

Abstract
Herein, we introduce the wavelength‐orthogonal crosslinking of hydrogel networks using two red‐shifted chromophores, i.e. acrylpyerene (AP, λ activation=410–490 nm) and styrylpyrido[2,3‐b]pyrazine (SPP, λ activation=400–550 nm), able to undergo [2+2] photocycloaddition in the visible‐light regime. The photoreactivity of the SPP moiety is pH‐dependent, whereby an acidic environment inhibits the cycloaddition. By employing a spiropyran‐based photoacid generator with suitable absorption wavelength, we are able to restrict the activation wavelength of the SPP moiety to the green light region (λ activation=520–550 nm), enabling wavelength‐orthogonal activation of the AP group. Our wavelength‐orthogonal photochemical system was successfully applied in the design of hydrogels whose stiffness can be tuned independently by either green or blue light.
Keywords: Hydrogels, Photoacid Generator, Photocycloaddition, Visible Light, Wavelength-Orthogonal
It's So Clearly Visible: Hydrogels can be formed by exclusively visible‐light irradiation with two colours of light, with each colour allowing mechanical stiffening of the gel independent of the order in which each colour is applied. Driven by mild [2+2] cycloadditions with halochromic properties, the photoreactivity of the reactions in water can be controlled by a photoacid generator.
The development of photoresponsive hydrogels has gained significant momentum over the past decade due to their dynamic viscoelastic properties and aqueous composition that highly resemble soft tissues in living systems. [1] Applying light‐mediated chemistries to modulate hydrogel networks allows for fine‐tuning of their chemical and physical properties both in time and space. Such light‐sensitive materials are programmable and adaptable, enabling highly defined control over signal presentation, mechanics and therapeutics’ release. [1a] Indeed, photoresponsive hydrogels have proven to be ideal candidates for applications in advanced cell culture,[ 1c , 2 ] controlled drug delivery, [3] and soft actuators. [4]
Significant efforts have been dedicated to the construction and investigation of photoresponsive hydrogels in various fields of biomaterials science. In particular, photodegradable hydrogels based on o‐nitrobenzyl photochemistry have been used for the differentiation of linear‐specified cells [5] and the fabrication of synthetic microvasculature. [6] Photo‐softening of hydrogel networks has been employed to deliver stem cells [7] as well as to control cell morphology, [8] while photo‐stiffening of hydrogels has enabled tracking cellular responses to mechanobiological signals.[ 7b , 9 ] Recent developments in photochemistry have led to chromophores with red‐shifted photoreactivity, enabling modulation of the materials by long wavelength (low energy) visible light. For example, photocycloadditions of red‐shifted triazole anthracene [10] and styrylpyrene [11] have been used in hydrogelation by visible light (400–500 nm) for 3D encapsulation of stem cells and conjugation of bioactive molecules. Visible light absorbing, photo‐cleavable moieties such as coumarin and perylene have been incorporated into hydrogel networks for photodegradation of the materials by blue (470 nm) or green light (530 nm). [12] Furthermore, ruthenium complexes have been used as crosslinkers in hydrogelation to afford photodegradation of the materials by long wavelength (500–600 nm) visible light. [13] More recently, we employed halochromic chromophores such as styrylquinoxaline, which can undergo photocycloaddition at ≤510 nm, to introduce additional triggers, such as pH, for modulating the properties of hydrogels. [14]
Presently, most light‐sensitive hydrogels respond only to a specific wavelength regime. Advanced hydrogels that can respond to several wavelengths will enable selective control of multiple functionalities, or orthogonal tuning of two or more properties in the same system.[ 1a , 1d ] The dynamic adaptation of material stiffness in a well‐defined controlled manner by discrete wavelengths of visible light offers a powerful handle for studying the dynamic cell‐materials interactions in biology research. [1b] Our team has exploited wavelength‐selective photochemistries in engineering hydrogels networks, and fabricating photoresponsive materials, whose stiffness can be modulated by different colors of light, independently of each other. [15] The utility of such hydrogel platforms has been demonstrated in cell culture studies, where the cell behavior was influenced by irradiation with specific wavelengths. Specifically, the λ‐selective stiffening of substrate induced cell detachment, [15a] while softening enhanced cell elongation and spreading, was explored. [15b] Nevertheless, the therein demonstrated λ‐selectivity can only proceed in one direction, from longer wavelength (lower energy) to shorter wavelength (higher energy).
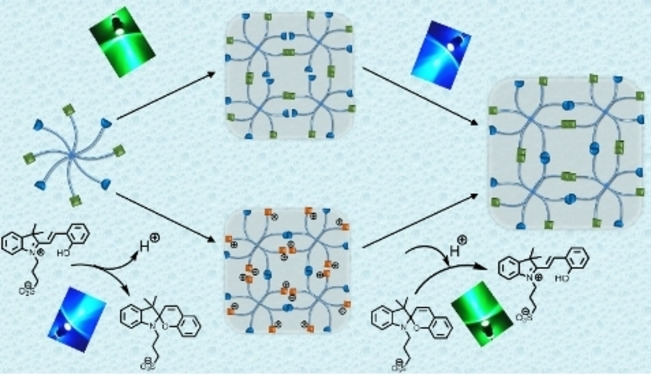
Herein, we pioneer the λ‐orthogonal crosslinking of polymers via [2+2] photocycloadditions of two red‐shifted chromophores, namely acrylpyerene (AP, λ activation=410–490 nm) and styrylpyrido[2,3‐b]pyrazine (SPP, λ activation=400–550 nm). While the photochemistry of the AP moiety has been established by our team, the photocycloaddition and halochromic properties of the SPP moiety are investigated herein for the first time. The photoreactivity of the SPP moiety in the blue light region can be inhibited by mixing the polymer with a spiropyran‐based photoacid generator (PAG) (λ activation=400–510 nm, Figure 1A). The photoreactive AP group is unaffected by the PAG, thus in the presence of the PAG the photocycloaddition of either AP or SPP can be activated by two discreet wavelengths, 445 nm (blue) or 525 nm (green) respectively, independently of each other. The chemical design was translated into a hydrogel network, affording a hydrogel platform of which stiffness can be orthogonally influenced by irradiation with blue or green light (Figure 1B).
Figure 1.

Restricting photoreactivity of halochromic styrylpyridopyrazine (SPP) using a photoacid generator and application in hydrogel design. A) MeO‐PEG‐SPP can undergo photocycloaddition by visible light at 400–550 nm and pH≥7. In the presence of a PAG (λ activation=400–510 nm) that lowers the pH (≤6), the photoreactivity of the SPP is inhibited; however, green light at 520–550 nm can still activate the photocycloaddition. Thus, the PAG effectively restricts the photoreactivity of MeO‐PEG‐SPP in the 520–550 nm window. B) The SPP group is attached to an 8‐arm PEG together with a non‐halochromic acrylamidylpyrene (AP), which can undergo photocycloaddition by blue light (400–470 nm), and the photoreactivity of the AP is not affected when mixed with the PAG. In the presence of the PAG, the 8‐arm PEG‐AP‐SPP can crosslink by either blue or green light via photocycloaddition of the AP or SPP group, respectively.
We previously introduced the styrylquinoxaline moiety, which can undergo [2+2] photocycloadditions induced by green light at λ≤520 nm. Inspired by this advance, we sought to critically red‐shift the photoreactivity of the styryl moiety by replacing the quinoxaline with a pyrido[2,3‐b]pyrazine. Styrylpyrido[2,3‐b]pyrazine (SPP) was readily prepared from pyridine‐2,3‐diamine starting material following a three‐step synthesis procedure with an overall yield of 23 % (refer to the Supporting Information). The chromophore was designed to feature a carboxylic acid handle for attaching to a molecule of interest, enabling its conjugation to the polymers, e.g. for use in polymer coupling and crosslinking. Subsequently, we prepared a poly(ethylene glycol) with SPP end‐group (MeO‐PEG‐SPP, Figure 1A), via N‐hydroxysuccinimide (NHS) ester and amine coupling (refer to the Supporting Information section 3, Scheme S1), and investigated its photoreactivity in water. The formation of the dimerized (MeO‐PEG‐SPP)2 was monitored by size exclusion chromatography (SEC). The SEC trace of the solution post‐irradiation indicates the formation of a polymer with double molecular weight compared to the molecular weight of the starting material (Figure 2A). The presence of the (MeO‐PEG‐SPP)2 dimer is further confirmed by high resolution mass spectrometry hyphenated to SEC (Figure 2B, S15, and S16), in which the mass spectrum obtained at 16.4 min shows multiple patterns of the PEG repeating units with a difference of m/z=44.0264/7, corresponding to the molecular weight of one PEG repeating unit. The obtained m/z values agree well with the simulated isotopic patterns of the photocycloaddition (MeO‐PEG‐SPP)2 product.
Figure 2.

Characterization of the SPP cycloaddition product. A) SEC data (calibrated against PMMA) of MeO‐PEG‐SPP and (MeO‐PEG‐SPP)2 after laser irradiation at 480 nm (6.32±0.18×1019 photons which result in a pulse energy of 848 μJ) in water (10 mg mL−1). B) SEC‐ESI‐MS analysis of the dimerization of MeO‐PEG‐SPP after irradiation at 480 nm. Zoom into the MS spectra of experimental and simulated isotopic patterns, indicating their excellent agreement. Detailed SEC‐ESI‐MS analysis is appended in the Supporting Information, Figure S16, S17. C) UV/Vis spectrum of MeO‐PEG‐SPP (solid black line) and the corresponding action plot (red circles) for the cycloaddition in water (10 mg mL−1). Experimental details are provided in Table S1
The wavelength‐dependent reactivity was thoroughly examined using a tunable laser as monochromatic light source, providing the same number of photons (6.32×1019) at each discreet wavelengths in the range between 400 and 600 nm. The data were subsequently presented in the form of an action plot, displaying the conversion as a function of wavelength (Figure 2C). Similar to previous reports on action plots of photochemical reactions in solution, [16] we observed a red‐shift (ca. 100 nm) in the photoreactivity of the SPP chromophore compared to its absorption spectrum, indicating a maximum absorption at 402 nm. The photoreactivity of the SPP moiety extends well into the green light region (up to 540 nm), which—to the best of our knowledge—is the longest wavelength employed in [2+2] photocycloaddition in water. Further, the absorptivity of water in this wavelength region is negligible, dismissing any heating effects of the solvent and attributing the higher reactivity predominantly to the SPP moiety.
The presence of the pyrido[2,3‐b]pyrazine induces a significant halochromic response on the SPP moiety, as seen by the bathochromic shift in the UV/Vis absorbance of the compound even in slightly acidic solvent such as chloroform (Figure S13). The pK a of the pyrazinium moiety was measured (by titration) as pK a=1.15, and we suggest that the protonation must occur on the pyridinium nitrogen of the pyrido subunit. [17] In comparison, the pK a of quinoxalinium in water is 0.6. [18] In aqueous environments, mild acidic condition (pH 3–5) induce a slight bathochromic shift in the absorbance of the MeO‐PEG‐SPP compared to the absorbance at neutral pH (Figure 3A). The change in UV/Vis absorbance is also associated with a significant reduction in the photoreactivity of the SPP moiety, as observed from the action‐plots of MeO‐PEG‐SPP at different pH values. The conversion to dimer product decreases by more than half, from ca. 50 % to ca. 20 %, when the pH of the solution is changed to acidic condition (pH 3–5). A further decrease in pH (≤1) completely inhibits the photoreactivity of the SPP moiety, and no cycloaddition product is observed. The inhibition of the photoreactivity correlates with a change in the UV/Vis absorbance of the solution, with a red‐shift in the maximum absorbance from 405 nm at pH 7 to 450 nm at pH 1.
Figure 3.
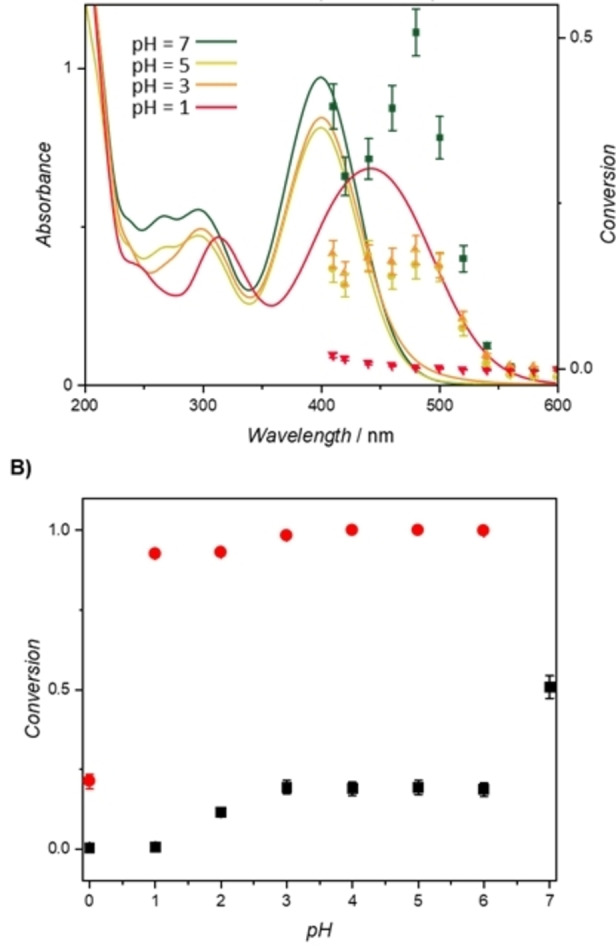
pH‐Dependent reactivity. A) UV/Vis spectra of MeO‐PEG‐SPP and corresponding action plots under various pH conditions (green squares: aqueous, yellow circles: pH 5, orange triangles: pH 3, red diamonds: pH 1). In all experiments, approximately 105 μmol of photons were deposited (Table S2–S4). B) Conversion to cycloaddition products as a function of pH for MeO‐PEG‐SPP (black squares) and MeO‐PEG‐AP (red circles); irradiation conditions: λ=480 nm (6.32±0.18×1019 photons) for MeO‐PEG‐SPP, and λ=420 nm (6.38±0.36×1019 photons) for MeO‐PEG‐AP.
As a reference, we examined the effect of pH on the photoreactivity of a non‐halochromic polymer, PEG with acrydylpyrene endgroup (MeO‐PEG‐AP, refer to Figure 1B for the chemical structure of the AP moiety), which can undergo efficient cycloaddition at λ≤470 nm (Figure 3B). [15a] As expected, the photocycloaddition of the AP moiety is unaffected by the pH of the solution when subjected to irradiation conditions identical to those of employed for MeO‐PEG‐SPP. Notably, the absence of the photocycloaddition adduct detected by SEC at pH 0 under blue light irradiation is likely due to the acid‐catalyzed hydrolysis of the styryl group, following a reverse Claisen‐Schmidt condensation (Scheme S2).
Having established the pH‐dependent photoreactivity of the SPP moiety, we implement an internal pH regulator to restrict the photoactivation of the SPP group to a specific light regime. Thus, we prepared a water soluble spiropyran‐based photoacid generator (PAG), which can reversibly reduce the pH of the solution under blue light (400–500 nm) irradiation (Figure 1A and Figure S18A). [19] A solution of PAG (c=1 mM) in deionized water has a pH of 5.5. Thus, we adjusted the pH of the PAG solution to ca 6.8, using saturated NaHCO3 solution, to apply the setting in the photocycloaddition of MeO‐PEG‐SPP. Inspection of the photoswitching property of the PAG by UV/Vis absorption spectroscopy reveals that the molecule rapidly isomerizes from merocyanine form (MEH) to spiropyran form (SP) under blue light irradiation (λ=445 nm, I=3 W cm−2, t=3 min). Here, the absorbance band of the MEH rapidly decreases, and a new absorbance band characteristic of the SP is observed (Figure S17B). [19] The change in UV/Vis absorbance was accompanied by a decrease in pH from 6.8 to 5.1. When placing the solution in the dark or irradiation with green light (525 nm), the absorption intensity of the PAG solution rapidly recovers (ca. 5 min) to the same absorbance band before blue light irradiation, and the pH value returns to 6.8. The photo‐induced switching is reversible and we observed up to six cycles of switching without significant loss of the photo‐isomerization ability of the PAG (Figure S18C).
Investigation of the photocycloaddition of MeO‐PEG‐SPP in the presence of the PAG suggests that the photoreactivity of the SPP group is inhibited in the blue light region. Specifically, irradiation (λ=445 nm, I=3 W cm−2, t=1 h) of the MeO‐PEG‐SPP solution resulted in the disappearance of the absorbance characteristic of the SPP group (Figure 4A); however, this absorbance band remains when irradiated with the addition of the PAG (Figure 4B). The inhibition of the [2+2] photocycloaddition of the SPP moiety by the PAG was further confirmed by NMR study, indicating very low formation of the dimer (ca. 5 %) when a solution of MeO‐PEG‐SPP and PAG was irradiated with blue light (Figure 4C). In contrast, green light (λ=425 nm, I=3 W cm−2, t=1.5 h) irradiation triggers the photocycloaddition, as seen by the disappearance of the resonances corresponding to the styryl protons at 7 ppm and 7.5 ppm, even when PAG was present in the reaction. We also undertook an action plot analysis for the mixture of MeO‐PEG‐SPP and PAG and observed an inhibition effect; however, because the number of photons supplied to the solution in action plot study is defined, the low conversion could be the result of competing absorption of the PAG compound. Indeed, both compounds have very similar absorbance bands in water, and PAG has a higher molar absorptivity (39 000 L mol−1 cm−1) compared to MeO‐PEG‐SPP (15 900 L mol−1 cm−1) at 460 nm.
Figure 4.

Ceasing SPP photoreactivity using a PAG. A) UV/Vis absorbance of the MeO‐PEG‐SPP in water before and after irradiation with blue light at λ=445 nm. B) UV/Vis absorbance of the MeO‐PEG‐SPP and spiropyran‐based PAG in water before and after irradiation with blue light at λ=445 nm. C) 1H NMR spectra (600 MHz, CDCl3) of MeO‐PEG‐SPP before (top spectrum) and after irradiation by green light at 525 nm with PAG, blue light at λ=445 nm, and blue light at λ=445 nm with PAG (bottom spectrum, irradiation conditions: I=3 W cm−2, t=1 h); the highlighted area indicates the resonances from styryl CH=CH protons.
We subsequently translated the findings from the halochromic photochemical ligations and PAG inhibition into materials engineering, specifically the modulation of hydrogels stiffness by light‐mediated polymer crosslinking. We first prepared an 8‐arm PEG containing SPP endgroup (Figure 5A), PEG‐(SPP)8 and examined the crosslinking of polymer solution (c=5 mM) under light irradiation (445 nm, I=20 mW cm−2) at pH 6.8, 5 and 3 by in situ rheological measurements. In dynamic rheology, the kinetics of the crosslinking process can be monitored by detection of the elastic moduli (G′) and viscous (G′′) moduli, with minimal disruption to the chemical reaction. [21] In our setup, the photopolymerizable material was characterized in a rheometer‐LED light source set up where the sample was irradiated directly while being measured, which captures the transition from sol to gel. Under neutral pH, the rheological data show an increase in the G′ values upon light irradiation, indicating the formation of a hydrogel network, and complete gelation occurred after ca. 25 min of irradiation (Figure 5B) when no further increase in the G′ value was observed. [22] Acidic conditions (pH≤5) fully suppress the photocrosslinking and no hydrogel was formed. Here, the halochromic effect of the SPP group is more profound compared to the styrylquinoxaline (SQ) group whereby the photocrosslinking of PEG‐(SQ)8 can effectively occur at pH 4–5. [14] In comparison, 8‐arm PEG with non‐halochromic AP endgroup, PEG‐(AP)8, displays efficient photocrosslinking under both neutral and acidic (pH 3–5) conditions (Figure 5B). Notably, the photocycloaddition of the AP group is more efficient than the SPP entity, as seen by the rapid photocrosslinking and complete gelation within 15 min of irradiation. This result is in agreement with the action plot study in which complete conversion of the MeO‐PEG‐AP was achieved, whilst only ca. 52 % conversion of the MeO‐PEG‐SPP was recorded under similar irradiation conditions, indicating a slower reaction kinetics for the dimerization of the SPP group, and thus a slower gelation kinetics. In both systems, the G′ values at complete gelation were similar and in the range of 5–7 kPa.
Figure 5.

λ‐Orthogonal crosslinking for hydrogelation. A) Schematic presentation of PEG‐(SPP)8 crosslinking under light irradiation, and the photocrosslinking is suppressed in acidic environments. B) Rheology data of PEG‐(SPP)8 solution (c=5 mM) show the evolution of storage modulus under blue light (445 nm, I=20 mW cm−2), which indicates the formation of hydrogel network, no gelation was seen when pH values of the solution was adjusted to 3 and 5. C) Rheology data of PEG‐(SPP)8 solution (c=5 mM, 10 wt %) showing the fast hydrogelation process, which is not affected by acidic (pH 3 and 5) conditions. D) and E) Rheology data of PEG‐(SPP)3(AP)4 solution (c=5 mM, 10 wt %) displaying λ‐orthogonal crosslinking of the polymer by either blue (445 nm) or green (525 nm) light (I=20 mW cm−2). F) Schematic presentation of experiments demonstrating the spatial control over the stiffening process by changing the light colour and a photomask. G) Strain sweep rheological data of two different parts of the hydrogel, initially formed by green light and selectively stiffened by blue light. H) Strain sweep rheological data of two different parts of the hydrogel, first formed by blue light and selectively stiffened by green light.
To develop a precursor platform for the λ‐orthogonal crosslinking of hydrogel networks, we prepared an eight‐arm PEG containing both the SPP and AP endgroups. Taking advantage of the efficient nature of the NHS ester and amine coupling, we reacted PEG‐(NH2)8 with SPP‐NHS and AP‐NHS in a one‐pot reaction with a stoichiometric molar ratio of the NH2 to the NHS group (Scheme S1). Analysis of the NMR spectrum of the resultant polymer indicates that a SPP/AP ratio of 3/4 was achieved, henceforth the polymer is referred to as PEG‐(SPP)3(AP)4. It is noted that repeating the synthesis of the PEG‐(SPP)3(AP)4 produced a polymer with the endgroups SPP/AP ratio of 3.3/4. We expect that any variation around the equivalent ratio is potentially due to the error in the preparation step, particularly the weighing of the compounds. The photocrosslinking of PEG‐(SPP)3(AP)4 solution (c=5 mM, pH 6.8) in the presence of the PAG (c=1 mM) was subsequently investigated by rheology. Green light (525 nm) irradiation caused an initial rise in the G′ value, reaching a plateau at G′=600 Pa after ca. 25 min and no further change in the G′ value was observed with prolonged irradiation (Figure 5D). The crosslinking only resumed when the wavelength was switched to blue light (445 nm), reaching a plateau value of G′=5.1 kPa. The λ‐orthogonality is demonstrated in another experiment, where the solution of PEG‐(SPP)3(AP)4 and PAG was first irradiated with blue light to achieve the first crosslinking of the AP endgroups (Figure 5E), reaching a plateau G′ value of 1.1 kPa. A second photo crosslinking step was induced when switching the light color to green, producing a network with G′=6.8 kPa. In both experiments, the recorded G′ values for the initial crosslinking are 8–10 times lower than the G′ values observed during the second crosslinking, despite a near equivalent amount of the photoreactive chain termini being consumed in each photocrosslinking step. The much stiffer gel formed after the second network formation may be due to enhancement effects associated with the physical interactions within the hydrophobic crosslinked structure. The increase of physical interactions within the network structure upon an increase in the number of crosslinks has been reported before for eight‐arm PEG‐based hydrogels crosslinked by Michael addition reactions. [20] In the absence of the PAG, the wavelength‐control photocrosslinking only proceeds in one direction, and complete gelation was achieved when the solution was first irradiated with blue light (Figure S18).
To further highlight the temporal control introduced by the λ‐orthogonality, we performed experiments where gels were initially formed by one colour of light, either blue (λ=445 nm) or green (λ=525 nm), followed by irradiation on selected areas by a different wavelength (Figure 5F and S19). As expected, each area on the hydrogels displayed well‐defined mechanical properties, with the regions irradiated by two wavelengths featuring much higher storage moduli compared to single wavelength irradiation. In particular, initial irradiation with green or blue enabled the formation of hydrogels with moduli of 980 Pa and 1020 Pa, respectively. When certain areas are exposed to the other wavelength, the moduli of those areas increased to 5.2 kPa (green then blue) and 4.8 kPa (blue then green). The slight difference in the moduli compared to the values from time sweep measurements is due to the difference in the irradiation periods.
In conclusion, we introduce a chemical design for the full λ‐orthogonal activation of two highly red‐shifted [2+2] photocycloaddition reactions, based on a halochromic styrylpyrido[2,3‐b]pyrazine moiety and a non‐halochromic acrylamidylpyrene function. By using a photoacid generator with suitable activation wavelength, we restrict the photoreactivity of the halochromic SPP to green light only, enabling its photocycloaddition to proceed independently of the photoactivation of the AP. Subsequently, we demonstrate the bidirectional photo‐stiffening of a hydrogel network, using either blue or green light. While the PAG was used as an additive, we envisage that all chromophores can be installed into a single chemical entity for the wavelength‐selective dynamic alteration of its materials properties. Critically, the exclusive use of visible light herein shows promise for applications in biomaterials engineering, particularly mechanotransduction studies of cells that are sensitive to UV light. [23]
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
C. B.‐K. acknowledges funding from the Australian Research Council (ARC) in the form of a Laureate Fellowship (FL170100014) enabling his photochemical research program as well as continued key support from the Queensland University of Technology (QUT). C. B.‐K. is grateful to the Volkswagen Foundation for support in the context of the project ‘Molecularly Engineered Light‐Adaptive Bioinspired Nanocomposites’. C.B.‐K. and J.B. acknowledge additional support via an ARC Discovery grant targeted at red‐shifting photoligation chemistry. Additional support by the Karlsruhe Institute of Technology (KIT) is gratefully acknowledged. Open access publishing facilitated by Queensland University of Technology, as part of the Wiley ‐ Queensland University of Technology agreement via the Council of Australian University Librarians.
V. X. Truong, J. Bachmann, A.-N. Unterreiner, J. P. Blinco, C. Barner-Kowollik, Angew. Chem. Int. Ed. 2022, 61, e202113076; Angew. Chem. 2022, 134, e202113076.
Contributor Information
Dr. Vinh X. Truong, Email: truongvx@qut.edu.au.
Prof. Christopher Barner‐Kowollik, Email: christopher.barnerkowollik@qut.edu.au, Email: christopher.barner-kowollik@kit.edu.
References
- 1.
- 1a. Li L., Scheiger J. M., Levkin P. A., Adv. Mater. 2019, 31, 1807333; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Rapp T. L., DeForest C. A., Adv. Healthcare Mater. 2020, 9, 1901553; [DOI] [PubMed] [Google Scholar]
- 1c. Dong Y., Jin G., Hong Y., Zhu H., Lu T. J., Xu F., Bai D., Lin M., ACS Appl. Mater. Interfaces 2018, 10, 12374–12389; [DOI] [PubMed] [Google Scholar]
- 1d. Lu P., Ahn D., Yunis R., Delafresnaye L., Corrigan N., Boyer C., Barner-Kowollik C., Page Z. A., Matter 2021, 4, 2172–2229. [Google Scholar]
- 2. Kloxin A. M., Tibbitt M. W., Anseth K. S., Nat. Protoc. 2010, 5, 1867–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Wang R., Yang Z., Luo J., Hsing I. M., Sun F., Proc. Natl. Acad. Sci. USA 2017, 114, 5912; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Nehls E. M., Rosales A. M., Anseth K. S., J. Mater. Chem. B 2016, 4, 1035–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ionov L., Mater. Today 2014, 17, 494–503. [Google Scholar]
- 5. Chen A. Y., Deng Z., Billings A. N., Seker U. O. S., Lu Michelle Y., Citorik R. J., Zakeri B., Lu T. K., Nat. Mater. 2014, 13, 515–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Brandenberg N., Lutolf M. P., Adv. Mater. 2016, 28, 7450–7456; [DOI] [PubMed] [Google Scholar]
- 6b. Arakawa C. K., Badeau B. A., Zheng Y., DeForest C. A., Adv. Mater. 2017, 29, 1703156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Brown T. E., Marozas I. A., Anseth K. S., Adv. Mater. 2017, 29, 1605001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Griffin D. R., Kasko A. M., J. Am. Chem. Soc. 2012, 134, 13103–13107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ji H., Xi K., Zhang Q., Jia X., RSC Adv. 2017, 7, 24331–24337. [Google Scholar]
- 9. Günay K. A., Ceccato T. L., Silver J. S., Bannister K. L., Bednarski O. J., Leinwand L. A., Anseth K. S., Angew. Chem. Int. Ed. 2019, 58, 9912–9916; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 10017–10021. [Google Scholar]
- 10. Truong V. X., Li F., Forsythe J. S., ACS Macro Lett. 2017, 6, 657–662. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Truong V. X., Li F., Ercole F., Forsythe J. S., ACS Macro Lett. 2018, 7, 464–469; [DOI] [PubMed] [Google Scholar]
- 11b. Doi T., Kawai H., Murayama K., Kashida H., Asanuma H., Chem. Eur. J. 2016, 22, 10533–10538; [DOI] [PubMed] [Google Scholar]
- 11c. Frisch H., Menzel J. P., Bloesser F. R., Marschner D. E., Mundsinger K., Barner-Kowollik C., J. Am. Chem. Soc. 2018, 140, 9551–9557. [DOI] [PubMed] [Google Scholar]
- 12. Truong V. X., Li F., Forsythe J. S., ACS Appl. Mater. Interfaces 2017, 9, 32441–32445. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Rapp T. L., Wang Y., Delessio M. A., Gau M. R., Dmochowski I. J., RSC Adv. 2019, 9, 4942–4947; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13b. Rapp T. L., Highley C. B., Manor B. C., Burdick J. A., Dmochowski I. J., Chem. Eur. J. 2018, 24, 2328–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kalayci K., Frisch H., Truong V. X., Barner-Kowollik C., Nat. Commun. 2020, 11, 4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Kalayci K., Frisch H., Barner-Kowollik C., Truong V. X., Adv. Funct. Mater. 2020, 30, 1908171; [Google Scholar]
- 15b. Pelloth J. L., Tran P. A., Walther A., Goldmann A. S., Frisch H., Truong V. X., Barner-Kowollik C., Adv. Mater. 2021, 33, 2102184; [DOI] [PubMed] [Google Scholar]
- 15c. Irshadeen I. M., Walden S. L., Wegener M., Truong V. X., Frisch H., Blinco J. P., Barner-Kowollik C., J. Am. Chem. Soc. 2021, 143, 21113–211126. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Kamm P. W., Blinco J. P., Unterreiner A.-N., Barner-Kowollik C., Chem. Commun. 2021, 57, 3991–3994; [DOI] [PubMed] [Google Scholar]
- 16b. Irshadeen I. M., De Bruycker K., Micallef A. S., Walden S. L., Frisch H., Barner-Kowollik C., Polym. Chem. 2021, 12, 4903–4909. [Google Scholar]
- 17. Albert A., Barlin G. B., J. Chem. Soc. 1963, 5737–5741. [Google Scholar]
- 18. Pereira J. A., Pessoa A. M., Cordeiro M. N. D. S., Fernandes R., Prudêncio C., Noronha J. P., Vieira M., Eur. J. Med. Chem. 2015, 97, 664–672. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Shi Z., Peng P., Strohecker D., Liao Y., J. Am. Chem. Soc. 2011, 133, 14699–14703; [DOI] [PubMed] [Google Scholar]
- 19b. Ou R., Zhang H., Truong V. X., Zhang L., Hegab H. M., Han L., Hou J., Zhang X., Deletic A., Jiang L., Simon G. P., Wang H., Nat. Sustainability 2020, 3, 1052–1058. [Google Scholar]
- 20. Jeong K. J., Panitch A., Biomacromolecules 2009, 10, 1090–1099. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Bonino C. A., Samorezov J. E., Jeon O., Alsberg E., Khan S. A., Soft Matter 2011, 7, 11510–11517; [Google Scholar]
- 21b. O'Connell C. D., Zhang B., Onofrillo C., Duchi S., Blanchard R., Quigley A., Bourke J., Gambhir S., Kapsa R., Di Bella C., Choong P., Wallace G. G., Soft Matter 2018, 14, 2142–2151. [DOI] [PubMed] [Google Scholar]
- 22. Li M., Dove A. P., Truong V. X., Angew. Chem. Int. Ed. 2020, 59, 2284–2288; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 2304–2308. [Google Scholar]
- 23. Richards D., Swift J., Wong L. S., Richardson S. M. in Cell Biology and Translational Medicine, Vol. 5: Stem Cells: Translational Science to Therapy (Ed.: Turksen K.), Springer International Publishing, Cham, 2019, pp. 53–69. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information