

Abstract
Introduction:
Acute non-traumatic convexity subarachnoid haemorrhage (cSAH) is increasingly recognized in cerebral amyloid angiopathy (CAA). We investigated: (a) the overlap between acute cSAH and cortical superficial siderosis–a new CAA haemorrhagic imaging signature; and (b) whether acute cSAH presents with particular clinical symptoms in probable CAA patients without lobar intracerebral haemorrhage.
Methods:
MRI scans of 130 consecutive patients meeting modified Boston criteria for probable CAA were analysed for cortical superficial siderosis (focal, ≤3 sulci; disseminated, ≥4 sulci), and key small vessel disease markers. We compared clinical, imaging and cortical superficial siderosis topographical mapping data between subjects with vs. without acute cSAH, using multivariable logistic regression.
Results:
We included 33 probable CAA patients presenting with acute cSAH and 97 without cSAH at presentation. Acute cSAH patients were more commonly presenting with transient focal neurological episodes (76% vs. 34%; p<0.0001) compared to CAA patients without cSAH. Acute cSAH patients were also more often clinically presenting with transient focal neurological episodes compared to cortical superficial siderosis-positive but cSAH-negative CAA subjects (76% vs. 30%; p<0.0001). Cortical superficial siderosis prevalence (but no other CAA severity markers) was higher among patients with cSAH vs. those without, especially disseminated cortical superficial siderosis (49% vs 19%; p<0.0001). In multivariable logistic regression, cortical superficial siderosis burden (OR: 5.53; 95%CI: 2.82–10.8, p<0.0001) and transient focal neurological episodes (OR: 11.7; 95%CI: 2.70–50.6, p=0.001) were independently associated with acute cSAH.
Conclusions:
This probable CAA cohort provides additional evidence for distinct disease phenotypes, determined by the presence of cSAH and cortical superficial siderosis.
Keywords: MRI, cerebral amyloid angiopathy, cerebral small vessel disease, cortical superficial siderosis, convexity subarachnoid haemorrhage
Introduction
Sporadic cerebral amyloid angiopathy (CAA) is a small vessel disease of the brain characterised by progressive amyloid-β deposition in the small cortical and leptomeningeal arterioles.1 2 CAA is a frequent neuropathological finding in older people, an important cause of symptomatic lobar intracerebral haemorrhage (ICH) and a contributor to cognitive impairment.1–3 The clinical spectrum of CAA, however, seems to be more variable. It includes not only major lobar ICH, but also diverse syndromes such as transient focal neurologic episodes (TFNEs) and rapidly progressive or chronic cognitive impairment.1 4
CAA is also associated with brain MRI markers of small vessel disease including multiple strictly lobar cerebral microbleeds (the hallmark of the Boston criteria for CAA diagnosis),5 6 white matter hyperintensities and enlarged perivascular spaces (EPVS) in the centrum semiovale.1 7 The radiologic spectrum of CAA-related brain injury extends beyond these lesions to include cortical superficial siderosis (cSS) and acute convexity subarachnoid haemorrrhage (cSAH) – among cardinal haemorrhagic signatures of the disease.8 cSS quite characteristically affects the cerebral convexities, and its imaging manifestation reflects blood-breakdown residues, including haemosiderin, that line the outermost surface of the cortex or lie in the subarachnoid space. cSS has recently gained enormous interest since it is important as both a diagnostic marker of CAA9 and a predictor of future ICH10 and might provide insights into CAA pathophysiology and different disease phenotypes.8 11 Acute cSAH is increasingly recognized in CAA and appears not to represent aneurysmal or traumatic bleeding like other forms of SAH.8 11 Previous case series and case-control studies on the topic, had both CAA patients presenting with acute cSAH with or without a history of previous lobar ICH,12 and hence, by definition, patients had different disease severity and diagnostic certainty. Also, the spectrum of clinical-imaging markers of CAA patients presenting with cSAH compared with other symptomatic CAA cases presenting without lobar ICH in the appropriate clinical context, remain somewhat limited12 13 and further studies are needed. Thus, many questions of clinical relevance remain unanswered in the field and CAA presentations with acute cSAH are not clearly appreciated as a disease phenotype. In most instances, it is hypothesized that acute cSAH in the context of CAA reflects episodes of leaking from brittle amyloid-loaded leptomeningeal or very superficial cortical small vessels,14 resulting in cSS at the chronic stage.8 14 15
To test this hypothesis of the inter-relation between cSS and acute cSAH8 and gain further insights into different CAA subtypes, we performed a systematic analysis of symptomatic CAA patients presenting without any lobar ICH at a tertiary centre Stroke Clinic. We specifically aimed to investigate the overlap between acute cSAH with cSS as well as similarity of other risk factors (such as APOE ε2) and if cSAH in the acute phase presents with particular clinical symptoms that differ from the symptoms CAA patients without ICH that present without acute cSAH at baseline.
Patients and Methods
Case selection and study population
We analyzed prospectively collected data from consecutive patients meeting modified Boston criteria for probable CAA in the absence of ICH (symptomatic or asymptomatic) seen at Massachusetts General Hospital Stroke service (including stroke unit and outpatient clinics) between 1994–2015. Detailed inclusion criteria included: 1) diagnosis of probable CAA by modified Boston criteria; 2) clinical presentation other than hemorrhagic stroke, and 3) available MR images (including T2*-weighted/SWI, diffusion-weighted imaging, T2- weighted and FLAIR sequences). Patients with history of ICH at baseline or inflammatory CAA were excluded. Patients without acute, chronic (symptomatic or asymptomatic) or previous history of ICH but with strictly deep CMBs, mixed (deep and lobar) and exclusively cerebellar CMBs, or single strictly lobar CMBs were not considered in this cohort. A flow-chart of patient selection is provided in Figure 1.
Figure 1.
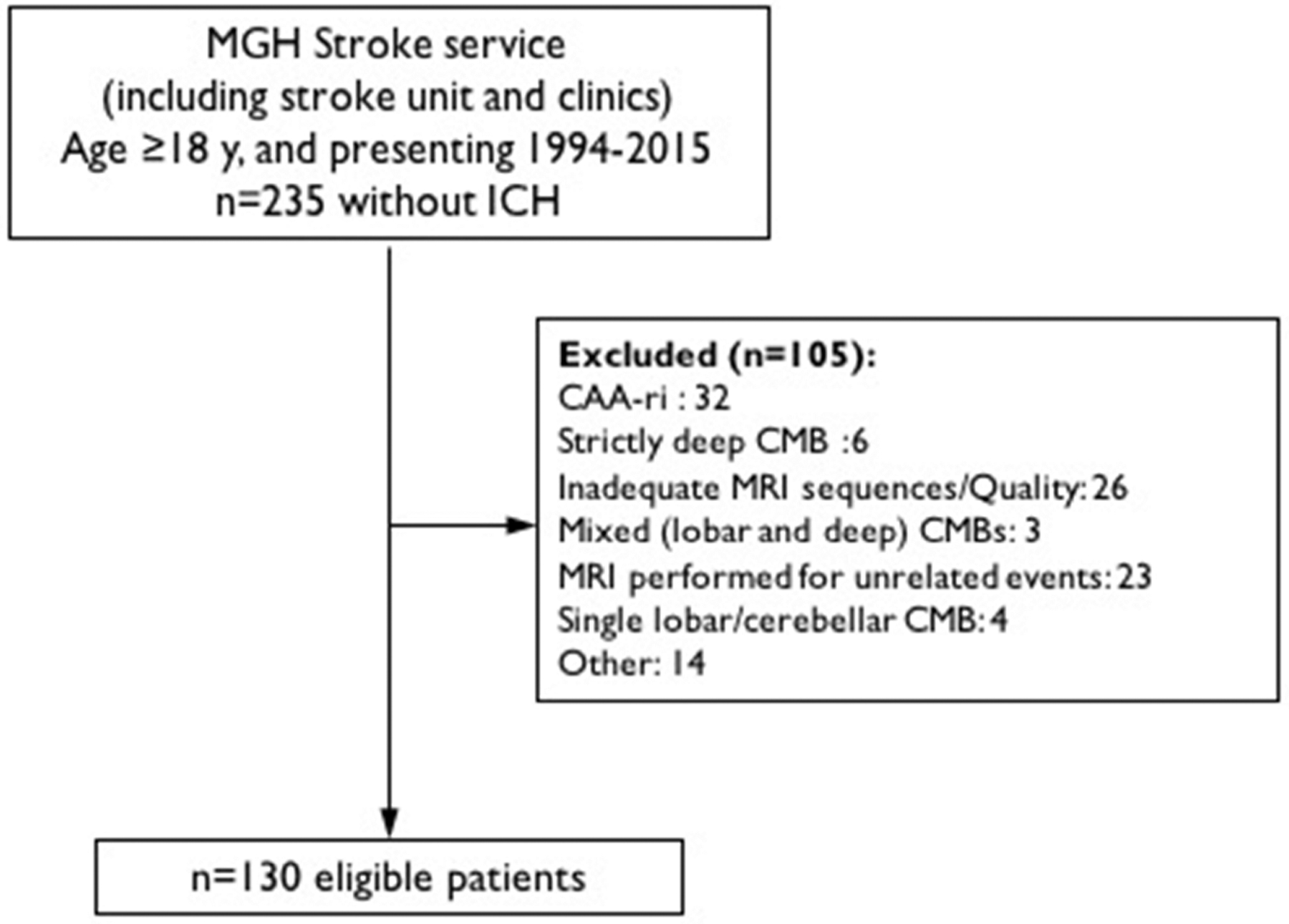
Cortical superficial siderosis (cSS) topographical mapping pipeline and methodology.
Clinical and APOE data
Full medical history, including demographic and clinical information and data on medication, was obtained at presentation through in-person interview with patient or surrogate using standardised data collection forms. Baseline neurological examination was performed as part of standard of care and symptoms recorded prospectively.
APOE genotype was determined in a subset of patients who provided blood samples and consented to genetic testing as previously described,16–18 and without knowledge of clinical or neuroimaging data.
Detailed symptoms of the baseline clinical presentations were collected blinded to imaging findings using pre-specified forms. Presentations were reviewed independently by two investigators and were classified based on all available clinical information but blinded to small vessel disease markers on MRI as: TFNEs (episodes of positive or negative neurological symptoms lasting for minutes with subsequent complete resolution, without a cause other that CAA after adequate evaluation, including angiography studies and carotid imaging, as previously suggested),19 20 cognitive complaints (e.g. mainly memory impairment, acute worsening of known cognitive impairment and mental status changes) or other non-focal neurological symptoms (including dizziness, confusion and gait impairment). Cases of disagreement were resolved by consensus.
Standard protocol approvals, registrations, and patient consents
This study was performed in accordance with the guidelines and with approval of the institutional review boards at our institution.
Neuroimaging data acquisition and analysis
Images were obtained using a 1.5 Tesla MR scanner and included whole brain T2-weighted, T2*-weighted gradient-recalled echo (T2*-GRE; echo time [TE] 750/50ms, 5mm slice thickness, 1mm inter-slice gap) and fluid attenuated inversion recovery (FLAIR; TR/TE 10000/140ms, inversion time 2,200 msec, 1 number of excitations, 5mm slice thickness, 1mm inter-slice gap). For a subset of patients (n=24) the paramagnetic MRI sequences included susceptibility Weighted Imaging (SWI, TR/TE 27/20ms, 1.5mm slice thickness). All MR images were reviewed blinded to clinical and genetic data by two trained observers, according to the STandards for ReportIng Vascular changes on nEuroimaging (STRIVE).21 MRIs were in general performed 1–3 days after the onset of symptoms in patients presenting with TFNEs.
CMBs presence and number were evaluated on axial T2*-GRE or SWI images using current consensus criteria7 and categorized according to previously validated MARS scale.22 For purposes of statistical analyses, the number of lobar CMBs was categorized using cut-points as defined previously (0, 1, 2–4, or ≥5).17
cSS and acute cSAH were defined and assessed in line with recent consensus recommendations.8 cSS was defined as curvilinear hypointensities following the cortical surface, distinct from the vessels, and was assessed on axial blood-sensitive sequences according to a validated scale: absent, focal (restricted to ≤3 sulci) or disseminated (affecting 4 or more sulci).9 23 cSS was also rated for multifocality (i.e. taking into account cSS presence at spatially separate foci in each hemisphere) using a protocol developed in our group as: (a) 0 – none; (b) 1 −1 sulcus or up to 3 immediately adjacent sulci with cSS; or (c) 2 – 2 or more non-adjacent sulci or more than 3 adjacent sulci with cSS. Based on the total score (i.e. adding the right and left hemisphere scores, 0–4): 0–no cSS, 1 – unifocal cSS, while ≥2 multifocal cSS. This method showed excellent interrater reliability (k=0.87). Acute cSAH was defined as linear hypointensity in the subarachnoid space affecting one or more cortical sulci on T2*-GRE/SWI sequences with corresponding hyperintensity in the subarachnoid space on T1-weighted or FLAIR images.7 23 The interrater reliability for acute cSAH definition was excellent (k=0.93 between two raters). All cSS and acute cSAH rating were jointly performed by two experienced raters.
EPVS were assessed on axial T2-weighted MR images, in the basal ganglia (BG) and centrum semiovale (CSO), using a validated 4-point visual rating scale (0=no PVS, 1=<10 PVS, 2=11–20 PVS, 3=21–40 PVS and 4=>40 PVS).24–26 We pre-specified a dichotomised classification of EPVS degree as high (score >2) or low (score ≤2) in line with previous studies.27 28 26
WMH volumes were calculated on axial FLAIR sequences with a previously described semi-automated planimetric method using MRICron software.29 Periventricular and deep WMH were also classified using the 0–3 Fazekas scale.30 The antero-posterior ratio of WMH lesions’ distribution was computed using a validated approach.31 As previously shown using this method, a lower score reflects more posteriorly distributed WMH lesions.31
Global atrophy was rated on axial brain T1-weighted imaging according to a previously validated 0–3 scale32 were 3 represents severe atrophy. We dichotomized patients into those with no or mild atrophy (0–1) and those with moderate to severe atrophy (2–3). Lacunes were defined according to STRIVE criteria,21 as round or ovoid fluid-filled cavities, between 3–15mm in diameter.
cSS topographical mapping
cSS outlines were obtained from blood-sensitive sequences (T2*-GRE or SWI) using a semi-automated sequential approach in MRI Cron33 from lesions first jointly characterized as cSS. This method involved a first manual outline followed by a threshold filtering higher values of the dynamic range (Figure 1). Visual check was performed to control for accuracy of the outline of each affected area.
For each subject, a surface map was constructed corresponding to the projection of each cSS site on the cortical mantle. All individual surface maps were then registered to a common space, and an average surface heat map was generated (Figure 1). The software suite FreeSurfer (https://surfer.nmr.mgh.harvard.edu, v5.3.0) was used for the aforementioned analyses.34–36
Statistics
Categorical variables were analysed using Pearson’s χ2 or Fisher exact test, and continuous variables by the 2-sample t test (for normal distributions), and Wilcoxon rank sum (for non-normal distributions). We compared demographic, clinical, imaging and APOE data of CAA patients with vs. without acute cSAH. A multivariable logistic regression analysis model was used to look for independent associations with acute cSAH, including clinical presentation and other CAA MRI signatures and correcting for potential confounders identified in the univariable analysis or based on potential biological significance. As sensitivity analyses, we further adjusted these models for different blood-sensitive MRI sequences (e.g. T2*-GRE vs. SWI). In a post-hoc analysis, we assessed the relationship between cSS (presence or burden) and other markers of CAA and APOE genotype. APOE genotype was analyzed using two categorical variables indicating presence of any ε2 or ε4 alleles. Significance level was set at 0.05. All tests of significance were two-tailed. Stata software (Version 13, StataCorp.) was used. The manuscript was prepare with reference to the STROBE guidelines.37
Results
Our final sample included 130 consecutive patients with probable CAA presenting to our center without any ICH (Figure 2). Review of all available clinical data of these cases yielded the following predominant baseline presentations: TFNEs (58/130; 45%), cognitive complains including non-focal neurological symptoms (72/130; 55%). Seven patients (5%) presented with a combination of these symptom clusters. MRI was performed within 1–3 days of symptoms onset in all patients. None of the included cases had a history of acute or previous history of head trauma. A total of 24/130 (19%) had SWI sequences available, while the rest of the patient cohort had T2*-GRE. Patients who underwent SWI vs. T2*-GRE were not different in demographic, clinical or neuroimaging characteristics (data not shown).
Figure 2.
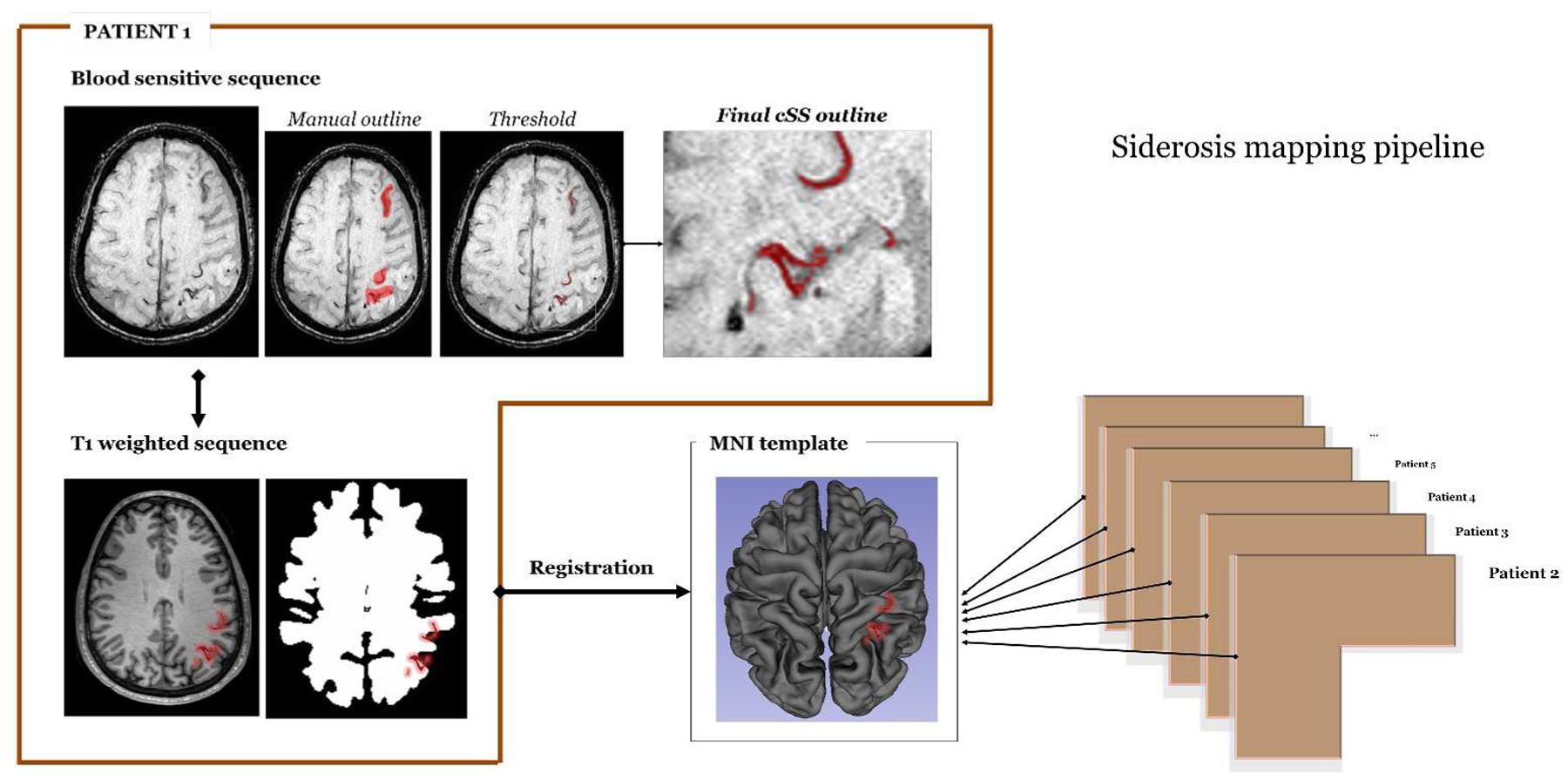
Flow-chart of patient selection.
Fifty-three percent of patients with TFNEs experienced sensory symptoms, typically consisting of (‘aura-like’, ‘migraine-like’ or ‘seizure-like’) stereotyped positive spreading paraesthesias lasting a few minutes to one hour. Transient visual symptoms (positive or negative phenomena) were seen in 16% of patients with TFNEs, while transient focal weakness and episodes of language impairment in 22% and 21% respectively. Patients with cognitive complaints had subacute memory impairment over months-years with a combination of symptoms, prompting referral to our Stroke service and brain MRI scan (CDR median: 1). A clinical diagnosis of dementia before the baseline presentation was made in 57% (95%CI: 41%−71%) patients with cognitive complaints. Only 8% (95%CI: 3%−19%) of patients presenting with TFNEs had dementia diagnosis.
Table 1 presents the clinical and imaging characteristics of the whole cohort and the comparisons between CAA patients with vs. without acute cSAH. No differences were found in demographic characteristics and vascular risk factors (Table 1). TFNE were common among cSAH patients (76% vs. 34%; p<0.0001). In these cases, all TFNEs could be anatomically correlated with the location of acute cSAH. In cases presenting with TFNEs but without acute cSAH on baseline MRI, patient’s transient symptoms were in general anatomically correlated with areas of cSS. Acute cSAH patients were also more often clinically presenting with TFNEs compared to cSS-positive but cSAH-negative CAA subjects (76% vs. 30%; p<0.0001).
Table 1.
Clinical, imaging and genetic characteristics for CAA patients presenting with vs. without acute cSAH.
| Whole CAA cohort (N = 130) | CAA with cSAH (N = 33) | CAA without cSAH (N = 97) | p-value | |
|---|---|---|---|---|
| Age at MRI, mean (95% CI), years | 74.2 (72.6–75.8) | 73.7 (70.2–77.3) | 74.4 (72.6–76.2) | 0.715 |
| Sex, female n (%) | 50 (38) | 16 (48) | 17 (18) | 0.171 |
| Hypertension, n (%) | 87 (67) | 22 (67) | 65 (67) | 0.971 |
| Atrial fibrillation, n (%) | 24 (18) | 3 (9) | 21 (22%) | 0.222 |
| Hypercholesterolemia, n (%) | 71 (55) | 21 (64) | 50 (52) | 0.228 |
| Warfarin use, n (%) | 12 (9) | 1 (3) | 11 (11) | 0.298 |
| Aspirin use, n (%) | 44 (34) | 10 (30) | 34 (35) | 0.619 |
| Statin use, n (%) | 57 (44) | 17 (52) | 40 (41) | 0.304 |
| Dementia diagnosis, n (%) | 39 (30) | 5 (15) | 34 (35) | 0.031 |
| TFNEs, n (%) | 58 (45) | 25 (76) | 33 (34) | <0.0001 |
| * APOE ε2 (1+ copies), n (%) | 17 (27) | 6 (35) | 11 (24) | 0.393 |
| * APOE ε4 (1+ copies), n (%) | 31 (50) | 8 (47) | 23 (51) | 0.776 |
| Severe (Fazekas 5–6) WMH, n (%) | 47 (36) | 10 (30) | 37 (38) | 0.418 |
| WMH volume, median (IQR), cc | 21.7 (13.4–32.3) | 20.3 (12.6–32.4) | 24.6 (13.9–43.7) | 0.229 |
| WMH antero-posterior ratio, (95%CI) | 10.3 (8.4–12.2) | 11.2 (8.1–14.3) | 10 (7.6–12.4) | 0.581 |
| Lacunes presence, n (%) | 44 (34) | 8 (24) | 36 (37) | 0.177 |
| Moderate/Severe atrophy, n (%) | 94 (72) | 22 (67) | 72 (74) | 0.402 |
| High grade CSO-EPVS (>20), n (%) | 74 (57) | 21 (64) | 53 (55) | 0.367 |
| Lobar CMBs presence, n (%) | 121 (93) | 30 (91) | 91 (94) | 0.570 |
| Lobar CMBs count, median (IQR) | 8 (3–40) | 6 (2–14) | 10 (3–48) | 0.115 |
| Logarithm of CMBs count | 2.52 (2.22–2.81) | 2.02 (1.56–2.49) | 2.68 (2.33–3.04) | 0.05 |
| Presence of cSS, n (%) | 58 (45) | 27 (82) | 31 (32) | <0.0001 |
| Focal cSS, n (%) | 24 (18) | 11 (33) | 13 (13) | <0.0001 |
| Disseminated cSS, n (%) | 34 (26) | 16 (48) | 18 (19) | |
| Multifocal cSS, n (%) | 38 (29) | 18 (55) | 20 (21) | <0.0001 |
APOE genotype available in 62 patients.
The prevalence of cSS was significantly higher among cSAH patients compared to CAA patients without acute cSAH, especially disseminated (48% vs 19%; p<0.0001) and multifocal cSS (55% vs 21%; p<0.0001) (Table 1 and Figure 3). There was no difference in the two groups in the prevalence or burden of all the other small vessel disease imaging markers, WMH (volume, Fazekas or posterior distribution), CSO-EPVS, atrophy and lacunes (Table 1). There was a trend for patients with acute cSAH to have lower lobar CMBs counts (Table 1). Patients with acute SAH more ofter had the APOE ε2 allele but this was not statistically significant. (35% vs. 24%).
Figure 3.
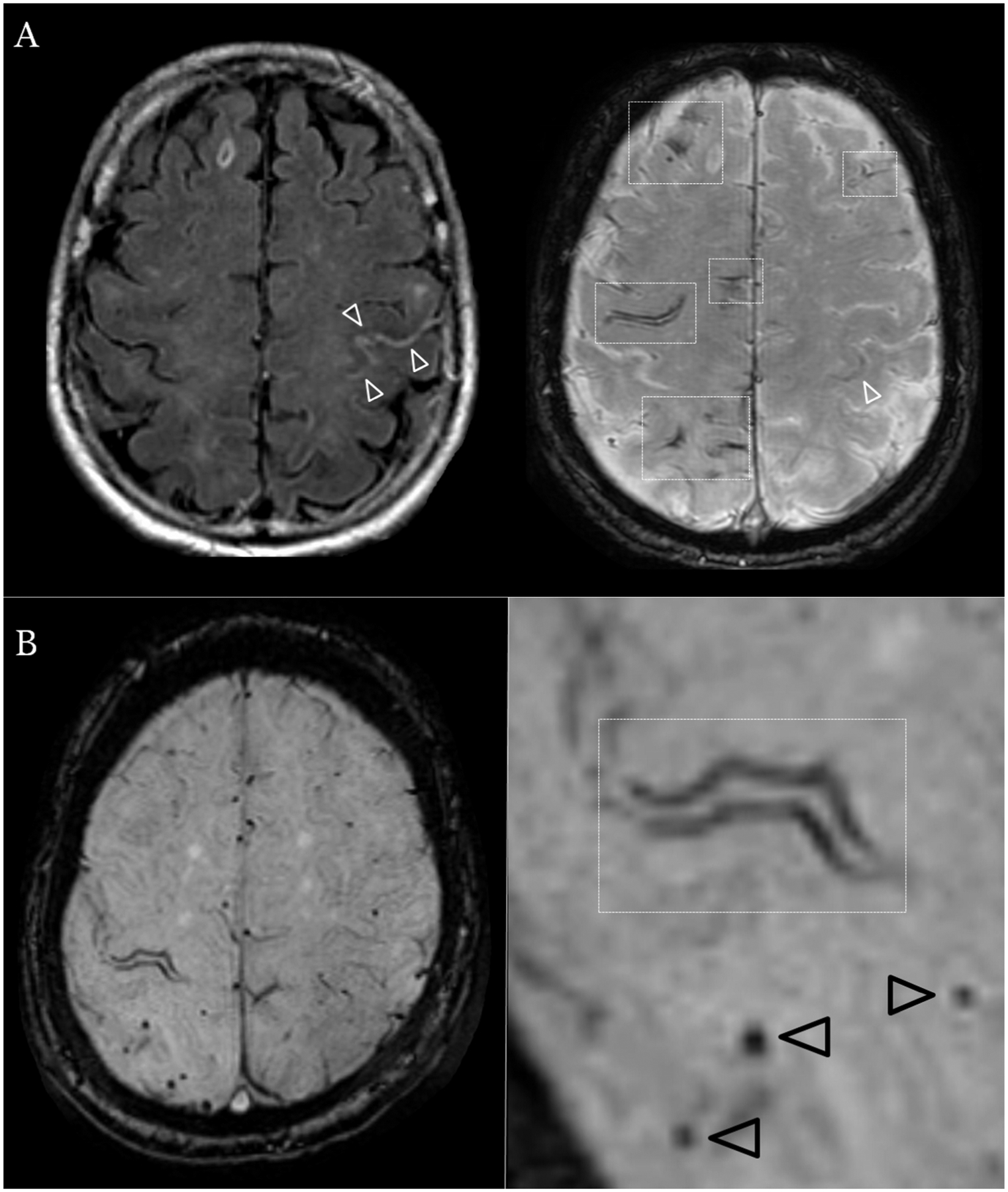
Representative patients with and without cSAH.
1.5 Tesla axial brain MRI sequences (A) FLAIR (left) sequence and SWI (right) axial sections in an 82-year old male evaluated for rapidly progressing right sided tingling resolved in 15 minutes after onset. No relevant medical history. Imaging demonstrates acute subarachnoid blood in the central sulcus (white arrowheads) and bilateral disseminated foci of cortical superficial siderosis (white rectangles). (B) SWI axial sequence in a 75-year old female with repeated stereotyped episodes of dizziness and left sided hand tingling. Imaging demonstrates chronic focal cortical superficial siderosis affecting the right central sulcus (white rectangle) as well as strictly lobar cerebral microbleeds (black arrowheads).
In multivariable logistic regression cSS burden (OR: 5.53; 95%CI: 2.82–10.8, p<0.0001) and TFNEs (OR: 11.7; 95%CI: 2.70–50.6, p=0.001) were independent correlates of acute cSAH (Table 2). Models remained consisted and of similar effect size in sensitivity analyses further adjusting for SWI vs. T2*-GRE sequences. Figure 4 presents the overall cSS topographical heat maps in patients with and without acute cSAH, demonstrating more extensive cSS involvement in multiple brain regions in the probable CAA patient group presenting with acute cSAH.
Table 2.
Multivariable logistic regression analysis for acute cSAH presence.
| OR (95%CI) | p-value | |
|---|---|---|
| cSS (no, focal or disseminated) | 5.53 (2.82–10.8) | <0.0001 |
| * Multifocal cSS (vs. unifocal) | 4.08 (1.56–10.6) | 0.004 |
| WMH volume (per cc increase) | 1.01 (0.98–1.03) | 0.671 |
| Severe CSO-PVS (Yes vs. No) | 1.66 (0.55–5.01) | 0.374 |
| Lobar CMBs burden | 0.81 (0.53–1.22) | 0.309 |
| Clinical TFNEs symptoms (vs. other presentations) | 11.7 (2.70–50.6) | 0.001 |
Alternative model using multifocal (vs. unifocal) cSS – all other estimated remain consistent.
Models remained consisted and of similar effect size after further adjusting for SWI/T2*-GRE sequence parameters.
Figure 4.
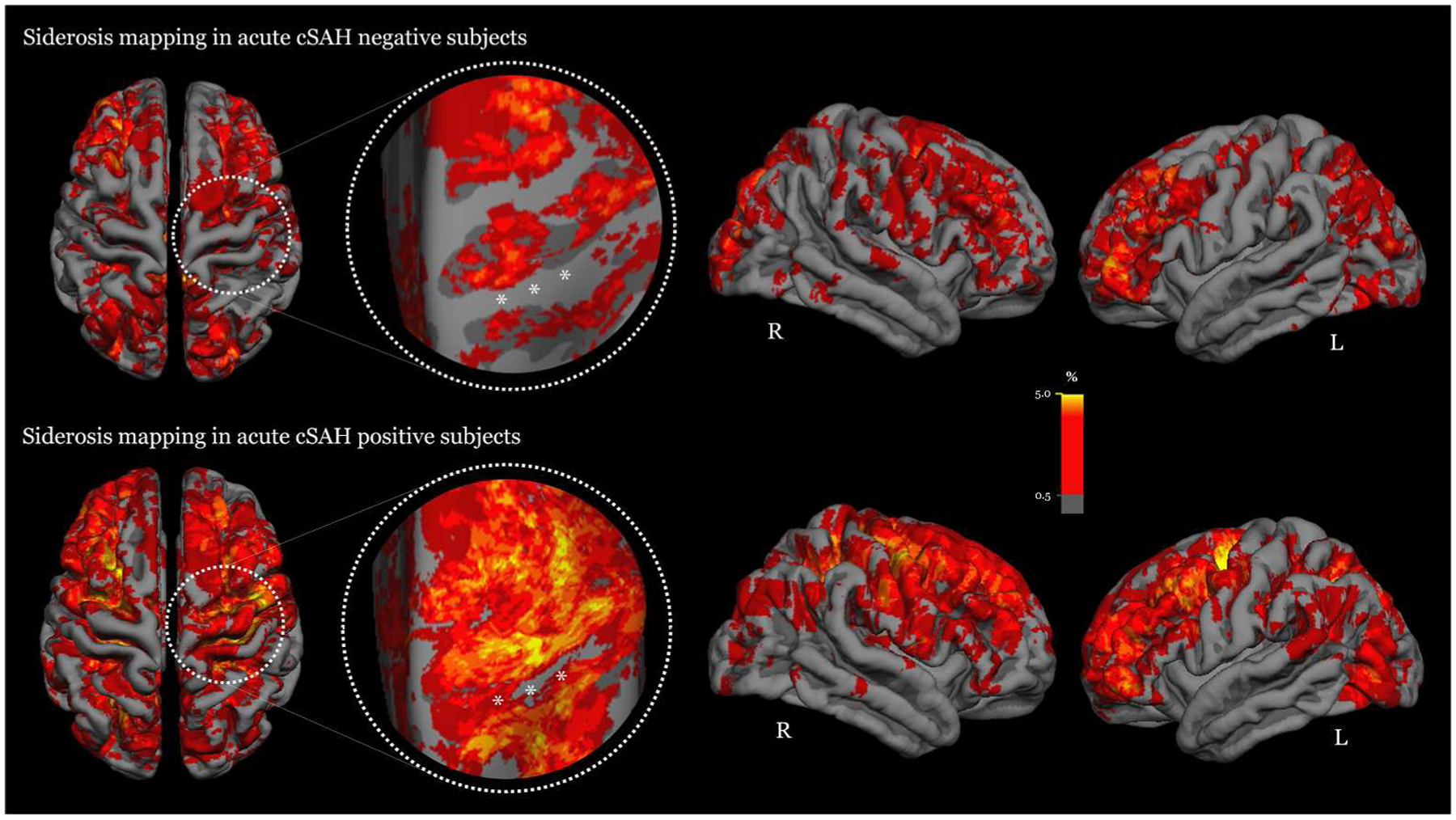
Averaged surface heat maps showcasing the whole brain distribution and frequency of cortical superfical siderosis sites in the two patient study groups: (a) probable CAA patients without acute cSAH (i.e. cSAH negative group, top panel); and (b) probable CAA patients presenting with acute cSAH (cSAH positive group, lower panel). The areas in the circle include the pre-central and central sulcus for illustration purposes. Compared to patients without acute cSAH (top panel), the patient group with acute cSAH in the lower panel demonstrates more extensive involvement by cSS – more brain areas are affected with higher degree of cSS per region. This is especially evident in the pre-central and central sulcus (circled areas), in which multiple cSS sites are covering the whole sulci. These eloquent areas are particular symptomatogenic and the occurrence of cSS (or cSAH in the acute stange) might underpin transient focal neurological episodes often seen in CAA patients (“amyloid spells”). The color scale bar indicates the degree of cSS involvement in different brain areas in our cohort.
cSS and APOE in the whole CAA group
cSS (presence and burden) was not associated with any of the other markers of CAA, including WMH, lobar CMBs, CSO-PVS and atrophy (all p>0.2). Among subjects with available genetic testing (n=62), APOE ε2 (but not ε4) allele was overrepresented in cases with cSS (40% vs. 16%; p=0.032), especially disseminated cSS (56% vs. 17% vs. 16% in those with disseminated vs. focal vs. no cSS, p=0.006 across the three groups). These results remained consistent and of similar effect size in a sensitivity multivariable logistic regression analysis adjusting for age and sex (OR: 2.75; 95%CI: 1.33–5.68; p=0.006 for the association between cSS burden and APOE ε2). There was no association between cSS (burden or presence) and APOE ε4. There was no association between CMBs counts and APOE genotype (p>0.2).
Discussion
The major findings from this study show that symptomatic CAA patients without ICH presenting at a tertiary stroke service with acute cSAH are more likely to have cSS (particularly disseminated) and a trend for less lobar CMBs counts compared to CAA patients without acute cSAH. The prevalence and burden of other small vessel disease markers are comparable between the two groups. While CMBs burden is associated with WMH, cSS demonstrated no associations with any other CAA markers in the whole group. In addition, there was an overall higher representation of the APOE ε2 in patients with cSS.
cSS and acute cSAH has been recently identified as key imaging signatures of CAA.8 15 38 Although the clinical-imaging features of CAA-related lobar ICH are well established, little is known about the specific patterns of symptomatic CAA presenting without major ICH,4 39 especially CAA-related cSAH. Our results provide new insights into the clinical and imaging spectrum of sporadic CAA, pointing to potentially different disease phenotypes according to acute cSAH presence.40 The most distinctive neuroimaging feature between the two groups was the much higher prevalence of disseminated and multifocal cSS in CAA patients with acute cSAH. In line with a recent study,15 the reported prevalence of cSS in this group, is much higher compared to histopathology-confirmed CAA-ICH9 and the reported prevalence in a previous imaging study of CAA-ICH.23 The high prevalence of cSS among CAA-related acute cSAH patients and the high prevalence of TFNE in CAA patients with either cSAH or cSS suggest a link between cSAH and cSS.8 41 Of note, patients with vs. without acute cSAH had a very similar profile of putative neuroimaging biomarkers of CAA severity, including lobar CMBs, WMH and CSO-PVS. It is hence possible that factors other than overall CAA pathological severity might be driving a more superficial haemorragic subtype of the disease, such as more predominant leptomeningeal pathology (with a trend for sparing cortical arteries) or specific associations with superficial vessel fragility partly driven by the APOE ε2 allele. Further neuropathological studies are needed to directly test this hypothesis.
Although the pathophysiological basis underlying acute cSAH and cSS in CAA remain poorly understood, the prevailing view is that fragile leptomeningeal or very superficial cortical vessels severely affected by amyloid deposition might lead to blood-leaking episodes into the subarachnoid space and thus cSAH in some patients.39, 40 These mechanisms could be linked with the neuropathological observation of more severe leptomeningeal CAA compared to cortical parenchymal in some cases.42 Depending on the location, if acute cSAH occurs in eloquent areas, such as the pre-central or central sulci, they will trigger TNFEs.41 43 In other locations, these episodes might be clinically silent and the breakdown of blood products will ultimately lead to cSS in the chronic stage,11 a process which needs the mobilization of macrophages and inflammatory pathways.44 Hence, it is possible that patients presenting with acute cSAH have had multiple previous episodes of asymptomatic cSAH, but only when such an episode happens in an eloquent brain area they come to medical attention and get an MRI. This hypothesis might explain our observations in the cSS topographical mapping analysis. In this analysis, we observed more involvement by cSS in multiple brain regions in probable CAA patients presenting with acute cSAH compared to patients without, likely representing multiple bleeding episodes in the subarachnoid space. This was particularly evidet in the precentral and central sulcus, symptomatogenic eloquent areas often responsible for TFNEs in CAA patients. Appropriate imaging can potentially reveal different time points of the same pathophysiological processes, providing information about the timing, as well as pattern, of cSAH/cSS bleeding events (i.e. dissemination in time and space). This hypothesis is also supported by a number of small imaging studies showing evolution of acute cSAH into cSS in follow-up scans.11 15 38 Large systematic studies assessing the evolution from acute cSAH through cSS need to be performed.
Our post-hoc analysis in the whole cohort, provide some evidence for the idea that cSS seems to be a stand-alone independent marker of the disease, indicating distinct pathophysiological mechanisms. APOE ε2 seems to be more strongly linked with cSS and bleeding, in line with a previous study on cSAH in CAA.13 This was a consistent finding in a separate cohort of CAA defined by clinico-radiographic criteria,45 as well as in a study of pathologically confirmed CAA.39 The APOE ε2 allele is known to be associated with CAA-related vasculopathic changes and lobar ICH,46 47 though any mechanistic links with cSS might be complex and need to be treated with caution.39
Notable strengths of our study include a large cohort of consecutive probable CAA cases without ICH (hence excluding the possibility of a ‘secondary’ mechanism of cSAH and cSS due to leakage of a lobar ICH into the subarachnoid space), with in-depth evaluation of MRIs by trained raters using validated scales. MR images were rated for a comprehensive range of imaging markers of small vessel disease that capture aspects of CAA severity, with volumetric assessment of total WMH burden and using cSS definitions and ratings according to recently suggested consensus standards.8 The detailed mapping of each individual cSS lesion allowed us to confirm the robustness of the findings. A number of previous papers have focussed on acute cSAH related to CAA in the last few years.12 However, previous studies on the topic were relatively small case series, typically including all possible and probable CAA patients presenting with and without a history of previous lobar ICH.12 Hence, previously reported cases were partly confounded by the different disease severity, duration, predominant phenotype (ICH vs. non-ICH CAA) and diagnostic certainty for CAA. No studies, to the best of our knowledge, explored the whole spectrum of clinical-imaging markers of CAA in acute cSAH compared with other symptomatic CAA cases presenting without lobar ICH in the appropriate clinical context, a question of clinical relevance for accurately phenotyping the disease. Also previous studies have alluded to the potential relationship between cSAH and APOE ε2 genotype, and between cSAH and TNFEs,12 but these data are rather limited.13 Finally, the relationship from acute cSAH to cSS (i.e. chronic) in a given patient has been proposed in previous reports and a recent consensus paper on the topic,8 but many aspects of this relationship have not been systematically explored at the group level and across different CAA subtypes without ICH at baseline.
We note the potential selection bias in our cohort due to our center’s expertise in CAA and the requirement for MRI performed as part of routine clinical care. Another limitation is the lack of APOE data in a substantial proportion of patients included, limiting the statistical power for detecting an association with acure cSAH. An additional limitation is that is impossible to score cSS without some unblinding to other imaging findings, though raters were blinded to MRI markers on the non-blood-sensitive sequences, including acute cSAH where possible and to all clinical data. Finally, the cross-sectional design of the current study did not allow us to assess potential causality of the reported associations and the clinical relevance of acute cSAH and cSS as risk factors for future symptomatic haemorrhage, key topics for further research.
Results from this cohort of probable CAA patients without ICH provide additional evidence for distinct disease phenotypes, determined by the presence of acute cSAH. Our study emphasizes the widening spectrum of CAA with presentations reflecting different neuroimaging and genetic features and suggests a crucial role for cSS. These findings require external validation in larger CAA cohorts. The natural history of patients presenting without ICH who have acute cSAH and/or cSS, and the risk of future ICH requires further investigation in prospective cohorts.
Funding and disclosures
The authors report no disclosures relevant to this work.
Study Funding:
This work was supported by NIH grants R01-AG026484 (S.M. Greenberg). Gregoire Boulouis was supported by a J. William Fulbright Scholarship and a Monahan Foundation Biomedical Research Grant. Andreas Charidimou receives postdoctoral support from the Bodossaki Foundation. This study is not industry sponsored.
References
- 1.Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry 2012;83(2):124–37. doi: 10.1136/jnnp-2011-301308 [published Online First: 2011/11/08] [DOI] [PubMed] [Google Scholar]
- 2.Viswanathan A, Greenberg SM. Cerebral amyloid angiopathy in the elderly. Annals of neurology 2011;70(6):871–80. doi: 10.1002/ana.22516 [published Online First: 2011/12/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987;18(2):311–24. [published Online First: 1987/03/01] [DOI] [PubMed] [Google Scholar]
- 4.Greenberg SM, Vonsattel JP, Stakes JW, et al. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993;43(10):2073–9. [published Online First: 1993/10/01] [DOI] [PubMed] [Google Scholar]
- 5.Martinez-Ramirez S, Romero JR, Shoamanesh A, et al. Diagnostic value of lobar microbleeds in individuals without intracerebral hemorrhage. Alzheimer’s & dementia: the journal of the Alzheimer’s Association 2015;11(12):1480–88. doi: 10.1016/j.jalz.2015.04.009 [published Online First: 2015/06/17] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knudsen KA, Rosand J, Karluk D, et al. Clinical diagnosis of cerebral amyloid angiopathy: validation of the Boston criteria. Neurology 2001;56(4):537–9. [published Online First: 2001/02/27] [DOI] [PubMed] [Google Scholar]
- 7.Greenberg SM, Vernooij MW, Cordonnier C, et al. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol 2009;8(2):165–74. doi: S1474–4422(09)70013–4 [pii] 10.1016/S1474-4422(09)70013-4 [published Online First: 2009/01/24] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Charidimou A, Linn J, Vernooij MW, et al. Cortical superficial siderosis: detection and clinical significance in cerebral amyloid angiopathy and related conditions. Brain 2015;138(Pt 8):2126–39. doi: 10.1093/brain/awv162 [published Online First: 2015/06/28] [DOI] [PubMed] [Google Scholar]
- 9.Linn J, Halpin A, Demaerel P, et al. Prevalence of superficial siderosis in patients with cerebral amyloid angiopathy. Neurology 2010;74(17):1346–50. doi: 74/17/1346 [pii] 10.1212/WNL.0b013e3181dad605 [published Online First: 2010/04/28] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Charidimou A, Peeters AP, Jager R, et al. Cortical superficial siderosis and intracerebral hemorrhage risk in cerebral amyloid angiopathy. Neurology 2013;81(19):1666–73. doi: 10.1212/01.wnl.0000435298.80023.7a [published Online First: 2013/10/11] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linn J, Herms J, Dichgans M, et al. Subarachnoid hemosiderosis and superficial cortical hemosiderosis in cerebral amyloid angiopathy. AJNR Am J Neuroradiol 2008;29(1):184–6. doi: ajnr.A0783 [pii] 10.3174/ajnr.A0783 [published Online First: 2007/10/20] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson D, Hostettler IC, Ambler G, et al. Convexity subarachnoid haemorrhage has a high risk of intracerebral haemorrhage in suspected cerebral amyloid angiopathy. Journal of neurology 2017;264(4):664–73. doi: 10.1007/s00415-017-8398-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez-Lizana E, Carmona-Iragui M, Alcolea D, et al. Cerebral amyloid angiopathy-related atraumatic convexal subarachnoid hemorrhage: an ARIA before the tsunami. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 2015. doi: 10.1038/jcbfm.2015.25 [published Online First: 2015/03/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beitzke M, Enzinger C, Wunsch G, et al. Contribution of convexal subarachnoid hemorrhage to disease progression in cerebral amyloid angiopathy. Stroke 2015;46(6):1533–40. doi: 10.1161/STROKEAHA.115.008778 [published Online First: 2015/05/09] [DOI] [PubMed] [Google Scholar]
- 15.Calviere L, Cuvinciuc V, Raposo N, et al. Acute Convexity Subarachnoid Hemorrhage Related to Cerebral Amyloid Angiopathy: Clinicoradiological Features and Outcome. Journal of stroke and cerebrovascular diseases: the official journal of National Stroke Association 2016. doi: 10.1016/j.jstrokecerebrovasdis.2015.11.010 [published Online First: 2016/03/01] [DOI] [PubMed] [Google Scholar]
- 16.Biffi A, Shulman JM, Jagiella JM, et al. Genetic variation at CR1 increases risk of cerebral amyloid angiopathy. Neurology 2012;78(5):334–41. doi: 10.1212/WNL.0b013e3182452b40 [published Online First: 2012/01/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biffi A, Halpin A, Towfighi A, et al. Aspirin and recurrent intracerebral hemorrhage in cerebral amyloid angiopathy. Neurology 2010;75(8):693–8. doi: 10.1212/WNL.0b013e3181eee40f [published Online First: 2010/08/25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brouwers HB, Biffi A, McNamara KA, et al. Apolipoprotein E genotype is associated with CT angiography spot sign in lobar intracerebral hemorrhage. Stroke 2012;43(8):2120–5. doi: 10.1161/STROKEAHA.112.659094 [published Online First: 2012/05/25] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Charidimou A, Peeters A, Fox Z, et al. Spectrum of transient focal neurological episodes in cerebral amyloid angiopathy: multicentre magnetic resonance imaging cohort study and meta-analysis. Stroke 2012;43(9):2324–30. doi: 10.1161/STROKEAHA.112.657759 [published Online First: 2012/07/17] [DOI] [PubMed] [Google Scholar]
- 20.Charidimou A, Law R, Werring DJ. Amyloid “spells” trouble. Lancet 2012;380(9853):1620. doi: 10.1016/S0140-6736(12)61333-6 [published Online First: 2012/11/06] [DOI] [PubMed] [Google Scholar]
- 21.Wardlaw JM, Smith EE, Biessels GJ, et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 2013;12(8):822–38. doi: 10.1016/S1474-4422(13)70124-8 [published Online First: 2013/07/23] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gregoire SM, Chaudhary UJ, Brown MM, et al. The Microbleed Anatomical Rating Scale (MARS): reliability of a tool to map brain microbleeds. Neurology 2009;73(21):1759–66. doi: 73/21/1759 [pii] 10.1212/WNL.0b013e3181c34a7d [published Online First: 2009/11/26] [DOI] [PubMed] [Google Scholar]
- 23.Charidimou A, Jager RH, Fox Z, et al. Prevalence and mechanisms of cortical superficial siderosis in cerebral amyloid angiopathy. Neurology 2013;81(7):626–32. doi: 10.1212/WNL.0b013e3182a08f2c [published Online First: 2013/07/19] [DOI] [PubMed] [Google Scholar]
- 24.Doubal FN, MacLullich AM, Ferguson KJ, et al. Enlarged perivascular spaces on MRI are a feature of cerebral small vessel disease. Stroke 2010;41(3):450–4. doi: STROKEAHA.109.564914 [pii] 10.1161/STROKEAHA.109.564914 [published Online First: 2010/01/09] [DOI] [PubMed] [Google Scholar]
- 25.Maclullich AM, Wardlaw JM, Ferguson KJ, et al. Enlarged perivascular spaces are associated with cognitive function in healthy elderly men. J Neurol Neurosurg Psychiatry 2004;75(11):1519–23. doi: 75/11/1519 [pii] 10.1136/jnnp.2003.030858 [published Online First: 2004/10/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charidimou A, Jaunmuktane Z, Baron JC, et al. White matter perivascular spaces: An MRI marker in pathology-proven cerebral amyloid angiopathy? Neurology 2014;82(1):57–62. doi: 10.1212/01.wnl.0000438225.02729.04 [published Online First: 2013/11/29] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu YC, Tzourio C, Soumare A, et al. Severity of dilated Virchow-Robin spaces is associated with age, blood pressure, and MRI markers of small vessel disease: a population-based study. Stroke 2010;41(11):2483–90. doi: 10.1161/strokeaha.110.591586 [published Online First: 2010/09/25] [DOI] [PubMed] [Google Scholar]
- 28.Martinez-Ramirez S, Pontes-Neto OM, Dumas AP, et al. Topography of dilated perivascular spaces in subjects from a memory clinic cohort. Neurology 2013;80(17):1551–6. doi: 10.1212/WNL.0b013e31828f1876 [published Online First: 2013/04/05] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rorden C, Karnath HO, Bonilha L. Improving lesion-symptom mapping. Journal of cognitive neuroscience 2007;19(7):1081–8. doi: 10.1162/jocn.2007.19.7.1081 [published Online First: 2007/06/23] [DOI] [PubMed] [Google Scholar]
- 30.Fazekas F, Chawluk JB, Alavi A, et al. MR signal abnormalities at 1.5 T in Alzheimer’s dementia and normal aging. AJR American journal of roentgenology 1987;149(2):351–6. doi: 10.2214/ajr.149.2.351 [published Online First: 1987/08/01] [DOI] [PubMed] [Google Scholar]
- 31.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology 2014;83(9):794–800. doi: 10.1212/WNL.0000000000000732 [published Online First: 2014/07/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pasquier F, Leys D, Weerts JG, et al. Inter- and intraobserver reproducibility of cerebral atrophy assessment on MRI scans with hemispheric infarcts. Eur Neurol 1996;36(5):268–72. [published Online First: 1996/01/01] [DOI] [PubMed] [Google Scholar]
- 33.Rorden C, Brett M. Stereotaxic display of brain lesions. Behavioural neurology 2000;12(4):191–200. [published Online First: 2001/09/25] [DOI] [PubMed] [Google Scholar]
- 34.Dale AM, Fischl B, Sereno MI. Cortical surface-based analysis. I. Segmentation and surface reconstruction. NeuroImage 1999;9(2):179–94. doi: 10.1006/nimg.1998.0395 [published Online First: 1999/02/05] [DOI] [PubMed] [Google Scholar]
- 35.Fischl B, Sereno MI, Dale AM. Cortical surface-based analysis. II: Inflation, flattening, and a surface-based coordinate system. NeuroImage 1999;9(2):195–207. doi: 10.1006/nimg.1998.0396 [published Online First: 1999/02/05] [DOI] [PubMed] [Google Scholar]
- 36.Segonne F, Dale AM, Busa E, et al. A hybrid approach to the skull stripping problem in MRI. NeuroImage 2004;22(3):1060–75. doi: 10.1016/j.neuroimage.2004.03.032 [published Online First: 2004/06/29] [DOI] [PubMed] [Google Scholar]
- 37.von Elm E, Altman DG, Egger M, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 2007;370(9596):1453–7. doi: 10.1016/S0140-6736(07)61602-X [published Online First: 2007/12/08] [DOI] [PubMed] [Google Scholar]
- 38.Raposo N, Viguier A, Cuvinciuc V, et al. Cortical subarachnoid haemorrhage in the elderly: a recurrent event probably related to cerebral amyloid angiopathy. Eur J Neurol 2011;18(4):597–603. doi: 10.1111/j.1468-1331.2010.03214.x [published Online First: 2010/11/03] [DOI] [PubMed] [Google Scholar]
- 39.Charidimou A, Martinez-Ramirez S, Shoamanesh A, et al. Cerebral amyloid angiopathy with and without hemorrhage: Evidence for different disease phenotypes. Neurology 2015;84(12):1206–12. doi: 10.1212/WNL.0000000000001398 [published Online First: 2015/02/27] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charidimou A, Jager HR. Developing biomarkers for cerebral amyloid angiopathy trials: do potential disease phenotypes hold promise? Lancet Neurol 2014;13(6):538–40. doi: 10.1016/S1474-4422(14)70096-1 [published Online First: 2014/05/23] [DOI] [PubMed] [Google Scholar]
- 41.Ni J, Auriel E, Jindal J, et al. The characteristics of superficial siderosis and convexity subarachnoid hemorrhage and clinical relevance in suspected cerebral amyloid angiopathy. Cerebrovasc Dis 2015;39(5–6):278–86. doi: 10.1159/000381223 [published Online First: 2015/04/15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kovari E, Herrmann FR, Hof PR, et al. The relationship between cerebral amyloid angiopathy and cortical microinfarcts in brain ageing and Alzheimer’s disease. Neuropathology and applied neurobiology 2013;39(5):498–509. doi: 10.1111/nan.12003 [published Online First: 2012/11/21] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Charidimou A Convexity subarachnoid hemorrhage in cerebral amyloid angiopathy: the saga continues. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 2015;35(5):707–9. doi: 10.1038/jcbfm.2015.24 [published Online First: 2015/02/19] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koeppen AH, Michael SC, Li D, et al. The pathology of superficial siderosis of the central nervous system. Acta neuropathologica 2008;116(4):371–82. doi: 10.1007/s00401-008-0421-z [published Online First: 2008/08/13] [DOI] [PubMed] [Google Scholar]
- 45.Shoamanesh A, Ramirez SM, Oliveira-Filho J, et al. Interrelationship of Cortical Superficial Siderosis and Microbleeds in Cerebral Amyloid Angiopathy. Neurology In press, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Greenberg SM, Vonsattel JP, Segal AZ, et al. Association of apolipoprotein E epsilon2 and vasculopathy in cerebral amyloid angiopathy. Neurology 1998;50(4):961–5. [published Online First: 1998/05/05] [DOI] [PubMed] [Google Scholar]
- 47.Nicoll JA, Burnett C, Love S, et al. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann Neurol 1997;41(6):716–21. doi: 10.1002/ana.410410607 [published Online First: 1997/06/01] [DOI] [PubMed] [Google Scholar]