

Abstract
The intradermal lipopolysaccharide (LPS) challenge in healthy volunteers has proven to be a valuable tool to study local inflammation in vivo. In the current study the inhibitory effects of oral and topical corticosteroid treatment on intradermal LPS responses were evaluated to benchmark the challenge for future investigational drugs. Twenty‐four healthy male volunteers received a two‐and‐a‐half‐day twice daily (b.i.d.) pretreatment with topical clobetasol propionate 0.05% and six healthy volunteers received a two‐and‐a‐half‐day b.i.d. pretreatment with oral prednisolone at 0.25 mg/kg body weight per administration. Participants received one injection regimen of either 0, 2, or 4 intradermal LPS injections (5 ng LPS in 50 µL 0.9% sodium chloride solution). The LPS response was evaluated by noninvasive (perfusion, skin temperature, and erythema) and invasive assessments (cellular and cytokine responses) in suction blister exudate. Both corticosteroids significantly suppressed the clinical inflammatory response (erythema P = 0.0001 for clobetasol and P = 0.0016 for prednisolone; heat P = 0.0245 for clobetasol, perfusion P < 0.0001 for clobetasol and P = 0.0036 for prednisolone). Clobetasol also significantly reduced the number of monocytes subsets, dendritic cells, natural killer cells, and T cells in blister exudate. A similar effect was observed for prednisolone. No relevant corticosteroid effects were observed on the cytokine response to LPS. We successfully demonstrated that the anti‐inflammatory effects of corticosteroids can be detected using our intradermal LPS challenge model, validating it for evaluation of future investigational drugs, as an initial assessment of the anti‐inflammatory effects of such compounds in a minimally invasive manner.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ In humans, intradermal lipopolysaccharide (LPS) administration causes an acute, localized, and transient inflammatory reaction that is safe and well tolerated. As such, the intradermal LPS challenge may be a valuable model for human clinical pharmacology studies.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study evaluated the inhibitory effect of known potent anti‐inflammatory drugs (topical and systemic corticosteroids) on the LPS‐induced dermal inflammatory response.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This is the first study that thoroughly benchmarks the intradermal LPS model with known anti‐inflammatory drugs. Importantly, based on the model, observations were made that may change the mechanistic thinking on corticosteroid effects in inflammatory skin diseases.
HOW MIGHT THIS CHANGE CLINICAL PHARMA‐COLOGY OR TRANSLATIONAL SCIENCE?
☑ Our findings support the use of the intradermal LPS model for future proof of mechanism studies on anti‐inflammatory compounds.
One of the major hurdles in anti‐inflammatory drug development is the translation of preclinical findings into early clinical studies, mainly because the huge differences in immune system between animals and humans. Further, healthy volunteers only sporadically suffer from inflammatory conditions and therefore the pharmacological target of novel investigational medicinal products are not activated or expressed. That is why commonly inflammatory challenge models are required for the evaluation of anti‐inflammatory drugs in healthy volunteers. One of the best known and widely used challenge models is the intravenous lipopolysaccharide (LPS) challenge. 1 , 2 Although intravenous LPS challenge has been proven to be valuable in clinical pharmacology and human physiology studies, it has several limitations. Firstly, intravenous LPS elicits primarily a systemic inflammatory response, meaning that the model is less suitable for measuring local effects of anti‐inflammatory investigational compounds in peripheral tissues. 2 Though there are contributions from peripheral tissues to the systemic inflammatory response, it is difficult to assess the exact role of the microvasculature or stromal cells in the intravenous LPS challenge model. Secondly, exposure to systemic LPS leads to a prolonged state of immunological hyporesponsiveness to LPS due to innate memory which hampers the possibility of rechallenging the same individual. 3 , 4 We and others have published a local inflammatory challenge with intradermal LPS. 5 , 6 In our comprehensive characterization we showed that intradermal administration of 5 ng of LPS induces a transient local inflammatory response, characterized by an increase in local perfusion, heat, erythema, immune cell attraction, and cytokine production. 6 As a next step, we conducted a study evaluating the effects of corticosteroid treatment on the LPS‐driven inflammatory response of the skin in heathy volunteers to validate our previously established intradermal LPS model. Corticosteroids were selected as these drugs have been used for more than 70 years as topical or oral treatment for inflammatory skin diseases. Surprisingly, systematic evaluations of the specific dermal anti‐inflammatory activities of corticosteroids are not readily available in the public domain, let alone data on the effect sizes of the suppression. There is evidence that corticosteroids exert different anti‐inflammatory effects in the systemic blood compartment than in peripheral tissue. 7 , 8 For our study we selected topical clobetasol propionate 0.05% and oral prednisolone, allowing a differentiation between the anti‐inflammatory effects of systemic and local treatment.
MATERIALS AND METHODS
This study was conducted from October 2018 to January 2019 at the Centre for Human Drug Research (CHDR) and according to the Dutch Act on Medical Research involving Human Subjects (WMO). The study protocol was registered in the EudraCT database (number 2018‐003510‐41). The study protocol was approved by a Medical Ethics Committee (Stichting Beoordeling Ethiek Biomedisch Onderzoek, Assen, The Netherlands) prior to the start of the clinical phase. Participants gave written informed consent before any study‐related procedures were undertaken.
Study design and participants
This was an open‐label interventional study. In total 30 nonsmoking, healthy males (Fitzpatrick skin type I‐III), aged 18 to 45 years, were included. Participants with any immune disease or recent infection were excluded. Twenty‐four (24) participants received a two‐and‐a‐half‐day twice daily (b.i.d.) pretreatment with topical clobetasol propionate 0.05% ointment on a designated area on the volar forearm prior to LPS administration (5 ng LPS in 50 µL 0.9% sodium chloride (NaCl) per injection), and 6 participants received a two‐and‐a‐half‐day b.i.d. pretreatment with oral prednisolone dosed at 0.25 mg/kg body weight per administration prior to LPS administration. On the day of LPS administration participants received the final administration of clobetasol or prednisolone. The contralateral volar forearm of the participants treated with clobetasol served as untreated control. Participants received one treatment regimen of either 0, 2, or 4 intradermal LPS injections (5 ng LPS in 50 µL 0.9% NaCL solution). See Figure 1 for a graphical display of the study design. In order to minimize any possible effects of the circadian rhythm on the response to LPS, LPS injections took place between 9:00 and 11:00 for clobetasol‐treated participants, and between 9:30 and 10:00 (3 participants) and 12:20 and 12:50 (3 participants) for prednisolone treated participants.
Figure 1.
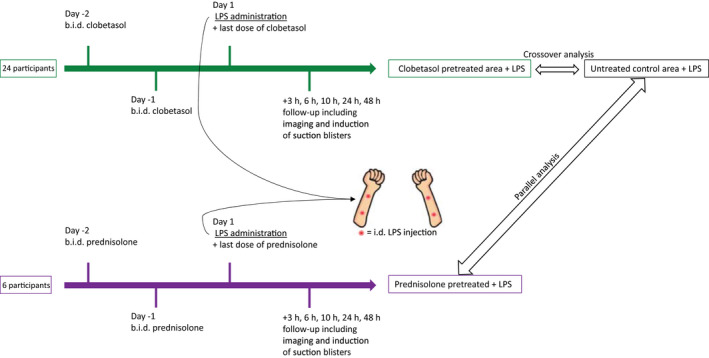
Graphical display of the study design. Participants were pretreated with either topical clobetasol or oral prednisolone at 0.25 mg/kg body weight for two‐and‐a‐half days prior to LPS administration. Follow‐up took place 3, 6, 10, 24, and 48 hours after LPS administration. b.i.d., twice daily; i.d., intradermal; LPS, lipopolysaccharide.
Skin assessments
The skin was evaluated before dosing and at 3, 6, 10, 24, and 48 hours after LPS administration. Erythema was assessed by multispectral photo analysis (Antera 3D, Miravex, Ireland), perfusion by laser speckle contrast imaging (LSCI; PeriCam PSI System, Perimed Jäfälla, Sweden), and temperature by thermography camera FLIR X6540sc (FLIR Systems Inc., Breda, The Netherlands). All skin assessments were performed in a climate‐controlled room with temperature between 19 and 21 degrees Celsius. At the indicated timepoints (Table S1 ) a suction blister was raised over the marked injection site or untreated (baseline) area. The induction of suction blisters was performed according to the method published by Motwani et al. 9 The performance of a suction blister would disqualify that area for further follow‐up with noninvasive measurements.
Suction blisters
Blister fluid was collected in a V‐bottom plate containing 50 μL 3% sodium citrate (Sigma, St. Louis, Missouri) in phosphate‐buffered saline (PBS) (Gibco, Waltham, Massachusetts) and kept on ice. The plate was centrifuged, and supernatant was weighed to estimate the volume and then frozen at −80°C for cytokine analysis (Meso Scale Discovery, Rockville, Maryland); the following cytokines were analyzed: interleukin 1β (IL‐1β), IL‐6, IL‐8, IL‐10, interferon‐γ (IFN‐γ), and tumor necrosis factor (TNF). Samples below the lower limit of quantification (LLOQ) were replaced by 0.5 × LLOQ. Please refer to Table S2 for an overview of samples above LLOQ. The pellet was resuspended in RoboSep buffer (Stemcell, Vancouver, Canada). A cocktail of fluorescent antibodies for cell surface markers was added to the cells and incubated for 30 minutes on ice. Stained samples were washed with PBS and measured with a MACSQuant 10 (Miltenyi Biotec GmbH, Bergisch Gladbach, Germany). Flow cytometry data were analyzed with Flowlogic 7.1 (Inivai, Mentone, Australia). Parallel to the blister fluid, peripheral blood was collected by venepuncture in a sodium heparin vacutainer (BD, Franklin Lakes, New Jersey). One hundred microliters whole blood was treated with red blood cell lysis buffer (eBioscience, Waltham, Massachusetts) and washed with PBS and resuspended in RoboSep buffer. Staining was similar to previously mentioned blister cells. The following antibodies were used: CD4 PerCP (clone OKT4 catalog (cat) No. 317432, BioLegend, San Diego, California), CD8 BV510 (clone SK1 cat No. 344732, BioLegend), CD56 PE‐Cy7 (clone MEM‐188, cat No. 304628, BioLegend), CD14 BV421 (clone M5E2, cat No. 301830, BioLegend), CD16 APC‐Cy7 (clone 3G8, cat No. 302018, BioLegend), CD19 FITC (clone HIB19, cat No. 302206, BioLegend), CD20 FITC (clone 2H7, cat No. 302304, BioLegend), HLA‐DR PE (clone REA805, cat No. 130‐111‐789, Miltenyi Biotec). Cell populations were classified based on the following profile: SSChighHLA‐DR‐CD16+ neutrophils, HLA‐DR+CD14+CD16‐ classical monocytes, HLA‐DR+CD14+CD16+ intermediate monocytes, HLA‐DR+CD14‐CD16+ nonclassical monocytes, HLA‐DR+CD14‐CD16‐ dendritic cells, HLA‐DR‐CD56+ NK Cells, HLA‐DR‐CD3+ T cells, and HLA‐DR+CD19+CD20+ B cells. An overview of the gating strategy can be found in Figure S1.
Statistics
Repeatedly measured end points were analyzed with a mixed model of variance with fixed factors treatment, time after LPS, and treatment by time after LPS, and random factor subject. If applicable (nonblister parameters) the baseline value (prior to LPS administration) was used as covariate. The following contrasts were calculated within the models: clobetasol propionate vs. untreated control (crossover), and prednisolone vs. untreated control (parallel). All calculations were performed using SAS for windows V9.4 (SAS Institute, Inc., Cary, NC). Outcomes were not adjusted for multiple testing due to the exploratory nature of this study.
RESULTS
Topical and systemic corticosteroids suppress the clinical response to intradermal LPS injection
To investigate whether two potent anti‐inflammatory corticosteroids (oral prednisolone and topical clobetasol propionate) are able to suppress the inflammatory response to intradermal LPS (Figure 2 ), participants received either a two‐and‐a‐half‐day pretreatment with b.i.d. oral prednisolone at 0.25 mg/kg per dose or b.i.d. topical application of clobetasol propionate 0.05% at a designated skin area. All treatments were administered under supervision at the clinical research unit, guaranteeing treatment compliance. Prednisolone treatment resulted in a statistically significant reduction of the LPS‐driven inflammatory response from baseline through 48 hours (erythema 95% confidence interval (CI): −0.10, −0.03; temperature 95% CI: −0.37, 0.02); perfusion 95% CI: −19.22, −4.10) when compared with LPS injection on untreated skin (“control”), (Figure 3a–c ). In addition, clobetasol treatment also statistically significant reduced the LPS‐driven inflammatory response of the skin from baseline through 48 hours (erythema 95% CI: −0.10, −0.03; temperature 95% CI: −0.18, −0.01; perfusion 95% CI: −10.46, −3.71), compared with control), albeit the reduction was less pronounced when compared with prednisolone (Figure 3a–c ). Table 1 provides an overview of the statistical analysis of skin temperature, perfusion, and erythema.
Figure 2.
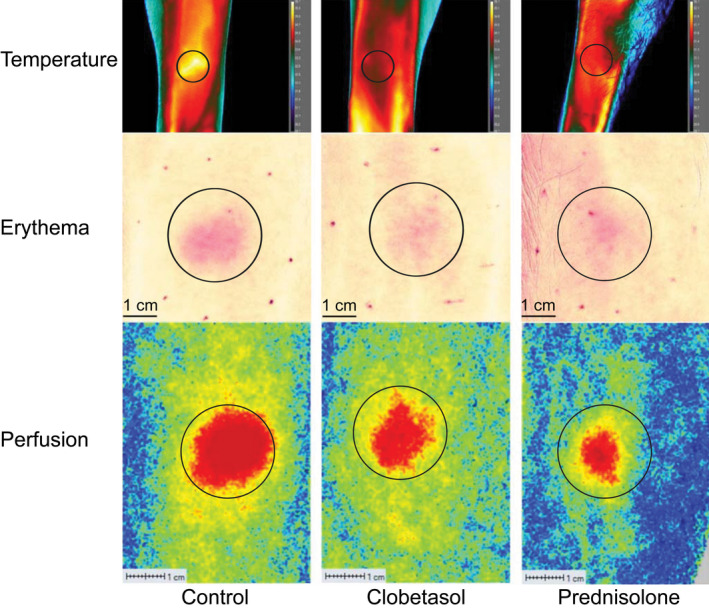
Representative images of skin temperature, erythema, and perfusion analysis 24 hours after intradermal LPS administration. Skin temperature was analyzed by thermography camera (FLIR X6540sc). Skin erythema was analyzed by multispectral photo analysis (Antera 3D). Skin perfusion was analyzed by laser speckle contrast imaging (PeriCam PSI System). LPS, lipopolysaccharide.
Figure 3.
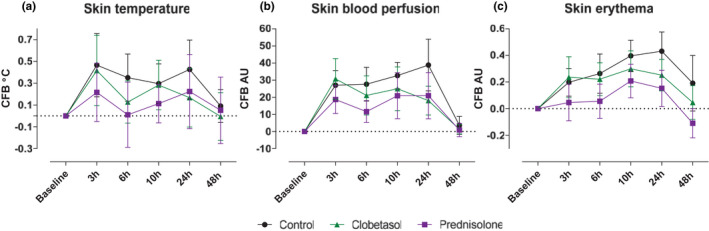
Topical clobetasol or oral prednisolone successfully reduced the inflammatory response to intradermal LPS. The inflammatory response was analyzed by quantifying (a) temperature, (b) skin blood perfusion, and (c) skin erythema. All data are presented as mean ± SD. Because the induction of a suction blister disqualified the area for further follow‐up, the sample size decreased over time. The sample size was as follows: baseline, 24 measurements; 3 hours, 18 measurements; 6 hours, 18 measurements; 10 hours, 12 measurements; 24 hours, 12 measurements; and 48 hours, 6 measurements. AU, arbitrary units; CFB, change from baseline; LPS, lipopolysaccharide.
Table 1.
Overview of the statistical analysis of skin temperature, perfusion, and erythema
| Control | Clobetasol | Prednisolone | |
|---|---|---|---|
| Skin temperature estimated mean (°C) | 0.41 | 0.31 | 0.23 |
| Estimated difference and (95% confidence interval) | – | −0.10 (−0.18, −0.01) | −0.18 (−0.37, 0.02) |
| P value | – | P = 0.0245 | P = 0.0688 |
| Skin blood perfusion estimated mean (AU) | 26.72 | 19.23 | 14.71 |
| Estimated difference and (95% confidence interval) | – | −7.09 (−10.46, −3.71) | −11.66 (−19.22, −4.10) |
| P value | – | P < 0.0001 | P = 0.0036 |
| Skin erythema estimated mean (AU) | 1.01 | 0.95 | 0.87 |
| Estimated difference and (95% confidence interval) | – | −0.07 (−0.10, −0.03) | −0.14 (−0.23, −0.06) |
| P value | – | P = 0.0001 | P = 0.0016 |
AU, arbitrary units; —, not applicable.
Topical and systemic corticosteroids reduce LPS‐driven immune cell infiltration
We have previously shown that the cellular response to intradermal LPS can be successfully studied using suction blisters. In this study, suction blisters were induced at the indicated timepoints according to Table S1 . On average, each blister contained ~ 20 to 40 microliters of blister fluid (data not shown). An overview of the different subsets characterized by flow cytometry is shown in Table S3 . In the absence of corticosteroid treatment, LPS administration led to a rapid influx of neutrophils (SSChighHLA‐DR‐CD16+) 6 hours after LPS administration (Figure 4a ), which declined to almost baseline levels at 48 hours. CD14+CD16‐ mononuclear phagocytes (“classical monocytes”) and CD14+CD16+ mononuclear phagocytes (“intermediate monocytes”) peaked at 24 hours (Figure 4b,c ), followed by CD14‐CD16+ mononuclear phagocytes (“nonclassical monocytes”) and dendritic cells (HLA‐DR+CD14‐CD16‐) which were already present at 24 hours but peaked 48 hours after LPS administration (Figure 4d,e ). NK‐cell (HLA‐DR‐CD56+) and T‐cell (HLA‐DR‐CD3+) influx was highest 24 hours after LPS but remained relatively high 48 hours after LPS (Figure 4f,g ). LPS did not cause a significant B‐cell (HLA‐DR+CD19+CD20+) response (Figure 4h ). Pretreatment with clobetasol significantly suppressed the influx of all studied inflammatory cells (Figure 4b–g ) except for neutrophils (Figure 4a ) and B cells (Figure 4h ). Prednisolone showed a comparable anti‐inflammatory effect, although not statistically significant, most likely because of a lack of power due to the parallel statistical analysis (compared with crossover statistical analysis for clobetasol) and fewer participants. The neutrophil influx was unaffected by prednisolone treatment (Figure 4a ). Prednisolone pretreatment led to a statistically significant increase (P < 0.0001), in contrast to other immune cell subsets, in amount of B cells found in the blister exudate (Figure 4h ). B cells were already present in high levels at baseline and were not affected by LPS administration. It is unknown whether there is a true physiological basis behind the B‐cell influx.
Figure 4.
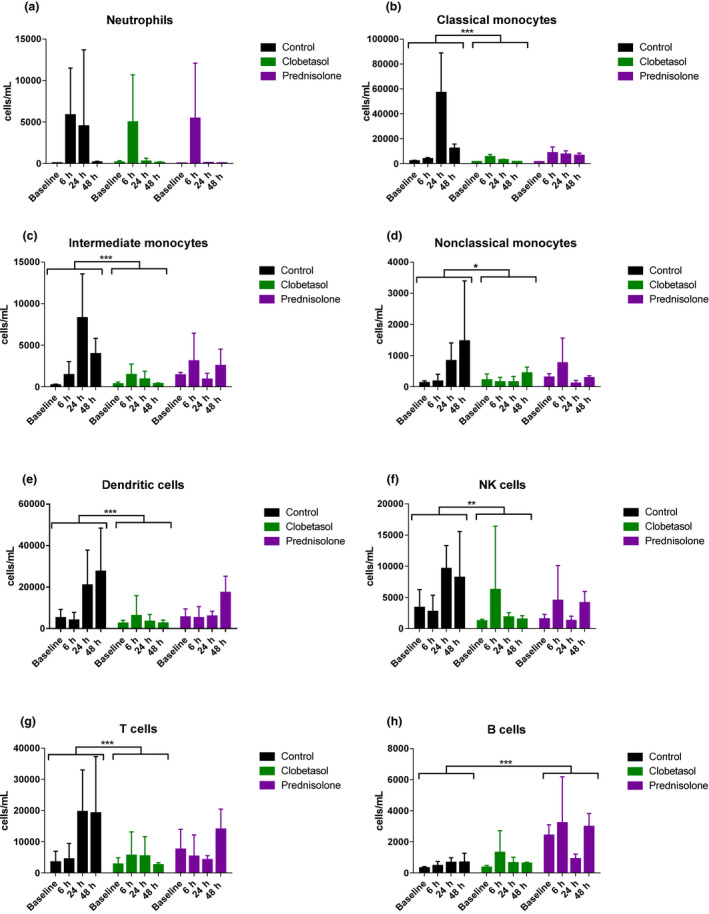
Topical and systemic corticosteroids reduced the LPS‐driven immune cell infiltration. (a) Neutrophils (b) Classical monocytes (c) Intermediate monocytes (d) Nonclassical monocytes (e) Dendritic cells (f) Natural killer cells (g) T cells (h) B cells. Immune cells were quantified in blister exudate by flow cytometry at different timepoints after LPS administration. Data are presented as mean ± SEM. *P < 0.05, **P = 0.001, and ***P < 0.0001. Prednisolone showed a comparable anti‐inflammatory effect, although not statistically significant, likely because of a lack of power due to the parallel analysis and fewer participants than clobetasol vs. control. LPS, lipopolysaccharide; NK, natural killer.
Topical or systemic corticosteroids do not affect LPS‐driven cytokine responses
In addition to the dermal cellular immune response, cytokine levels were measured in blister exudate. LPS administration drove a strong cytokine response, detectable 6 hours after LPS administration, consisting of IL‐1β, IL‐6, IL‐8, IL‐10, IFN‐γ, and TNF (Figure 5a–f ). At 48 hours after LPS administration, cytokine levels were back at baseline levels (Figure 5a–f ). Clobetasol and prednisolone pretreatment did not substantially alter the cytokine response to LPS administration (Figure 5a–f ). No statistical analysis was performed for the contrast between control, clobetasol, and prednisolone due to the large number of samples with cytokine levels below the LLOQ, at baseline, and at the 24 and 48 hour timepoints. A visual inspection of the data suggests that prednisolone and clobetasol suppressed the IFN‐γ response to LPS (Figure 5e ), whereas clobetasol caused a trend toward an increase in IL‐1β and IL‐6 response to LPS (Figure 5a–b ).
Figure 5.
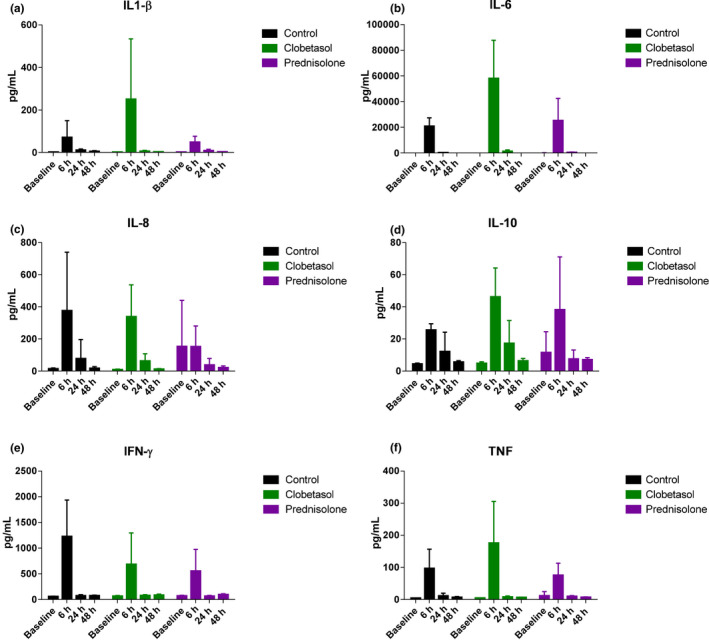
Topical and systemic corticosteroids do not affect the LPS‐driven cytokine responses. (a) IL‐1β (b) IL‐6 (c) IL‐8 (d) IL‐10 (e) IFN‐γ (f) TNF. Cytokine concentrations in blister exudate were analyzed by MSD. Data are presented as mean ± SD. IL, interleukin; IFN‐γ, interferon‐γ; MSD, meso scale discovery; TNF, tumor necrosis factor.
DISCUSSION
The aim of this study was to validate our previously established intradermal LPS model with the known anti‐inflammatory corticosteroids administered as topical clobetasol propionate 0.05% ointment and oral prednisolone, and to provide mechanistic insights into the local mode of action of both corticosteroids. Both anti‐inflammatory drugs suppressed the objectified clinical response to LPS in terms of local skin temperature, perfusion, and erythema. A power calculation was performed which showed that in a parallel study design a sample size of eight participants per treatment group with five post‐LPS measurements would provide 80% power for a minimum detectable effect size of 11.6 arbitrary units (AU) for LSCI. Likewise, a sample size of 12 would provide 80% power for a minimum detectable effect size of 9.2 AU. The maximum LPS response was 40.9 AU ± 15.1 (mean and SD) as measured by LSCI and occurred at 24 hours after injection. The LPS‐induced clinical response is likely partially mediated through inducible nitric oxide synthase (iNOS) leading to nitric oxide production and subsequent dermal vasodilation. 10 , 11 Corticosteroids are known inhibitors of iNOS, 12 and Faurschou et al. demonstrated in a clinical trial that topical corticosteroids reduce iNOS‐mediated vasodilation. 13 , 14 Both glucocorticoids also inhibited the dermal inflammatory cell influx in response to LPS, measured in suction blister fluid: the attraction of monocytes, dendritic cells, NK cells, and T cells was suppressed under corticosteroid treatment. Interestingly, the neutrophil influx remained unaffected by topical or systemic glucocorticoid pretreatment. Glucocorticoids exert both pro‐inflammatory and anti‐inflammatory effects on neutrophils which, as summarized by Ronchetti et al., can sometimes seem contradictory. 15 Glucocorticoids enhance the mobilization of neutrophils from the bone marrow into the circulation, and reduce the expression of adhesion molecules such as L‐selectin (CD62L) on neutrophils and adhesion molecules such as ICAM‐1 on the endothelium. 15 , 16 The net result is neutrophilia which was also present at baseline in our participants receiving prednisolone (data not shown). In line with this, glucocorticoids may impair the capacity of neutrophils to extravasate into inflamed tissue, 7 but in the present study the recruitment of neutrophils was unaffected (Figure 4a ). Neutrophils, as we have already shown during the earlier characterization of the intradermal LPS model, are the first cells to arrive in response to LPS, 5 , 6 and it is known that neutrophils play a central role in orchestrating the acute immune response. 17 The fact that we did not observe a reduction in neutrophil infiltration but did observe a reduction of all other immune cell subsets gave rise to our hypothesis that corticosteroid pretreatment did not affect the capacity of stromal cells and local immune cells to initiate neutrophil attraction but did lead to a reduced capacity of arriving neutrophils to further drive the acute immune response. A future study should focus not only on neutrophil recruitment and cell count but also investigate neutrophil function and activation status at the site of inflammation. Another finding was that pretreatment with clobetasol or prednisolone did not result in an obvious reduction in the LPS‐induced cytokine response (Figure 5 ). One could argue that the absence of such an inhibiting effect may relate to sample timing. The earliest blister assessment was made at 6 hours after LPS, which in theory may have been too late to cover the acute neutrophil response. We did not include an earlier timepoint that possibly could have shown a reduced response, or that could have shown a delay in peak response, whereas the clinical outcome measures (skin temperature, skin perfusion, and erythema) show a corticosteroid effect as early as 3 hours after LPS injection. However, our findings are supported by the work of Bartko et al., who observed that dexamethasone suppressed the systemic response to instilled LPS but was unable to significantly suppress the local release of IL‐6, IL‐8, and TNF in humans. 8 Another possible explanation could be that due to the small blister volume and low cytokine concentrations, results are more likely to fall below the detection limit, which makes it difficult to observe subtle changes between treatment and control. Our data demonstrate that attracted monocytes and lymphocytes are probably minor contributors to the local cytokine response, and supposedly skin‐resident immune cells such as dendritic cells, macrophages, keratinocytes, and fibroblasts are the primary source of LPS‐driven cytokines. Interestingly, all these cell types have been described to be sensitive to the anti‐inflammatory effects of corticoids in vitro; 18 , 19 , 20 , 21 therefore it is intriguing to find in our study that corticosteroid treatment did not suppress cytokine production in vivo. Given the fact that we used conventional therapeutic corticosteroid doses that, in the case of prednisolone, led to systemic anti‐inflammatory activity, 22 , 23 and based on the significant inhibition of local perfusion, erythema, and temperature, it is unlikely that the local drug exposure was too low to suppress cytokine responses. Our observations suggest that follow‐up investigations on the mechanisms behind the LPS‐driven tissue response, and the effects of corticosteroids herein should be explored further. In particular, the apparent discrepancy between the convincing corticosteroid effects on clinical symptoms (LPS‐driven perfusion, erythema, and temperature increase) and immune cell attraction (monocytes, DCs, NK cells, and lymphocytes) on one hand, and the lack of a significant inhibitory effect on local cytokine production and neutrophil influx on the other hand, is an important observation that may change the mechanistic thinking on corticosteroid effects in inflammatory skin diseases.
In conclusion, in this present study we have successfully demonstrated that both topical and systemic corticosteroid pretreatment are effective in suppressing the classical hallmarks of LPS‐induced dermal inflammation (erythema, heat, and perfusion) and that the suppressed inflammatory response can also be quantified by a reduction in inflammatory cell attraction. These findings support the use of the intradermal LPS model for future proof‐of‐mechanism studies as well as profiling studies of novel anti‐inflammatory compounds.
FUNDING
Cutanea Life Science, Inc., Wayne, Pennsylvania, USA.
CONFLICT OF INTEREST
Gary Feiss was employed by Cutanea Life Sciences during the execution of the study. All other authors declared no competing interests for this work.
AUTHOR CONTRIBUTIONS
T.P.B., M.A.A.J., and M.M. wrote the manuscript. M.M., T.P.B., G.F., R.R., D.W.G., and P.H. designed the research. T.P.B., J.B., F.C.v.H., I.M.E.J., W.t.V., P.H., H.W.G., and M.S. performed the research. D.W.G., M.A.A.J., P.H., M.M., M.L.d.K., and T.P.B analyzed the data.
Supporting information
Figure S1
Table S1
Table S2
Table S3
[Correction added on 18 January 2022, after first online publication: The author name Pieter Hameeteman has been corrected to Pieter W. Hameeteman in this version].
- 1. van Poelgeest, E.P. et al. Characterization of immune cell, endothelial, and renal responses upon experimental human endotoxemia. J. Pharmacol. Toxicol. Methods 89, 39–46 (2018). [DOI] [PubMed] [Google Scholar]
- 2. Brooks, D. , Barr, L.C. , Wiscombe, S. , McAuley, D.F. , Simpson, A.J. & Rostron, A.J. Human lipopolysaccharide models provide mechanistic and therapeutic insights into systemic and pulmonary inflammation. Eur. Respir. J. (2020). 56, 1901298. [DOI] [PubMed] [Google Scholar]
- 3. Seeley, J.J. & Ghosh, S. Molecular mechanisms of innate memory and tolerance to LPS. J. Leukoc. Biol. 101, 107–119 (2017). [DOI] [PubMed] [Google Scholar]
- 4. Greisman, S.E. & Woodward, W.E. Mechanisms of Endotoxin tolerance. 3. The refractory state during continuous intravenous infusion of endotoxin. J. Exp. Med. 121, 911–933 (1965). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Basran, A. et al. Roles of neutrophils in the regulation of the extent of human inflammation through delivery of IL‐1 and clearance of chemokines. J. Leukoc. Biol. 93, 7–19 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buters, T.P. et al. Intradermal lipopolysaccharide challenge as an acute in vivo inflammatory model in healthy volunteers. Br. J. Clin. Pharmacol. (2021). https://doi.org/ 10.1111/bcp.14999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Michel, O. et al. Evaluation of oral corticosteroids and phosphodiesterase‐4 inhibitor on the acute inflammation induced by inhaled lipopolysaccharide in human. Pulm. Pharmacol. Ther. 20, 676–683 (2007). [DOI] [PubMed] [Google Scholar]
- 8. Bartko, J. et al. Dissociation between systemic and pulmonary anti‐inflammatory effects of dexamethasone in humans. Br. J. Clin. Pharmacol. 81, 865–877 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Motwani, M.P. et al. Novel translational model of resolving inflammation triggered by UV‐killed E. coli. J. Pathol. Clin. Res. 2, 154–165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dauphinee, S.M. & Karsan, A. Lipopolysaccharide signaling in endothelial cells. Lab. Invest. 86, 9–22 (2006). [DOI] [PubMed] [Google Scholar]
- 11. Cals‐Grierson, M.M. & Ormerod, A.D. Nitric oxide function in the skin. Nitric Oxide 10, 179–193 (2004). [DOI] [PubMed] [Google Scholar]
- 12. Ahluwalia, A. Topical glucocorticoids and the skin–mechanisms of action: an update. Mediators Inflamm. 7, 183–193 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Faurschou, A. & Wulf, H.C. Topical corticosteroids in the treatment of acute sunburn: a randomized, double‐blind clinical trial. Arch. Dermatol. 144, 620–624 (2008). [DOI] [PubMed] [Google Scholar]
- 14. Rhodes, L.E. , Belgi, G. , Parslew, R. , McLoughlin, L. , Clough, G.F. & Friedmann, P.S. Ultraviolet‐B‐induced erythema is mediated by nitric oxide and prostaglandin E2 in combination. J. Invest. Dermatol. 117, 880–885 (2001). [DOI] [PubMed] [Google Scholar]
- 15. Ronchetti, S. , Ricci, E. , Migliorati, G. , Gentili, M. & Riccardi, C. How Glucocorticoids affect the neutrophil life. Int. J. Mol. Sci. 19, 4090 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Filep, J.G. , Delalandre, A. , Payette, Y. & Földes‐Filep, É. Glucocorticoid receptor regulates expression of L‐selectin and CD11/CD18 on human neutrophils. Circulation 96, 295–301 (1997). [DOI] [PubMed] [Google Scholar]
- 17. Rosales, C. Neutrophil: a cell with many roles in inflammation or several cell types? Front. Physiol. 9, 113 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Moser, M. et al. Glucocorticoids down‐regulate dendritic cell function in vitro and in vivo . Eur. J. Immunol. 25, 2818–2824 (1995). [DOI] [PubMed] [Google Scholar]
- 19. Ehrchen, J.M. , Roth, J. & Barczyk‐Kahlert, K. More than suppression: glucocorticoid action on monocytes and macrophages. Front. Immunol. 10, 2028 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kirnbauer, R. et al. Regulation of epidermal cell interleukin‐6 production by UV light and corticosteroids. J. Invest. Dermatol. 96, 484–489 (1991). [DOI] [PubMed] [Google Scholar]
- 21. Waage, A. , Slupphaug, G. & Shalaby, R. Glucocorticoids inhibit the production of IL6 from monocytes, endothelial cells and fibroblasts. Eur. J. Immunol. 20, 2439–2443 (1990). [DOI] [PubMed] [Google Scholar]
- 22. de Kruif, M.D. et al. Prednisolone dose‐dependently influences inflammation and coagulation during human endotoxemia. J. Immunol. 178, 1845–1851 (2007). [DOI] [PubMed] [Google Scholar]
- 23. Kauh, E. et al. Prednisone affects inflammation, glucose tolerance, and bone turnover within hours of treatment in healthy individuals. Eur. J. Endocrinol. 166, 459–467 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Table S1
Table S2
Table S3