, V. , Volpicelli‐Daley, L. , & West, A. (2017). Alpha‐synuclein fibril‐induced inclusion spread in rats and mice correlates with dopaminergic neurodegeneration. Neurobiology of Disease, 105, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almandoz‐Gil, L. , Ingelsson, M. , & Bergstrom, J. (2018). Generation and characterization of stable alpha‐Synuclein oligomers. Methods in Molecular Biology, 1779, 61–71. [DOI] [PubMed] [Google Scholar]
- Ashrafi, A. , Garcia, P. , Kollmus, H. , Schughart, K. , Del Sol, A. , Buttini, M. , & Glaab, E. (2017). Absence of regulator of G‐protein signaling 4 does not protect against dopamine neuron dysfunction and injury in the mouse 6‐hydroxydopamine lesion model of Parkinson's disease. Neurobiology of Aging, 58, 30–33. [DOI] [PubMed] [Google Scholar]
- Ayers, J. I. , Brooks, M. M. , Rutherford, N. J. , Howard, J. K. , Sorrentino, Z. A. , Riffe, C. J. , & Giasson, B. I. (2017). Robust central nervous system pathology in transgenic mice following peripheral injection of alpha‐synuclein fibrils. Journal of Virology, 91(2), e02095–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellucci, A. , Mercuri, N. B. , Venneri, A. , Faustini, G. , Longhena, F. , Pizzi, M. , Missale, C. , & Spano, P. (2016). Review: Parkinson's disease: From synaptic loss to connectome dysfunction. Neuropathology and Applied Neurobiology, 42(1), 77–94. [DOI] [PubMed] [Google Scholar]
- Bendor, J. T. , Logan, T. P. , & Edwards, R. H. (2013). The function of alpha‐synuclein. Neuron, 79(6), 1044–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengoa‐Vergniory, N. , Roberts, R. F. , Wade‐Martins, R. , & Alegre‐Abarrategui, J. (2017). Alpha‐synuclein oligomers: A new hope. Acta Neuropathologica, 134(6), 819–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini, Y. H. Y. (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society. Sries B (Methodological), 57(1), 289–300. [Google Scholar]
- Bhattacharya, A. , & Biber, K. (2016). The microglial ATP‐gated ion channel P2X7 as a CNS drug target. Glia, 64(10), 1772–1787. [DOI] [PubMed] [Google Scholar]
- Biber, K. , Owens, T. , & Boddeke, E. (2014). What is microglia neurotoxicity (not)? Glia, 62(6), 841–854. [DOI] [PubMed] [Google Scholar]
- Bougnaud, S. , Golebiewska, A. , Oudin, A. , Keunen, O. , Harter, P. N. , Mader, L. , Azuaje, F. , Fritah, S. , Stieber, D. , Kaoma, T. , Vallar, L. , Brons, N. H. C. , Daubon, T. , Miletic, H. , Sundstrom, T. , Herold‐Mende, C. , Mittelbronn, M. , Bjerkvig, R. , & Niclou, S. P. (2016). Molecular crosstalk between tumour and brain parenchyma instructs histopathological features in glioblastoma. Oncotarget, 7(22), 31955–31971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak, H. , & Braak, E. (1995). Staging of Alzheimer's disease‐related neurofibrillary changes. Neurobiology of Aging, 16(3), 278–284. [DOI] [PubMed] [Google Scholar]
- Braak, H. , Del Tredici, K. , Rub, U. , de Vos, R. A. , Jansen Steur, E. N. , & Braak, E. (2003). Staging of brain pathology related to sporadic Parkinson's disease. Neurobiology of Aging, 24(2), 197–211. [DOI] [PubMed] [Google Scholar]
- Braak, H. , Ghebremedhin, E. , Rub, U. , Bratzke, H. , & Del Tredici, K. (2004). Stages in the development of Parkinson's disease‐related pathology. Cell and Tissue Research, 318(1), 121–134. [DOI] [PubMed] [Google Scholar]
- Breitling, R. , Armengaud, P. , Amtmann, A. , & Herzyk, P. (2004). Rank products: A simple, yet powerful, new method to detect differentially regulated genes in replicated microarray experiments. FEBS Letters, 573(1–3), 83–92. [DOI] [PubMed] [Google Scholar]
- Brochard, V. , Combadiere, B. , Prigent, A. , Laouar, Y. , Perrin, A. , Beray‐Berthat, V. , & Hunot, S. (2009). Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. The Journal of Clinical Investigation, 119(1), 182–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browaeys, R. , Saelens, W. , & Saeys, Y. (2020). NicheNet: Modeling intercellular communication by linking ligands to target genes. Nature Methods, 17(2), 159–162. [DOI] [PubMed] [Google Scholar]
- Brundin, P. , Ma, J. , & Kordower, J. H. (2016). How strong is the evidence that Parkinson's disease is a prion disorder? Current Opinion in Neurology, 29(4), 459–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brundin, P. , & Melki, R. (2017). Prying into the prion hypothesis for Parkinson's disease. The Journal of Neuroscience, 37(41), 9808–9818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burre, J. , Sharma, M. , & Sudhof, T. C. (2018). Cell biology and pathophysiology of alpha‐Synuclein. Cold Spring Harbor Perspectives in Medicine, 8(3), a024091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini, M. , Masliah, E. , Barbour, R. , Grajeda, H. , Motter, R. , Johnson‐Wood, K., Khan, K., Seubert, P., Freedman, S., Schenk, D., & Games, D. (2005). Beta‐amyloid immunotherapy prevents synaptic degeneration in a mouse model of Alzheimer's disease. The Journal of Neuroscience, 25(40), 9096–9101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttini, M. , Orth, M. , Bellosta, S. , Akeefe, H. , Pitas, R. E. , Wyss‐Coray, T. , Mucke, L. , & Mahley, R. W. (1999). Expression of human apolipoprotein E3 or E4 in the brains of Apoe−/− mice: Isoform‐specific effects on neurodegeneration. The Journal of Neuroscience, 19(12), 4867–4880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabeza‐Arvelaiz, Y. , Fleming, S. M. , Richter, F. , Masliah, E. , Chesselet, M. F. , & Schiestl, R. H. (2011). Analysis of striatal transcriptome in mice overexpressing human wild‐type alpha‐synuclein supports synaptic dysfunction and suggests mechanisms of neuroprotection for striatal neurons. Molecular Neurodegeneration, 6, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun, M. E. , Jucker, M. , Martin, L. J. , Thinakaran, G. , Price, D. L. , & Mouton, P. R. (1996). Comparative evaluation of synaptophysin‐based methods for quantification of synapses. Journal of Neurocytology, 25(12), 821–828. [DOI] [PubMed] [Google Scholar]
- Chiti, F. , & Dobson, C. M. (2017). Protein misfolding, amyloid formation, and human disease: A summary of progress over the last decade. Annual Review of Biochemistry, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- Chu, Y. , & Kordower, J. H. (2010). Lewy body pathology in fetal grafts. Annals of the New York Academy of Sciences, 1184, 55–67. [DOI] [PubMed] [Google Scholar]
- Chu, Y. , Muller, S. , Tavares, A. , Barret, O. , Alagille, D. , Seibyl, J. , Tamagnan, G. , Marek, K. , Luk, K. C. , Trojanowski, J. Q. , Lee, V. M. Y. , & Kordower, J. H. (2019). Intrastriatal alpha‐synuclein fibrils in monkeys: Spreading, imaging and neuropathological changes. Brain, 142(11), 3565–3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croisier, E. , Moran, L. B. , Dexter, D. T. , Pearce, R. K. , & Graeber, M. B. (2005). Microglial inflammation in the parkinsonian substantia nigra: Relationship to alpha‐synuclein deposition. Journal of Neuroinflammation, 2, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotti, A. , & Ransohoff, R. M. (2016). Microglial physiology and pathophysiology: Insights from genome‐wide transcriptional profiling. Immunity, 44(3), 505–515. [DOI] [PubMed] [Google Scholar]
- Del Carratore, F. , Jankevics, A. , Eisinga, R. , Heskes, T. , Hong, F. , & Breitling, R. (2017). RankProd 2.0: A refactored bioconductor package for detecting differentially expressed features in molecular profiling datasets. Bioinformatics, 33(17), 2774–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra, A. A. , Ingrassia, A. , de Menezes, R. X. , van Kesteren, R. E. , Rozemuller, A. J. , Heutink, P. , & van de Berg, W. D. (2015). Evidence for immune response, axonal dysfunction and reduced endocytosis in the substantia nigra in early stage Parkinson's disease. PLoS One, 10(6), e0128651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra, A. A. , Voorn, P. , Berendse, H. W. , Groenewegen, H. J. , Netherlands Brain, B. , Rozemuller, A. J. , & van de Berg, W. D. (2014). Stage‐dependent nigral neuronal loss in incidental Lewy body and Parkinson's disease. Movement Disorders, 29(10), 1244–1251. [DOI] [PubMed] [Google Scholar]
- Doane, F. W. A. N. (1987). Electron microscopy in diagnostic virology. Cambridge University Press. [Google Scholar]
- Doorn, K. J. , Lucassen, P. J. , Boddeke, H. W. , Prins, M. , Berendse, H. W. , Drukarch, B. , & van Dam, A. M. (2012). Emerging roles of microglial activation and non‐motor symptoms in Parkinson's disease. Progress in Neurobiology, 98(2), 222–238. [DOI] [PubMed] [Google Scholar]
- Doorn, K. J. , Moors, T. , Drukarch, B. , van de Berg, W. , Lucassen, P. J. , & van Dam, A. M. (2014). Microglial phenotypes and toll‐like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathologica Communications, 2, 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubbelaar, M. L. , Kracht, L. , Eggen, B. J. L. , & Boddeke, E. (2018). The kaleidoscope of microglial phenotypes. Frontiers in Immunology, 9, 1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy, M. F. , Collier, T. J. , Patterson, J. R. , Kemp, C. J. , Luk, K. C. , Tansey, M. G. , Paumier, K. L. , Kanaan, N. M. , Luke‐Fischer, D. , Polinksi, N. K. , Barth, O. L. , Howe, J. W. , Vaikath, N. N. , Majbour, N. K. , El‐Agnaf, O. M. A. , & Sortwell, C. E. (2018). Lewy body‐like alpha‐synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. Journal of Neuroinflammation, 15(1), 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espay, A. J. , & Marras, C. (2019). Clinical Parkinson disease subtyping does not predict pathology. Nature Reviews. Neurology, 15(4), 189–190. [DOI] [PubMed] [Google Scholar]
- Fellner, L. , Irschick, R. , Schanda, K. , Reindl, M. , Klimaschewski, L. , Poewe, W. , Wenning, G. K. , & Stefanova, N. (2013). Toll‐like receptor 4 is required for alpha‐synuclein dependent activation of microglia and astroglia. Glia, 61(3), 349–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo‐Pereira, M. E. , Corwin, C. , & Babich, J. (2016). Prostaglandin J2: A potential target for halting inflammation‐induced neurodegeneration. Annals of the New York Academy of Sciences, 1363, 125–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippini, A. , Gennarelli, M. , & Russo, I. (2019). Alpha‐Synuclein and glia in Parkinson's disease: A beneficial or a detrimental duet for the endo‐lysosomal system? Cellular and Molecular Neurobiology, 39(2), 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, R. , Shen, Q. , Xu, P. , Luo, J. J. , & Tang, Y. (2014). Phagocytosis of microglia in the central nervous system diseases. Molecular Neurobiology, 49(3), 1422–1434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, K. A. , Ostaszewski, M. , Matsuoka, Y. , Ghosh, S. , Glaab, E. , Trefois, C. , Crespo, I. , Perumal, T. M. , Jurkowski, W. , Antony, P. M. , Diederich, N. , Buttini, M. , Kodama, A. , Satagopam, V. P. , Eifes, S. , Del Sol, A. , Schneider, R. , Kitano, H. , & Balling, R. (2014). Integrating pathways of Parkinson's disease in a molecular interaction map. Molecular Neurobiology, 49(1), 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawron, P. , Ostaszewski, M. , Satagopam, V. , Gebel, S. , Mazein, A. , Kuzma, M. , Zorzan, S. , McGee, F. , Otjacques, B. , Balling, R. , & Schneider, R. (2016). MINERVA‐a platform for visualization and curation of molecular interaction networks. NPJ Systems Biology and Applications, 2, 16020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhard, A. , Pavese, N. , Hotton, G. , Turkheimer, F. , Es, M. , Hammers, A. , Eggert, K. , Oertel, W. , Banati, R. B. , & Brooks, D. J. (2006). In vivo imaging of microglial activation with [11C](R)‐PK11195 PET in idiopathic Parkinson's disease. Neurobiology of Disease, 21(2), 404–412. [DOI] [PubMed] [Google Scholar]
- Glaab, E. , & Schneider, R. (2015). Comparative pathway and network analysis of brain transcriptome changes during adult aging and in Parkinson's disease. Neurobiology of Disease, 74, 1–13. [DOI] [PubMed] [Google Scholar]
- Goedert, M. (2015). Neurodegeneration. Alzheimer's and Parkinson's diseases: The prion concept in relation to assembled Abeta, tau, and alpha‐synuclein. Science, 349(6248), 1255555. [DOI] [PubMed] [Google Scholar]
- Grozdanov, V. , Bousset, L. , Hoffmeister, M. , Bliederhaeuser, C. , Meier, C. , Madiona, K. , Pieri, L. , Kiechle, M. , McLean, P. J. , Kassubek, J. , Behrends, C. , Ludolph, A. C. , Weishaupt, J. H. , Melki, R. , & Danzer, K. M. (2019). Increased immune activation by pathologic alpha‐synuclein in Parkinson's disease. Annals of Neurology, 86(4), 593–606. [DOI] [PubMed] [Google Scholar]
- Hakim, R. , Zachariadis, V. , Sankavaram, S. R. , Han, J. , Harris, R. A. , Brundin, L. , Enge, M. , & Svensson, M. (2021). Spinal cord Injury induces permanent reprogramming of microglia into a disease‐associated state which contributes to functional recovery. The Journal of Neuroscience, 41(40), 8441–8459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, T. R. , Dufort, C. , Dissing‐Olesen, L. , Giera, S. , Young, A. , Wysoker, A. , Walker, A. J. , Gergits, F. , Segel, M. , Nemesh, J. , Marsh, S. E. , Saunders, A. , Macosko, E. , Ginhoux, F. , Chen, J. , Franklin, R. J. M. , Piao, X. , McCarroll, S. A. , & Stevens, B. (2019). Single‐cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell‐state changes. Immunity, 50(1), 253–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, T. R. , Marsh, S. E. , & Stevens, B. (2019). Immune signaling in neurodegeneration. Immunity, 50(4), 955–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms, A. S. , Delic, V. , Thome, A. D. , Bryant, N. , Liu, Z. , Chandra, S. , Jurkuveniate, A. , & West, A. B. (2017). Alpha‐synuclein fibrils recruit peripheral immune cells in the rat brain prior to neurodegeneration. Acta Neuropathologica Communications, 5(1), 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayat, M. A. M. , & Miller, S. E. (1990). Negative Staining. McGraw‐Hill. [Google Scholar]
- Helwig, M. , Klinkenberg, M. , Rusconi, R. , Musgrove, R. E. , Majbour, N. K. , El‐Agnaf, O. M. , Ulusoy, A. , & Di Monte, D. A. (2016). Brain propagation of transduced alpha‐synuclein involves non‐fibrillar protein species and is enhanced in alpha‐synuclein null mice. Brain, 139(3), 856–870. [DOI] [PubMed] [Google Scholar]
- Henrich, M. T. , Geibl, F. F. , Lakshminarasimhan, H. , Stegmann, A. , Giasson, B. I. , Mao, X. , Dawson, V. L. , Dawson, T. M. , Oertel, W. , & Surmeier, D. J. (2020). Determinants of seeding and spreading of alpha‐synuclein pathology in the brain. Science Advances, 6(46), eabc2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch, E. C. , & Standaert, D. G. (2020). Ten unsolved questions about neuroinflammation in Parkinson's disease. Movement Disorders, 36, 16–24. [DOI] [PubMed] [Google Scholar]
- Hoksza, D. , Gawron, P. , Ostaszewski, M. , Smula, E. , & Schneider, R. (2019). MINERVA API and plugins: Opening molecular network analysis and visualization to the community. Bioinformatics, 35(21), 4496–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmqvist, S. , Chutna, O. , Bousset, L. , Aldrin‐Kirk, P. , Li, W. , Bjorklund, T. , Wang, Z.‐Y. , Roybon, L. , Melki, R. , & Li, J. Y. (2014). Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathologica, 128(6), 805–820. [DOI] [PubMed] [Google Scholar]
- Holtman, I. R. , Raj, D. D. , Miller, J. A. , Schaafsma, W. , Yin, Z. , Brouwer, N. , Wes, P. D. , Moller, T. , Orre, M. , Kamphuis, W. , Hol, E. M. , Boddeke, E. W. G. M. , & Eggen, B. J. (2015). Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co‐expression meta‐analysis. Acta Neuropathologica Communications, 3, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, F. , Breitling, R. , McEntee, C. W. , Wittner, B. S. , Nemhauser, J. L. , & Chory, J. (2006). RankProd: A bioconductor package for detecting differentially expressed genes in meta‐analysis. Bioinformatics, 22(22), 2825–2827. [DOI] [PubMed] [Google Scholar]
- Hsieh, Y. C. , Mounsey, R. B. , & Teismann, P. (2011). MPP(+)‐induced toxicity in the presence of dopamine is mediated by COX‐2 through oxidative stress. Naunyn‐Schmiedeberg's Archives of Pharmacology, 384(2), 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes, C. D. , Choi, M. L. , Ryten, M. , Hopkins, L. , Drews, A. , Botia, J. A. , Iljina, M. , Rodrigues, M. , Gagliano, S. A. , Gandhi, S. , Bryant, C. , & Klenerman, D. (2019). Picomolar concentrations of oligomeric alpha‐synuclein sensitizes TLR4 to play an initiating role in Parkinson's disease pathogenesis. Acta Neuropathologica, 137(1), 103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeger, C. , Glaab, E. , Michelucci, A. , Binz, T. M. , Koeglsberger, S. , Garcia, P. , Trezzi, J.‐P. , Ghelfi, J. , Balling, R. , & Buttini, M. (2015). The mouse brain metabolome: Region‐specific signatures and response to excitotoxic neuronal injury. The American Journal of Pathology, 185(6), 1699–1712. [DOI] [PubMed] [Google Scholar]
- Jager, C. , Hiller, K. , & Buttini, M. (2016). Metabolic profiling and quantification of neurotransmitters in mouse brain by gas chromatography‐mass spectrometry. Current Protocols in Mouse Biology, 6(3), 333–342. [DOI] [PubMed] [Google Scholar]
- Janda, E. , Boi, L. , & Carta, A. R. (2018). Microglial phagocytosis and its regulation: A therapeutic target in Parkinson's disease? Frontiers in Molecular Neuroscience, 11, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jellinger, K. A. (2009a). A critical evaluation of current staging of alpha‐synuclein pathology in Lewy body disorders. Biochimica et Biophysica Acta, 1792(7), 730–740. [DOI] [PubMed] [Google Scholar]
- Jellinger, K. A. (2009b). Formation and development of Lewy pathology: A critical update. Journal of Neurology, 256(3), 270–279. [DOI] [PubMed] [Google Scholar]
- Joers, V. , Tansey, M. G. , Mulas, G. , & Carta, A. R. (2017). Microglial phenotypes in Parkinson's disease and animal models of the disease. Progress in Neurobiology, 155, 57–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker, M. , & Walker, L. C. (2018). Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nature Neuroscience, 21(10), 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannarkat, G. T. , Boss, J. M. , & Tansey, M. G. (2013). The role of innate and adaptive immunity in Parkinson's disease. Journal of Parkinson's Disease, 3(4), 493–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keren‐Shaul, H. , Spinrad, A. , Weiner, A. , Matcovitch‐Natan, O. , Dvir‐Szternfeld, R. , Ulland, T. K. , David, E. , Baruch, K. , Lara‐Astaiso, D. , Toth, B. , Itzkovitz, S. , Colonna, M. , Schwartz., M., & Amit, I. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell, 169(7), 1276–1290. [DOI] [PubMed] [Google Scholar]
- Kim, C. , Ho, D. H. , Suk, J. E. , You, S. , Michael, S. , Kang, J. , Lee, S. J. , Masliah, E. , Hwang, D. , Lee, H. J. , & Lee, S. J. (2013). Neuron‐released oligomeric alpha‐synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nature Communications, 4, 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, C. , Lee, H. J. , Masliah, E. , & Lee, S. J. (2016). Non‐cell‐autonomous neurotoxicity of alpha‐synuclein through microglial toll‐like receptor 2. Exp Neurobiol, 25(3), 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, C. , Spencer, B. , Rockenstein, E. , Yamakado, H. , Mante, M. , Adame, A. , Fields, J. A. , Masliah, D. , Iba, M. , Lee, H. J. , Rissman, R. A. , Lee, S. J. , & Masliah, E. (2018). Immunotherapy targeting toll‐like receptor 2 alleviates neurodegeneration in models of synucleinopathy by modulating alpha‐synuclein transmission and neuroinflammation. Molecular Neurodegeneration, 13(1), 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konishi, H. , & Kiyama, H. (2018). Microglial TREM2/DAP12 signaling: A double‐edged sword in neural diseases. Frontiers in Cellular Neuroscience, 12, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer, M. L. , & Schulz‐Schaeffer, W. J. (2007). Presynaptic alpha‐synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with Lewy bodies. The Journal of Neuroscience, 27(6), 1405–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lema Tome, C. M. , Tyson, T. , Rey, N. L. , Grathwohl, S. , Britschgi, M. , & Brundin, P. (2013). Inflammation and alpha‐synuclein's prion‐like behavior in Parkinson's disease–is there a link? Molecular Neurobiology, 47(2), 561–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, M. K. , & Farrer, M. J. (2014). Genetics and genomics of Parkinson's disease. Genome Medicine, 6(6), 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk, K. C. , Kehm, V. , Carroll, J. , Zhang, B. , O'Brien, P. , Trojanowski, J. Q. , & Lee, V. M. (2012). Pathological alpha‐synuclein transmission initiates Parkinson‐like neurodegeneration in nontransgenic mice. Science, 338(6109), 949–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luna, E. , & Luk, K. C. (2015). Bent out of shape: Alpha‐synuclein misfolding and the convergence of pathogenic pathways in Parkinson's disease. FEBS Letters, 589(24), 3749–3759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, M. W. , Wang, J. , Zhang, Q. , Wang, R. , Dhandapani, K. M. , Vadlamudi, R. K. , & Brann, D. W. (2017). NADPH oxidase in brain injury and neurodegenerative disorders. Molecular Neurodegeneration, 12(1), 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaplate‐Armand, C. , Florent‐Bechard, S. , Youssef, I. , Koziel, V. , Sponne, I. , Kriem, B. , Leininger‐Muller, B. , Olivier, J. L. , Oster, T. , & Pillot, T. (2006). Soluble oligomers of amyloid‐beta peptide induce neuronal apoptosis by activating a cPLA2‐dependent sphingomyelinase‐ceramide pathway. Neurobiology of Disease, 23(1), 178–189. [DOI] [PubMed] [Google Scholar]
- Mathys, H. , Adaikkan, C. , Gao, F. , Young, J. Z. , Manet, E. , Hemberg, M. , De Jager, P. , Ransohoff, R. M. , Regev, A. , & Tsai, L. H. (2017). Temporal tracking of microglia activation in neurodegeneration at single‐cell resolution. Cell Reports, 21(2), 366–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezias, C. , Rey, N. , Brundin, P. , & Raj, A. (2020). Neural connectivity predicts spreading of alpha‐synuclein pathology in fibril‐injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiology of Disease, 134, 104623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelucci, A. , Mittelbronn, M. , & Gomez‐Nicola, D. (2018). Microglia in health and disease: A unique immune cell population. Frontiers in Immunology, 9, 1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milber, J. M. , Noorigian, J. V. , Morley, J. F. , Petrovitch, H. , White, L. , Ross, G. W. , & Duda, J. E. (2012). Lewy pathology is not the first sign of degeneration in vulnerable neurons in Parkinson disease. Neurology, 79(24), 2307–2314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, R. M. , Kiser, G. L. , Kaysser‐Kranich, T. , Casaceli, C. , Colla, E. , Lee, M. K. , Palaniappan, C. , & Federoff, H. J. (2007). Wild‐type and mutant alpha‐synuclein induce a multi‐component gene expression profile consistent with shared pathophysiology in different transgenic mouse models of PD. Experimental Neurology, 204(1), 421–432. [DOI] [PubMed] [Google Scholar]
- Mrdjen, D. , Pavlovic, A. , Hartmann, F. J. , Schreiner, B. , Utz, S. G. , Leung, B. P. , Lelios, I. , Heppner, F. L. , Kipnis, J. , Merkler, D. , Greter, M. , & Becher, B. (2018). High‐dimensional single‐cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity, 48(2), 380–395. [DOI] [PubMed] [Google Scholar]
- Olanow, C. W. , Savolainen, M. , Chu, Y. , Halliday, G. M. , & Kordower, J. H. (2019). Temporal evolution of microglia and alpha‐synuclein accumulation following foetal grafting in Parkinson's disease. Brain, 142(6), 1690–1700. [DOI] [PubMed] [Google Scholar]
- Osterberg, V. R. , Spinelli, K. J. , Weston, L. J. , Luk, K. C. , Woltjer, R. L. , & Unni, V. K. (2015). Progressive aggregation of alpha‐synuclein and selective degeneration of lewy inclusion‐bearing neurons in a mouse model of parkinsonism. Cell Reports, 10(8), 1252–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paumier, K. L. , Luk, K. C. , Manfredsson, F. P. , Kanaan, N. M. , Lipton, J. W. , Collier, T. J. , Steece‐Collier, K. , Kemp, C. J. , Celano, S. , Schulz, E. , Sandoval, I. M. , Fleming, S. , Dirr, E. , Polinksi, N. K. , Trojanowski, J. Q. , Lee, V. M. , & Sortwell, C. E. (2015). Intrastriatal injection of pre‐formed mouse alpha‐synuclein fibrils into rats triggers alpha‐synuclein pathology and bilateral nigrostriatal degeneration. Neurobiology of Disease, 82, 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos, G. , & Franklin, K. (2008). The mouse brain atlas in stereotactic coordinates (3rd ed.). Elsevier Academic Press. [Google Scholar]
- Peelaerts, W. , Bousset, L. , Van der Perren, A. , Moskalyuk, A. , Pulizzi, R. , Giugliano, M. , Van den Haute, C. , Melki, R. , & Baekelandt, V. (2015). Alpha‐synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature, 522(7556), 340–344. [DOI] [PubMed] [Google Scholar]
- Poewe, W. , Seppi, K. , Tanner, C. M. , Halliday, G. M. , Brundin, P. , Volkmann, J. , Schrag, A. E. , & Lang, A. E. (2017). Parkinson disease. Nature Reviews Disease Primers, 3, 17013. [DOI] [PubMed] [Google Scholar]
- Ransohoff, R. M. (2016). A polarizing question: Do M1 and M2 microglia exist? Nature Neuroscience, 19(8), 987–991. [DOI] [PubMed] [Google Scholar]
- Ransohoff, R. M. , & El Khoury, J. (2015). Microglia in health and disease. Cold Spring Harbor Perspectives in Biology, 8(1), a020560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, N. L. , Bousset, L. , George, S. , Madaj, Z. , Meyerdirk, L. , Schulz, E. , Steiner, J. A. , Melki, R. , & Brundin, P. (2019). Alpha‐synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathologica Communications, 7(1), 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey, N. L. , George, S. , & Brundin, P. (2016). Review: Spreading the word: Precise animal models and validated methods are vital when evaluating prion‐like behaviour of alpha‐synuclein. Neuropathology and Applied Neurobiology, 42(1), 51–76. [DOI] [PubMed] [Google Scholar]
- Roberts, R. F. , Wade‐Martins, R. , & Alegre‐Abarrategui, J. (2015). Direct visualization of alpha‐synuclein oligomers reveals previously undetected pathology in Parkinson's disease brain. Brain, 138(6), 1642–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, E. M. , De Miranda, B. , & Sanders, L. H. (2018). Alpha‐synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson's disease. Neurobiology of Disease, 109, 249–257. [DOI] [PubMed] [Google Scholar]
- Rodriguez, L. , Marano, M. M. , & Tandon, A. (2018). Import and export of misfolded alpha‐synuclein. Frontiers in Neuroscience, 12, 344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, C. A. , & Poirier, M. A. (2004). Protein aggregation and neurodegenerative disease. Nature Medicine, 10, 10–17. [DOI] [PubMed] [Google Scholar]
- Sacino, A. N. , Brooks, M. , Thomas, M. A. , McKinney, A. B. , Lee, S. , Regenhardt, R. W. , McGarvey, N. H. , Ayers, J. I. , Notterpek, L. , Borchelt, D. R. , Golde, T. , & Giasson, B. I. (2014). Intramuscular injection of alpha‐synuclein induces CNS alpha‐synuclein pathology and a rapid‐onset motor phenotype in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America, 111(29), 10732–10737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheckel, C. , & Aguzzi, A. (2018). Prions, prionoids and protein misfolding disorders. Nature Reviews. Genetics, 19(7), 405–418. [DOI] [PubMed] [Google Scholar]
- Scheiblich, H. , Dansokho, C. , Mercan, D. , Schmidt, S. V. , Bousset, L. , Wischhof, L. , Eikens, F. , Odainic, A. , Spitzer, J. , Grieb, A. , Schwartz, S. , Dano, D. , Latz, E. , Melki, R. , & Heneka, M. T. (2021). Microglia jointly degrade fibrillar alpha‐synuclein cargo by distribution through tunneling nanotubes. Cell, 184(20), 5089–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe, D. J. (2003). Folding proteins in fatal ways. Nature, 426(6968), 900–904. [DOI] [PubMed] [Google Scholar]
- Seok, J. , Warren, H. S. , Cuenca, A. G. , Mindrinos, M. N. , Baker, H. V. , Xu, W. , Richards, D.R , Honari, S. , Moore, E. E. , Minei, J. P. Cuschieri, J. , Bankey, P. E. , Johnson, J. L. , Sperry, J. , Nathens, A. B. , Billiar, T. R. , West, M. A. , Jeschke, M. G. , Klein, M. B. , … Host Response to Injury, Large Scale Collaborative Research Program . (2013). Genomic responses in mouse models poorly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America, 110(9), 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon, P. , Markiel, A. , Ozier, O. , Baliga, N. S. , Wang, J. T. , Ramage, D. , Amin, N. , Schwikowski, B. , & Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13(11), 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton, A. , & Hardy, J. (2019). Progress in the genetic analysis of Parkinson's disease. Human Molecular Genetics, 28(2), 215–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini, M. G. , Crowther, R. A. , Jakes, R. , Hasegawa, M. , & Goedert, M. (1998). Alpha‐synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with Lewy bodies. Proceedings of the National Academy of Sciences of the United States of America, 95(11), 6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini, M. G. , & Goedert, M. (2018). Neurodegeneration and the ordered assembly of alpha‐synuclein. Cell and Tissue Research, 373(1), 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian, A. , Tamayo, P. , Mootha, V. K. , Mukherjee, S. , Ebert, B. L. , Gillette, M. A. , Paulovich, P. , Pomeroy, S. L. , Golub, T. R. , Lander, E. S. , & Mesirov, J. P. (2005). Gene set enrichment analysis: A knowledge‐based approach for interpreting genome‐wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America, 102(43), 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier, D. J. , Obeso, J. A. , & Halliday, G. M. (2017a). Parkinson's disease is not simply a prion disorder. The Journal of Neuroscience, 37(41), 9799–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier, D. J. , Obeso, J. A. , & Halliday, G. M. (2017b). Selective neuronal vulnerability in Parkinson disease. Nature Reviews. Neuroscience, 18(2), 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takao, K. , & Miyakawa, T. (2015). Genomic responses in mouse models greatly mimic human inflammatory diseases. Proceedings of the National Academy of Sciences of the United States of America, 112(4), 1167–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, E. K. , Chao, Y. X. , West, A. , Chan, L. L. , Poewe, W. , & Jankovic, J. (2020). Parkinson disease and the immune system ‐ associations, mechanisms and therapeutics. Nature Reviews. Neurology, 16, 303–318. [DOI] [PubMed] [Google Scholar]
- Tansey, M. G. , & Romero‐Ramos, M. (2019). Immune system responses in Parkinson's disease: Early and dynamic. The European Journal of Neuroscience, 49(3), 364–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay, T. L. , Sagar Dautzenberg, J. , Grun, D. , & Prinz, M. (2018). Unique microglia recovery population revealed by single‐cell RNAseq following neurodegeneration. Acta Neuropathologica Communications, 6(1), 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teismann, P. , Tieu, K. , Choi, D. K. , Wu, D. C. , Naini, A. , Hunot, S. , Vila, M. , Jackson‐Lewis, V. , & Przedborski, S. (2003). Cyclooxygenase‐2 is instrumental in Parkinson's disease neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America, 100(9), 5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terada, T. , Yokokura, M. , Yoshikawa, E. , Futatsubashi, M. , Kono, S. , Konishi, T. , Miyajima, H. , Hashizume, T. , & Ouchi, Y. (2016). Extrastriatal spreading of microglial activation in Parkinson's disease: A positron emission tomography study. Annals of Nuclear Medicine, 30(8), 579–587. [DOI] [PubMed] [Google Scholar]
- Trifilieff, P. , Rives, M. L. , Urizar, E. , Piskorowski, R. A. , Vishwasrao, H. D. , Castrillon, J. , Schmauss, C. , Slattman, M. , Gullberg, M. , & Javitch, J. A. (2011). Detection of antigen interactions ex vivo by proximity ligation assay: Endogenous dopamine D2‐adenosine A2A receptor complexes in the striatum. BioTechniques, 51(2), 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy, A. , Musgrove, R. E. , Rusconi, R. , Klinkenberg, M. , Helwig, M. , Schneider, A. , & Di Monte, D. A. (2015). Neuron‐to‐neuron alpha‐synuclein propagation in vivo is independent of neuronal injury. Acta Neuropathologica Communications, 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulusoy, A. , Rusconi, R. , Perez‐Revuelta, B. I. , Musgrove, R. E. , Helwig, M. , Winzen‐Reichert, B. , & Di Monte, D. A. (2013). Caudo‐rostral brain spreading of alpha‐synuclein through vagal connections. EMBO Molecular Medicine, 5(7), 1119–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriarte Huarte, O. , Kyriakis, D. , Heurtaux, T. , Pires‐Afonso, Y. , Grzyb, K. , Halder, R. , Buttini, M. , Skupin, A. , Mittelbronn, M. , & Michelucci, A. (2021). Single‐cell transcriptomics and in situ morphological analyses reveal microglia heterogeneity across the nigrostriatal pathway. Frontiers in Immunology, 12, 639613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uriarte Huarte, O. , Richart, L. , Mittelbronn, M. , & Michelucci, A. (2021). Microglia in health and disease: The strength to be diverse and reactive. Frontiers in Cellular Neuroscience, 15, 660523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaikath, N. N. , Hmila, I. , Gupta, V. , Erskine, D. , Ingelsson, M. , & El‐Agnaf, O. M. A. (2019). Antibodies against alpha‐synuclein: Tools and therapies. Journal of Neurochemistry, 150(5), 612–625. [DOI] [PubMed] [Google Scholar]
- Vasili, E. , Dominguez‐Meijide, A. , & Outeiro, T. F. (2019). Spreading of alpha‐synuclein and tau: A systematic comparison of the mechanisms involved. Frontiers in Molecular Neuroscience, 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker, L. C. , & Jucker, M. (2015). Neurodegenerative diseases: Expanding the prion concept. Annual Review of Neuroscience, 38, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, D. M. , & Selkoe, D. J. (2004). Oligomers on the brain: The emerging role of soluble protein aggregates in neurodegeneration. Protein and Peptide Letters, 11(3), 213–228. [DOI] [PubMed] [Google Scholar]
- Walsh, D. M. , & Selkoe, D. J. (2016). A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nature Reviews. Neuroscience, 17(4), 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan, Y. W. , Al‐Ouran, R. , Mangleburg, C. G. , Perumal, T. M. , Lee, T. V. , Allison, K. , Swarup, V. , Funk, C. C. , Gaiteri, C. , Allen, M. , Wang, M. , Neuner, S. M. , Kaczorowski, C. C. , Philip, V. M. , Howell, G. R. , Martini‐Stoica, H. , Zheng, H. , Mei, H. , Zhing, X. , … Logsdon, B. A. (2020). Meta‐analysis of the Alzheimer's disease human brain transcriptome and functional dissection in mouse models. Cell Reports, 32(2), 107908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, M. B. , Richter, F. , Lee, S. K. , Gabby, L. , Wu, J. , Masliah, E. , Effros, R. B. , & Chesselet, M. F. (2012). Regionally‐specific microglial activation in young mice over‐expressing human wildtype alpha‐synuclein. Experimental Neurology, 237(2), 318–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen, A. , Liu, Y. , Arndt, J. W. , Huy, C. , Quan, C. , Smith, B. A. , Baeriswyl, J. L. , Cavegn, N. , Senn, L. , Su, L. , Marsh, G. , Auluck, P. K. , Montrasio, F. , Nitsch, R. M. , Hirst, W. D. , Cedarbaum, J. M. , Pepinsky, R. B. , Grimm, J. , & Weinreb, P. H. (2019). Development of an aggregate‐selective, human‐derived alpha‐synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson's disease models. Neurobiology of Disease, 124, 276–288. [DOI] [PubMed] [Google Scholar]
- Wolf, S. A. , Boddeke, H. W. , & Kettenmann, H. (2017). Microglia in physiology and disease. Annual Review of Physiology, 79, 619–643. [DOI] [PubMed] [Google Scholar]
- Wu, Q. , Takano, H. , Riddle, D. M. , Trojanowski, J. Q. , Coulter, D. A. , & Lee, V. M. (2019). Alpha‐synuclein (alphaSyn) preformed fibrils induce endogenous alphaSyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. The Journal of Neuroscience, 39(26), 5080–5094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, S. S. , Beyreuther, K. , & Schmitt, H. P. (1993). Quantitative assessment of the synaptophysin immuno‐reactivity of the cortical neuropil in various neurodegenerative disorders with dementia. Dementia, 4(2), 66–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Upper panel: Western blot characterization of α‐syn moities used for intrastriatal injections. Loading of the gel was as follows: lanes 1, 6, 14 – ladder; lanes 5, 10,11‐ blank; lanes 2,3,4 – α‐syn oligomer, 10, 100, and 500 ng respectively; lanes 7, 8, 9 – α‐syn PFFs, not sonicated, 10,100, 500 ng respectively; lanes 11, 12, 13 – α‐syn PFFs (sonicated), 10,100, 500 ng respectively. Bands from corresponding to the MW of α‐syn monomers, dimers, and trimmers are circled (red, blue, orange, respectively). Note the presence of high molecular weight moities (visible as a smear) after loading of non‐sonicated PFFs. The sonication process of PFFs appears to reduce all higher molecular weight species of α‐syn and the amount of dimers and monomers, since smears and monomer/dimer bands were visible after loading 100 ng of non‐sonicated PFFs, but not after loading the same amount of sonicated PFFs. Lower panels: Electron microscopy of α‐syn moities. A: Oligomers; B: unsonicated α‐syn PFFs; C: sonicated α‐syn PFFs. Oligomers were primarily composed of small globular moities. Unsonicated PFFs contained many large assemblies of α‐syn fibrils, whereas sonicated PFFs were composed mainly of small fibrillar fragments, resulting form the sonication‐induced disruption of the large assemblies. Scale bar = 152 nm.
Figure S2 Minimal presence of α‐syn inclusions in different brain regions 13 DPI after intrastriatal injection of α‐syn PFFs. Only few α‐syn inclusions were seen, ipsi‐ and contralaterally, in the frontal cortex, amygdala, amd ipsilaterally, but not contralaterally, in the striatum and SN. Scale bar = 130 μm.
Figure S3 No nigro‐striatal degeneration and microgliosis 13 dpi after intrastriatal injection of α‐syn PFFs. No loss of striatal TH‐positive axons (first row), striatal DAT‐positive synaptic terminals (second row), nigral TH‐positive neurons (third row) was observed 13 dpi after injection of PFFs. No increase of Iba1‐positive microglial reaction in the SN was observed (last row). Microphotographs show examples of PBS‐injected control brains (ipsilateral) and ipsilateral α‐syn PFF‐injected brains. Scale bars = 25 μm (striatal panels), 200 μm (Subst. Nigra panels).
Figure S4 Venn diagrams of DEGs induced in the ventral midbrain of mice at 13 dpi and 90 dpi after striatal injections of α‐syn PFFs. Comparisons for each time point were made between DEGs in ipsilateral ventral midbrain of PFF‐injected mice versus those in the ipsilateral ventral midbrain for PBS‐injected mice (ipsi PFF vs. ipsi PBS, n = 6/group; upper 4 panels), and between DEGs in ipsilateral ventral midbrain versus the contralateral ventral midbrains of PFF‐injected mice (ipsi PFF vs. contra PFF, n = 6/group; lower 4 panels). Diagrams on the left show Venn diagrams with numbers of DEGs with a significance level of p < .05 (n = 6 mice/group). Because some gene products have an effect within biological pathways while the expression of their genes may only change minimally, we show DEGs that emerge with this low level of statistical stringency. In ipsi PFF vs. ipsi PBS, the number of DEGs was 2631 at 13 DPI, and 2584 at 90 DPI, with 985 DEGs common to both time points. In ipsi PFF vs. 3584 at 13 DPI, and 2990 at 90 DPI, with 960 DEGs common to both time points. In ipsi PFF vs. contra PFF, the number of DEGs was 4268 at 13 DPI, and 3976 at 90 DPI, with 1312 DEGs common to both time points. Panels on the right show Venn diagrams with the number of DEGs after adjusting for multiple hypothesis testing at pfp < 0.1. In ipsi PFF vs. ipsi PBS, the number of DEGs was 308 at 13 DPI, and 94 at 90 DPI, with 37 DEGs common to both time points. In ipsi PFF vs. contra PFF, the number of DEGs was 674 at 13 DPI, and 688 at 90 DPI, with 210 DEGs common to both time points. The Venn diagrams below the main ones indicate the number of DEGs that show enhanced (“up”) versus decreased (“down”), as well as the overlaps, in the different comparisons.
Figure S5 Heatmaps illustrating the ventral midbrain gene expression patterns at 13 dpi and 90 dpi after intrastriatal injection of α‐syn PFFs. Heatmaps are shown for p‐values <0.05 or pfp < 0.1. Scaled row expression values (row Z‐score) in red indicate higher, in blue lower, and in white unchanged gene expression (see Materials and Methods for details). Comparisons of ipsi PFF versus ipsi PBS (upper panel row) and ipsi PBS versus contra PFF (lower panel row) reveal distinctive gene expression patterns at 13 dpi for both comparisons (whether higher, pfp < 0.1, or lower, p < 0.05, statistical stringency is used), whereas a distinctive pattern at 90 dpi only appears in the ipsi PFF versus contra PFF, but not in the ipsi PFF versus ipsi PBS, comparisons.
Figure S6 Venn diagrams of DEGs, induced in the ventral midbrain of mice at 13 dpi and 90 dpi after striatal injections of α‐syn PFFs, overlapping with DEGs of isolated microglia from different neurological disease models. APP stands for Alzheimer model mice, ERRCC1 mice are a model of premature aging, SOP1 mice are a model for amyothrophic lateral sclerosis, and LPS are mice injected with endotoxin. See main text for details and literature reference.
Figure S7 Venn diagrams of DEGs, induced in the ventral midbrain of mice at 13 dpi and 90 dpi after striatal injections of α‐syn PFFs, overlapping with DEGs from PD databases. No clear pattern was observed at this level of analysis. See man text for details and literature references.
Table S1 Antibodies used.
Table S2 Softwares used.
Table S3 Overlapping genes of the datasets of the α‐syn seeding/spreading model at 13dpi and 90 dpi with genes of microglial subtypes isolated from the nigro‐striatal circuit. See main text for details.
Table S4 Overlapping genes of the α‐syn seeding/preadig model at 13dpi and 90 dpi with genes of a disease‐associated microglia core signature (see main text for details).
Table S5 Unusual microglial gene expression signature in the α‐syn seeding/spreading model at 13dpi and 90 dpi. The bold higlighted genes may be involved in inducing neurodegeneration (see main text for details).
Table S6 Molecular astrocyte and peripheral immune cell genes after striatal PFF injection.
Data Availability Statement
Original datasets and list of genes from comparative analyses are available upon any reasonable request to the correspondence author (manuel.buttini@uni.lu). The GEO accession number of the microarray expression data is GSE15571. (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE155716).