

Abstract
Pathogenic variants in heterogeneous nuclear ribonucleoprotein U (HNRNPU) results in a novel neurodevelopmental disorder recently delineated. Here, we report on 17 previously unpublished patients carrying HNRNPU pathogenic variants. All patients were found to harbor de novo loss‐of‐function variants except for one individual where the inheritance could not be determined, as a parent was unavailable for testing. All patients had seizures which started in early childhood, global developmental delay, intellectual disability, and dysmorphic features. In addition, hypotonia, behavioral abnormalities (such as autistic features, aggression, anxiety, and obsessive–compulsive behaviors), and cardiac (septal defects) and/or brain abnormalities (ventriculomegaly and corpus callosum thinning/agenesis) were frequently observed. We have noted four recurrent variants in the literature (c.1089G>A p.(Trp363*), c.706_707del p.(Glu236Thrfs*6), c.847_857del p.(Phe283Serfs*5), and c.1681dels p.(Gln561Serfs*45)).
Keywords: global developmental delay, HNRNPU, intellectual disability, seizures
1. INTRODUCTION
Heterogeneous nuclear ribonucleoproteins (HNRNPs) are a large family of RNA‐binding proteins that play a role in controlling the maturation of newly formed heterogeneous nuclear RNAs into messenger RNAs, and have roles in RNA splicing, polyadenylation, capping, modification, export, localization, translation, and turnover (Glisovic et al., 2008; Keene, 2007; Wu et al., 2018). Through interacting with ribonucleoproteins (RNPs), HNRNPs are directly involved in every stage of mRNA formation and processing as well as overseeing its entire development (Chen et al., 2013; Lim et al., 2016). The ability to do this shows that the HNRNP family comprises a number of incredibly complex and versatile proteins. In fact, it is thought that the functional flexibility the HNRNP family possesses can be explained by their ability to produce multiple alternatively spliced isoforms and their ability to form complexes with other HNRNP members (Geuens et al., 2016).
HNRNPs have also been linked to various diseases, including cancer, neurodevelopmental disorders, spinal muscular atrophy, amyotrophic lateral sclerosis, congenital myasthenic syndrome, multiple sclerosis, Alzheimer's disease, and fronto‐temporal lobe dementia (Low et al., 2021). Their key roles in regulating transcriptional and post‐transcriptional gene expression and their links to numerous diseases mean that unsurprisingly they have attracted much attention by researchers.
Most HNRNP proteins are predominantly present in the nucleus during steady state and translocate into the cytoplasm upon post‐translational stimulation or when recruited by other HNRNPs (Han et al., 2010). This too is true of HNRNPU—the largest of the HNRNP proteins, its role largely involves transcription and alternative splicing (among other functions) (Geuens et al., 2016).
Patients with variants in HNRNPU have been known to present with seizures, global developmental delay, and intellectual disability (ID). To date, several studies have reported probands of patients with deletions encompassing or pathogenic variants of HNRNPU (Bramswig et al., 2017; Caliebe et al., 2010; Depienne et al., 2017; Durkin et al., 2020; Leduc et al., 2017; Song et al., 2021; Thierry et al., 2012; Yates et al., 2017). In this study, we expand on previously unpublished data from our group and further describe unreported n = 17 probands who have de novo loss‐of‐function variants in HNRNPU. We provide a comprehensive overview of published literature with specific emphasis on the seizure phenotype which has not been expanded previously and examine the genotype data available from both this study and previous studies.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
Informed consent for inclusion and publication was obtained from all participants involved in this study.
The DDD study has UK Research Ethics Committee approval (10/H0305/83, granted by the Cambridge South REC, and GEN/284/12 granted by the Republic of Ireland REC).
2.2. Method
In this study, we describe 17 probands with previously unreported likely pathogenic variants. We used the American College Medical Genetics (ACMG) guidelines for variant classification (Richards et al., 2015). Proband 6 was identified through identifying previously unpublished patients on DECIPHER (http://decipher.sanger.ac.uk.) (Firth et al., 2009). Proband 5, 7 and 8 were identified by contacting clinicians who have uploaded HNRNPU submissions onto GeneMatcher (https://genematcher.org/statistics/). The rest of the probands (1–4 and 9–17) were identified through contacting individuals who are part of an online support group for families supporting individuals with HNRNPU variants. This meant that for these patients (patients 1–4 and 9–17) anonymized data on a standardized clinical proforma was collected from the affected individual's carer/parent and where possible, additional data was collated by contacting the clinician responsible for these patients.
For the literature review, a systematic method was used to perform an initial search of the relevant literature. The databases used include Web of Science, MEDLINE via Ovid, PubMED, and The Cochrane Library. Searches were undertaken using the keyword “HNRNPU.” Publications were used based on their title, then the abstract and further publications were identified through the references of included papers.
3. RESULTS
3.1. Proband 1
Patient 1 was a 4‐year‐old male born to nonconsanguineous white European parents with no family history of intellectual disability or epilepsy. The pregnancy was complicated by pre‐eclampsia and abnormalities detected on foetal scans. Borderline ventriculomegaly, mild bilateral hydronephrosis, and a short femur length were detected antenatally. At 36 weeks, intra‐uterine growth restriction (IUGR) and abnormal umbilical artery dopplers were detected, so the decision was made to induce labor, and baby was delivered at 36 + 6 weeks gestation by emergency caesarean section. He required phototherapy for mild jaundice. There was no history of maternal illnesses and no significant exposures during the pregnancy.
His birth weight was on the 3rd centile. He was born with feeding difficulties and had mild bilateral hydronephrosis which resolved spontaneously at birth. He was found to have ostium secundum atrial septal defect (ASD) of moderate size and mild branch pulmonary stenosis at birth, both of which resolved spontaneously. He also developed neonatal jaundice which resolved with phototherapy.
He developed a seizure phenotype at 6 months. His seizures characteristically started as absence seizures and then moved on to became focal, and if they continued for a significant amount of time became tonic–clonic. He had five admissions to the intensive care unit (ICU)—four of which were for status epilepticus and one from respiratory arrest due to a benzodiazepine overdose. He has been seizure free for over a year. He remained on topiramate and sodium valproate for epilepsy and clobazam was added in when he was unwell. Before the age of 3 years, he suffered from breakthrough seizures despite medications, these occurred every few weeks to every few months and were triggered by high fevers.
He has moderate intellectual disability and global developmental delay. He spoke his first word at 2 years, and this remained the only word he could speak. He was able to sit unsupported at 10 months and took his first steps at 30 months.
Dysmorphic features included a bilateral and a long columella. He was also born with four limb post‐axial polydactyly and a convergent strabismus. He had a left sided inguinal hernia at 2 months. Polydactyly was removed, and the hernia was repaired at 2 months.
3.2. Proband 2
Patient 2 was a 34‐year‐old female, third child born to nonconsanguineous parents. The family history was unremarkable other than one paternal first cousin with a history of epilepsy. There was no history of maternal illnesses or significant exposures during pregnancy. There were no abnormalities noted on scans during pregnancy.
She was born at 39 weeks gestation with a birth weight on the 56th centile. There was hypotonia noted during the neonatal period and she remained hypotonic. She developed a seizure phenotype at 12 months and was diagnosed with epilepsy with tonic–clonic seizures. She had several breakthrough seizures in early childhood and during adolescence despite being on medication—but since starting lacosamide she has remained seizure free.
She has global developmental delay. She was able to sit unsupported at 12 months, spoke her first word at 24 months and took her first steps at 3.5 years. She has moderate intellectual disability. Dysmorphic features included a thin upper vermillion, slight frontal bossing and prominent eyebrows. She also had strabismus.
She was also diagnosed with autism, obsessive compulsive disorder (OCD), and anxiety.
3.3. Proband 3
Patient 3 was an 11‐year‐old female. Her maternal grandparents are cousins. There was a family history of maternal multiple sclerosis. The mother took interferon beta‐1a (Rebif) injections for her MS for the first 5 weeks of pregnancy but stopped this as soon as she found out she was pregnant. There was no family history of intellectual disability or epilepsy. There were no abnormalities detected antenatally, however the child's mother noted that there were reduced foetal movements compared to her first pregnancy.
She was born by elective caesarean at 37 + 6 weeks gestation, birth weight on the 6th centile, head circumference on the 31st centile and height on the 5th centile. She was born hypotonic and had feeding difficulties during the neonatal period and remained hypotonic.
She was diagnosed with epilepsy at 2 years of age and had therapy‐refractory focal and secondary generalized seizures, tonic seizures, as well as absence seizures with eyelid myoclonus. She was on clobazam, lamotrigine, perampanel, and brivaracetam for her epilepsy, yet still had between 5 and 12 breakthrough seizures a month.
She had severe intellectual disability and global developmental delay. Developmental abnormalities were first noticed at 3 months of age when it was noted that she rarely smiled, had poor eye contact and poor drinking. She was able to sit unsupported at 12 months and took her first steps at 3 years. She is aphasic. She was also diagnosed with autism spectrum disorder and has secondary microcephaly.
3.4. Proband 4
Patient 4 was a 7‐year‐old female born to nonconsanguineous, white European parents. She was the oldest of four children, all her siblings were fit and well with no medical conditions. There was a family history of childhood and adult epilepsy affecting family members on her paternal side. There were no maternal illnesses or significant exposures during the pregnancy. The pregnancy was normal apart from vanishing twin syndrome at 7 weeks gestation.
She was born at 42 weeks with a birth weight on the 2nd centile and head circumference on the 23rd centile. She was hypotonic and had feeding difficulties at birth; the hypotonia has remained.
She experienced her first tonic–clonic seizure at 14 months, although it was suspected she was having absence seizures prior to this. She suffered from tonic–clonic and focal seizures which were controlled with medication, however, she experienced breakthrough seizures when her medication dose was not closely monitored.
She has global developmental delay and severe intellectual disability. She could sit unsupported at 12 months and took her first steps at 22 months.
Dysmorphic features included a small nose, a narrow mouth and frontal bossing. She also has a diagnosis of autism spectrum disorder and strabismus.
A brain MRI noted mesial sclerosis of the hippocampus and enlarged temporal lobe, there were no other abnormalities noted on MRI.
3.5. Proband 5
Patient 5 was a 26‐year‐old female born at 39 weeks gestation on the 19th centile for weight. She did not experience hypotonia or feeding difficulties during the neonatal period.
She has epilepsy and took lamotrigine. She had previously taken sodium valproate and vigabatrin which was unable to control the seizures completely.
She has severe intellectual disability and global developmental delay. She was aphasic, was unable to sit unsupported and took her first steps at 30 months. She also displayed auto‐aggressive behavior, depression, and anxieties related to hyperventilation. There was no hypotonia.
Dysmorphic features included a prominent nose, low set ears, a prominent epicanthus, a low set hairline, hallux valgux, and alopecia. She also had scoliosis.
3.6. Proband 6
Patient 6 was a 9‐year‐old female. She was born by caesarean section at term of gestation complicated by IUGR with a birth weight of 2330 gr (10th centile), length of 46 cm (10th‐25th centile), and occipitofrontal circumference of 31.5 cm (5th centile). Her perinatal history was unremarkable, and she was neither hypotonic nor had feeding problems in the neonatal period. At 15 months of age, she had her first tonic–clonic generalized seizure with abnormal EEG and she was started on valproate. She had several other seizures always during fever episodes.
She walked independently at 18 months and said her first words at 10 months, but her language development was delayed and by the age of 6 years she could say about 100 words and she spoke in short sentences of 1–2 words. By the age of 9 years, her language further improved, and she was able to speak by simple sentences and to make simple calculations. She had good social skills. She developed hyperphagia and gained weight by the age of about 9 years. On physical examination at the age of 9 years, her weight was 26.5 kg (25th centile), her height was 116.5 cm (SDS −2.9), her BMI was 19.5 (89th centile) and her occipitofrontal circumference was 49.6 cm (SDS −3.1).
She was noted to have dysmorphic features including a thin upper vermillion and bilateral II‐III toe syndactyly. She was also found to have cyclical neutropenia with an otherwise normal hematological work‐up. An MRI of her brain showed ventriculomegaly and a cyst on the pineal gland.
3.7. Proband 7
Patient 7 was a 5‐year‐old male. He was born at 40 + 1 weeks and weighed on 27th centile. He suffered from hypotonia in the neonatal period as well as feeding problems and reflux. He does not have epilepsy, however, has had seven febrile seizures – the first of which was at 12 months of age.
He has moderate intellectual disability and global developmental delay. He first sat unsupported at 12 months and said his first word and took his first steps both at 24 months.
Dysmorphic features included hypermetropia, and a thin upper vermillion. He had a large atrial septal defect which required surgery. He also had autism, strabismus and hyperlaxity.
A brain MRI showed medio‐posterior atrophy of the corpus callosum, occipital atrophy, and global cerebral atrophy.
3.8. Proband 8
Patient 8 was a 23‐year‐old female, born at 40 weeks on the 49th centile for weight. She had hypotonia and feeding difficulties in the neonatal period. She remained hypotonic.
She suffered from epilepsy for which she took levetiracetam and lamotrigine. She had moderate intellectual disability and global developmental delay. She took her first steps at 2 years and spoke her first word at 3 years.
Dysmorphic features included a long columella, a high‐arched palate, synophrys, and hallux valgus.
3.9. Proband 9
Patient 9 was a 3‐year‐old male, born to nonconsanguineous parents with no significant family history. There was no history of maternal illnesses nor significant exposures during the pregnancy. He was born at 40 weeks and weighed on the 62nd centile. He did not suffer feeding difficulties or hypotonia during the neonatal period.
Since birth, he developed hypotonia and epilepsy with EEG abnormalities, he took levetiracetam for epilepsy which remains well controlled. He experienced his first febrile seizure at 11 months and his first afebrile seizure at 18 months. His seizures were originally focal seizures but have since progressed to generalized tonic–clonic seizures.
He has global developmental delay. He was able to first sit unsupported at 14 months, spoke his first word at 23 months and took his first steps at 3 years.
He had a ventricular septal defect (VSD) repair at 4 months and a bilateral orchidopexies at 3 years 4 months for cryptorchidism. Dysmorphic features included a sacral dimple and single palmar crease.
3.10. Proband 10
Patient 10 was a 3‐year‐old male born to white European, nonconsanguineous parents. There was no history of maternal illnesses, significant exposures, or abnormal scans during the pregnancy. The family history was unremarkable.
He was born at 39 weeks and weighed on the 44th centile. He was born with hypotonia (which remained) and feeding difficulties.
At 10 months, he experienced his first possible febrile seizure. At 12 months, he had his first absence seizure. He was on medication for his epilepsy, but still experienced breakthrough seizures when his levetiracetam dose was adjusted.
He has intellectual disability and global developmental delay. He was able to sit unsupported at 11 months, said his first word at 12 months and took his first steps at 19 months. He has autism spectrum disorder.
3.11. Proband 11
Patient 11 was a 14‐year‐old female born to nonconsanguineous, white European parents. She was born at 39 weeks gestation with a head circumference on the 32nd centile. She experienced hypotonia during the neonatal period (which has remained) and her feeding difficulties led to her to being gastrostomy fed as a neonate.
She had her first seizure at 11 months. Her epilepsy consisted of tonic–clonic, absence and atonic seizures. Her absence seizures are refractory to treatment with sodium valproate and lamotrigine.
She has severe intellectual disability and global developmental delay. She was able to sit unsupported at 18 months, took her first steps at 6 years and said her first word at 12 years.
Dysmorphic features included a small nose. She also had scoliosis, joint hypermobility, a hirsute back, clinodactyly of the Vth finger bilaterally and has grommet insertion in both ears.
She was diagnosed with autism spectrum disorder and displayed aggressive behavior.
3.12. Proband 12
Patient 12 was an 8‐year‐old male. Like his brother, he was born at 36 weeks by caesarean section because of pre‐eclampsia and IUGR. He was born with a weight on the 14th centile, height on the 8th centile and head circumference on the 10th centile. He had hypotonia, jaundice and hypoglycaemia soon after birth and needed total parenteral nutrition. He had haematemesis during the neonatal period.
He has intellectual disability and epilepsy for which he took sodium valproate. He has global developmental delay; he said his first word at 18 months and gained sphincteric control at 5 years old.
Dysmorphic features included a depressed nasal bridge, a high forehead, synophrys, diastasis of the upper incisors, bilateral single palmer crease, clinodactyly of the Vth finger bilaterally, preaxial polydactyly of right foot, syndactyly II‐III toe of left foot and proportionate short stature.
3.13. Proband 13
Patient 13 was an 18‐year‐old male born to nonconsanguineous parents with an unremarkable family history. There were no abnormalities on antenatal scans, or significant illnesses or exposures during pregnancy. He was born at 40 weeks and his birthweight was on the 54th centile. He did not experience hypotonia during the neonatal period, but he did have feeding difficulties.
He has epilepsy and experienced his first seizure at 12 months of age. He had tonic–clonic seizures for which he took sodium valproate, but these remained refractory to treatment. An EEG showed diffuse slowing, L epileptiform discharges.
He was diagnosed with severe intellectual disability and global developmental delay; he was aphasic. He first sat unsupported at 11 months and took his first steps at 24 months.
Dysmorphic features included hypertelorism, bilateral epicanthic folds, a broad nasal root, a high and narrow palate, dysplastic pinnae, posteriorly rotated and slightly low set ears, bitemporal narrowing, a prominent forehead, II‐III toe syndactyly with prominent and large central incisors.
He displays autistic features but is not formally diagnosed with autism spectrum disorder. He also has behavioral issues. He displays hypertonia in the extremities but axial hypotonia and has been diagnosed with spastic diplegic cerebral palsy. Brain MRIs at 6 months and at 6 years were both normal.
3.14. Proband 14
Patient 14 was a 3‐year‐old male born to nonconsanguineous parents. There were no significant maternal exposures or illnesses during pregnancy and no abnormalities were detected on antenatal scans. There was no significant family history. He was born at 40 + 2 weeks and weighed on the 62nd centile. There was no hypotonia or feeding difficulties during the neonatal period.
He experienced his first febrile seizure at 11 months. His first tonic–clonic seizure occurred at 18 months. His epilepsy is well controlled on levetiracetam. He has moderate–severe intellectual disability and global developmental delay. He first sat upright at 14 months, spoke his first word at 23 months and took his first steps at 3 years.
Dysmorphic features included a thin upper vermillion, a single palmar crease, and a sacral dimple. He had a ventricular septal defect repair at 4 months and a bilateral orchidopexies at 3 years 4 months for cryptorchidism.
3.15. Proband 15
Patient 15 was a 2‐year‐old female born to nonconsanguineous parents with no significant family history. There were no significant maternal exposures or illnesses during pregnancy. IUGR was noted on antenatal the scans, and her mother reported that this baby had reduced foetal movements compared to her previous pregnancy.
She was born at 37 weeks and weighed on the 0.4th centile. She was hypotonic during the neonatal period but did not have feeding difficulties. She has epilepsy with EEG abnormalities and experienced her first seizure at 18 months. She had focal seizures and occasionally had refractory seizures despite treatment with levetiracetam.
She has moderate intellectual disability and global developmental delay. She has not taken her first steps nor spoken her first word yet at 2 years of age but sat unsupported at 12 months.
3.16. Proband 16
Patient 16 was a 36‐year‐old male born to nonconsanguineous parents. The pregnancy was normal apart from hospitalisation at 7 weeks due to hyperemesis. There were no abnormalities observed on antenatal scans. He was born at 40 weeks on the 63rd centile.
He was hypotonic and had feeding difficulties during the neonatal period. He did not have epilepsy but at 18 months had the first of many febrile seizures. He has mild intellectual disability and global developmental delay yet. He first sat upright at 6.5 months, spoke his first word at 9.5 months and took his first steps at 10.5 months.
He displayed autistic features and anxiety as well as aggressive behavior and has been diagnosed with OCD. He had swallowing problems, constipation, and an incisor with two roots and has suffered from chronic ear infections. He had ear surgery on a perforated ear drum and used a hearing aid.
He is not diabetic but has low insulin levels and experienced low blood sugars. He also struggled to gain weight despite eating a high calorific diet.
3.17. Proband 17
Patient 17 was a 3‐year‐old female born to nonconsanguineous parents with no significant family history. There were no abnormalities observed on antenatal scans and there were no significant maternal illnesses/ exposures. She was born at 39 + 4 weeks and weighed on the 53rd centile.
She was hypotonic in the neonatal period (which remains) but experienced no feeding difficulties. She has epilepsy with no EEG abnormalities and experienced her first seizure at 10 months. She had both tonic–clonic and absence seizures which remained well controlled with sodium valproate.
She has mild intellectual disability and global developmental delay. She first sat unsupported at 9 months, spoke her first word at 12 months, and took her first steps at 2 years and 3 months.
Brain imaging showed asymmetry of lateral ventricles (left bigger than right) and she was born with an atrial septal defect. She also takes levothyroxine for hypothyroidism.
4. LITERATURE REVIEW AND DISCUSSION
4.1. HNRNPU ‐related neurodevelopmental disorder
The HNRNPU gene, located on 1q44, is a DNA and RNA‐binding protein (Durkin et al., 2020). It is involved in nuclear chromatin organization, telomere‐length regulation, mRNA alternative splicing and stability, Xist‐mediated transcriptional silencing, mitotic cell cycle regulation, negatively regulates glucocorticoid‐mediated transcriptional activation and participates in circadian regulation (https://www.genecards.org/cgi-bin/carddisp.pl?gene=HNRNPU). Thierry et al. (2012) showed that HNRNPU is expressed in at least six different tissues: adult brain, heart, kidney, liver, cerebellum tissues, and foetal brain tissue, with the strongest expression in the cerebellum.
Previous research has found that deletions encompassing or de novo loss‐of‐function variants of HNRNPU can lead to a neurodevelopmental disorder characterised by variable degree but usually moderate to severe intellectual disability, seizures, behavioral abnormalities, and characteristic craniofacial dysmorphism and agenesis of the corpus callosum (Ballif et al., 2012; Bramswig et al., 2017; Durkin et al., 2020; Leduc et al., 2017; Yates et al., 2017). The phenotypic variability is possibly attributable to the variable size and gene content between patients (Caliebe et al., 2010). Additionally, a study by Caliebe et al. (2010) in which whole exome sequencing was used suggested that haploinsufficiency was the main mechanism of pathogenicity in HNRNPU variants.
A comprehensive review of the literature published so far on HNRNPU found eight papers which described clinical features of effected individuals. The detailed findings of our 17 probands can be found in Table 1, and the collated findings of this literature review are reported in Table 2. Table 3 depicts the association between gene variant, type of variant and phenotype. Variant nomenclature in Tables 1 and 3 is according to gene transcript NM_031844.2.
TABLE 1.
Phenotypic data patients 1–17
| Patient number | Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | Patient 7 | Patient 8 | Patient 9 |
|---|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | Female | Female | Male | Female | Male |
| Age (in years) | 3.9 | 34.83 | 7.4 | 7.9 | 26.5 | 9.83 | 5.5 | 23 | 3.83 |
| Consanguinity in the family | No | No | Grandparents are cousins | No | – | No | – | – | No |
| Family history of intellectual disability or epilepsy | No | First cousin has epilepsy | No | Several have epilepsy | – | First cousin of the father and paternal grandmother who has mengioma | – | – | No |
| Heterozygous cDNA change | c.878‐9T>G | c.508C>T | c.2270_2271delCT | c.893 A>G | c.1617dup | c.2304_2305del | c.2425‐2A>G | c.730_731delAG | – |
| Amino acid change | – | p.Gln170 | p.Pro757Argfs*7 | p.(H298R) | p.(Ala540Serfs*10) | p.(Gly769Glufs*83) | – | p.(Arg244Glyfs*3) | – |
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo | De novo |
| Additional genetic defect | – | No | No | m.15266A>G | No | No | No | No | No |
| Gestational age at birth (weeks) | 36+6 | 39 | 35 | 42 | 39 | 38 | 40+1 | 40 | 40 |
| Birth weight (kg) | 2.3 | 3.20 | 2.77 | 2.78 | 2.86 | 2.33 | 3.28 | 3.4 | 3.7 |
| Maternal illnesses or significant exposures | Pre‐eclampsia | No | Rebif for first 5 weeks | No | – | No | – | – | No |
| Abnormalities/abnormal scans | Ventriculomegaly, bilateral hydronephrosis, short femurs, IUGR | No | No | Vanishing twin syndrome at 7 weeks | – | IUGR | – | – | – |
| Neonatal hypotonia | No | Yes | Yes | Yes | No | No | Yes | Yes | No |
| Neonatal feeding difficulties | Yes | Yes | Yes | Yes | No | No | Yes | Yes | No |
| Other neonatal problems | Jaundice | No | No | No | No | No | Reflux | No | No |
| Intellectual disability | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | – |
| Severity of intellectual disability | Moderate | Moderate | Moderate to severe | Severe | Severe | Moderate | Moderate | Moderate | – |
| Global developmental delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Language delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Age of first words | 2 – only has one word | 24 months | Aphasic | Aphasic | Aphasic | 10 months | 2 years | 3 years | 23 months |
| Motor delay | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Age first sat unsupported | 10 months | 12 months | 12 months | 12 months | – | – | 12 months | – | 14 months |
| Age of first steps | 30 months | 3.5 years | 36 months | 22 months | 30 months | 18 months | 24 months | 2 years | 3 years |
| Any psychiatric diagnosis? | No | Autism, OCD, Anxiety | Autism | Autism | No | No | Autism | No | No |
| Other psychopathology | No | No | No | No | Aggressive behavior, depression, anxiety | Hyperphagia | No | No | No |
| Hypotonia | Yes | Yes | Yes | Yes | No | No | Yes | Yes | Yes |
| Epilepsy | Yes | Yes | Yes | Yes | Yes | Yes | No but had seven febrile seizures | Yes | Yes |
| EEG abnormality | No | No | – | Yes | No | Yes | – | – | Yes |
| Delayed myelinisation | No | – | No | No | No | No | No | – | No |
| Corpus callosum abnormalities | No | – | No | – | No | No | Medio‐posterior atrophy | – | No |
| Colpocephaly | No | – | No | No | No | No | No | – | No |
| Ventriculomegaly | Borderline ventriculomegaly | – | No | No | No | Yes | No | – | No |
| Other | No | – | No | Mesial sclerosis of hippocampus, enlarged temporal lobes | No | Cyst on pineal gland | Occipital atrophy, global cerebral atrophy | – | No |
| Age of first seizure | 6 months | 12 months | <24 months | 14 months | – | 15 months | 12 months febrile | – | 11 months febrile, 18 months epileptic |
| Seizure types | Absence, focal, tonic–clonic | Tonic–clonic | Focal, tonicclonic, tonic, absences, myoclonus | Absence, focal, tonic–clonic | – | Tonic–clonic | – | – | Focal, tonic–clonic |
| Refractory seizures | No longer | No | Yes | Yes | – | No | – | – | No |
| Dysmorphic features: eyes | Bilateral convergent squint | Strabismus | – | Strabismus | Prominent epicanthus | Normal | Strabismus, hypermetropia | Synophris | – |
| Nose | Long columella | – | – | Small | Prominent nose | Normal | Normal | Long columella | – |
| Mouth | – | Thin upper vermillion | – | Narrow | Cleft palate | Thin upper vermillion | Thin upper vermillion | No | – |
| Ear | – | – | – | – | Low set | Normal | Normal | No | – |
| Face | – | Oblong, prominent eyebrows | Aspecific dysmorphism | Round, small | – | Normal | – | No | – |
| Forehead | – | Slight frontal bossing | – | Frontal bossing | Low‐set hairline | Normal | – | No | – |
| Other | – | – | Tapering fingers, cutis marmorata | – | Scoliosis, hallux valgus, short hand, big phalanx | Short stature, II‐III toe syndactyly | – | No | – |
| Other clinical features | Four limb post‐axial polydactyly, inguinal hernia, moderate ASD secundum, mild pulm. stenosis | – | – | – | Alopecia, eczema | Cyclic neutropenia | Large ASD, hyperlaxity | Arched palate, hallux valgus | Sacral dimple, single palmar crease, VSD, Bilateral undescended testes |
| Past tests: Karyotyping | 46XY | – | – | – | Yes | – | No | No | No |
| CGH‐array | – | – | Normal | Yes | Yes | Yes | Normal | Normal | Yes |
| Single gene tests | – | – | No | No | Yes | Yes | Yes | No | No |
| Metabolic testing | – | – | Normal | Yes | Yes | No | Normal | No | Yes |
| Current medical treatment | Topiramate, sodium valproate, clobazam | Lacosamide | Clobazam, lamotrigine, perampanel and brivaracetam | – | Lamotrigine, omeprazole, trimebutine, hydroxyzine, betamethasone | Sodium valproate | No | Keppra, lamotrigine | Keppra |
| Medical treatment used in the past | – | – | – | – | Sodium valproate, vigabatrin, forlax | – | – | – | – |
| Patient 10 | Patient 11 | Patient 12 | Patient 13 | Patient 14 | Patient 15 | Patient 16 | Patient 17 |
|---|---|---|---|---|---|---|---|
| Male | Female | Male | Male | Male | Female | Male | Female |
| 3.3 | 14 | 8.75 | 18.08 | 3.75 | 2.1 | 36.7 | 3.16 |
| No | No | – | No | No | No | No | No |
| No | – | – | No | No | No | – | No |
| – | – | c.1665_1666delGT | c.1834G>A | Del exons 1‐11 | c.742dup | c.804‐9_804‐6del | c.1243del |
| – | – | – | p.Asp612Asn | – | pARG248Lysfs*12 | – | p. (Asp415metfster3) |
| De novo | De novo | De novo | – | De novo | De novo | De novo | De novo |
| – | – | – | Loss of clone RP11‐38O23 at Xp11.23 | No | No | No | Duplication (16p13.11p12.3) |
| 39 | 39 | 36 | 40 | 40+2 | 37 | 40 | 39+4 |
| 3.49 | – | 2.26 | 3.6 | 3.7 | 2 | 3.71 | 3.44 |
| No | – | Pre‐eclampsia | No | No | No | Hyperemesis at 7 weeks | No |
| No | – | IUGR | No | No | IUGR, few foetal movements | No | No |
| Yes | Yes | Yes | No | No | Yes | Yes | Yes |
| Yes | Yes | Yes | Yes | No | No | Yes | No |
| No | No | Haemateme sis, jaundice | No | No | No | – | No |
| Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| – | Severe | – | Severe | Moderate | Moderate | Mild | Mild |
| Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| Yes | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| 12 months | 12 years | 18 months | Aphasic | 23 months | Aphasic | 5.5 months | 12 months |
| – | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| 11 months | 18 months | – | 11 months | 14 months | 12 months | 6 months | 9 months |
| 19 months | 6 years | – | 24 months | 3 years | Not achieved | 10.5 months | 2 years 3 months |
| Autism | Autism | – | Likely autism | – | – | Autistic features, OCD | No |
| No | Aggressive behavior | – | Behavioral issues | No | No | Aggressive behaviors, anxiety | No |
| Yes | Yes | Yes | Hypertonic in extremities, axial hypotonia | Yes | Yes | Yes | Yes |
| Yes | Yes | Yes | Yes | Yes | Yes | No (but febrile seizures) | Yes |
| – | Yes | – | Diffuse slowing, l epileptiform discharges | Yes | Yes | As a child | No |
| – | – | – | – | – | – | No | No |
| – | – | – | No | No | – | No | No |
| – | – | – | Normal | No | – | No | No |
| – | – | – | No | No | – | No | Asymmetry (left>right) |
| – | – | – | No | No | – | No | No |
| 10 months febrile, 12 months absence | 11 months | – | 12 months | 11 months febrile, 18 months epileptic | 18 months | 18 months febrile | 10 months |
| Absence | Tonic–clonic, absence, atonic | – | Tonic–clonic | Tonic–clonic | Focal seizures | – | Tonic–clonic, absence |
| Yes | Yes | – | Yes | Yes | Yes | – | No |
| Strabismus | – | Synophris | Hypertelorism, bilateral epicanthal folds | No | No | Thinning outer eyebrows | No |
| – | Small | Depressed nasal bridge | Broad nasal root | No | No | Depressed nasal bridge | No |
| – | – | Diastasis of upper incisors | High and narrow palate | Thin upper vermillion | Thin upper vermillion | Slightly high palate | No |
| – | – | – | Dysplastic pinnae, posteriorly rotated, low set | No | No | – | No |
| – | – | – | Bitemporal narrowing | No | No | – | No |
| – | – | High forehead | Prominent | No | No | – | No |
| – | Clinodactyly of Vth finger bilaterally | Clinodactyly of Vth finger bilaterally, preaxial polydactyly of right foot, syndactyly II–III toe of left foot | II–III toe syndactyly | Single palmar crease, sacral dimple | No | Swallowing problems, constipation, chronic ear infections. | No |
| – | Hypermobile, hairy back, grommet's | Single palmar crease bilaterally, short stature | Prominent and large central incisors, Spastic diplegia | Hypermetropia, VSD, bilateral undescended testes | – | An incisor is a double tooth with two roots | ASD |
| – | – | Yes | 46XY | No | – | Yes | – |
| – | Yes | – | Yes | No | Yes | No | Inherited duplicati |
| – | – | – | No | No | Yes | No | Nono |
| – | – | – | Yes | No | No | Yes | Yes |
| Levetiracetam | Sodium valproate, lamotrigine | Sodium valproate | Sodium valproate | Keppra | Levetiracetam | CBD with low THC. manganese Linaclotide, Melatonin, B12 | Sodium valproate, levothyroxine |
| – | – | – | Carbamazepine, Levetiracetam | – | Grommets, Toe alignment splint | Phenobarbital, phenytoin. Bilateral hearing aids. Ear surgery. Citalopram, risperidone | No |
Note: Variant nomenclature is according to gene transcript NM_031844.2. “–” indicates no data was available.
TABLE 2.
Phenotype data from literature review
| Our study | Durkin et al. (2020) | Thierry et al. (2012) | Yates et al. (2017) | Bramswig et al. (2017) | Leduc et al. (2017) | Depienne et al. (2017) | Caliebe et al. (2010) | Song et al. (2021) | Overall | |
|---|---|---|---|---|---|---|---|---|---|---|
| Neonatal hypotonia | 11/17 | n/r | n/r | 2/7 | 2/7 | 1/4 | n/r | n/r | n/r | 16/35 (45.7%) |
| Neonatal feeding difficulties | 10/17 | 10/15 | n/r | 1/6 | 3/7 | n/r | n/r | n/r | n/r | 21/37 (56.8%) |
| Intellectual disability | 16/17 | 11/21 | 11/11 | 7/7 | 6/6 | 4/4 | 7/7 | 4/4 | 1/2 | 69/79 (87.3%) |
| Global developmental delay | 17/17 | 20/20 | 11/11 | 7/7 | 5/5 | 4/4 | 7/7 | 4/4 | 2/2 | 75/75 (100%) |
| Autistic features | 8/16 | 3/21 | 4/9 | 1/5 | 4/6 | 1/4 | 1/7 | n/r | 1/2 | 23/70 (32.9%) |
| Hypotonia | 15/17 | n/r | 4/9 | 2/7 | 6/7 | 2/4 | 4/5 | 3/4 | n/r | 36/45 (80.0%) |
| Seizures | 17/17 | 20/21 | 11/11 | 5/7 | 6/6 | 4/4 | 6/7 | 4/4 | 2/2 | 75/79 (94.9%) |
| Seizures before 24 months | 12/12 | 9/10 | 10/11 | 5/6 | 5/6 | 3/4 | 6/6 | n/r | 2/2 | 52/57 (91.2%) |
| Seizure type | ||||||||||
| Tonic–clonic | 8/11 | 7/16 | n/r | 1/6 | 4/6 | 3/4 | 5/5 | n/r | 2/2 | 30/50 (60.0%) |
| Absence | 6/11 | 5/16 | n/r | 2/6 | 3/6 | 2/4 | 4/5 | n/r | 0/2 | 22/50 (44.0%) |
| Refractory | 6/11 | n/r | 1/11 | 1/6 | 2/5 | n/r | 6/6 | 1/4 | 1/2 | 19/42 (45.2%) |
| Dysmorphism | 16/17 | 21/21 | 10/11 | 6/7 | 7/7 | 4/4 | n/r | 3/3 | 2/2 | 69/72 (95.8%) |
| Thin upper vermillion | 7/15 | 3/20 | n/r | 6/7 | 3/6 | 0/4 | n/r | 2/3 | 1/2 | 22/58 (37.9%) |
| Strabismus | 5/15 | 3/20 | 4/8 | 1/7 | 2/6 | 2/4 | n/r | 2/3 | 1/2 | 20/57 (35.1%) |
| Abnormality on brain MRI | 6/12 | 5/15 | 8/11 | 3/6 | 5/6 | 3/4 | 4/6 | 4/4 | 0/2 | 37/64 (57.8%) |
| Cardiac abnormality | 7/16 | 3/14 | 1/10 | 1/6 | 4/6 | 0/4 | n/r | 1/4 | 2/2 | 19/62 (30.6%) |
TABLE 3.
Genotype–phenotype data from the literature
| Variants | Neonatal feeding difficulties | Intellectual disability | Autistic features | Hypotonia | Tonic–clonic seizures | Absence seizures | Refractory seizures | Brain abnormalities | Cardiac abnormalities | Variant identified by |
|---|---|---|---|---|---|---|---|---|---|---|
| Splice site variants | ||||||||||
| c.692‐1G>A p.? | − | + | − | − | − | − | − | − | − | Depienne et al. (2017) |
| c.2425‐3C>A p.? | − | + | + | − | + | + | − | + | − | Depienne et al. (2017) |
| c.1117+1G>A p.? | − | + | − | + | − | − | − | − | − | Yates et al. (2017) |
| c.1743+1G>C p.? | + | + | − | − | + | + | − | − | − | Durkin et al. (2020) |
| c.692‐1G>A p.? | − | − | − | − | − | − | − | − | + | Durkin et al. (2020) |
| c.2425‐2A>G p.? | + | + | − | + | − | − | − | + | + | This study |
| c.143‐149del7 p.? | − | − | − | − | + | − | − | − | + | Song et al. (2021) |
| c.878‐9T>G p.? | + | + | + | − | + | + | − | + | + | This study |
| c.804‐9_804‐6del p.? | + | + | + | + | − | − | − | − | − | This study |
| Nonsense variants | ||||||||||
| c.508C>T p.(Gln170*) | − | + | + | − | − | − | − | − | − | This study |
| c.619C>T p.(Gln207*) | + | − | − | − | − | + | − | − | − | Durkin et al. (2020) |
| c.1450C>T p.(Arg484*) | − | − | − | − | − | − | − | − | − | Durkin et al. (2020) |
| c.67C>T p.(Arg23*) | + | − | − | − | − | − | − | − | − | Durkin et al. (2020) |
| c.1088G>A p.(Trp363*) | − | + | − | − | − | − | − | − | + | Durkin et al. (2020) |
| c.1801C>T p.(Arg601*) | − | + | + | − | − | − | − | − | − | Durkin et al. (2020) |
| c.1089G>A p.(Trp363*) | − | + | + | + | + | + | + | + | + | Durkin et al. (2020) |
| c.960G>A p.(Trp320*) | − | + | − | + | + | − | − | − | − | Yates et al. (2017) |
| c.1714C>T p.(Arg572*) | − | + | − | + | + | − | − | + | − | Leduc et al. (2017) |
| c.1089G>A p.(Trp363*) | − | + | + | + | + | + | + | + | + | Leduc et al. (2017) |
| c.817C>T p.(Gln273*) | + | + | − | + | − | − | − | − | + | Bramswig et al. (2017) |
| c.511C>T p.(Gln171*) | + | + | − | + | + | − | − | + | + | Bramswig et al. (2017) |
| c.523C>T p.(Gln175*) | − | + | − | − | + | + | − | − | − | Bramswig et al. (2017) |
| c.1681C>T p.(Gln561*) | − | + | − | + | − | + | − | − | − | Depienne et al. (2017) |
| Missense variants | ||||||||||
| c.893A>G p.(His298Arg) | + | + | + | + | + | + | + | + | − | This study |
| c.418G>A p.(Glu140Lys) | + | + | + | − | − | − | − | − | + | Yates et al. (2017) |
| c.970A>G p.(Arg324Gly) | + | + | − | + | − | − | + | + | + | Bramswig et al. (2017) |
| c.1132T>C p.(Ser378Pro) | − | + | − | + | + | + | + | + | − | Bramswig et al. (2017) |
| c.1834G>A p.(Asp612Asn) | + | + | + | + | + | − | + | − | − | This study |
| In‐frame deletions | ||||||||||
| c.837_839del p.(Glu279del) | + | + | − | − | − | − | − | − | − | Durkin et al. (2020) |
| c.1744_1767del p.(Thr582_Gln589del) | − | + | + | + | + | − | − | − | − | Bramswig et al. (2017) |
| Frameshift duplications | ||||||||||
| c.1617dup p.(Ala540Glyfs*4) | − | + | − | − | − | − | − | − | − | This study |
| c.1868dup p.(Glu624Argfs*24) | − | + | − | + | + | − | − | − | − | Depienne et al. (2017) |
| c.742dup p.(Arg248Lysfs*12) | − | + | − | + | − | − | + | − | − | This study |
| Frameshift deletions | ||||||||||
| c.1836del p.(Tyr613Ilefs*11) | − | + | − | − | + | + | − | − | + | Durkin et al. (2020) |
| c.1641del p.(Asp548Ilefs*5) | − | + | − | − | + | + | − | − | − | Durkin et al. (2020) |
| c.1681del p.(Gln561Serfs*45) | − | + | + | + | + | + | + | + | + | Durkin et al. (2020) |
| c.23del p.(Val8Glufs*4) | − | + | − | − | − | − | − | − | − | Yates et al. (2017) |
| c.1664del p.(Leu555Argfs*51) | − | + | − | − | − | − | − | − | + | Yates et al. (2017) |
| c.1681del p.(Gln561Serfs*45) | + | + | + | + | + | + | + | + | + | Depienne et al. (2017) |
| c.1282delC p.(Gly429Alafs*53) | − | + | + | − | + | − | + | − | + | Song et al. (2021) |
| c.16delinsATT p.(Val6Ilefs*4) | − | + | − | + | + | − | − | + | − | Depienne et al. (2017) |
| c.1243del p.(Asp415Metfs*3) | − | + | − | + | + | + | − | + | + | This study |
| c.2304_2305del p.(Gly769Glufs*83) | − | + | − | − | − | − | − | + | − | This study |
| c.730_731delAG p.(Arg244Glyfs*3) | + | + | − | + | − | − | − | − | − | This study |
| c.1665_1666delGT p.(Leu556Alafs*12) | + | + | − | − | − | − | − | − | − | This study |
| c.2270_2271delCT p.(Pro757Argfs*7) | + | + | + | + | + | + | + | − | − | Leduc et al. (2017) |
| c.1925_1926del p.(Leu642Profs*5) | + | + | − | − | − | − | − | − | + | Durkin et al. (2020) |
| c.395_401del p.(Asn132Thrfs*63) | + | − | − | − | + | − | − | − | − | Durkin et al. (2020) |
| c.2083_2084del p.(Ser695Trpfs*6) | − | − | − | − | + | − | − | − | − | Durkin et al. (2020) |
| c.706_707del p.(Glu236Thrfs*6) | + | + | + | + | + | + | + | + | + | Durkin et al. (2020) |
| c.454_466del p.(Ala152Thrfs*41) | − | + | + | − | − | + | − | − | − | Durkin et al. (2020) |
| c.706_707del p.(Glu236Thrfs*6) | − | + | + | + | + | + | + | + | + | Durkin et al. (2020) |
| c.712_715del p.(Lys238Alafs*100) | − | − | + | − | − | − | − | − | − | Durkin et al. (2020) |
| c.1626_1627insA p.(Lys544Glufs*25) | − | + | − | − | − | + | − | − | − | Yates et al. (2017) |
| c.1424_1425insTC p.(Ile476Profs*7) | − | + | + | − | − | + | + | − | − | Yates et al. (2017) |
| c.651_660del p.(Gly218Alafs*118) | − | + | − | + | + | − | − | + | − | Leduc et al. (2017) |
| c.2270_2271del p.(Pro757Argfs*7) | − | + | − | + | − | + | − | − | − | This study |
| c.2299_2302del p.(Asn767Glufs*66) | − | + | − | + | − | − | − | + | − | Depienne et al. (2017) |
| c.847_857del p.(Phe283Serfs*5) | − | + | + | + | + | + | + | + | + | Durkin et al. (2020) |
| Multi‐exon deletion | ||||||||||
| c.2167+35_*4156del p.? | − | + | − | − | − | − | − | − | + | Durkin et al. (2020) |
Note: Variant nomenclature in each table is according to gene transcript NM_031844.2.
In the following discussion, the numbers quoted for individual phenotypes includes both data collected from this study and the data collated from the literature.
Our research has further delineated common phenotypes of individuals with HNRNPU variants. Our data shows 11/17 individuals had neonatal hypotonia and 10/17 had neonatal feeding difficulties. Previous research by our group found that 11/21 individuals had feeding difficulties during the neonatal period and 2/7 had neonatal hypotonia (Durkin et al., 2020; Yates et al., 2017). The literature review found that 46% of individuals with HNRNPU variants experienced neonatal hypotonia and 57% neonatal feeding difficulties. 80% of individuals had hypotonia after the neonatal period.
Fourteen of seventeen patients had moderate or severe intellectual disability, two had mild ID. ID was suspected but not confirmed in the other case. Every individual (17/17) had global developmental delay. This is in keeping with published literature, which shows 100% of patients with HNRNPU had global developmental delay, and 87% have intellectual disability. Evidently, both phenotypes—particularly global developmental delay are common phenotypes associated with variants in this gene.
Seizures are another very widely reported feature of HNRNPU variants, and it is no surprise that our study further describes this. In the literature, 95% of patients had seizure phenotype—very few of these were febrile seizures and not epileptic. Interestingly, 91% of patients with epilepsy had their first epileptic seizure before their second birthday—and one patient had it days after their second birthday. The most common seizure type in our study was tonic–clonic seizures, with 8/11 individuals who reported seizure type describing this. Six of eleven described absence seizures and 6/11 described seizures which were refractory to anti‐epileptic treatments. Once again, these findings further emphasize findings from the literature where 60% of patients experienced tonic–clonic seizures and 44% absence seizures. Our review found 45% of individuals reported on experiencing seizures refractory to treatment.
Thirty‐three percent of patients from the literature review have either a diagnosis of autism spectrum disorder or autistic features. The incidence of autism/autistic features in our cohort was higher than this with half of our patients (8/16) meeting these criteria.
Our previous research has described the craniofacial dysmorphisms associated with HNRNPU variants. In this study, it should be noted that only 6/17 of individuals in our cohort were described by clinicians, the other 9/16 were described by parents of the affected individual so the descriptions for some of these patients are likely to be not all encompassing. Sixteen of seventeen of our cohort were described with a degree of craniofacial dysmorphism. The most frequent features described in our study were 5/15 had strabismus, 7/15 had a thin upper vermillion, 4/15 had frontal bossing, 4/17 had clinodactyly of the V finger, 4/17 had a long columella, 2/17 had hallux valgus, and 3/17 had syndactyly of the II‐III finger. In the literature, we found 96% had a dysmorphism, the most frequent being strabismus 35% and 38% had a thin upper vermillion. Previous work by us suggested that the most common dysmorphic features were palpebral fissure abnormalities, microcephaly, and wide spaced teeth (Durkin et al., 2020). In this cohort, 1 patient was described with palpebral fissure abnormalities and 1 with wide spaced teeth (another of our patient's described a double‐rooted incisor), none of the other features were described.
We found 6/12 patients had abnormalities on brain MRI, the most common abnormality being ventriculomegaly (3/11). Fifty‐eight percent of individuals in the literature also had nonspecific abnormalities noted on brain MRI.
Seven of sixteen of our patients had a cardiac abnormality—all six were septal defects, four atrial septal defects, and three ventricular septal defect. Once again, this reflects the literature where 31% of individuals had cardiac abnormalities, ASD being the most common followed by VSD, PDA and then jointly tricuspid atresia, tetralogy of Fallot, aortic dilatation, and transposition of the great arteries.
Yates et al. (2017) described two patients in their study displaying aggressive outbursts and Epi4K‐Consortium and Epilepsy Phenome/Genome Project (2013) also described such a patient (who only described one patient with very limited clinical information and therefore, excluded from our literature review). Interestingly, three patients in our cohort were described as displaying aggressive behavior. Also noteworthy is that in our previous research Durkin et al. (2020) described one patient with hyperphagia. Similarly, one patient in this study also displays this phenotype.
Our study found two patients with diagnosed obsessive compulsive disorder. This is not a newly reported finding in HNRNPU as Bramswig et al. (2017), Durkin et al. (2020), Depienne et al. (2017), and Leduc et al. (2017) all reported patients who displayed obsessive compulsive behaviors, so there are six cases now reported in the literature.
Other observations from our study include our reporting of three patients with II‐III toe syndactyly—previously unreported in the literature. We also reported two cases of polydactyly which adds to the two cases we reported previously. And we report two cases of clinodactyly of the V finger, again furthering on from our previous research (Thierry et al., 2012).
We reported two cases of cryptorchidism, rendering a total of seven cases of this in the literature (Durkin et al., 2020: two patients, Thierry et al., 2012: two patients, and Bramswig et al., 2017: one patient).
Finally, one of our patient's has cyclical neutropenia which was previously unreported in association HNRNPU.
4.2. Recurring genotypes
Out of the 57 genetic variants in the literature, we found four recurrent variants. The c.1089G>A p.(Trp363*), c.706_707del p.(Glu236Thrfs*6), c.847_857del p.(Phe283Serfs*5), and c.1681dels p.(Gln561Serfs*45) variants have all been documented on two occasions, and an additional missense variant has been reported occurring at the 1681 nucleotide (c.1681C>T p.(Gln561*)). Additionally, there have been two reported splice site variants occurring at the 2452 nucleotide (c.2425‐2A>G p.? and c.2425‐3C>A p.?).
Figure 1 shows that many variants occur in the first half of exon 2 and exons 3, 9, and 10 of HNRNPU. Variant nomenclature is according to gene transcript NM_031844.2. Yet, no variants have occurred in exons 8 or 13. The remaining variants are scattered sparsely through the rest of the gene.
FIGURE 1.
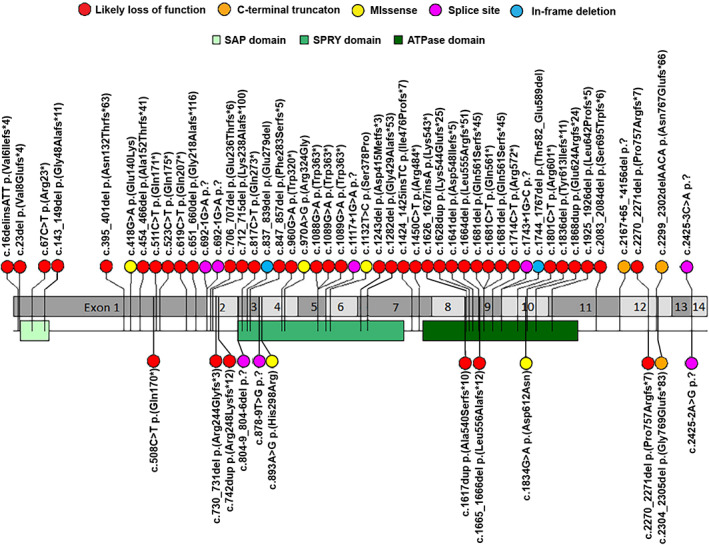
Variant interpretation plot for all probands with pathogenic HNRNPU variants in this cohort and published literature. Variant nomenclature is according to gene transcript NM_031844.2. Refer to Table 3 regarding references for the variants
5. CONCLUSION
In this study, we add to our published literature regarding HNRNPU‐related neurodevelopmental disorder. We have documented phenotypes previously unreported for individuals with HNRNPU including II‐III toe syndactyly and cyclical neutropenia. And we have identified phenotypes sparsely documented in the literature including hyperphagia, obsessive compulsive behaviors, polydactyly, clinodactyly of the V finger, and cryptorchidism.
Finally, due to 58% of individuals having abnormalities on brain MRI and 31% of individuals having cardiac abnormalities, we recommend that individuals diagnosed with HNRNPU should be evaluated with brain imaging (if refractory to seizure medications) and echocardiogram to screen for anatomical abnormalities (as baseline screening following diagnosis) There is not enough data published to determine on the efficacy of each anti‐epileptic medication in controlling seizures. In the literature, the most frequently used medication is sodium valproate which usually works. However, when seizures are refractory to sodium valproate there is no consensus in the literature on the next medication(s) to use.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
AUTHOR CONTRIBUTIONS
James Taylor wrote the manuscript and collected the data. Meena Balasubramanian supervised the project. Michael Spiller made Figure 1. All other authors contributed to data collection and approved the final manuscript.
ACKNOWLEDGMENTS
This study makes use of data generated by the DECIPHER community. A full list of centers who contributed to the generation of the data is available from https://deciphergenomics.org/about/stats and via email from contact@deciphergenomics.org. Funding for the DECIPHER project was provided by Wellcome. This work was supported by Telethon Foundation, Telethon Undiagnosed Diseases Program (TUDP, GSP15001) and was in part generated within the European Reference Network ITHACA. This study also makes use of GeneMatcher. We would also like to thank all the families for consenting to this publication.
Taylor, J. , Spiller, M. , Ranguin, K. , Vitobello, A. , Philippe, C. , Bruel, A.‐L. , Cappuccio, G. , Brunetti‐Pierri, N. , Willems, M. , Isidor, B. , Park, K. , & Balasubramanian, M. (2022). Expanding the phenotype of HNRNPU ‐related neurodevelopmental disorder with emphasis on seizure phenotype and review of literature. American Journal of Medical Genetics Part A, 188A:1497–1514. 10.1002/ajmg.a.62677
Funding information Telethon Foundation, Grant/Award Number: GSP15001
DATA AVAILABILITY STATEMENT
Open access Decipher entries (www.deciphergenomics.org).
REFERENCES
- Ballif, B. C. , Rosenfeld, J. A. , Traylor, R. , Theisen, A. , Bader, P. I. , Ladda, R. L. , Sell, S. L. , Steinraths, M. , Surti, U. , McGuire, M. , Williams, S. , Farrell, S. A. , Filiano, J. , Schnur, R. E. , Coffey, L. B. , Tervo, R. C. , Stroud, T. , Marble, M. , Netzloff, M. , Hanson, K. , … Shaffer, L. G. (2012). High‐resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Human Genetics, 131(1), 145–156. [DOI] [PubMed] [Google Scholar]
- Bramswig, N. C. , Lüdecke, H. J. , Hamdan, F. F. , Altmüller, J. , Beleggia, F. , Elcioglu, N. H. , Freyer, C. , Gerkes, E. H. , Demirkol, Y. K. , Knupp, K. G. , Kuechler, A. , Li, Y. , Lowenstein, D. H. , Michaud, J. L. , Park, K. , Stegmann, A. , Veenstra‐Knol, H. E. , Wieland, T. , Wollnik, B. , Engels, H. , … Wieczorek, D. (2017). Heterozygous HNRNPU variants cause early onset epilepsy and severe intellectual disability. Human Genetics, 136(7), 821–834. [DOI] [PubMed] [Google Scholar]
- Caliebe, A. , Kroes, H. Y. , van der Smagt, J. J. , Martin‐Subero, J. I. , Tönnies, H. , van 't Slot, R. , Nievelstein, R. A. , Muhle, H. , Stephani, U. , Alfke, K. , Stefanova, I. , Hellenbroich, Y. , Gillessen‐Kaesbach, G. , Hochstenbach, R. , Siebert, R. , & Poot, M. (2010). Four patients with speech delay, seizures and variable corpus callosum thickness sharing a 0.440 Mb deletion in region 1q44 containing the HNRPU gene. European Journal of Medical Genetics, 53(4), 179–185. [DOI] [PubMed] [Google Scholar]
- Chen, Q. , Jin, M. , Zhu, J. , Xiao, Q. , & Zhang, L. (2013). Functions of heterogeneous nuclear ribonucleoproteins in stem cell potency and differentiation. BioMed Research International, 2013, 623978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depienne, C. , Nava, C. , Keren, B. , Heide, S. , Rastetter, A. , Passemard, S. , Chantot‐Bastaraud, S. , Moutard, M. L. , Agrawal, P. B. , VanNoy, G. , Stoler, J. M. , Amor, D. J., Billette de Villemeur, T., Doummar, D., Alby, C., Cormier‐Daire, V., Garel, C., Marzin, P., Scheidecker, S., de Saint‐Martin, A., … Mignot, C. (2017). Genetic and phenotypic dissection of 1q43q44 microdeletion syndrome and neurodevelopmental phenotypes associated with mutations in ZBTB18 and HNRNPU. Human Genetics, 136, 463–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durkin, A. , Albaba, S. , Fry, A. E. , Morton, J. E. , Douglas, A. , Beleza, A. , Williams, D. , Volker‐Touw, C. , Lynch, S. A. , Canham, N. , Clowes, V. , Straub, V. , Lachlan, K. , Gibbon, F. , El Gamal, M. , Varghese, V. , Parker, M. J. , Newbury‐Ecob, R. , Turnpenny, P. D. , Gardham, A. , … Balasubramanian, M. (2020). Clinical findings of 21 previously unreported probands with HNRNPU‐related syndrome and comprehensive literature review. American Journal of Medical Genetics Part A, 182(7), 1637–1654. [DOI] [PubMed] [Google Scholar]
- Epi4K‐Consortium & Epilepsy Phenome/Genome Project . (2013). De novo mutations in epileptic encephalopathies. Nature, 501, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firth, H. V. , Richards, S. M. , Bevan, A. P. , Clayton, S. , Corpas, M. , Rajan, D. , Van Vooren, S. , Moreau, Y. , Pettett, R. M. , & Carter, N. P. (2009). DECIPHER: Database of chromosomal imbalance and phenotype in humans using ensembl resources. American Journal of Human Genetics, 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geuens, T. , Bouhy, D. , & Timmerman, V. (2016). The hnRNP family: Insights into their role in health and disease. Human Genetics, 135, 851–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glisovic, T. , Bachorik, J. L. , Yong, J. , & Dreyfuss, G. (2008). RNA‐binding proteins and post‐transcriptional gene regulation. FEBS Letters, 582(14), 1977–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han, S. P. , Tang, Y. H. , & Smith, R. (2010). Functional diversity of the hnRNPs: Past, present and perspectives. Biochemical Journal, 430, 379–392. [DOI] [PubMed] [Google Scholar]
- Keene, J. D. (2007). RNA regulons: Coordination of post‐transcriptional events. Nature Reviews. Genetics, 8(7), 533–543. [DOI] [PubMed] [Google Scholar]
- Leduc, M. S. , Chao, H. T. , Qu, C. , Walkiewicz, M. , Xiao, R. , Magoulas, P. , Pan, S. , Beuten, J. , He, W. , Bernstein, J. A. , Schaaf, C. P. , Scaglia, F. , Eng, C. M. , & Yang, Y. (2017). Clinical and molecular characterization of de novo loss of function variants in HNRNPU. American Journal of Medical Genetics Part A, 173(10), 2680–2689. [DOI] [PubMed] [Google Scholar]
- Lim, I. , Jung, Y. , Kim, D. Y. , & Kim, K. T. (2016). HnRNP Q has a suppressive role in the translation of mouse Cryptochrome1. PLoS One, 11(7), e0159018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low, Y. H. , Asi, Y. , Foti, S. C. , & Lashley, T. (2021). Heterogeneous nuclear ribonucleoproteins: Implications in neurological diseases. Molecular Neurobiology, 58(2), 631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , Grody, W. W. , Hegde, M. , Lyon, E. , Spector, E. , Voelkerding, K. , Rehm, H. L. , & ACMG Laboratory Quality Assurance Committee (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Z. , Zhang, Y. , Yang, C. , Yi, Z. , Li, F. , Xue, J. , Yang, X. , & Li, B. (2021). De novo frameshift variants of HNRNPU in patients with early infantile epileptic encephalopathy: Two case reports and literature review. International Journal of Developmental Neuroscience, 81, 663–668. [DOI] [PubMed] [Google Scholar]
- Thierry, G. , Bénéteau, C. , Pichon, O. , Flori, E. , Isidor, B. , Popelard, F. , Delrue, M. A. , Duboscq‐Bidot, L. , Thuresson, A. C. , van Bon, B. W. , Cailley, D. , Rooryck, C. , Paubel, A. , Metay, C. , Dusser, A. , Pasquier, L. , Béri, M. , Bonnet, C. , Jaillard, S. , Dubourg, C. , … Le Caignec, C. (2012). Molecular characterization of 1q44 microdeletion in 11 patients reveals three candidate genes for intellectual disability and seizures. American Journal of Medical Genetics Part A, 158(7), 1633–1640. [DOI] [PubMed] [Google Scholar]
- Wu, B. , Su, S. , Patil, D. P. , Liu, H. , Gan, J. , Jaffrey, S. R. , & Ma, J. (2018). Molecular basis for the specific and multivariant recognitions of RNA substrates by human hnRNP A2/B1. Nature Communications, 9(1), 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates, T. M., Vasudevan, P. C., Chandler, K. E., Donnelly, D. E., Stark, Z., Sadedin, S., Willoughby, J., Broad Center for Mendelian Genomics, DDD study, & Balasubramanian, M. (2017). De novo mutations in HNRNPU result in a neurodevelopmental syndrome. American Journal of Medical Genetics Part A, 173(11), 3003–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Open access Decipher entries (www.deciphergenomics.org).