

Abstract
Over 10,000 rare genetic diseases have been identified, and millions of newborns are affected by severe rare genetic diseases each year. A variety of Human Phenotype Ontology (HPO)‐based clinical decision support systems (CDSS) and patient repositories have been developed to support clinicians in diagnosing patients with suspected rare genetic diseases. In September 2017, we released PubCaseFinder (https://pubcasefinder.dbcls.jp), a web‐based CDSS that provides ranked lists of genetic and rare diseases using HPO‐based phenotypic similarities, where top‐listed diseases represent the most likely differential diagnosis. We also developed a Matchmaker Exchange (MME) application programming interface (API) to query PubCaseFinder, which has been adopted by several patient repositories. In this paper, we describe notable updates regarding PubCaseFinder, the GeneYenta matching algorithm implemented in PubCaseFinder, and the PubCaseFinder API. The updated GeneYenta matching algorithm improves the performance of the CDSS automated differential diagnosis function. Moreover, the updated PubCaseFinder and new API empower patient repositories participating in MME and medical professionals to actively use HPO‐based resources.
Keywords: API, HPO, matching algorithm, Matchmaker Exchange, PubCaseFinder
In September 2017, we released PubCaseFinder (https://pubcasefinder.dbcls.jp), a web‐based clinical decision support system that provides ranked lists of genetic and rare diseases using Human Phenotype Ontology‐based phenotypic similarities, where top‐listed diseases represent the most likely differential diagnosis. In this paper, we describe notable updates regarding PubCaseFinder, the GeneYenta matching algorithm implemented in PubCaseFinder, and the PubCaseFinder API.
1. INTRODUCTION
At present, over 10,000 rare diseasecs, ~80% of which are genetic in origin, have been identified (Haendel et al., 2020), and millions of newborns each year are affected by severe rare genetic diseases (Posey et al., 2019). Unfortunately, up to 60% of patients affected by rare genetic diseases never receive a diagnosis (Boycott et al., 2019), which hinders the optimization of clinical management and early intervention (Yu & Zhang, 2015). In addition to next‐generation sequencing (NGS)‐based analysis, a variety of clinical decision support systems (CDSS) and patient repositories have been developed to support clinicians in diagnosing patients with suspected rare genetic diseases (Azzariti & Hamosh, 2020; Faviez et al., 2020).
In NGS‐based analysis, gene‐prioritizing systems complement a labor‐intensive process (Faviez et al., 2020). Whole‐exome sequencing is often performed to identify candidate causative genes, resulting in a relatively high (currently 40%) diagnostic yield (Boycott et al., 2019). Trained experts can spend hours looking for literature to support a single candidate causative gene that best explains a patient's symptoms and signs, collectively called “phenotypes” (Birgmeier et al., 2020). Gene‐prioritizing systems use gene−phenotype associations (GPA) to prioritize candidate causative genes based on phenotypic similarities. Although diagnostic rates have improved with whole‐exome analysis, more than 50% of patients with suspected rare genetic diseases remain undiagnosed. Clinicians can collect reported phenotypes from trusted medical sources (e.g., textbooks, literature, and databases) for candidate diseases to determine which diseases show phenotypic overlap with undiagnosed patients’ phenotypes (Wise et al., 2019). Recently, phenotype‐driven differential diagnosis systems have been implemented to simplify and accelerate the time‐consuming differential diagnosis process and improve diagnostic rates (Faviez et al., 2020). Many of these systems utilize disease−phenotype associations (DPA) to rank diseases based on phenotypic similarities.
Even in cases where NGS‐based analysis and differential diagnosis processes do not lead to a diagnosis, patient repositories may facilitate diagnoses by identifying new diseases and causative genes among the exomes or genomes of unrelated patients with similar phenotypes. However, discovering multiple unrelated patients with similar genotypes and phenotypes can be difficult because they are scattered in different hospitals and countries. To enable centralized access, many patient repositories have developed web portals with contact mechanisms to encourage collaboration and information exchange between users around the world (Azzariti & Hamosh, 2020).
Recently, the Human Phenotype Ontology (HPO)‐based CDSS and patient repositories have expanded, and clinical applications have grown (Kohler et al., 2021). An ontology, human‐ and machine‐readable format, is designed to provide a standard and controlled vocabulary to represent knowledge in the domain and several biomedical ontologies such as HPO, the Orphanet Rare Disease Ontology (ORDO) (Nguengang Wakap et al., 2020), and the Mondo Disease Ontology (Mondo) (Shefchek et al., 2020) have been constructed. Since its initial construction in 2008, HPO has been curated by domain experts to provide a standardized vocabulary for describing phenotypic abnormalities that are widely observed in human genetic diseases. The Monarch Initiative (Shefchek et al., 2020) and Orphadata (Nguengang Wakap et al., 2020) have gathered manually annotated GPA and DPA that are extracted from literature and databases, such as Online Mendelian Inheritance in Man (OMIM) and Orphanet, and encoded those phenotypes with the corresponding HPO terms. Using these HPO‐based annotations and biomedical ontologies, many gene‐prioritizing systems, such as the Automated Mendelian Literature Evaluation (Birgmeier et al., 2020), Phen2Gene (Zhao et al., 2020), and Variant Interpretation using Biomedical Literature Evidence (van der Velde et al., 2020), and phenotype‐driven differential diagnosis systems, such as Phenomizer (Köhler et al., 2009), Genetic Disease Diagnosis based on Phenotypes (Chen et al., 2019), and Likelihood Ratio Interpretation of Clinical Abnormalities (LIRICAL) (Robinson et al., 2020) have been implemented. In addition, most patient repositories participating in the Matchmaker Exchange (MME) (Azzariti & Hamosh, 2020), an international collaborative project for matchmaking of unrelated patients through a standardized application programming interface (API), also use HPO to encode patient phenotypes.
In September 2017, we released PubCaseFinder (https://pubcasefinder.dbcls.jp), a web‐based CDSS to assist in prioritizing causative genes and the differential diagnosis process (Fujiwara et al., 2018). PubCaseFinder provides HPO‐based ranked lists of 7848 genetic diseases, 3619 rare diseases, 4025 causative genes, and 18,893 open‐sharing cases adopting the GeneYenta matching algorithm (Gottlieb et al., 2015). We developed the PubCaseFinder API to enable querying using HPO terms and developed the MME API (Buske, Schiettecatte, et al., 2015) as a secondary querying option to enable the use of PubCaseFinder by the Initiative on Rare and Undiagnosed Disease (IRUD) (Adachi et al., 2017), PhenomeCentral (Buske, Girdea, et al., 2015), and RD‐Connect (Thompson et al., 2014). To make PubCaseFinder convenient for more patient repositories and medical professionals, we implemented a set of updates for PubCaseFinder, the GeneYenta matching algorithm, and the PubCaseFinder API. The updated PubCaseFinder facilitates phenotyping to provide a more precise record of patient phenotypic abnormalities and enables the filtering of ranked lists using causative genes, modes of inheritance, and disease names. The previous GeneYenta matching algorithm was not robust when users incorrectly or imprecisely specified the patient phenotype. The automated differential diagnosis performance of PubCaseFinder has been improved by the updated GeneYenta matching algorithm. Moreover, we aim to implement a convenient new API for patient repositories. We believe that these updates will ultimately contribute to improving rare genetic disease diagnostic rates.
2. METHODS
2.1. Data sources
To develop PubCaseFinder, we used several biomedical ontologies and annotations. We downloaded the HPO file (releases/2021‐08‐02) from https://hpo.jax.org/app/. The file for Mondo (releases/2021‐06‐01), an ontology that integrates existing sources of disease definitions, such as OMIM, Orphanet, and Disease Ontology (Schriml et al., 2019), was downloaded from https://mondo.monarchinitiative.org. The Foundational Model of Anatomy (FMA) file (ver.5.0.0) (Golbreich et al., 2013), a standardized vocabulary to represent a coherent body of explicit declarative knowledge about human anatomy, was downloaded from https://bioportal.bioontology.org/ontologies/FMA. HPO contains a set of 16,544 terms that were integrated with 13,520 textual definitions and 21,183 synonyms, and 20,464 “is–a” (parent–child) relationships were established among HPO terms. Mondo contains a set of 24,486 terms integrated with 15,535 textual definitions, 99,446 synonyms, and 37,948 “is–a” relationships and provides connections with other resources (e.g., OMIM and Orphanet). We also downloaded HPO‐Japanese containing a set of 10,182 Japanese terms and Mondo‐Japanese containing a set of 3756 Japanese terms from https://github.com/ogishima/HPO-Japanese and https://github.com/aidrd/mondo-japanese, respectively. FMA contains a set of 104,859 terms integrated with 56,855 synonyms and 104,779 “is–a” relationships were established between FMA terms. We downloaded 248,150 NCBI Gene GPAs and 152,014 OMIM and Orphanet DPAs on August 1, 2021, from https://hpo.jax.org/app/download/annotation. We retrieved 18,893 open‐sharing cases from DECIPHER (https://www.deciphergenomics.org/) (Bragin et al., 2014), MyGene2 (https://mygene2.org/MyGene2/) (MyGene2.org, 2016), and the Undiagnosed Diseases Program (https://undiagnosed.hms.harvard.edu/) (Macnamara et al., 2020). Each case contains a set of HPO terms as a phenotype profile and a set of candidate genes for the genetic condition. We equipped PubCaseFinder with an automatic update system to maintain ontology and annotation updates.
2.2. Mapping HPO terms to OBJ files of BodyParts3D/Anatomography
To map HPO terms to FMA terms, we used the external links in HPO and the Uber Anatomy Ontology (UBERON) (Mungall et al., 2012). We retrieved links between HPO terms and UBERON terms from the HPO file. Then, we downloaded the UBERON file (releases/2021‐07‐06) from https://bioportal.bioontology.org/ontologies/UBERON. The UBERON file contains links between UBERON terms and FMA terms. Using these links, we mapped 3642 HPO terms to 991 FMA terms. Using the relationships between the FMA terms and the OBJ files of BodyParts3D/Anatomography, which were manually mapped by the BodyParts3D/Anatomography project (Mitsuhashi et al., 2009), we mapped 991 FMA terms to 2536 BodyParts3D/Anatomography OBJ files and constructed a human 3D model using those OBJ files.
2.3. Improvement of the GeneYenta matching algorithm
To calculate semantic similarities between two sets of HPO terms, PubCaseFinder uses GeneYenta, a user‐weighted matching algorithm, to set a matching weight for each phenotype. The algorithm computes similarities ranging from 0% (no phenotypic overlap) to 100% (complete phenotypic overlap). By using the algorithm, the semantic similarity SIM(c,d) between a case and a disease is calculated: c is the set of HPO terms related to a case and d is the set of HPO terms related to a disease. The updated GeneYenta algorithm uses the following formulas to calculate SIM(c,d).
The algorithm begins by determining the information content (IC t ) of each HPO term t. First, we collected case reports from PubMed to use the following query: “case reports” [Publication Type] OR “case reports” [ti] OR “case report” [ti]. We found 1,264,571 case reports that had both titles and abstracts (as of August 1, 2021). We annotated the case report titles and abstracts with HPO terms using ConceptMapper (Tanenblatt et al., 2010). P(t) is the probability of occurrence of an HPO term t in a set of case reports. The IC t of the HPO term t is defined as follows:
where annot all is the total number of annotations of all HPO terms in case reports, and annot t is the total number of annotations of the HPO term t and all its descendants in case reports. That is, for the root node, P(t) is 1, and IC t is 0. There is an inverse relation between IC and the total number of annotations of an HPO term t. The IC t of the most informative common ancestor of the two HPO terms was assigned as the similarity sim(t,t') between two HPO terms, which is defined as follows:
where A t is the HPO term t and all ancestral HPO terms of t, and a t is the HPO term of the intersection of A t and A t'. The similarity SIM(c,d) between c and d assesses the resemblance between their sets of HPO terms and is defined as follows:
where R t is a weight in accordance with how important a term t is. If the difference between IC t and is large, it means that t and t' are far apart in the HPO hierarchy. For example, of the pair of t 1 and t' 1 (Figure 1b) is larger than that of the pair t 2 and t' 1 (Figure 1c) because t 1 is farther away from t' 1 than t 2. If there are several HPO terms with small , such as t 2,t 3,t 4, and t 5, we assumed that the HPO terms with large , such as t 1, should be noises or imprecisions (Figure 1a). To improve the robustness of the GeneYenta algorithm for managing such noisy and imprecise inputs, we introduced three new parameters E, K, and W. Among the set of HPO terms related to a case, if the number of HPO terms whose is less than or equal to E is more than K, then the weights of those HPO terms are set to 1. The weights for the remaining HPO terms are set to W, which is less than 1, because they should be noises or imprecisions.
Figure 1.
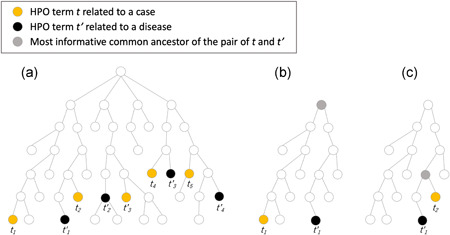
Phenotype hierarchy. (a) Sets of HPO terms related to a case and a disease mapped on the phenotype hierarchy. (b) The most informative common ancestor of the pair of t 1 and t' 1. (c) The most informative common ancestor of the pair of t 2 and t' 1
C(c,d) is the number of t related to c if the difference between IC t and is less than E. This is defined as follows:
The weight R t for t related to c is 1 if C(c,d) is less than K or the difference between IC t and is less than E. If these conditions are not met, the weight R t is W, which is defined as follows:
Finally, PubCaseFinder provides a ranked list of diseases according to SIM(c,d).
2.4. Fine‐tuning of the updated GeneYenta algorithm parameters
We performed a grid search to find the optimal hyperparameters (E, K, and W) for the updated GeneYenta algorithm to manage noisy and imprecise input. To perform the grid search, we created 500 simulated cases by choosing 5−10 HPO terms at random from the DPAs related to OMIM. For example, we created a simulated case by randomly selecting 8 terms from the 20 HPO terms that annotated Gaucher disease type I. To simulate noise and imprecision in measuring or recording phenotypic abnormalities, we added one to three terms randomly selected from all HPO terms to some cases. In addition, we randomly replaced original HPO terms with their parent HPO term or grandparent HPO term in some cases. We set up a grid of hyperparameter values with E values [1, 2, 3, 4, 5, 6, 7, 8, 9, 10], K values [1, 2, 3, 4, 5], and W values [0.1, 0.2, 0.3, 0.4, 0.5]. For each combination, we obtained the ranked lists of diseases for the set of 500 simulated cases. To find the optimal hyperparameters, we calculated the “recall at ranks” (i.e., the proportion of cases where the correct diagnosis appeared in the top 10 listed diseases) for each combination. The combination of E: 4, K: 4, and W: 0.1 resulted in the best top 10 recall number (Supporting Information Table S2).
2.5. Performance evaluation of the updated GeneYenta algorithm
We used evaluation data set 1, consisting of 384 case reports collected to evaluate LIRICAL (Robinson et al., 2020), to evaluate the performance of the updated GeneYenta algorithm. All case reports were annotated with phenotypes and diagnoses; the former was represented by HPO terms, and the latter was represented by OMIM IDs (https://www.cell.com/cms/10.1016/j.ajhg.2020.06.021/attachment/7574984e-81ad-435a-ab13-4d354f016181/mmc1.pdf). To imitate noisy and inaccurate cases, we randomly added two terms from all HPO terms to each case in data set 1 to make data set 2. In addition, we randomly replaced some HPO terms in each case in data set 2 with their parent HPO term to make data set 3. Further, we randomly replaced some HPO terms in each case in data set 2 with their grandparent HPO term to make data set 4. We then calculated the top 1, top 2, top 3, top 5, and top 10 recall rates using data set 1, data set 2, data set 3, and data set 4 in the original GeneYenta algorithm and the updated GeneYenta algorithm to compare their performance.
3. RESULTS
The number of queries to PubCaseFinder has been increasing every year (Figure 2). The PubCaseFinder web application, GeneYenta matching algorithm, and PubCaseFinder API updates are described below.
Figure 2.
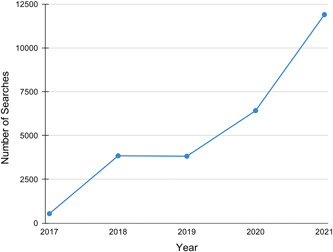
The number of times the PubCaseFinder search button has been clicked each year from its 2017 launch through 2021
3.1. PubCaseFinder update
PubCaseFinder provides several functions (Figure 3) to help users enter a large number of HPO terms, which facilitate precise patient phenotyping. The text search function enables users to select appropriate HPO terms from the list of hits in a partial match, including synonyms. The hierarchical structure view function enables users to easily identify higher and lower concept terms of a given HPO term. For example, users can find the HPO term “microcephaly” by searching for its synonym “small head” (Figure 3a) and then replace the term with its lower concept “congenital microcephaly” using the hierarchical structure view function (Figure 3b). We have also newly implemented a human 3D model function and a textual annotation function. The human 3D model function is useful for medical professionals who may not be familiar with specific medical terminology. For example, by selecting the mandible region in the human 3D model, users can find HPO terms related to the mandible that do not include the word “mandible,” such as retrognathia, micrognathia, narrow jaw, and broad jaw (Figure 3c). The text search function (Figure 3a) does not enable users to automatically extract HPO terms from free‐text, while the textual annotation function (Figure 3d) allows users to easily extract HPO terms from free‐text‐based Japanese clinical notes and use them to query PubCaseFinder. Moreover, PubCaseFinder has been linked with Doc2Hpo (Liu et al., 2019), an interactive web application for semi‐automatically extracting HPO terms from English clinical text, allowing users to query PubCaseFinder using HPO terms extracted using Doc2Hpo.
Figure 3.
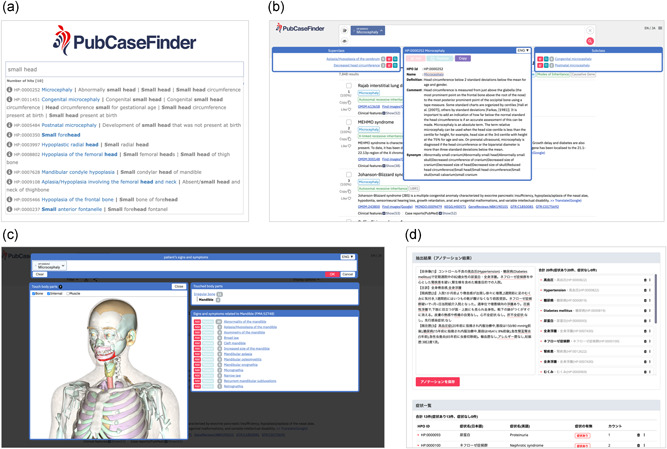
Web‐based phenotyping functions in PubCaseFinder. (a) The text search function. (b) The hierarchical structure view function. (c) The human 3D model function. Users can zoom in and out of the 3D model by scrolling, move the 3D model by dragging, and select parts of the 3D model by clicking. (d) The textual annotation function
Using the updated GeneYenta algorithm, a set of HPO terms entered by the user can be compared with genetic diseases, rare diseases, causative genes, and open‐sharing cases on the basis of phenotypic similarity (Figure 4a). A higher phenotypic similarity will show a higher probability for a candidate differential diagnosis, causative gene, or similar case. We have newly implemented a filter function that enables users to filter ranked lists in PubCaseFinder to specify a National Center for Biotechnology Information (NCBI) Gene ID as a causative gene, an HPO term as a mode of inheritance, and/or a Monarch Disease Ontology (Mondo) (Shefchek et al., 2020) term as a disease name. When an HPO or Mondo term is specified, all its lower concepts are also subject to the filter. Moreover, this filter supports search expressions using logical operators. For example, when specifying “autosomal dominant inheritance (HP:0000006),” the ranked list is filtered to 3328 genetic diseases. When specifying “autoimmune disease (MONDO:0007179),” the list is filtered to 56 genetic diseases. When both “autosomal dominant inheritance (HP:0000006)” and “autoimmune disease (MONDO:0007179)” are specified using “AND” as a logical operator, the ranked list of genetic diseases is filtered to 30 (Figure 4b).
Figure 4.
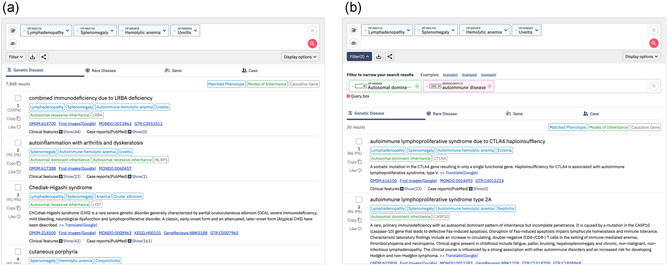
Ranked lists and the filtering function in PubCaseFinder. (a) A ranked list of genetic diseases. (b) A ranked list of genetic diseases filtered by “autosomal dominant inheritance (HP:0000006)” and “autoimmune disease (MONDO:0007179)” using “AND” as a logical operator
3.2. GeneYenta matching algorithm update
We updated the GeneYenta matching algorithm to manage noise and imprecision in measuring or recording HPO terms as phenotypic abnormalities. To evaluate the performance of the updated GeneYenta algorithm, we compared the performance of PubCaseFinder and PubCaseFinder+, which use the GeneYenta algorithm and the updated GeneYenta algorithm, respectively (Supporting Information Table S1). Figure 5 shows the recall rates by PubCaseFinder and PubCaseFinder+ using data set 1, data set 2, data set 3, and data set 4. The top‐10 recall rate of PubCaseFinder+ is 55% (Figure 5a), indicating a correct diagnosis is obtained in the top 10 of the ranked list of diseases for approximately one in every two cases. The PubCaseFinder+ recall rates are higher than those of PubCaseFinder for data set 1 (Figure 5a), data set 2 (Figure 5b), data set 3 (Figure 5c), and data set 4 (Figure 5d). Especially in data set 2, data set 3, and data set 4, there are large differences in recall rates. These results show that the updated GeneYenta algorithm manages with noise and imprecision better than the original algorithm.
Figure 5.
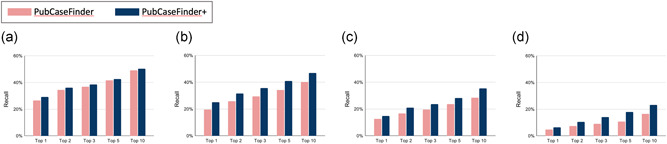
PubCaseFinder and PubCaseFinder+ performance comparison. Recall rates for data set 1 (a), data set 2 (b), data set 3 (c), and data set 4 (d)
3.3. PubCaseFinder API update
We updated the PubCaseFinder API using a fixed URL syntax that translates a standard set of input parameters to return the requested data (Table 1).
Table 1.
Input parameters for the PubCaseFinder application programming interface (API)
| Parameter name | Value |
|---|---|
| RANKED_LIST_TARGET | “omim” |
| “orphanet” | |
| “gene” | |
| RANKED_LIST_FORMAT | “json” |
| “tsv” | |
| HPO_ID | HPO IDs with commas |
| RECORD_TARGET | “omim” |
| “orphanet” | |
| “gene” | |
| RECORD_ID | OMIM ID or ORPHA ID or NCBI Gene ID |
| DISEASE_TARGET | “omim” |
| “orphanet” | |
| DISEASE_ID | OMIM ID or ORPHA ID |
| GENE_ID | NCBI Gene ID |
| MONDO_ID | MONDO ID |
Users can obtain any ranked lists by indicating the [RANKED_LIST_TARGET], [RANKED_LIST_FORMAT], and [HPO_ID] (Table 1) in the following format: https://pubcasefinder.dbcls.jp/api/get_ranked_list?target=[RANKED_LIST_TARGET]&format=[RANKED_LIST_FORMAT]&hpo_id=[HPO_ID]
For example, a ranked list of genetic diseases using the HPO IDs “HP:0002089” and “HP:0001998” in JavaScript Object Notation (JSON) format can be obtained using the following URL: https://pubcasefinder.dbcls.jp/api/get_ranked_list?target=omim&format=json&hpo_id=HP:0002089,HP:0001998
Each data record in ranked lists of PubCaseFinder can be identified by OMIM ID, ORPHA ID, or NCBI Gene ID; therefore, records can be obtained by indicating the [RECORD_TARGET] and [RECORD_ID] (Table 1) in the following format: https://pubcasefinder.dbcls.jp/api/get_data_record?target=[RECORD_TARGET]&id=[RECORD_ID]
For example, the URL to retrieve the data record for OMIM ID “612690” in JSON format can be represented as follows: https://pubcasefinder.dbcls.jp/api/get_data_record?target=omim&id=612690
DPAs for each disease in OMIM and Orphanet can be obtained by indicating the [DISEASE_TARGET] and [DISEASE_ID] (Table 1) in the following format: https://pubcasefinder.dbcls.jp/api/get_dpa?target=[DISEASE_TARGET]&id=[DISEASE_ID]
For example, DPAs for the OMIM ID “612690” can be obtained using the following URL: https://pubcasefinder.dbcls.jp/api/get_dpa?target=omim&id=612690
GPAs for each gene can be obtained by indicating the [GENE_ID] (Table 1) in the following format: https://pubcasefinder.dbcls.jp/api/get_gpa?id=[GENE_ID]
For example, GPAs for the NCBI Gene ID “1723” can be obtained using the following URL: https://pubcasefinder.dbcls.jp/api/get_gpa?id=1723
Users can obtain case reports related to rare genetic diseases in PubMed by indicating the [MONDO_ID] (Table 1) in the following format: https://pubcasefinder.dbcls.jp/api/get_case_report?id=[MONDO_ID]
For example, case reports for the Mondo ID “0008752” can be obtained using the following URL: https://pubcasefinder.dbcls.jp/api/get_case_report?id=MONDO:0008752
4. CONCLUSION AND FUTURE WORK
PubCaseFinder was released in September 2017, and the number of queries to PubCaseFinder has been increasing every year. This increase in queries can be attributed to the increase in HPO‐based resources established by various projects, such as MME and HPO translation. Many Japanese cases with suspected rare genetic diseases have been HPO‐encoded through the IRUD, which participates in MME. Moreover, HPO‐Japanese, which is translated by Japanese physicians, has contributed to the spread of HPO‐based resources in Japan. To make these resources available to more patient repositories and medical professionals, we have updated PubCaseFinder, the GeneYenta matching algorithm, and the PubCaseFinder API. We have also equipped PubCaseFinder with an automatic update system to maintain updated resources. To contribute to the development of HPO, we plan to propose the HPO project for new synonyms and layperson synonyms which will be requests submitted by PubCaseFinder users.
We believe that the multilingualization of PubCaseFinder will play an important role in promoting the use of HPO‐based resources via PubCaseFinder. PubCaseFinder already supports two languages, English and Japanese, and is designed to support multiple languages. Therefore, we can easily extend the languages supported by PubCaseFinder using HPO and Mondo translations.
Additionally, we are working to make PubCaseFinder fully compatible with Phenopackets (https://github.com/phenopackets), an open standard for sharing lists of disease and patient phenotypic abnormalities, including details about age, sex, onset, and evidence. The textual annotation function of PubCaseFinder is already compatible with Phenopackets, and we plan to update other phenotyping functions and the PubCaseFinder API to support Phenopackets, which will contribute to improving the convenience of PubCaseFinder.
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
WEB RESOURCES
BodyParts3D/Anatomography: https://lifesciencedb.jp/bp3d/; DECIPHER: https://www.deciphergenomics.org/; Foundational Model of Anatomy: https://bioportal.bioontology.org/ontologies/FMA; HPO‐based Annotations: https://hpo.jax.org/app/download/annotation; Human Phenotype Ontology: https://hpo.jax.org/app/; Matchmaker Exchange: https://www.matchmakerexchange.org; Mondo Disease Ontology: https://mondo.monarchinitiative.org; MyGene2: https://mygene2.org/MyGene2/; OMIM: https://omim.org/; Orphanet: https://www.orpha.net/; Phenopackets: http://phenopackets.org; PubCaseFinder: https://pubcasefinder.dbcls.jp; Undiagnosed Diseases Program: https://undiagnosed.hms.harvard.edu/
Supporting information
Supporting information.
ACKNOWLEDGMENTS
We are grateful to all members of the Matchmaker Exchange working group for steering our effort. This development of PubCaseFinder was supported by the National Bioscience Database Center (NBDC) of the Japan Science and Technology Agency (JST) and by “Challenging Exploratory Research Projects for the Future” grant from Research Organization of Information and Systems (ROIS). We are very grateful to Prof. Peter Robinson for providing the evaluation data set. We also thank Prof. Kousaku Okubo, Prof. Kenjiro Kosaki, Dr. Shoko Kawamoto, Dr. Yasunori Yamamoto, and Dr. Orion Buske for their help.
Fujiwara, T. , Shin, J.‐M. , & Yamaguchi, A. (2022). Advances in the development of PubCaseFinder, including the new application programming interface and matching algorithm. Human Mutation, 43, 734–742. 10.1002/humu.24341
DATA AVAILABILITY STATEMENT
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.
REFERENCES
- Adachi, T. , Kawamura, K. , Furusawa, Y. , Nishizaki, Y. , Imanishi, N. , Umehara, S. , Izumi, K. , & Suematsu, M. (2017). Japan's initiative on rare and undiagnosed diseases (IRUD): Towards an end to the diagnostic odyssey. European Journal of Human Genetics, 25(9), 1025–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzariti, D. R. , & Hamosh, A. (2020). Genomic data sharing for novel Mendelian disease gene discovery: The matchmaker exchange. Annual Review of Genomics and Human Genetics, 21, 305–326. 10.1146/annurev-genom-083118-014915 [DOI] [PubMed] [Google Scholar]
- Birgmeier, J. , Haeussler, M. , Deisseroth, C. A. , Steinberg, E. H. , Jagadeesh, K. A. , Ratner, A. J. , Guturu, H. , Wenger, A. M. , Diekhans, M. E. , Stenson, P. D. , Cooper, D. N. , Ré, C. , Beggs, A. H. , Bernstein, J. A. , & Bejerano, G. (2020). AMELIE speeds Mendelian diagnosis by matching patient phenotype and genotype to primary literature. Science Translational Medicine, 12(544), eaau9113. 10.1126/scitranslmed.aau9113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boycott, K. M. , Hartley, T. , Biesecker, L. G. , Gibbs, R. A. , Innes, A. M. , Riess, O. , Belmont, J. , Dunwoodie, S. L. , Jojic, N. , Lassmann, T. , Mackay, D. , Temple, I. K. , Visel, A. , & Baynam, G. (2019). A diagnosis for all rare genetic diseases: The horizon and the next frontiers. Cell, 177(1), 32–37. 10.1016/j.cell.2019.02.040 [DOI] [PubMed] [Google Scholar]
- Bragin, E. , Chatzimichali, E. A. , Wright, C. F. , Hurles, M. E. , Firth, H. V. , Bevan, A. P. , & Swaminathan, G. J. (2014). DECIPHER: Database for the interpretation of phenotype‐linked plausibly pathogenic sequence and copy‐number variation. Nucleic Acids Research, 42(Database issue), D993–D1000. 10.1093/nar/gkt937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske, O. J. , Girdea, M. , Dumitriu, S. , Gallinger, B. , Hartley, T. , Trang, H. , Misyura, A. , Friedman, T. , Beaulieu, C. , Bone, W. P. , Links, A. E. , Washington, N. L. , Haendel, M. A. , Robinson, P. N. , Boerkoel, C. F. , Adams, D. , Gahl, W. A. , Boycott, K. M. , & Brudno, M. (2015). PhenomeCentral: A portal for phenotypic and genotypic matchmaking of patients with rare genetic diseases. Human Mutation, 36(10), 931–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buske, O. J. , Schiettecatte, F. , Hutton, B. , Dumitriu, S. , Misyura, A. , Huang, L. , Hartley, T. , Girdea, M. , Sobreira, N. , Mungall, C. , & Brudno, M. (2015). The Matchmaker Exchange API: Automating patient matching through the exchange of structured phenotypic and genotypic profiles. Human Mutation, 36(10), 922–927. 10.1002/humu.22850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, J. , Xu, H. , Jegga, A. , Zhang, K. , White, P. S. , & Zhang, G. (2019). Novel phenotype‐disease matching tool for rare genetic diseases. Genetics in Medicine, 21(2), 339–346. 10.1038/s41436-018-0050-4 [DOI] [PubMed] [Google Scholar]
- Faviez, C. , Chen, X. , Garcelon, N. , Neuraz, A. , Knebelmann, B. , Salomon, R. , Lyonnet, S. , Saunier, S. , & Burgun, A. (2020). Diagnosis support systems for rare diseases: A scoping review. Orphanet Journal of Rare Diseases, 15(1), 94. 10.1186/s13023-020-01374-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara, T. , Yamamoto, Y. , Kim, J. D. , Buske, O. , & Takagi, T. (2018). PubCaseFinder: A case‐report‐based, phenotype‐driven differential‐diagnosis system for rare diseases. The American Journal of Human Genetics, 103(3), 389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golbreich, C. , Grosjean, J. , & Darmoni, S. J. (2013). The foundational model of anatomy in OWL 2 and its use. Artificial Intelligence in Medicine, 57(2), 119–132. 10.1016/j.artmed.2012.11.002 [DOI] [PubMed] [Google Scholar]
- Gottlieb, M. M. , Arenillas, D. J. , Maithripala, S. , Maurer, Z. D. , Tarailo Graovac, M. , Armstrong, L. , Patel, M. , van Karnebeek, C. , & Wasserman, W. W. (2015). GeneYenta: A phenotype‐based rare disease case matching tool based on online dating algorithms for the acceleration of exome interpretation. Human Mutation, 36(4), 432–438. 10.1002/humu.22772 [DOI] [PubMed] [Google Scholar]
- Haendel, M. , Vasilevsky, N. , Unni, D. , Bologa, C. , Harris, N. , Rehm, H. , Hamosh, A. , Baynam, G. , Groza, T. , McMurry, J. , Dawkins, H. , Rath, A. , Thaxon, C. , Bocci, G. , Joachimiak, M. P. , Köhler, S. , Robinson, P. N. , Mungall, C. , & Oprea, T. I. (2020). How many rare diseases are there? Nature Reviews Drug Discovery, 19(2), 77–78. 10.1038/d41573-019-00180-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohler, S. , Gargano, M. , Matentzoglu, N. , Carmody, L. C. , Lewis‐Smith, D. , Vasilevsky, N. A. , Danis, D. , Balagura, G. , Baynam, G. , Brower, A. M. , Callahan, T. J. , Chute, C. G. , Est, J. L. , Galer, P. D. , Ganesan, S. , Griese, M. , Haimel, M. , Pazmandi, J. , Hanauer, M. , … Robinson, P. N. (2021). The Human Phenotype Ontology in 2021. Nucleic Acids Research, 49(D1), D1207–D1217. 10.1093/nar/gkaa1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler, S. , Schulz, M. H. , Krawitz, P. , Bauer, S. , Dölken, S. , Ott, C. E. , Mundlos, C. , Horn, D. , Mundlos, S. , & Robinson, P. N. (2009). Clinical diagnostics in human genetics with semantic similarity searches in ontologies. American Journal of Human Genetics, 85(4), 457–464. 10.1016/j.ajhg.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Peres Kury, F. S. , Li, Z. , Ta, C. , Wang, K. , & Weng, C. (2019). Doc2Hpo: A web application for efficient and accurate HPO concept curation. Nucleic Acids Research, 47(W1), W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macnamara, E. F. , D'Souza, P. , Undiagnosed Diseases Network , & Tifft, C. J. (2020). The undiagnosed diseases program: Approach to diagnosis. Translational Science of Rare Diseases, 4(3−4), 179–188. 10.3233/TRD-190045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuhashi, N. , Fujieda, K. , Tamura, T. , Kawamoto, S. , Takagi, T. , & Okubo, K. (2009). BodyParts3D: 3D structure database for anatomical concepts. Nucleic Acids Research, 37(Database issue), D782–D785. 10.1093/nar/gkn613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mungall, C. J. , Torniai, C. , Gkoutos, G. V. , Lewis, S. E. , & Haendel, M. A. (2012). Uberon, an integrative multi‐species anatomy ontology. Genome Biology, 13(1), 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MyGene2.org. (2016). Website aims to accelerate gene discovery, diagnosis, treatment: MyGene2.org fosters open sharing among families, researchers, and clinicians. American Journal of Medical Genetics, Part A, 170(6), 1388–1389. 10.1002/ajmg.a.37746 [DOI] [PubMed] [Google Scholar]
- Nguengang Wakap, S. , Lambert, D. M. , Olry, A. , Rodwell, C. , Gueydan, C. , Lanneau, V. , Murphy, D. , Le Cam, Y. , & Rath, A. (2020). Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. European Journal of Human Genetics: EJHG, 28(2), 165–173. 10.1038/s41431-019-0508-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posey, J. E. , O'Donnell‐Luria, A. H. , Chong, J. X. , Harel, T. , Jhangiani, S. N. , Coban Akdemir, Z. H. , Buyske, S. , Pehlivan, D. , Carvalho, C. , Baxter, S. , Sobreira, N. , Liu, P. , Wu, N. , Rosenfeld, J. A. , Kumar, S. , Avramopoulos, D. , White, J. J. , Doheny, K. F. , Witmer, P. D. , … Centers for Mendelian Genomics . (2019). Insights into genetics, human biology and disease gleaned from family based genomic studies. Genetics in Medicine, 21(4), 798–812. 10.1038/s41436-018-0408-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, P. N. , Ravanmehr, V. , Jacobsen, J. , Danis, D. , Zhang, X. A. , Carmody, L. C. , Gargano, M. A. , Thaxton, C. L. , UNC Biocuration Core , Karlebach, G. , Reese, J. , Holtgrewe, M. , Köhler, S. , McMurry, J. A. , Haendel, M. A. , & Smedley, D. (2020). Interpretable clinical genomics with a likelihood ratio paradigm. American Journal of Human Genetics, 107(3), 403–417. 10.1016/j.ajhg.2020.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schriml, L. M. , Mitraka, E. , Munro, J. , Tauber, B. , Schor, M. , Nickle, L. , Felix, V. , Jeng, L. , Bearer, C. , Lichenstein, R. , Bisordi, K. , Campion, N. , Hyman, B. , Kurland, D. , Oates, C. P. , Kibbey, S. , Sreekumar, P. , Le, C. , Giglio, M. , & Greene, C. (2019). Human Disease Ontology 2018 update: Classification, content and workflow expansion. Nucleic Acids Research, 47(D1), D955–D962. 10.1093/nar/gky1032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefchek, K. A. , Harris, N. L. , Gargano, M. , Matentzoglu, N. , Unni, D. , Brush, M. , Keith, D. , Conlin, T. , Vasilevsky, N. , Zhang, X. A. , Balhoff, J. P. , Babb, L. , Bello, S. M. , Blau, H. , Bradford, Y. , Carbon, S. , Carmody, L. , Chan, L. E. , Cipriani, V. , … Osumi‐Sutherland, D. (2020). The Monarch Initiative in 2019: An integrative data and analytic platform connecting phenotypes to genotypes across species. Nucleic Acids Research, 48(D1), D704–D715. 10.1093/nar/gkz997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenblatt, M. , Coden, A. , & Sominsky, I. (2010). The ConceptMapper approach to named entity recognition. In Proceedings of the seventh international conference on language resources and evaluation (LREC'10), 546–551. [Google Scholar]
- Thompson, R. , Johnston, L. , Taruscio, D. , Monaco, L. , Béroud, C. , Gut, I. G. , Hansson, M. G. , 't Hoen, P. B. , Patrinos, G. P. , Dawkins, H. , Ensini, M. , Zatloukal, K. , Koubi, D. , Heslop, E. , Paschall, J. E. , Posada, M. , Robinson, P. N. , Bushby, K. , & Lochmüller, H. (2014). RD‐Connect: An integrated platform connecting databases, registries, biobanks and clinical bioinformatics for rare disease research. Journal of General Internal Medicine, 29(3), 780–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velde, K. J. , van den Hoek, S. , van Dijk, F. , Hendriksen, D. , van Diemen, C. C. , Johansson, L. F. , Abbott, K. M. , Deelen, P. , Sikkema‐Raddatz, B. , & Swertz, M. A. (2020). A pipeline‐friendly software tool for genome diagnostics to prioritize genes by matching patient symptoms to literature. Advanced Genetics, 1(1), e10023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise, A. L. , Manolio, T. A. , Mensah, G. A. , Peterson, J. F. , Roden, D. M. , Tamburro, C. , Williams, M. S. , & Green, E. D. (2019). Genomic medicine for undiagnosed diseases. Lancet, 394(10197), 533–540. 10.1016/S0140-6736(19)31274-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , & Zhang, V. W. (2015). Precision medicine for continuing phenotype expansion of human genetic diseases. BioMed Research International, 2015, 745043. 10.1155/2015/745043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, M. , Havrilla, J. M. , Fang, L. , Chen, Y. , Peng, J. , Liu, C. , Wu, C. , Sarmady, M. , Botas, P. , Isla, J. , Lyon, G. J. , Weng, C. , & Wang, K. (2020). Phen2Gene: Rapid phenotype‐driven gene prioritization for rare diseases. NAR Genomics and Bioinformatics, 2(2), lqaa032. 10.1093/nargab/lqaa032 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article and its supplementary materials.