

Abstract
Aims
Filgotinib is a potent, oral, JAK1‐preferential inhibitor for the treatment of rheumatoid arthritis (RA). This report describes exposure‐response (ER) analyses of filgotinib for dose confirmation based on three phase 3 and two phase 2 studies in moderate to severe RA patients.
Methods
The pharmacokinetic exposures used in ER analyses were derived from population pharmacokinetic analysis. The exposure‐efficacy relationships were assessed for efficacy endpoints (ACR20/50/70 and DAS28) over effective area under curve (AUCeff), the combined exposures of filgotinib and GS‐829845 (major, active metabolite), with nonlinear logistic regression models developed. Also, a t‐test was performed to compare the exposure between subjects who achieved response and those who did not. For the ER analyses of safety, exposures were examined between subjects who experienced and who did not experience the evaluated safety events, which was conducted separately for filgotinib and GS‐829845.
Results
The nonlinear logistic regression showed increasing response with increasing exposure, with exposures at 200 mg dose primarily residing on the curve plateau. Also, AUCeff was significantly higher in the subjects who achieved responses compared to those who did not (10 900 vs 9900 h*ng/mL for ACR20, P value < .0001). For exposure‐safety analyses, filgotinib and GS‐829845 exposures were similar irrespective of the presence/absence of the evaluated safety endpoints, indicating no exposure‐safety relationship for common treatment‐emergent adverse events (TEAEs)/laboratory abnormalities and serious TEAEs/infections.
Conclusions
ER analyses confirmed that filgotinib produced more robust therapeutic effects across the exposure range observed at 200 mg once daily compared to lower doses, and collectively with the lack of exposure‐safety relationship, the 200 mg once daily dose was supported for commercialization.
Keywords: drug safety; PK‐PD; randomized controlled trial; rheumatoid arthritis; therapeutics; Specialties: filgotinib, JAK inhibitors, exposure‐response
What is already known about this subject
Filgotinib is an oral, potent JAK1 preferential inhibitor, which has demonstrated clinical improvement in moderate to severe rheumatoid arthritis and has been granted marketing approval in the European Union and Japan.
Previous exposure‐response analyses based on two phase 2 studies supported selection of 200 and 100 mg once‐daily doses in the phase 3 studies.
What this study adds
Exposure‐response analyses confirm that filgotinib produced more robust therapeutic effects across the exposure range observed at 200 mg once daily compared to lower doses.
The positive exposure‐efficacy relationship and a lack of exposure‐safety relationship on the evaluated safety endpoints supported the 200 mg once‐daily dose for commercialization.
1. INTRODUCTION
Rheumatoid arthritis (RA) is a chronic, inflammatory joint disease that primarily involves the lining of synovial joints and can cause progressive disability. 1 Research efforts are now targeting Janus kinases (JAK)‐ signal transducer and activator of transcription proteins (STAT) signalling cascade as a therapeutic strategy. 2 The European Medicines Agency and the Pharmaceuticals and Medical Devices Agency have approved four oral, small‐molecule JAK inhibitors, tofacitinib, baricitinib, upadacitinib and filgotinib for the treatment of RA. 3 , 4 , 5 These agents differ in their in vitro selectivity profile for JAK subtypes. Newer generations of JAK inhibitors are more selective, specifically targeting inhibition of JAK1 while avoiding potential undesirable effects of inhibiting downstream signalling from JAK2 and JAK3 . 3 , 6 , 7
Filgotinib is an oral, once‐daily, potent JAK1 preferential inhibitor 8 that has demonstrated clinical improvement in RA and was granted marketing approval in the European Union and Japan for treatment of moderate to severe RA in adults. Filgotinib has met all the primary endpoints, ACR20 at week 12 or week 24, in its phase 3 clinical trials, FINCH 1, FINCH 2 and FINCH 3. 9 , 10 , 11 These studies showed that filgotinib was well tolerated and highly efficacious in patients with moderately to severely active RA by reducing the signs and symptoms of disease, improving function and slowing the progression of joint destruction. Filgotinib also showed clinical benefit in ulcerative colitis 12 , 13 and is currently being investigated for treatment of other chronic inflammatory diseases, such as Crohn's disease. 14
The pharmacokinetics of filgotinib have been well characterized through noncompartmental analyses across phase 1 studies. Filgotinib is primarily metabolized via carboxylesterase‐2 (CES2) to form GS‐829845, the major circulating metabolite of filgotinib. GS‐829845 is also a JAK1 preferential inhibitor but is approximately 10‐fold less potent than the parent and with a longer half‐life (approximately 19 hours for GS‐829845 vs 7 hours for filgotinib). 8 , 15 , 16 GS‐829845 is primarily cleared in renal elimination.
This report describes exposure‐efficacy and exposure‐safety analyses for filgotinib and its major active metabolite, GS‐829845, based on population pharmacokinetic (PopPK) model‐derived PK exposures, and efficacy and safety data from three phase 3 studies (FINCH 1, FINCH 2 and FINCH 3) and two phase 2 studies (DARWIN 1 and DARWIN 2) in patients with moderate to severe RA to support dose recommendation. 9 , 10 , 11 , 17 , 18 , 19
2. METHODS
2.1. Study design and population
The protocol and informed consent for each of the studies were approved by the local institutional review boards. All subjects provided written informed consent before study participation. All studies were conducted according to the principles of the Declaration of Helsinki and the Good Clinical Practice Guidelines of the International Conference of Harmonisation. The study design, patient eligibility, dose administration, statistical analyses and study outcome details for the studies included in this report have been previously published (FINCH 1: NCT02889796, FINCH 2: NCT02873936, FINCH 3: NCT02886728, DARWIN 1: NCT01888874, DARWIN 2: NCT01894516). 10 , 11 , 17 , 18 , 19
Brief summaries of the study design for each study are as follows. In the analyses, FINCH 1, FINCH 2 and FINCH 3 were three phase 3, randomized, double‐blind, placebo‐ and active‐controlled studies in adults with moderately to severely active RA with filgotinib 200 or 100 mg once daily in the treatment group, where subjects (N = 1759) had an inadequate response (IR) to methotrexate (MTX; MTX‐IR) in FINCH 1 up to 52 weeks, subjects (N = 449) had an inadequate response or were intolerant to at least one biologic disease‐modifying antirheumatic drug in FINCH 2 up to 24 weeks and subjects (N = 1252) were naïve to MTX therapy in FINCH 3 up to 52 weeks. 9 , 10 , 11 , 17 DARWIN 1 and DARWIN 2 were two phase 2, multicentre, 24‐week, double‐blind, placebo‐controlled studies in subjects with moderately to severely active RA who had an inadequate response to MTX alone, where filgotinib was add‐on in DARWIN 1 with once‐daily doses of 50, 100 and 200 mg and twice‐daily doses of 25, 50 and 100 mg (N = 594) and DARWIN 2 had filgotinib monotherapy with once daily doses of 50, 100 and 200 mg (N = 283). 18 , 19
2.2. Population PK analysis
PopPK was performed to provide model predicted individual PK parameter estimates for exposure‐response (ER) analyses. Data from seven phase 1 studies, four phase 2 studies and three phase 3 studies were analysed using nonlinear mixed‐effects modelling (NONMEM, version 7.3.0 or later; ICON Development Solutions, Ellicott City, MD) and Perl‐Speaks‐NONMEM (PsN; Uppsala University, Sweden). A summary of the data, including population, posology, sampling time, number of subjects and number of data points in the PopPK modelling, is shown in Supporting Information Tables S1 and S2. The bioanalytical methods in the included studies were previously described. 20 , 21
Filgotinib and GS‐829845 models were developed separately. Various structural models and random effect models were evaluated to reach the base models. Stepwise forward addition and backward elimination were implemented in the covariate model building process, with evaluated covariates including demographics (age, sex, body weight, race), pathophysiological factors (baseline estimated creatinine clearance [CLcr], baseline bilirubin, baseline alanine aminotransferase, baseline aspartate aminotransferase, RA disease status [subjects with RA vs healthy subjects], baseline C‐reactive protein [CRP] and RA duration) and fed status with rationale described in the Supporting Information (Rationale for Covariate Selection). Model selection was done based on a log‐likelihood ratio test at an acceptance P value of .01 (forward addition) or .001 (backward elimination). The difference in −2 times the log of the likelihood (−2LL) between a full and reduced model was assumed to have a χ2 asymptotic distribution with degrees of freedom equal to the difference in number of parameters between the two models. Model performance evaluation was based on goodness‐of‐fit evaluation, prediction corrected visual predictive check (pcVPC) and bootstrap resampling techniques. Individual PK parameter estimates were predicted from the final models for ER analyses.
2.3. Exposure‐efficacy analysis
The analysis dataset for filgotinib and its active metabolite GS‐829845 included all subjects from the three phase 3 and two phase 2 studies who (i) were randomized/enrolled and had received at least one dose of filgotinib at the randomized/enrolled phase and (ii) had at least one nonmissing PK parameter of interest estimated from a PopPK model (described above) for the analyte of interest. ER analyses for efficacy were performed following completion of phase 3 and phase 2 studies to support the dose for commercialization.
The primary objective of the included studies was to evaluate the effect of filgotinib for the treatment of RA as measured by the proportion of subjects achieving ACR20 (primary endpoint) at week 12 (DARWIN 1, DARWIN 2, FINCH 1 and FINCH 3) or week 24 (FINCH 2). Secondary efficacy endpoints included the proportion of subjects who achieved ACR50, ACR70 and Disease Activity Score (DAS) 28(CRP) ≤ 3.2 or DAS28(CRP) < 2.6 at week 12 or week 24, as applicable. Exposure‐efficacy analyses were conducted to assess the relationship between filgotinib exposures and the various efficacy endpoints based on pooled phase 2 and phase 3 data regardless of the RA population. Clopper‐Pearson is a common method for calculating binomial confidence intervals and was applied in the exposure‐efficacy analyses.
As both filgotinib and its metabolite, GS‐829845, contribute to efficacy via JAK1 inhibition, their PopPK‐derived exposures were combined by accounting for relative inhibition potency in the analyses for efficacy. AUC0‐24 of filgotinib and AUC0‐24 of its active metabolite GS‐829845 were combined into AUCeff. AUCeff was calculated using the equation:
where AUCFIL and AUCmet are the AUC0‐24 of filgotinib and GS‐829845, respectively, 1/10 is the difference in potency between parent and metabolite, 8 and 425.51 and 357.43 are the molecular weights of filgotinib and GS‐829845, respectively.
Subjects were grouped into octile subgroups based on the AUCeff in the filgotinib and GS‐829845 analysis set. For each subject, the determination of octile subgroup was based on the rankings of AUCeff values from all the subjects in the filgotinib and GS‐829845 analysis set with the number of observations approximately equally distributed within the eight octile subgroups. The relationship between exposure (AUCeff) and binary efficacy endpoints (ACR20, ACR50, ACR70, DAS28 (CRP) ≤ 3.2, and DAS28(CRP) < 2.6 at week 12) were described by nonlinear logistic regression using the equation:
where P is the probability of having a response, log[P/(1 − P)] is logit, E max is the maximum of logit, E min is the minimum of logit and EC50 is the AUCeff to achieve logit = 50% × E max + 50% × E min. Data in the treatment group and placebo group at week 12 were included in the regression. Exposure‐efficacy relationships were also evaluated by examining AUCeff in subjects who achieved and who did not achieve ACR20/50/70 or DAS28 responses in phase 2 and phase 3 studies across all the doses and a t‐test was performed to compare AUCeff. The exposure‐efficacy analysis was conducted using SAS version 9.4.
2.4. Exposure‐safety analysis
ER analyses for safety were pooled from all five studies across all the doses and were performed separately for filgotinib and GS‐829845 to characterize the individual safety profile of each analyte. Data were not included in the analysis if they were collected after subjects were rerandomized and were switched to a different treatment group. The evaluated safety endpoints included the five most frequent treatment‐emergent adverse events (TEAEs) (nausea, nasopharyngitis, upper respiratory tract infection, headache and hypertension) and grade 3/4 laboratory abnormalities (glucose increase, lymphocyte decrease, phosphate decrease, lipase increase and alanine transaminase [ALT] increase) that occurred in the filgotinib 200 mg once‐daily group based on phase 2 and phase 3 studies. Serious TEAEs and serious infections were also evaluated against the exposures. Exposures were compared based on the presence and absence of selected safety events.
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20. 22
3. RESULTS
Summaries of baseline characteristics in the ER analysis population stratified by study are shown in Table 1. The baseline characteristics were summarized by continuous variables (age, body weight, CLcr, CRP and RA duration) and categorical variables (sex and race). The baseline characteristics were as expected in a population with moderately to severely active RA.
TABLE 1.
Demographics and baseline characteristics of subjects with RA in the exposure‐response analysis dataset
| Subject characteristic | DARWIN 1 (n = 520) | DARWIN 2 (n = 257) | FINCH 1 (n = 926) | FINCH 2 (n = 290) | FINCH 3 (n = 796) | |
|---|---|---|---|---|---|---|
| Continuous variables, median (min, max) | ||||||
| Age (y) | 55 (19, 79) | 52 (18, 79) | 53 (18, 86) | 56 (20, 80) | 53 (18,85) | |
| Body weight (kg) | 72.3 (39, 157) | 72.0 (37, 181) | 68.3 (35.6, 143) | 78.4 (40.6, 157) | 71.0 (36.3, 148) | |
| CLcr (mL/min) | 110 (38.0, 312) | 115 (54.6, 264) | 107 (38.0, 291) | 113 (49.2, 298) | 109 (36.6, 311) | |
| CRP (mg/L) | 16.7 (1, 158) | 17.1 (1, 244) | 8.88 (0.19, 163) | 10.7 (0.34, 145) | 0.81 (0.02, 18.8) | |
| RA duration (y) | 6.2 (0.5, 43.2) | 6.9 (0.5, 49.6) | 5.3 (0.1, 45.4) | 10.1 (0.9, 49.7) | 0.4 (0.0, 47.6) | |
| Categorical variables, n (%) | ||||||
| Sex | Male | 99 (19.0%) | 48 (18.7%) | 174 (18.8%) | 59 (20.4%) | 175 (22.0%) |
| Female | 421 (81.0%) | 209 (81.3%) | 751 (81.2%) | 230 (79.6%) | 620 (78.8%) | |
| Race N (%) | White | 394 (75.8%) | 193 (75.1%) | 617 (66.7%) | 211 (73.0%) | 515 (64.8%) |
| Black | 3 (0.6%) | 3 (1.2%) | 14 (1.5%) | 26 (9.0%) | 31 (3.9%) | |
| Asian | 2 (0.4%) | 1 (0.4%) | 232 (25.1%) | 35 (12.1%) | 182 (22.9%) | |
| Other | 121 (23.3%) | 60 (23.3%) | 62 (6.7%) | 17 (5.9%) | 67 (8.4%) | |
Abbreviations: CLcr, creatinine clearance; CRP, C‐reactive protein; max, maximum; min, minimum; N, number of subjects; RA, rheumatoid arthritis.
3.1. Population PK analysis
The final PopPK model development dataset for filgotinib included 13 376 PK datapoints from 3125 subjects (details by study are provided in Supporting Information Tables S1 and S2). Plasma concentrations of filgotinib were best described by a two‐compartment model with a mixture model for absorption and linear elimination following a staged approach (Supporting Information Figure S1). The two subpopulations for absorption, rapid versus slower, were described, respectively, by a first‐order (with absorption rate constant [k a] being fixed to a high value to mimic an almost instantaneous absorption profile) and a sequential zero‐ then first‐order absorption. An inter‐individual variability (IIV) was included on oral clearance (CL/F), apparent central volume of distribution of the drug (V c/F), k a and duration of the zero‐order input (D1). The model included a difference in bioavailability (F) between tablets and capsules. Weight effects were included on CL/F, apparent intercompartmental clearance (Q/F), V c/F and peripheral volume of distribution (V p/F) using standard fixed allometric exponents of 0.75 for the clearance and 1 for the volume of distribution parameters. Moreover, baseline CRP and sex were identified as statistically significant covariates on filgotinib CL/F, whereas race (white and Asian vs black or African American vs other) was identified as a statistically significant covariate on V c/F (Supporting Information Table S3).
The covariate analysis was first performed in a forward addition and backward elimination process with all the coefficients estimated. As body weight was a significant covariate on both CL/F and V c/F, the impact of standard allometric scaling (fixed exponents of 0.75 for clearances and 1 for volume of distribution parameters) was evaluated. The model with fixed allometric scaling provided no change in overall model fit and increased stability due to the lower number of parameters to be estimated, so despite an increase in objective function value (OFV) of 30.8 points, it was accepted as the final model. The covariate impact on filgotinib PK was shown in a tornado plot for the final model (Supporting Information Figure S2); the difference in exposure was not clinically meaningful.
Although the final filgotinib model had a tendency to modestly underpredict C max (geometric mean ratios [GMRs] of 0.76‐0.79 based on different populations), the model was able to adequately predict AUCtau with GMR = 0.98, and thus could be used to predict individual exposures for ER analyses.
For GS‐829845, the final model development dataset included 14 896 PK datapoints from 3155 subjects (details by study are provided in Supporting Information Tables S1 and S2). Plasma concentrations of GS‐829845 were best described by a one‐compartment model with first‐order absorption and first‐order elimination. An IIV was included on CL/F, V c/F and k a. Statistically significant covariates included the effects of baseline CLcr, baseline CRP, patient status and sex on CL/F, Asian race versus non‐Asian race and duration of RA on V/F, and formulation on F and k a (Supporting Information Table S4). The covariate impact on GS‐829845 PK is shown in a tornado plot (Supporting Information Figure S3); the difference in exposure was not clinically meaningful.
The final models adequately described the plasma concentrations of filgotinib and GS‐829845 separately, as assessed by diagnostic plots/metrics including goodness‐of‐fit evaluation, pcVPC and bootstrap resampling (Supporting Information Tables S3 and S4, and Supporting Information Figures S4–S7). Thus, the predicted individual PK exposures were deemed adequate to be used in the ER analyses.
3.2. Exposure‐efficacy analysis
3.2.1. ER for efficacy supporting dose confirmation
The analysis set for exposure‐efficacy included pooled phase 2 and phase 3 subjects with RA who received filgotinib and had evaluable PopPK‐based exposure estimates (AUC0‐24) for both filgotinib and GS‐829845 (N = 2678).
Exposure‐efficacy analysis across the phase 2 and phase 3 programme confirmed that filgotinib produced robust therapeutic effects across the exposure range observed at 200 mg once daily at both week 12 and week 24 (Figures 1 and S8). In this ER analysis over a wide dose/exposure range, high response rates (approximately 75‐80% for ACR20, the primary endpoint) were demonstrated across octile groups associated with 200 mg in subjects with RA receiving filgotinib at week 12 (Figure 1). ACR20 responses in the filgotinib 200 mg group appeared to be on the plateau of the ER curve in the analysis (approximately 75‐80%), with the filgotinib 100 mg group being slightly lower than the curve plateau (approximately 70%). For multiple secondary efficacy endpoints including ACR50, ACR70, DAS28(CRP) ≤ 3.2 and DAS28(CRP) < 2.6, a trend of increasing response with increasing exposure was also observed over the first four octiles (corresponding to 100 mg and lower doses) and a plateau was observed over the last four octiles (corresponding to 200 mg exposures). The analysis on week 24 data showed similar findings to the week 12 analysis (Supporting Information Figure S8).
FIGURE 1.
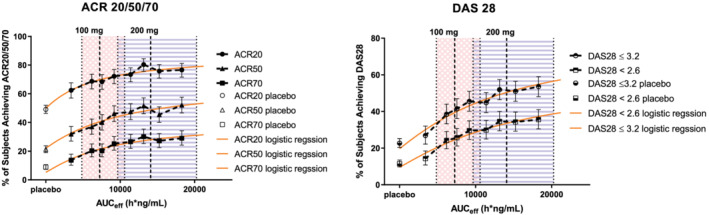
Exposure‐response relationship of AUCeff based on filgotinib and GS‐829845 against ACR and DAS28(CRP) responses at week 12 in subjects with rheumatoid arthritis (RA) in pooled phase 2 and phase 3 studies. The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies DARWIN 1, DARWIN 2, FINCH 1, FINCH 2 and FINCH 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. Each symbol represents the proportion of subjects achieving the ACR response, with the vertical line showing the 95% confidence interval within each group based on the Clopper‐Pearson method. Circles show ACR20, triangles show ACR50 and squares show ACR70 in the left panel. Semi‐hollow circles show DAS28 ≤ 3.2 and semi‐hollow triangles show DAS28 < 2.6 in the right panel. Orange solid lines are prediction curves based on nonlinear logistic regression. Shaded areas with blue stripes show median (dashed vertical line) and 5th and 95th percentiles (dotted vertical lines) of AUCeff for filgotinib 200 mg once daily in phase 2 and phase 3 subjects with RA. Shaded areas with a pink cross pattern show median (dashed vertical line) and 5th and 95th percentiles (dotted vertical lines) of AUCeff for filgotinib 100 mg once daily in phase 2 and phase 3 subjects with RA. AUCeff is based on the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
Nonlinear logistic regression models were developed and models adequately described the exposure‐efficacy relationships at week12 (Figure 1). The E max and E min estimates (Supporting Information Table S5) and the prediction curves (Figure 1) confirmed increasing response with increasing exposure observed for ACR20, ACR50, ACR70, DAS28(CRP) ≤ 3.2 and DAS28(CRP) < 2.6.
With the developed nonlinear logistic regression models, clinical relevance was evaluated for PK changes in filgotinib and GS‐829845 exposures due to the influence of intrinsic/extrinsic factors. The AUCeff reduction from the most influential covariates, including body weight for filgotinib and baseline CLcr (97% of subjects in the PopPK dataset had normal renal function or mild renal impairment) for GS‐829845, was approximately 20% and this lead to <4% reduction in probability in achieving a response across all the efficacy endpoints at 200 mg, thus the moderate PK changes from PopPK‐identified covariates are not clinically meaningful and do not warrant a dose adjustment in the evaluated population (ie, normal to mild renal function).
Exposure‐efficacy relationships were also evaluated by examining AUCeff in subjects who achieved and who did not achieve ACR20/50/70 or DAS28 responses in phase 2 and phase 3 studies across all the doses and a t‐test was performed. Figure 2 shows that subjects who achieved responses had significantly higher AUCeff compared with those who did not achieve responses at both week 12 and week 24. The difference was statistically significant across all the efficacy endpoints with P <.01. For ACR20 at week 12, the AUCeff was 10 900 h*ng/mL in the subjects who achieved ACR20 compared to 9900 h*ng/mL in those who did not achieve responses.
FIGURE 2.
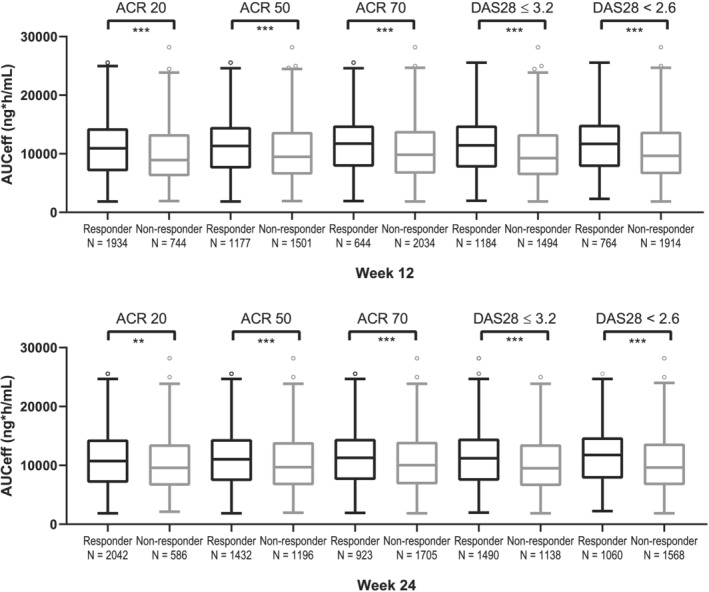
Boxplot of AUCeff in subjects who achieved and did not achieve ACR20/50/70 and DAS28(CRP) responses in pooled phase 2 and phase 3 studies. The pharmacokinetic (PK)/pharmacodynamic analysis set includes subjects with rheumatoid arthritis who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2 and FINCH 3, and had at least one nonmissing PK parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. ACR and DAS28 responses at week 12 are shown in the upper panel and ACR and DAS28 responses at week 24 are shown in the lower panel. **P < .01; ***P < .0001
Overall, filgotinib exposure‐efficacy analysis across the phase 2 and phase 3 programme confirmed that filgotinib produced more robust therapeutic effects across the exposure range observed at 200 mg once daily compared to lower doses. This analysis supported 200 mg for most adult patients and 100 mg for special populations who may have elevated PK exposures. Also, modest changes in filgotinib and GS‐829845 exposures due to the influence of intrinsic/extrinsic factors are not clinically meaningful.
3.3. Exposure‐safety analysis
The ER analyses for safety were based on the pooled population in the phase 2 and phase 3 programme across all the doses and were performed separately for filgotinib and GS‐829845 to characterize the individual safety profiles of each analyte.
3.3.1. The most frequent TEAEs
The five evaluated TEAEs were nausea, nasopharyngitis, upper respiratory tract infection, headache and hypertension. The analysis set for exposure safety included pooled phase 2 and phase 3 subjects with RA who received filgotinib and had evaluable PopPK‐based exposure estimates (AUC0‐24) (N = 2678 for filgotinib and N = 2707 for GS‐829845). Data were not included in the analysis if they were collected after subjects were rerandomized and were switched to a different treatment group. As shown in Figure 3 (filgotinib) and Figure 4 (GS‐829845), filgotinib or GS‐829845 exposures (AUC0‐24) in subjects with RA were similar regardless of the presence (black) or absence (gray) of the evaluated TEAEs up to week 52.
FIGURE 3.
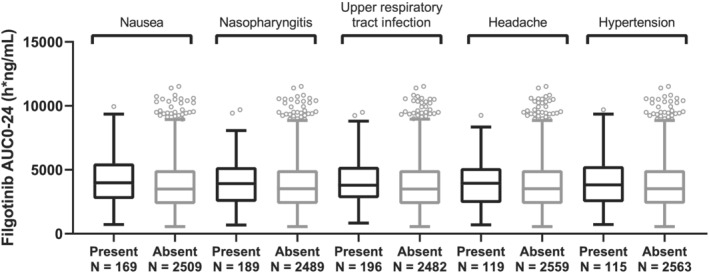
Filgotinib AUC0‐24 by the five most frequent treatment‐emergent adverse events in subjects with rheumatoid arthritis (RA) up to week 52 data. The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2, FINCH 3, DARWIN 1, DARWIN 2 and DARWIN 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. AUC0‐24 is the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
FIGURE 4.
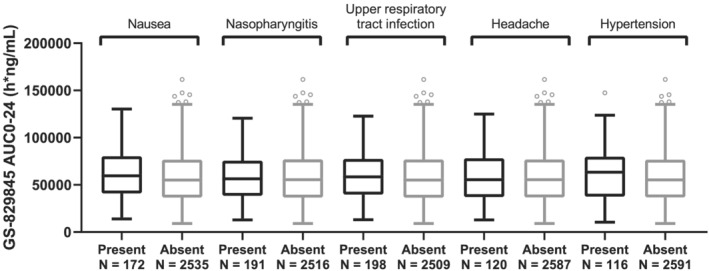
GS‐829845 AUC0‐24 by the five most frequent treatment‐emergent adverse events in subjects with rheumatoid arthritis (RA) up to week 52 data. The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2, FINCH 3, DARWIN 1, DARWIN 2 and DARWIN 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. AUC0‐24 is the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
3.3.2. The most frequent grade 3/4 laboratory abnormalities
The five evaluated grade 3/4 laboratory abnormalities were glucose increase, lymphocyte decrease, phosphate decrease, lipase increase and ALT increase. As shown in Figure 5 (filgotinib) and Figure 6 (GS‐829845), filgotinib or GS‐829845 exposures (AUC0‐24) were highly overlapping between subjects who experienced (black) and who did not experience (gray) the selected grade 3/4 laboratory abnormalities up to week 52.
FIGURE 5.
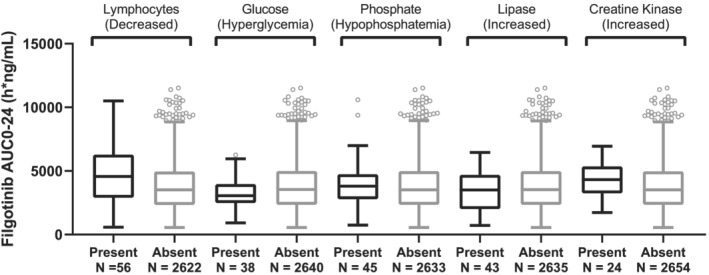
Filgotinib AUC0‐24 by the five most frequent grade 3/4 laboratory abnormalities in subjects with rheumatoid arthritis (RA). The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2, FINCH 3, DARWIN 1, DARWIN 2 and DARWIN 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. AUC0‐24 is the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
FIGURE 6.
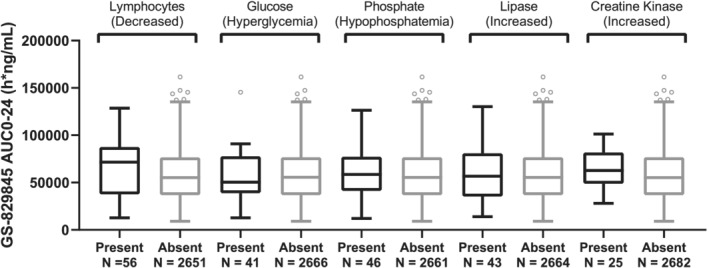
GS‐829845 AUC0‐24 by the five most frequent grade ¾ laboratory abnormalities in subjects with rheumatoid arthritis (RA). The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2, FINCH 3, DARWIN 1, DARWIN 2 and DARWIN 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. AUC0‐24 is the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
3.3.3. Serious TEAEs and serious infections
Serious TEAEs and serious infections are also evaluated against the filgotinib and GS‐829845 exposures and no exposure‐driven patterns were present based on the safety data up to week 52 (Supporting Information Figure S9).
Overall, the exposure‐safety relationships for either filgotinib and GS‐829845 demonstrate no trend toward increasing incidence of common TEAEs, common laboratory abnormalities, serious TEAEs or serious infections with increasing exposure.
4. DISCUSSION
In the PopPK analysis, plasma concentrations were adequately described by the final models and thus the predicted individual PK exposures were further used in the ER analyses. The final filgotinib model was able to adequately predict AUCtau with GMR = 0.98, but it still had a tendency to modestly underpredict C max (GMRs of 0.76‐0.79 based on different populations). Various efforts were made to address this issue in both the structural and stochastic models. However, none of these additional modifications to the PopPK model resulted in further improvement of the C max underprediction. Overall, the bias is acknowledged and taken into consideration when using model‐derived exposures in the context of ER analyses with particular attention to the use of C max. In all, estimated filgotinib AUCtau is adequate for the intended purpose to support ER analyses in subjects with RA. A joint parent‐metabolite modelling approached was considered and attempted as well, but separate modelling was more reasonable based on mechanism and model performance.
As a basis for the ER relationships, the PK‐pharmacodynamic relationship of filgotinib was studied previously. Dose‐dependent inhibition of the JAK1‐related interleukin‐6 (IL‐6)‐induced phosphorylated signal transducer and activator of transcription 1 (pSTAT1) by filgotinib was demonstrated at doses of 50 mg of filgotinib and higher with maximum inhibition of pSTAT1 (~78%) plateaued at or above 200 mg total daily dose and intermediate inhibition (~47%) observed at a total daily dose of 100 mg. 23
ER analyses based on phase 2 and phase 3 clinical studies were further conducted to confirm the dose. Exposure‐efficacy analyses consistently revealed high response rates (approximately 75‐80% for ACR20 at week 12) across the exposure range for the filgotinib 200 mg doses. Increasing response with increasing exposure was demonstrated by the nonlinear logistic regression for multiple efficacy endpoints with a plateau corresponding to filgotinib 200 mg exposures.
Exposure‐safety relationships established that filgotinib and GS‐829845 exposures (AUC0‐24) were similar regardless of the presence or absence of the most frequent TEAEs, the most frequent grade 3/4 laboratory abnormalities, serious TEAEs or serious infections, indicating no exposure‐safety relationship. Since the number of subjects with safety event presence was less than 10%, the assessment was just visual without performing a t‐test due to the unbalanced number of subjects. One limitation of the exposure‐safety analyses is that the grade 3/4 laboratory abnormalities were not treated as continuous data for further analyses and continuous data can contain more information compared to categorical data. Another limitation is that treatment duration was not fully evaluated, although the analyses were actually performed based on week 12 (data not shown), week 24 (data not shown) and week 52 data, and the results were consistent.
Other exposure metrics, such as C tau and C max, were also evaluated. C tau in the form of C tau‐eff (combination of C tau from both parent and metabolite) was tested in the exposure‐efficacy analyses and C max was evaluated in the exposure‐safety analyses. The results using C tau‐eff or C max were similar to those based on AUC.
With the identified ER relationship, clinical relevance was evaluated for PK changes in filgotinib and GS‐829845 exposures due to the influence of intrinsic/extrinsic factors. The moderate reduction in PK due to the most influential covariates only resulted in <4% reduction in response rate across all the efficacy endpoints for 200 mg; there are no safety concerns given the lack of an exposure‐safety relationship. Thus, these PK changes are not clinically meaningful and do not warrant a dose adjustment in the evaluated population (ie, normal to mild renal function). The limitation of current ER analyses is insufficient information to guide dosing in special populations, such as elderly subjects and subjects with moderate to severe renal impairment.
The special populations whose PK may not be best characterized in the pooled analyses (only 3% with ≥75 years of age and 3% with moderate to severe renal impairment) were covered in dedicated studies. Filgotinib and its metabolite were shown to be moderately higher (1.45‐ and 1.33‐fold, respectively) in elderly subjects (≥75 years) compared with younger subjects; in subjects with severe renal impairment (RI), filgotinib was 1.54 fold higher and 2.74 fold higher for the metabolite. 16 For hepatic impairment, there was no clinically relevant effect on filgotinib and GS‐829845 exposure (AUC and C max) in subjects with moderate hepatic impairment compared with matched healthy controls. It was overall suggested that modest changes in filgotinib and GS‐829845 exposures due to the influence of other intrinsic/extrinsic factors, such as moderate hepatic impairment and food intake, are not clinically meaningful. 20 , 24 However, for special populations who may have elevated PK exposures (eg, patients aged 75 years and older, and patients with moderate to severe RI) and whose clinical data was limited, 100 mg is recommended, as this dose was shown to provide efficacious exposures.
Collectively, the ER analyses indicate robust therapeutic effects across the exposure range observed at 200 mg once daily in subjects with moderately to severely active RA. The trend towards greater efficacy with higher exposures observed for the primary and secondary endpoints (ACR20, ACR50, ACR70, DAS28(CRP) ≤ 3.2 and DAS28(CRP) < 2.6) and a lack of exposure‐safety relationship based on the evaluated TEAEs and common laboratory abnormalities indicates an advantage to the 200 mg filgotinib dose relative to the 100 mg filgotinib dose for most adult patients.
5. CONCLUSIONS
Based on the exposure‐efficacy analyses, exposures associated with 200 mg once‐daily filgotinib corresponded to higher ACR responses compared with those associated with filgotinib 100 mg once daily or lower doses, showing a plateau over higher exposures corresponding to 200 mg for multiple efficacy endpoints (ACR20, ACR50, ACR70, DAS28(CRP) ≤ 3.2 and DAS28(CRP) < 2.6). For the safety analyses, it was shown that filgotinib was generally well tolerated with no exposure‐dependent effects on the evaluated safety endpoints. Overall, the ER analyses supported 200 mg once‐daily doses for commercialization.
COMPETING INTERESTS
A.Meng, K.A., C.N., L.N., S.M.C., B.K., B.B. and A.Mathias are/were employees and shareholders of Gilead Sciences, Inc. F.B., P.C. and C.C. were under a work contract with Gilead Sciences, Inc. when this research work was conducted.
CONTRIBUTORS
A.Meng designed and conducted the exposure‐response analysis. A.Meng, K.A., C.N., B.P.K., B.B. and A.Mathias interpreted the results. L.N. and S.‐M.C. conducted the statistical analyses. A.Meng, F.B., P.C. and C.C. conducted the population pharmacokinetic analyses. A.Meng wrote the manuscript.
Supporting information
Figure S1 Flow chart for the staged approach in filgotinib population pharmacokinetic (PopPK) model development. Due to the complexity in characterizing filgotinib absorption, a staged approach was implemented to develop the PopPK model: (1) phase 1 data only; (2) intensive PK data from all clinical studies; and (3) intensive and sparse PK data. This staged approach reduced some of the challenges introduced by the large heterogeneity present in the analysis dataset (different phases, doses, formulations and study designs) and therefore improved both the characterization of filgotinib absorption as well as the quantification of formulation effects on the exposure. Based on the analysis, including all intensive PK data (step 2), a two‐compartment structural model with a mixture absorption model and linear elimination, and a formulation effect on F were used to describe the filgotinib exposure data in the full analysis dataset (step 3). Due to the relatively limited absorption/formulation data available from the phase 3 studies, absorption parameters as well as the formulation effect on F were fixed to the estimates obtained when modelling only the intensive PK data
Figure S2 Covariate impact on filgotinib exposures. Base, as represented by the black vertical line and values, refers to the median post hoc AUCtau, C max or C tau of filgotinib. The black shaded bar with values at each end shows the 5th to 95th percentile exposure range across the entire population. Each blue shaded bar represents the influence of single or combined covariates on the steady‐state exposure. The label at the left end of the bar represents the covariate(s) being evaluated. The upper and lower values for each covariate capture 90% of the plausible range in the population. For the combined effect, the low case for AUCtau and C max is a 105 kg other‐race male with the following covariate values: 0.2 mg/L CRP. The high case is a 48 kg black‐race female with the following covariate values: 63.4 mg/L CRP. For C tau, race effects were inverted in the two combined cases due to fact that they only influence Vc/F. The length of each bar describes the potential impact of that covariate(s) on filgotinib exposure at steady state, with the percentage value in the parentheses at each end representing the percentage change of exposure from the base. The covariates are listed in descending order of influence. The plot for AUCtau is shown in the upper panel, the plot for C max is shown in the mid panel and the plot for C tau is shown in the lower panel. AUCtau, area under the concentration‐time curve over the dosing interval; BCRP, baseline C‐reactive protein; C max, maximum steady‐state concentration; CRP, C‐reactive protein; C tau, observed drug concentration at the end of the dosing interval; FORM, formulation; WT, baseline body weight
Figure S3 Covariate impact on GS‐829845 exposures. Base, as represented by the black vertical line and values, refers to the median post hoc AUCtau of GS‐829845, where the base case is a 71.8 kg white female who has had RA for 4 years taking 200 mg with the following covariate values: 7.2 mg/L CRP and 109.8 mL/min CLcr. The black shaded bar with values at each end shows the 5th to 95th percentile exposure range across the entire population. Each blue shaded bar represents the influence of single or combined covariates on the steady‐state exposure. The label at the left end of the bar represents the covariate(s) being evaluated. The upper and lower values for each covariate capture 90% of the plausible range in the population. For the combined effect, the Low case for AUCtau and C max is a 105 kg Asian male who has had RA for about a month with the following covariate values: 0.2 mg/L CRP and 186.3 mL/min CLcr. The High case is a 48 kg non‐Asian female who has had RA for 23 years with the following covariate values: 63.4 mg/L CRP and 64.2 mL/min CLcr. The length of each bar describes the potential impact of that covariate(s) on GS‐829845 exposure at steady state, with the percentage value in the parentheses at each end representing the percentage change of exposure from the base. The covariates are listed in descending order of influence. The plot for AUCtau is shown in the upper panel, the plot for C max is shown in the middle panel and Ctau is shown in the lower panel. AUCtau, area under the concentration‐time curve over the dosing interval; CLcr, creatinine clearance; C max, maximum steady‐state concentration; CRP, C‐reactive protein; C tau, observed drug concentration at the end of the dosing interval; RA, rheumatoid arthritis; WT, baseline body weight
Figure S4 Standard goodness‐of‐fit plots for the final PopPK model for filgotinib. Cond., conditional; CWRES, conditional weighted residuals; Ind., individual; Pop., population; PopPK, population pharmacokinetic. The circles represent individual data points, the grey lines represent loess smooth curves and the dashed lines represent the line of unity (y = x), the unit line at 0 (y = 0) or a|CWRES|of 6
Figure S5 Standard goodness‐of‐fit plots for the final PopPK model for GS‐829845. CWRES, conditional weighted residuals; Ind., individual; Pop., population; PopPK, population pharmacokinetic. The circles represent individual data points, the grey lines represent loess smooth curves and the dashed lines represent the line of unity (y = x), the unit line at 0 (y = 0) or a|CWRES|of 6
Figure S6 pcVPC of plasma concentration‐time profiles for filgotinib stratified by phase and formulation. CI, confidence interval; pcVPC, prediction‐corrected visual predictive check. pcVPC plots show the median (solid black lines) and spread (5th to 95th percentile, dashed black line) of the observed concentrations in all subjects. Observed data are shown in black circles. The red area is the 95% CI of the simulated median and the blue area is the 95% CI of the simulated 5th and 95th percentiles. pcVPC is stratified by phase and formulation
Figure S7 pcVPC of plasma concentration‐time profiles for GS‐829845 stratified by phase and formulation. CI, confidence interval; DV, observer concentrations; pcVPC, prediction‐corrected visual predictive check. The pcVPC plots show the median (solid black lines) and spread (5th to 95th percentile, dashed black line) of the DV in all subjects. Observed data are shown in black circles. The red area is the 95% CI of the simulated median and the blue area is the 95% CI of the simulated 5th and 95th percentiles
Figure S8 Exposure‐response relationship of AUCeff based on filgotinib and GS‐829845 against ACR and DAS28 (CRP) responses at week 24 in subjects with rheumatoid arthritis (RA) in pooled phase 2 and phase 3 studies. The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies DARWIN 1, DARWIN 2, FINCH 1, FINCH 2 and FINCH 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. Each symbol represents the proportion of subjects achieving the ACR response, with the vertical line showing the 95% confidence interval within each group based on the Clopper‐Pearson method. Circles show ACR20, triangles show ACR50 and squares show ACR70 in the left panel. Hollow circles show DAS28 ≤ 3.2 and hollow triangles show DAS28 < 2.6 in the right panel. Shaded areas with blue stripes show median (dashed vertical line) and 5th and 95th percentiles (dotted vertical lines) of AUCeff for filgotinib 200 mg once daily in phase 2 and phase 3 subjects with RA; shaded areas with pink cross pattern show median (dashed vertical line) and 5th and 95th percentiles (dotted vertical lines) of AUCeff for filgotinib 100 mg once daily in phase 2 and phase 3 subjects with RA. AUCeff is based on the population PK predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
Figure S9 Filgotinib (left) and GS‐829845 (right) AUC0‐24 by serious treatment‐emergent adverse events and serious infections in subjects with rheumatoid arthritis (RA). The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2 FINCH 3, DARWIN 1, DARWIN 2 and DARWIN 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. AUC0‐24 is the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
Table S1 Summary of studies included in the population pharmacokinetic analysis
Table S2 Summary of filgotinib and GS‐829845 data in the population pharmacokinetic datasets
Table S3 Comparison of final model estimates and bootstrap and SIR results for filgotinib
Table S4 Comparison of final model estimates and bootstrap results for GS‐829845
Table S5 Parameter estimates in nonlinear logistic regression for exposure‐efficacy relationships at week 12
ACKNOWLEDGMENTS
The authors thank the investigators and coordinators at the participating centres and the subjects who participated in these studies. The authors acknowledge Shringi Sharma and Yan Xin for their contribution to the analysis plan and Ahmed Othman for his support in finalizing this manuscript. Medical writing support was provided by Y. Sunila Reddy, Pharm.D. This study was funded by Gilead Sciences Inc.
These studies were funded by Gilead Sciences, Inc and Galapagos NV.
Meng A, Anderson K, Nelson C, et al. Exposure‐response relationships for the efficacy and safety of filgotinib and its metabolite GS‐829845 in subjects with rheumatoid arthritis based on phase 2 and phase 3 studies. Br J Clin Pharmacol. 2022;88(7):3211-3221. doi: 10.1111/bcp.15239
No principal investigators were involved in this analysis.
Funding information Galapagos NV; Gilead Sciences
DATA AVAILABILITY STATEMENT
Gilead is committed to full transparency of publications. Clinical trial registration numbers or other unique trial and/or study identifiers should be included in all publications and presentations that report results from clinical studies, as outlined in the CONSORT statement. Journal and congress rules concerning inclusion of clinical trial registration numbers/unique study identifiers should be followed. Upon journal request, in cases where the redacted protocol and analysis plan have not been publicly disclosed, Gilead may provide a copy of the protocol and prespecified data analysis plan for a submitted publication. Data may also be shared in a journal specified server, in a manner consistent with the Gilead Clinical Trial Transparency & Data Sharing Policy.
REFERENCES
- 1. Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J. Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res. 2018;6(1):1‐14. doi: 10.1038/s41413-018-0016-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gadina M. Janus kinases: an ideal target for the treatment of autoimmune diseases. J Investig Dermatol Symp Proc. 2013;16(1):S70‐S72. doi: 10.1038/jidsymp.2013.29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. El Jammal T, Gerfaud‐Valentin M, Sève P, Jamilloux Y. Inhibition of JAK/STAT signaling in rheumatologic disorders: The expanding spectrum. Joint Bone Spine. 2020;87(2):119‐129. doi: 10.1016/j.jbspin.2019.09.005 [DOI] [PubMed] [Google Scholar]
- 4. Jegatheeswaran J, Turk M, Pope JE. Comparison of Janus kinase inhibitors in the treatment of rheumatoid arthritis: a systemic literature review. Immunotherapy. 2019. Jun;11(8):737‐754. doi: 10.2217/imt-2018-0178. Epub 2019 Apr 8. PMID: 30955397 [DOI] [PubMed] [Google Scholar]
- 5. Tanaka Y, Kavanaugh A, Wicklund J, McInnes IB. Filgotinib, a novel JAK1‐preferential inhibitor for the treatment of rheumatoid arthritis: An overview from clinical trials [published online ahead of print, 2021 Apr 15. Mod Rheumatol. 2021;(1):1‐11. doi: 10.1080/14397595.2021.1902617 [DOI] [PubMed] [Google Scholar]
- 6. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK‐STAT Signaling as a target for inflammatory and autoimmune diseases: Current and future prospects [published correction appears in Drugs. 2017 May;77(8):939] [published correction appears in Drugs. 2017 Jun 12;]. Drugs. 2017;77(5):521‐546. doi: 10.1007/s40265-017-0701-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vainchenker W, Dusa A, Constantinescu SN. JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Semin Cell Dev Biol. 2008;19(4):385‐393. doi: 10.1016/j.semcdb.2008.07.002 [DOI] [PubMed] [Google Scholar]
- 8. Traves PG, Murray B, Campigotto F, Galien R, Meng A, Di Paolo JA. JAK selectivity and the implications for clinical inhibition of pharmacodynamic cytokine signalling by filgotinib, upadacitinib, tofacitinib and baricitinib. Ann Rheum Dis. 2021;80(7):865‐875. doi: 10.1136/annrheumdis-2020-219012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Combe B, Kivitz A, Tanaka Y, et al. LB0001 Efficacy and safety of filgotinib for patients with rheumatoid arthritis with inadequate response to methotrexate: FINCH 1 primary outcome results. Ann Rheum Dis. 2019;78:77‐78. [Google Scholar]
- 10. Genovese MC, Kalunian K, Gottenberg JE, et al. Effect of filgotinib vs placebo on clinical response in patients with moderate to severe rheumatoid arthritis refractory to disease‐modifying antirheumatic drug therapy: The FINCH 2 randomized clinical trial [published correction appears in JAMA. 2020 Feb 4;323(5):480]. Jama. 2019;322(4):315‐325. doi: 10.1001/jama.2019.9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Westhovens R, Rigby W, van der Heijde D, et al. Efficacy and safety of filgotinib for patients with rheumatoid arthritis naïve to methotrexate therapy: FINCH 3 primary outcome results. Eur Congress Rheumatol12‐15 June. 2019. Madrid, Spain; Abstract LB003. [Google Scholar]
- 12. Feagan BG, Loftus EV Jr, Danese S, et al. A15 efficacy and safety of filgotinib as induction therapy for patients with moderately to severely active ulcerative colitis: Results from the phase 2b/3 selection study. J Can Assoc Gastroenterol. 2021. Mar;4(Supplement_1):18‐20. [Google Scholar]
- 13. Peyrin‐Biroulet L, Loftus EV Jr, Danese S, et al. A17 efficacy and safety of filgotinib as maintenance therapy for patients with moderately to severely Active ulcerative colitis: Results from the phase 2b/3 selection study. J Can Assoc Gastroenterol. 2021. Mar;4(Supplement_1):21‐23.33644673 [Google Scholar]
- 14. Vermeire S, Schreiber S, Petryka R, et al. Clinical remission in patients with moderate‐to‐severe Crohn's disease treated with filgotinib (the FITZROY study): results from a phase 2, double‐blind, randomised, placebo‐controlled trial. Lancet. 2017;389(10066):266‐275. doi: 10.1016/S0140-6736(16)32537-5 [DOI] [PubMed] [Google Scholar]
- 15. Van Rompaey L, Galien R, van der Aar EM, et al. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol. 2013;191(7):3568‐3577. doi: 10.4049/jimmunol.1201348 [DOI] [PubMed] [Google Scholar]
- 16. Namour F, Fagard L, Van der Aa A, Harrison P, Xin Y, Tasset C. Influence of age and renal impairment on the steady state pharmacokinetics of filgotinib, a selective JAK1 inhibitor. Br J Clin Pharmacol. 2018;84(12):2779‐2789. doi: 10.1111/bcp.13726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Combe B, Kivitz A, Tanaka Y, et al. Filgotinib versus placebo or adalimumab in patients with rheumatoid arthritis and inadequate response to methotrexate: a phase III randomised clinical trial. Ann Rheum Dis. 2021;80(7):848‐858. doi: 10.1136/annrheumdis-2020-219214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Westhovens R, Taylor PC, Alten R, et al. Filgotinib (GLPG0634/GS‐6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose‐finding study (DARWIN 1). Ann Rheum Dis. 2017;76(6):998‐1008. doi: 10.1136/annrheumdis-2016-210104 [DOI] [PubMed] [Google Scholar]
- 19. Kavanaugh A, Kremer J, Ponce L, et al. Filgotinib (GLPG0634/GS‐6034), an oral selective JAK1 inhibitor, is effective as monotherapy in patients with active rheumatoid arthritis: results from a randomised, dose‐finding study (DARWIN 2). Ann Rheum Dis. 2017;76(6):1009‐1019. doi: 10.1136/annrheumdis-2016-210105 [DOI] [PubMed] [Google Scholar]
- 20. Anderson K, Zheng H, Kotecha M, et al. The relative bioavailability and effects of food and acid‐reducing agents on filgotinib tablets in healthy subjects. Clin Pharmacol Drug Dev. 2019;8(5):585‐594. doi: 10.1002/cpdd.659 [DOI] [PubMed] [Google Scholar]
- 21. Vanhoutte F, Mazur M, Voloshyn O, et al. Efficacy, safety, pharmacokinetics, and pharmacodynamics of filgotinib, a selective JAK‐1 inhibitor, after short‐term treatment of rheumatoid arthritis: results of two randomized phase IIA trials. Arthritis Rheumatol. 2017;69(10):1949‐1959. doi: 10.1002/art.40186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176(S1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Namour F, Diderichsen PM, Cox E, et al. Pharmacokinetics and pharmacokinetic/pharmacodynamic modeling of filgotinib (GLPG0634), a selective JAK1 inhibitor, in support of phase IIB dose selection. Clin Pharmacokinet. 2015;54(8):859‐874. doi: 10.1007/s40262-015-0240-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Anderson K, Zheng H, Medzihradsky O, et al. THU0117 Pharmacokinetics and short‐term safety of filgotinib, a selective janus kinase 1 inhibitor, in subjects with moderate hepatic impairment. Ann Rheum Dis. 2019;78:331. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Flow chart for the staged approach in filgotinib population pharmacokinetic (PopPK) model development. Due to the complexity in characterizing filgotinib absorption, a staged approach was implemented to develop the PopPK model: (1) phase 1 data only; (2) intensive PK data from all clinical studies; and (3) intensive and sparse PK data. This staged approach reduced some of the challenges introduced by the large heterogeneity present in the analysis dataset (different phases, doses, formulations and study designs) and therefore improved both the characterization of filgotinib absorption as well as the quantification of formulation effects on the exposure. Based on the analysis, including all intensive PK data (step 2), a two‐compartment structural model with a mixture absorption model and linear elimination, and a formulation effect on F were used to describe the filgotinib exposure data in the full analysis dataset (step 3). Due to the relatively limited absorption/formulation data available from the phase 3 studies, absorption parameters as well as the formulation effect on F were fixed to the estimates obtained when modelling only the intensive PK data
Figure S2 Covariate impact on filgotinib exposures. Base, as represented by the black vertical line and values, refers to the median post hoc AUCtau, C max or C tau of filgotinib. The black shaded bar with values at each end shows the 5th to 95th percentile exposure range across the entire population. Each blue shaded bar represents the influence of single or combined covariates on the steady‐state exposure. The label at the left end of the bar represents the covariate(s) being evaluated. The upper and lower values for each covariate capture 90% of the plausible range in the population. For the combined effect, the low case for AUCtau and C max is a 105 kg other‐race male with the following covariate values: 0.2 mg/L CRP. The high case is a 48 kg black‐race female with the following covariate values: 63.4 mg/L CRP. For C tau, race effects were inverted in the two combined cases due to fact that they only influence Vc/F. The length of each bar describes the potential impact of that covariate(s) on filgotinib exposure at steady state, with the percentage value in the parentheses at each end representing the percentage change of exposure from the base. The covariates are listed in descending order of influence. The plot for AUCtau is shown in the upper panel, the plot for C max is shown in the mid panel and the plot for C tau is shown in the lower panel. AUCtau, area under the concentration‐time curve over the dosing interval; BCRP, baseline C‐reactive protein; C max, maximum steady‐state concentration; CRP, C‐reactive protein; C tau, observed drug concentration at the end of the dosing interval; FORM, formulation; WT, baseline body weight
Figure S3 Covariate impact on GS‐829845 exposures. Base, as represented by the black vertical line and values, refers to the median post hoc AUCtau of GS‐829845, where the base case is a 71.8 kg white female who has had RA for 4 years taking 200 mg with the following covariate values: 7.2 mg/L CRP and 109.8 mL/min CLcr. The black shaded bar with values at each end shows the 5th to 95th percentile exposure range across the entire population. Each blue shaded bar represents the influence of single or combined covariates on the steady‐state exposure. The label at the left end of the bar represents the covariate(s) being evaluated. The upper and lower values for each covariate capture 90% of the plausible range in the population. For the combined effect, the Low case for AUCtau and C max is a 105 kg Asian male who has had RA for about a month with the following covariate values: 0.2 mg/L CRP and 186.3 mL/min CLcr. The High case is a 48 kg non‐Asian female who has had RA for 23 years with the following covariate values: 63.4 mg/L CRP and 64.2 mL/min CLcr. The length of each bar describes the potential impact of that covariate(s) on GS‐829845 exposure at steady state, with the percentage value in the parentheses at each end representing the percentage change of exposure from the base. The covariates are listed in descending order of influence. The plot for AUCtau is shown in the upper panel, the plot for C max is shown in the middle panel and Ctau is shown in the lower panel. AUCtau, area under the concentration‐time curve over the dosing interval; CLcr, creatinine clearance; C max, maximum steady‐state concentration; CRP, C‐reactive protein; C tau, observed drug concentration at the end of the dosing interval; RA, rheumatoid arthritis; WT, baseline body weight
Figure S4 Standard goodness‐of‐fit plots for the final PopPK model for filgotinib. Cond., conditional; CWRES, conditional weighted residuals; Ind., individual; Pop., population; PopPK, population pharmacokinetic. The circles represent individual data points, the grey lines represent loess smooth curves and the dashed lines represent the line of unity (y = x), the unit line at 0 (y = 0) or a|CWRES|of 6
Figure S5 Standard goodness‐of‐fit plots for the final PopPK model for GS‐829845. CWRES, conditional weighted residuals; Ind., individual; Pop., population; PopPK, population pharmacokinetic. The circles represent individual data points, the grey lines represent loess smooth curves and the dashed lines represent the line of unity (y = x), the unit line at 0 (y = 0) or a|CWRES|of 6
Figure S6 pcVPC of plasma concentration‐time profiles for filgotinib stratified by phase and formulation. CI, confidence interval; pcVPC, prediction‐corrected visual predictive check. pcVPC plots show the median (solid black lines) and spread (5th to 95th percentile, dashed black line) of the observed concentrations in all subjects. Observed data are shown in black circles. The red area is the 95% CI of the simulated median and the blue area is the 95% CI of the simulated 5th and 95th percentiles. pcVPC is stratified by phase and formulation
Figure S7 pcVPC of plasma concentration‐time profiles for GS‐829845 stratified by phase and formulation. CI, confidence interval; DV, observer concentrations; pcVPC, prediction‐corrected visual predictive check. The pcVPC plots show the median (solid black lines) and spread (5th to 95th percentile, dashed black line) of the DV in all subjects. Observed data are shown in black circles. The red area is the 95% CI of the simulated median and the blue area is the 95% CI of the simulated 5th and 95th percentiles
Figure S8 Exposure‐response relationship of AUCeff based on filgotinib and GS‐829845 against ACR and DAS28 (CRP) responses at week 24 in subjects with rheumatoid arthritis (RA) in pooled phase 2 and phase 3 studies. The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies DARWIN 1, DARWIN 2, FINCH 1, FINCH 2 and FINCH 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. Each symbol represents the proportion of subjects achieving the ACR response, with the vertical line showing the 95% confidence interval within each group based on the Clopper‐Pearson method. Circles show ACR20, triangles show ACR50 and squares show ACR70 in the left panel. Hollow circles show DAS28 ≤ 3.2 and hollow triangles show DAS28 < 2.6 in the right panel. Shaded areas with blue stripes show median (dashed vertical line) and 5th and 95th percentiles (dotted vertical lines) of AUCeff for filgotinib 200 mg once daily in phase 2 and phase 3 subjects with RA; shaded areas with pink cross pattern show median (dashed vertical line) and 5th and 95th percentiles (dotted vertical lines) of AUCeff for filgotinib 100 mg once daily in phase 2 and phase 3 subjects with RA. AUCeff is based on the population PK predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
Figure S9 Filgotinib (left) and GS‐829845 (right) AUC0‐24 by serious treatment‐emergent adverse events and serious infections in subjects with rheumatoid arthritis (RA). The analysis set includes subjects with RA who were enrolled/randomized, received at least one dose of filgotinib in studies FINCH 1, FINCH 2 FINCH 3, DARWIN 1, DARWIN 2 and DARWIN 3, and had at least one nonmissing pharmacokinetic (PK) parameter of interest. For each box, the bottom and top edges are located at the sample 25th (Q1) and 75th (Q3) percentiles, respectively, the centre horizontal line is drawn at the 50th percentile (median) and outliners (beyond 1.5 × the interquartile range) are displayed as small squares. AUC0‐24 is the population PK‐predicted exposure in phase 2 and phase 3 subjects with RA receiving filgotinib
Table S1 Summary of studies included in the population pharmacokinetic analysis
Table S2 Summary of filgotinib and GS‐829845 data in the population pharmacokinetic datasets
Table S3 Comparison of final model estimates and bootstrap and SIR results for filgotinib
Table S4 Comparison of final model estimates and bootstrap results for GS‐829845
Table S5 Parameter estimates in nonlinear logistic regression for exposure‐efficacy relationships at week 12
Data Availability Statement
Gilead is committed to full transparency of publications. Clinical trial registration numbers or other unique trial and/or study identifiers should be included in all publications and presentations that report results from clinical studies, as outlined in the CONSORT statement. Journal and congress rules concerning inclusion of clinical trial registration numbers/unique study identifiers should be followed. Upon journal request, in cases where the redacted protocol and analysis plan have not been publicly disclosed, Gilead may provide a copy of the protocol and prespecified data analysis plan for a submitted publication. Data may also be shared in a journal specified server, in a manner consistent with the Gilead Clinical Trial Transparency & Data Sharing Policy.