

Abstract
This review addresses questions on how to accomplish successful central nervous system (CNS) drug delivery (i.e., having the right concentration at the right CNS site, at the right time), by understanding the rate and extent of blood‐brain barrier (BBB) transport and intra‐CNS distribution in relation to CNS target site(s) exposure. To this end, we need to obtain and integrate quantitative and connected data on BBB using the Combinatory Mapping Approach that includes in vivo and ex vivo animal measurements, and the physiologically based comprehensive LEICNSPK3.0 mathematical model that can translate from animals to humans. For small molecules, slow diffusional BBB transport and active influx and efflux BBB transport determine the differences between plasma and CNS pharmacokinetics. Obviously, active efflux is important for limiting CNS drug delivery. Furthermore, liposomal formulations of small molecules may to a certain extent circumvent active influx and efflux at the BBB. Interestingly, for CNS pathologies, despite all reported disease associated BBB and CNS functional changes in animals and humans, integrative studies typically show a lack of changes on CNS drug delivery for the small molecules. In contrast, the understanding of the complex vesicle‐based BBB transport modes that are important for CNS delivery of large molecules is in progress, and their BBB transport seems to be significantly affected by CNS diseases. In conclusion, today, CNS drug delivery of small drugs can be well assessed and understood by integrative approaches, although there is still quite a long way to go to understand CNS drug delivery of large molecules.
RELEVANT QUESTIONS FOR SUCCESSFUL CENTRAL NERVOUS SYSTEM (TARGET SITE) DELIVERY
With the aim to have the right concentration at the right central nervous system (CNS) site, a number of questions are important:
How much drug is entering the CNS and how fast?
In relation to the necessary concentrations for appropriate drug effect at the CNS target site, be it extra‐ or intracellular in the brain, or in the spinal cord. Here, the focus should be on the unbound concentrations that are available for blood‐brain barrier (BBB) transport, intra‐brain distribution, and interaction with the CNS target site.
How can we address the interconnection of CNS target site delivery processes?
Drug distribution into and within the CNS is governed by several concentration and time‐dependent processes that are connected and at the same time independent of each other. Following (unbound) drug exposure in plasma we need to consider passive and active transport across the BBB, brain extracellular fluid (ECF) bulk flow, passive and active extracellular‐intracellular exchange, and cerebrospinal fluid (CSF) turnover. Different types of information on the above‐mentioned processes can be obtained by in silico, in vitro, ex vivo, and in vivo studies and should be further integrated, for which physiologically based approaches are best suitable.
What is available for target interaction?
This requires understanding of the relationship between the processes that govern CNS target site pharmacokinetics (PKs), as indicated above. The CNS target site interaction will depend on target site PK and binding kinetics. Integrative approaches lead to understanding how a specific drug should be administered and what dose is needed to achieve the plasma PK profile for optimal CNS target site distribution.
How to influence CNS drug delivery?
For drug properties, instead of what was earlier believed, more lipophilic drugs will tend to have more brain binding but will not improve CNS drug delivery in terms of unbound concentrations available for the target, while also increasing the risk of interaction with the most important efflux transporter at the BBB, P‐glycoprotein (P‐gp). Apart from the above‐mentioned change in CNS target site exposure by adapting plasma PK, changes in CNS target site exposure can be achieved by the use of nano formulations. For avoidance of CNS side effects, the drug should be a strong substrate for efflux transport at the BBB.
How to address the impact of diseases?
Measurement of CNS drug delivery in a diseased human brain can only be made with noninvasive imaging techniques, lacking information of the unbound drug, or via CSF lumbar puncture for which the relationship to brain target site concentrations is context depended. The second‐best option is to measure in and learn from animal models of disease.
All these questions are addressed in more detail below.
HOW MUCH DRUG IS ENTERING THE CNS AND HOW FAST?
The BBB and its properties
The BBB consists of the endothelial cells of the brain capillaries, separating blood from the brain parenchymal tissue. It is a part of the integrative neurovascular unit, where also astrocytes, pericytes, and the basement membrane, microglia, as well as the glycocalyx covering the blood side of the endothelial cells, are important players to support the crucial properties of the BBB. 1 , 2 The BBB is present throughout the brain parenchyma and therefore is the most efficient interface for CNS drug delivery. 3 However, many small and even more so larger drugs are hindered in their entry into the CNS. This may be caused by restricted passive BBB transport, and/or active efflux transporters that effectively limit the BBB transport. On the other hand, active influx transporters may be an opportunity to enhance the delivery. 4 , 5
Potential new therapeutics for many CNS diseases are large molecules/ biologics, including recombinant lysosomal enzymes, neurotrophins, decoy receptors, and therapeutic antibodies. These large molecules in general cross the BBB to an extremely low extent. The restrictive properties of the BBB include lower levels of vesicular trafficking than other vessels, that limit transcellular transport or transcytosis. 6 , 7 Thus, BBB targeting vectors have to be used. Receptor‐specific monoclonal antibodies cross the BBB via receptor‐mediated transport. Although a lot has been learned on the mechanisms that regulate transcytosis at the BBB, it is still to a large extent a black box, and the basic cell biology of transcytosis at the BBB is still elusive. 7 , 8 , 9
Three fundamental CNS drug delivery properties
CNS drug delivery can be described with three fundamental properties. The first is the rate of transport across the BBB (with associated parameters like clearance in, clearance out, permeability, PS product, etc.), that has been studied for many years both in vivo and in vitro. It is important not to confound this measure with the extent of delivery to the CNS, as these are two independent properties of the delivery. The extent of brain delivery describes the partitioning of drug across the BBB at steady‐state. When expressed as the ratio of the unbound drug in the brain over the unbound drug in plasma, it is called Kp ,uu,brain, where Kp ,uu values < 1 indicate net active efflux, Kp ,uu values ~ 1 indicate dominatingly passive transport, and Kp ,uu values > 1 indicate net transport into the brain. The third aspect is the nonspecific and specific binding of the drug to brain parenchymal tissue. Total brain concentrations as such are not informative, as only the unbound concentration is available for target interaction. Brain tissue binding can therefore be expressed with Vu ,brain, the unbound volume of distribution of a drug in the brain (further described below). The larger the brain tissue binding (larger Vu ,brain) the longer the time it takes to reach equilibrium between plasma and the brain.
The need to distinguish among rate, extent, and brain binding in understanding CNS drug delivery can be exemplified by results on opioids (Table 1 ). 10
Table 1.
Opioid rate and extent of BBB transport and brain binding to illustrate the independence of these parameters
| Drug | Extent | Rate | Brain binding | Reference |
|---|---|---|---|---|
| Kp ,uu,brain | Permeability clearance (µl*min‐1*brain‐1) |
Vu ,brain (ml*g brain‐1) |
||
| Oxycodone | 3.0 | 1910 | 2.2 | 4 |
| Codeine | 1.06 | ‐ | 3.6 | 102 |
| Morphine | 0.29 | 14 | 1.7 | 103, 104 |
| M6G | 0.29 | 0.35 | 0.19 | 105 |
| M3G | 0.08 | 0.11 | 0.25 | 106 |
| Loperamide | 0.007 | 99 | 370 | 107, 108 |
BBB, blood‐brain barrier.
These results clearly show that a drug can have a fast rate of BBB transport (permeability clearance) but have a low extent of BBB transport (Kp ,uu,brain; e.g., due to being a strong P‐gp (and/or BCRP) substrate), whereas also the opposite is possible, with a slow rate of BBB transport but a higher CNS delivery. This clearly illustrates that rate, extent, and binding are three independent properties. 4 , 10 , 11 More generally speaking, Kp ,uu,brain can differ 430‐fold, as shown in Table 1 . This clinically relevant range is smaller than what has been obtained for total brain concentrations (as log BB or Kp ), which has been shown to differ by up to at least 2,000‐fold, or for BBB permeability, which span an even larger range of up to at least 20,000‐fold, the 2 latter promising more than what can be accomplished clinically. 10 The extent of delivery of unbound drug is considered to be the most important measure, unless a fast rate (in and out) is essential (e.g., in achieving and recover from anesthesia).
ASSESSMENT OF BBB TRANSPORT AND INTRA‐BRAIN DISTRIBUTION
Different methods are used to assess the rate and/or the extent of BBB transport. These range from in silico, in vitro, to in vivo methods and from old to very recently developed methods. The choice of how to assess BBB transport should be based on the question to be answered.
In vitro methods
Cell models (rate and extent)
BBB cell models can be used to investigate mechanisms of BBB function in disease as well as rate and extent of drug transport. For transport studies, epithelial cell lines, such as MDCK cells from kidney origin, or Caco‐2 cells transfected with P‐gp, and/or BCRP, are often used instead of BBB cell models. Whereas valuable to test potential transporter substrates, they lack BBB‐specific properties. 12 , 13 Permeability is most often measured as permeability surface area product. The efflux ratio, measuring apical to basolateral and the opposite, is sometimes used as an in vitro correlate to the extent parameter Kp ,uu,brain. Quite good results have been reported to map Kp ,uu,brain in vitro. 14 However, more efforts are needed to further develop the cell models for comparisons with and ultimate prediction of in vivo drug transport.
The cell culture field sometimes still compare rate with extent (e.g., by correlating permeability to total partitioning (Kp )). 15 , 16 , 17 , 18 As both total concentration and permeability are connected to lipophilicity, a good correlation in these models is not of real clinical value.
Much progress has been made with the ability to produce BBB cells from human pluripotent stem cells and induced pluripotent stem cells. 19 , 20 These cells produce much tighter junctions than, for example, the very much used immortalized human hCMEC/D3 cells, 21 and the stem cell‐derived cells are today state‐of‐the‐art. 19 , 22
The latest promising progress in the field is using spheroids of the BBB and neurovascular unit, 23 , 24 but these methods are not yet validated regarding their ability to quantify drug transport. Other developments in the area are the dynamic and microfluidic models. These models are rather complicated and time‐consuming and are likely less suitable for high‐throughput studies. 25 In addition, in vitro membranes or other chemical methods are used to study (passive) permeability, like the PAMPA‐BBB method, that is extensively used. 26
Although all the above‐mentioned techniques have been applied to small molecules, understanding of BBB transport and intra‐brain distribution of large molecules is currently a challenge, and huge efforts are being made to further understand the BBB transport mechanisms associated with large molecules’ mainly receptor mediated transport. 6
In situ/ex vivo methods
The brain homogenate method
The brain homogenate method can be used for assessment of the extent of brain tissue binding and is needed as a complement to obtain unbound concentrations in the brain. It gives the nonspecific binding to brain tissue components, as the fraction of unbound drug in brain homogenate (fu ,brain). Brains can be obtained following drug administration at the end of an animal experiment, or can be obtained from naïve animals, with drug being administered to the brain homogenate. Diluted homogenate is used and recalculated back to nondiluted values. 27
In situ brain perfusion
The in situ brain perfusion technique can be used to measure the kinetics of the rate of BBB transport as brain uptake, from which transport or permeability constants can be calculated. It is based on direct perfusion of the internal carotid artery containing the drug, together with two calibrator compounds, one with negligible and the other with flow limited brain uptake. The brains of rats or mice can be harvested at different timepoints after the start of the perfusion, and the amount of compounds in the brain and the flow rate of the perfusion are used for calculations. 28 , 29 , 30
Brain slice
The brain slice technique can be used for assessment of the extent of brain distribution and binding. It measures the average uptake into brain cells, measured as the volume of distribution of unbound drug, Vu ,brain (mL/g brain). It measures the ratio of total drug concentration in the slice in relation to the concentration in the surrounding buffer, the latter representing unbound drug concentration. 31 , 32 As the slices are functional, the distribution describes binding, possible active uptake, and pH partitioning into subcellular structures, and is therefore more physiologically relevant than the brain homogenate method.
Quantitative proteomics
Quantitative proteomics can be used to quantitatively investigate transport systems at the BBB. It provides a means to relate transporter protein expression levels between animals and humans (as well as between in vitro and in vivo). Uchida and colleagues demonstrated, for example, that P‐gp functionality in in vivo BBB can be accurately predicted from data obtained from an in vitro system, in mice, monkeys, and humans (also in pathological conditions). 33 , 34 , 35
In vivo methods
CSF sampling
CSF sampling can be used to assess both rate and extent of drug distribution to the CNS. It is often used as an alternative to brainECF concentrations, and compared with plasma concentrations to have an indication of BBB transport. In animals, terminal CSF sampling is often used in rats. A time‐course of concentrations in CSF can be obtained through serial CSF sampling via a permanent cannula in the cisterna magna. However, serial CSF sampling withdraws fluid that can have a significant influence on the CSF volume in animals, and may therefore influence physiology, a concern that must be taken into account. Rat (cisterna magna) and human (lumbar) CSF concentration have also been questioned as good biomarkers for brain unbound target site concentrations. 36 Typically in humans, lumbar CSF is collected by a single lumbar puncture (at presumed steady‐state conditions). Reviewing this relationship, CSF drug concentrations provided a rather good indication, 37 but not a reliable measure for predicting brainECF concentrations. 36 Although CSF may not equal or closely resemble CNS target site concentrations, it does provide information on the extent of drug distribution between plasma and lumbar CSF, which may be useful to bridge preclinical and clinical CSF (Kp ,uu,CSF). 37 However, it leaves out information on the time‐dependency (rate). 36 Moreover, the relation between brainECF and lumbar CSF concentrations depends on the relative contribution of CNS processes, especially the CSF turnover rate and possible differences in the quantitative presence of transporters. 38
Microdialysis
Microdialysis is a technique that allows location‐dependent measurement of unbound drug concentrations as a function of time, and thereby provides information on both the rate and extent of BBB transport, while also giving information on unbound plasma PK (that also can be determined by microdialysis). To date, microdialysis is the only technique that provides such quantitative and time resolution information of the unbound drug in brainECF at the location of the semipermeable part of the tiny microdialysis probe. To that end, drug molecules need to be small enough to traverse the semipermeable membrane and go into the dialysate upon constant perfusion of the microdialysis probe. The dialysate is collected in fractions, and is subsequently analyzed. The dialysate concentrations need to be corrected for in vivo recovery of the drug molecules, as there will be a difference between dialysate and brainECF concentrations. 39 As microdialysis is a water‐based technique, it has limitations for very lipophilic compounds that tend to stick to tubing and probes. With these limitations in mind, it is an invaluable method for detailed investigation of CNS drug distribution. Although minimally invasive to the brain of rats, the technique is not widely accepted to be used in the human brain, where it may only be used under very special conditions. 40 , 41
Open flow microperfusion/large pore microdialysis
Cerebral open flow microperfusion 42 and large pore microdialysis 43 , 44 can be used to assess BBB transport of large molecules including nanobodies. These techniques resemble the classical microdialysis as described above, but with the absence of a semipermeable membrane or with large pores in the membrane. Therefore, a push‐pull system is needed to balance the fluid that goes in and what goes out from the brain tissue. By continuously sampling brainECF, it provides information on rate and extent of BBB transport for (larger) molecules. In addition, here, in vivo recovery should be determined.
Positron emission tomography
Positron emission tomography (PET) is a powerful method to obtain information on the rate and extent of CNS drug delivery and the influence of transporter functionality. 45 This imaging technique is based on the injection of a PET ligand that carries a positron‐emitting radionuclide (tracer), where the emitted positron annihilates with an electron to produce gamma rays that are detected by gamma cameras to form a 3D image, as a function of time. The time span is limited by the decay of the radiotracer, and is 60 minutes for 11C (3 half‐lives). While giving spatial information in addition, which is a plus on its own, this technique does not distinguish between bound and unbound molecules, nor between extra and intracellular concentrations. In addition, radioactive metabolites that enter the brain are included in the measurements. The BBB transport information can thus not be obtained separated from intra‐brain distribution processes. To bridge PET data to unbound concentrations, PET and microdialysis has been combined in an investigative study. 46 Results are promising, but more studies are needed to see if PET can be used as a translational tool in humans, something that would be very valuable. PET is also useful for large molecules, as shown by measurements on brain distribution of a bispecific antibody‐based PET radioligand for imaging of amyloid‐β. 47 As PET is noninvasive and can be used in humans, and it was used to evaluate prediction of human Kp,uu,brain via a proteomics‐informed relative expressive factor approach. 48 Using in vitro data on active P‐gp transporter functionality based on efflux ratios in a human P‐gp‐transfected MDCKII cell based Transwell assay, the Kp ,uu,brain of verapamil, N‐desmethyl loperamide, and metoclopramide could be adequately predicted within two‐fold of observed data.
In silico models
Many in silico studies has modeled the rate (permeability) and/or extent of BBB transport based on the total ratio of drug concentration in brain vs. plasma (logBB or Kp ,brain). In these studies, lipophilicity was found to be the main driver. 49 , 50 , 51
When studying the extent of BBB transport of the unbound drug, it was found that among many different physicochemical properties, Kp ,uu,brain was mainly dependent on the number of hydrogen bond acceptors, and not on lipophilicity. Hydrogen bonding acceptor properties could explain ~ 40% of the correlation between prediction and in vivo data. 37 , 52 , 53 , 54 These models are therefore mainly suitable for qualitative classification and are not yet capable of predicting the Kp ,uu,brain of CNS discovery compounds. 55 This is because incorporation of active transport at the BBB needs additional attention. Recent mathematical model efforts include incorporation of P‐gp efflux ratio 56 , 57 and BCRP. 58 The Brain Exposure Efficiency Score is another model approach for including influx and efflux active transporters. 59
CNS physiologically‐based PK (PBPK) models are more complex by taking into account many PK processes. The basis for these models is that the PK of a drug in CNS compartments is the result of the combination of CNS physiology and drug properties. Therefore, these models allow translation between CNS systems having their own individual CNS physiology (e.g., between species, between health and a particular disease, etc.). A number of CNS PBPK models have been developed for small molecules, 38 , 60 , 61 , 62 , 63 , 64 , 65 , 66 and a first model has been developed also for anti‐transferrin receptor antibody affinity variants in rats determined using microdialysis. 43 , 44 The LEICNSPK3.0 model is the most comprehensive and generic small molecule CNS PBPK model. It explicitly includes extracellular and intracellular brain compartments, the brain target sites, and will be discussed below in the section on integrative approaches. 38 , 66
INTEGRATIVE APPROACHES TO UNDERSTAND INTERDEPENDENCIES OF PROCESSES IN CNS TARGET SITE DELIVERY
Mastermind research approach
Transport of drugs into, within, and out of the CNS is governed by the BBB, the anatomy of the brain parenchyma and fluid spaces, and physiological processes, as well as drug‐specific properties. In combination, they determine the drug concentration‐time profiles in specific CNS compartments. The interest in CNS target site delivery has put much focus on BBB transport. However, we also need to look beyond the BBB and include intra‐brain distribution processes. When data are obtained at multiple locations and as a function of time from individual study subjects, mathematical modeling of these data can reveal the rate and extent of the underlying processes as well as their mutual interdependencies at best, because the data obtained from that subject are interconnected (smart data). This approach is called the “Mastermind Research Approach,” in allegory to the mastermind board game. 39 In this game, you have to study the position and color of all pins, as otherwise you will not be able to crack the code of the multiple pins with their colors and positions. In other words, the Mastermind Research Approach indicates to measure as many parameters as possible from a particular setting, and to relate all these parameters for understanding of their inter‐relationships. It is very powerful in (also) CNS drug research, to reveal the contributions and variability of individual processes on the causal path between drug dosing and CNS effect in animals. This can be converted to the human situation by replacement of the animal physiological parameters by those of humans.
Combinatory mapping approach
The Combinatory Mapping Approach (CMA) integrates the Kp ,uu,brain with intra‐brain distribution of drugs in steady‐state conditions (Figure 1 ). 67 With the CMA, the steady‐state relationship between plasma PK and CNS PK in different brain compartments can be obtained in a more high‐throughput mode, which makes it very useful in drug discovery. Combining all information, it provides early CNS development information on plasma and target site concentrations, to be linked to pharmacodynamics, with a possibility to give preclinical information on the dose needed to obtain central effects. Based on the target site concentrations needed for effect, corresponding plasma concentrations and dose requirements can be calculated.
Figure 1.
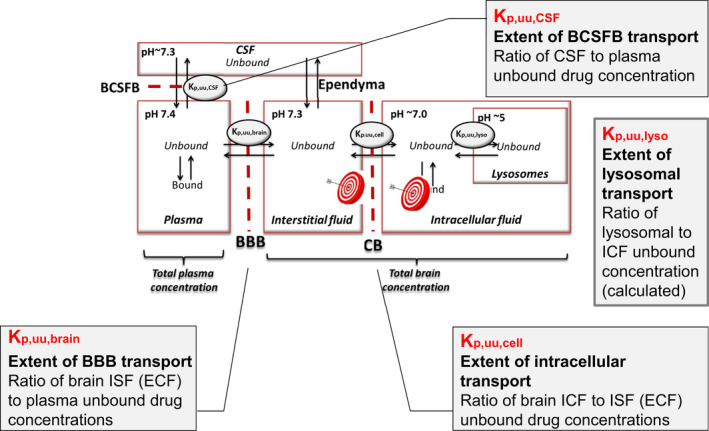
The Combinatory Mapping Approach. The combinatory Mapping Approach, providing partition coefficients of unbound drug (Kp ,uu values) between multiple CNS compartments: brain interstitial/extracellular fluid (brainECF = brainISF), brain intracellular fluid (brainICF), brain cell lysosomes, via measurements and/or calculations, also including compartment pH and pKa dependent neutral and charged drug molecules. The extent of transport between plasma and CSF is described by the blood‐cerebrospinal fluid barrier (BCSFB). BBB, blood‐brain barrier; CNS, central nervous system; CSF, cerebrospinal fluid.
Kp ,uu,cell represents cellular uptake in relation to brainECF concentrations. The Kp ,uu,cell value includes subcellular distribution and is influenced by the degree of lysosomal uptake and/or active uptake or efflux at the cellular barrier. Subcellular drug distribution can be estimated based on the pH partitioning theory. Thus, the cytosolic partitioning Kp ,uu,cyto (cytosolic to brainECF concentration ratio), and Kp ,uu,lyso (partitioning into acidic subcellular compartments in relation to cytosolic concentrations), can be predicted using pH and pKa values. The cellular partitioning Kp ,uu,cell, can also be predicted in the same way or be measured. In comparative studies, it was found that measured vs. predicted cellular partitioning with Kp ,uu,cell in some cases deviated quite extensively, promoting the use of the measured parameter with using Vu ,brain obtained by the brain slice technique, as this technique leaves intact the cellular compartments. 67 , 68
The PBPK LeiCNSPK3.0 model
The most comprehensive CNS PBPK model is the LeiCNSPK3.0 model that has been developed by the Mastermind Research Approach. 39 Smart data was generated by in‐house experiments in rats, measuring CNS drug distribution by microdialysis at multiple locations in individual rats for multiple drugs, with and without active transport blockers, and by using literature data on rat CNS physiology. By this approach, in one microdialysis experiment in each single freely moving animal, serial multilevel time course data are obtained that are per definition linked. Advanced mathematical modeling of such data can reveal the inter‐relationships of PK processes. This is a highly efficient approach for developing predictive PBPK models, while also contributing to refinement, reduction, and replacement of animal experiments. 62 , 63 , 64 , 65 The LEICNSPK3.0 model structure includes peripheral, plasma, and multiple CNS compartments, fluid flow, pH, and pKa dependent neutral and charged drug molecules, and brain membrane nonspecific binding (Figure 2a ). The predictions are shown for examples for rats (Figure 2b ) as well as humans (Figure 2c ), with predictions being within twofold error of actually observed data in humans from other studies. 38
Figure 2.
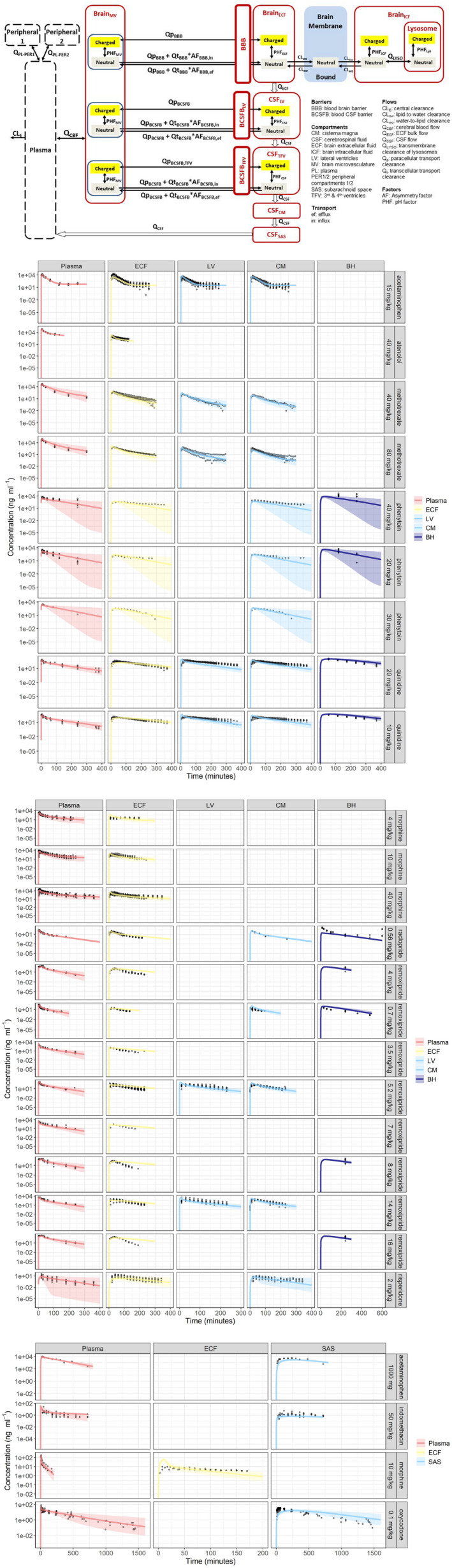
The LEICNSPK3.0 model. (a) The LEICNSPK3.0 model structure, including peripheral, plasma, and multiple CNS compartments (brainECF, brainICF, brain cell lysosomes, and CSF in LV = lateral ventricle, TFV = third and fourth ventricle, CM = cisterna magna; and SAS = subarachnoid space), (brainECF bulk flow and CSF flow), pH and pKa dependent neural and charged drug molecules, and brain membrane (nonspecific) binding. (b) Model predictions overlayed actual data observed in rats. (c) Model prediction and actual data observed in humans. 38 CNS, central nervous system; CSF, cerebrospinal fluid; ECF, extracellular fluid.
This model has been used as an “in silico” screening method for “what‐if” scenarios, in which physiological parameters reported to be changed in different CNS conditions were used to observe possible impact of such changes on the PK in different CNS compartments. 38 The model can as well be used to explore CNS distribution of new investigational drugs (preferable with input from Kp ,uu values as obtained via the CMA), and be used further during later stages of CNS drug development. It can also explore optimal plasma PK profiles for the intended CNS target site exposure.
Chang et al. developed a PBPK model to characterize brain disposition of mAbs in the mouse, rat, monkey, and in humans. 69 In addition to the LeiCNSPK3.0 model backbone, this model added FcRn mediated transport of mAbs. It was used to be the first model‐based characterization of published and in‐house PK data on the disposition of mAbs in the rat brain, including the data on PK of mAb in different regions of brain determined using microdialysis studies performed in‐house.
HOW TO CHANGE CNS DRUG DELIVERY?
Today, there is a much higher awareness of relevant methods and measurements to select the best compounds to maximize CNS effects, or the opposite—to select the ones that minimize side effects from the CNS. From drug synthesis and formulation perspectives, there are factors that can be thought of in order to reduce or enhance CNS exposure.
How to reduce?
To reduce CNS side effects the BBB can be used as an opportunity, where the most efficient method of reducing CNS drug delivery is that the drug is a strong P‐gp substrate (and/or BCRP substrate). This is obvious, for example, the second generation antihistamines with their low transport to the brain and thereby less sedation as a side effect. Loperamide is another excellent example of a drug with intended peripheral effects, being a very strong P‐gp substrate that hinders CNS effects. However, both antihistamines and loperamide were developed by serendipity and not by a planned strategy. Today, with the increased knowledge on BBB transport this knowledge can be used as a strategy. The PET study on species differences in BBB transport of the three PET ligands [11C]‐verapamil, [11C]‐GR205171, and [18F]‐altanserin exemplifies the impact of P‐gp on brain exposure in these species. 70
To be noted is that a study on regional differences in brain drug delivery in rats showed that antipsychotics exhibited quite large regional differences in their distribution across the BBB. Risperidone, being a strong P‐gp substrate exhibited the highest difference, with 5.4‐fold higher distribution into frontal cortex than to cerebellum (Kp ,uu,brain 0.28 vs. 0.05). 71 Thus, this can also influence possible effect vs. side effect relationships. The binding differed maximally twofold between regions.
How to enhance?
Drugs with moderate to high passive permeability and no significant interaction with active transporters (Kp ,uu,brain close to unity) are likely the least influenced by other delivery strategies than to enhance the dose.
For drugs with other properties, multiple approaches are available or in development. Here, we discuss only the noninvasive approaches.
Active influx transporters
Active influx transporters have been found at the BBB for a number of drugs to be active for morphine, 72 oxycodone, 4 diphenhydramine, 5 , 73 and naloxone. 74 Other drugs like L‐Dopa, melphalan, gabapentin, and ketoprofen 75 utilize the L‐amino acid transporter 1 (LAT1) to cross the BBB. Recent advances in LAT1‐mediated targeted CNS drug delivery have been reviewed. 76
Nanodelivery
Nanodelivery can be seen as a promising noninvasive technology with which to improve uptake across the BBB. 77 From a clinical perspective, biocompatibility and avoidance of adverse effects from the dosage form are of utmost importance. In this regard, liposomal formulations are the most well‐tested and safe alternatives, that are also in clinical use for other purposes. It should, however, be noted that liposomal administration adds another level of complexity in terms of additional PK processes, as the distribution and elimination of the liposome itself and release of the drug in the body and brain should be considered (Figure 3 ). For understanding and further prediction to humans, mechanism‐based in vivo (microdialysis) experiments are the only ones that can separate the liposomal encapsulated drug from the released drug, something needed to decipher the processes.
Figure 3.
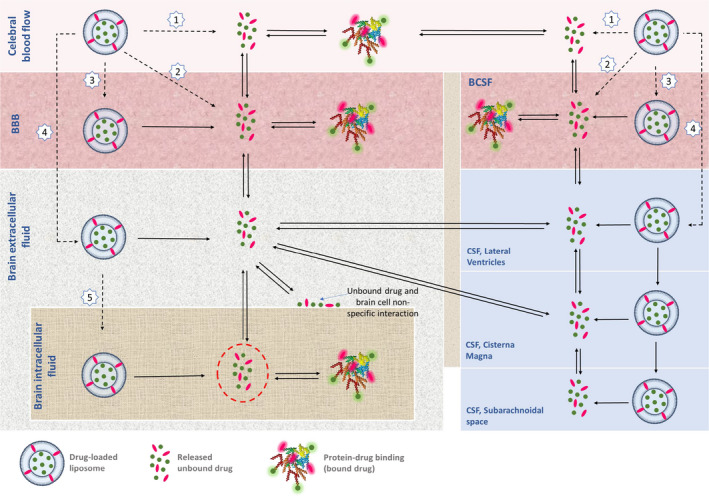
Schematic processes involved in the fate of the drug, using liposomal formulation for targeted brain drug delivery. Liposomal release of the drug in plasma, liposomal transport across the BBB, and liposomal release of the drug into brainECF should be considered as well as plasma PK, BBB transport, brain delivery, and intra‐brain distribution of the unbound drug itself. The drug can reach the target site via a liposome as a carrier (black dashed line) or as the released unbound drug (black full line). After intravenous administration of the liposome containing the drug, the following can happen: [1] The drug can be released from the liposome in the blood and reversibly bind to plasma proteins. It is only the unbound drug that can cross the BBB or BCSFB to reach the brainECF: [2] The liposome can fuse with the BBB cell membrane and release the drug into the cytosol of the BBB endothelial / BCSFB epithelial cells; [3] The liposome may undergo endocytosis in the BBB / BCSFB and release the drug into the intracellular fluid of the BBB endothelial / BCSFB epithelial cells; [4] The liposome can cross the BBB / BCSFB via transcytosis to reach the brainECF, followed by drug release into the brainECF / CSF. The unbound drug can exchange between brainECF and CSF, and between brainECF and brainICF; [5] The liposome may enter brainICF and can release the drug in the brainICF. Only the unbound drugs that reach the brainECF and/or brainICF (red dashed circles) are available for target site binding in these physiological compartments to induce their pharmacological effect. 78 BBB, blood‐brain barrier; BCSFB, blood‐cerebrospinal fluid barrier; CSF, cerebrospinal fluid; ECF, extracellular fluid; PK, pharmacokinetic.
Many studies have been performed in experimental animals on liposomal brain delivery. Whereas providing interesting data, such studies have room for improvement to provide sufficient mechanistic insight. 78 A number of animal studies using microdialysis for time‐course information on unbound drug in plasma and brain provide such mechanistic information. 78 , 79 , 80 , 81 , 82 , 83 , 84
Overall, these studies show that brain distribution of a drug administered as liposomal formulation depends on both drug properties and liposomal formulation characteristics (Table 2 ). It may indeed enhance BBB drug delivery for drugs that have limited BBB transport, such as methotrexate and DAMGO, but may also counteract active uptake, as is the case for diphenhydramine (Table 2 ) and quetiapine, where the latter used lipid core nanocapsules. 79 In general, the rather sparse quantitative data gathered so far seems to indicate that active transporters at the BBB, be it both influx and efflux transporters, can under certain conditions be circumvented by liposomal delivery. In other situations, no change is observed, which complicated the picture quite significantly. More quantitative studies are needed to provide an improved basis, including its prediction to humans from animal data. 78 , 84 , 85 , 86
Table 2.
Mechanism‐based (microdialysis) approaches used to determine Kp ,uu,brain values after administration of the free drug, as well as after administration of the free drug in the presence of empty liposomes, as well as after administration of the drug loaded in the liposomal formulation
| Liposomal formulation | Drug | Kp,uu, brain values | Drug delivery enhancement | Formulation | Administration details | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Free drug | Co‐administration free drug + liposomes | Drug‐loaded liposome | Free drug + liposomes/Free drug | Free drug/drug‐loaded liposome | |||||||||
| Kp,uu, free drug | Kp,uu, free drug + liposome | Kp,uu, drug in liposome | Kp,uu, free+ lipo control/Kp ,uu, drug free | Kp,uu, drug in lipo /Kpuu, free | EYPC | HSPC | Cholesterol | PEG | Loading dose | Constant infussion | |||
| GSH‐PEG liposomal (EYPC phospholipid) | DAMGO | 0.09 | NA | 0.2 | NA | 2.3 | 1,000 mM | NA | 750 mM | 18 mM mPEG2000‐DSPE (1 mol%) | 75 µg/minute/kg free DAMGO; 1250 μg; liposomal DAMGO/minute/kg for 10 minutes | 60 µg/minute/kg of free DAMGO and liposomal DAMGO for 2 hours | 109 |
| GSH‐PEG liposomal (EYPC phospholipid) | 0.05 | 0.05 | 0.1 | 1.0 | 2.0 | 100 mM | NA | 750 mM | 18 mM mPEG2000‐DSPE (1 mol%) | 75 µg/minute/kg free DAMGO, liposome emulsion (1250 μg DAMGO/minute/kg) for 10 minutes | 60 µg/minute/kg of free DAMGO and liposomal DAMGO for 2 hours | 85 | |
| PEG liposomal (EYPC phospholipid) | 0.05 | 0.05 | 0.08 | 1.0 | 1.6 | ||||||||
| PEG liposomal (EYPC phospholipid) | DPH | 3.00 | 2.3 | 1.50 | 0.8 | 0.5 | 100 mM | NA | 66 mM | 8.7 mM mPEG2000‐DSPE (5 mol%) | 4.5 mg/kg (150 μg/minute/kg) of PEG liposomal and 4.5 mg/kg free DPH for 30 minutes (a short infusion regiment) | 82 | |
| PEG liposomal (EYPC phospholipid)‐low dose of liposome | MTX | 0.10 | NA | 0.28 | NA | 2.8 | 100 mM | NA | 66 mM | 8.7 mM mPEG2000‐DSPE (5 mol%) | 2.3 mg/kg (77 mg/minute/kg) and free MTX of 7.2 µg/minute/kg for 30 minutes | 2.3 mg/kg (77 mg/minute/kg) and free MTX of 6 µg/minute/kg for 9.5 hours | 110 |
| PEG liposomal (EYPC phospholipid) ‐ High dose of liposome | 0.10 | NA | 0.32 | NA | 3.2 | 15 mg/kg (500 mg/minute/kg) and free MTX of 7.2 µg/minute/kg for 30 minutes | 15 mg/kg (500 mg/minute/kg) and free MTX of 6 µg/minute/kg for 9.5 hours | ||||||
| PEG liposomal (HSPC phospholipid) | 0.10 | NA | 0.11 | 9.8 | 1.1 | NA | 100 mM | 15 mg/kg (500 mg/minute/kg) and free MTX of 7.2 µg/minute/kg for 30 minutes | 15 mg/kg (500 mg/minute/kg) and free MTX of 6 µg/minute/kg for 9.5 hours | ||||
| PEG liposomal (EYPC phospholipid) | MTX | 0.10 | 0.09 | 1.50 | 0.9 | 15.0 | 100 mM | NA | 66 mM | 1.7 mM mPEG2000‐DSPE (1 mol%) | free MTX of 7.2 µg/minute/kg and liposomal formulation 15 mg/kg for 30 minutes | liposomal formulation of 15 mg/kg and free MTX of 6 µg/minute/kg for 9.5 hours | 81 |
| GSH‐PEG liposomal (EYPC phospholipid) | 0.10 | 0.09 | 0.53 | 0.9 | 5.3 | ||||||||
| PEG liposomal (HSPC phospholipid) | 0.10 | 0.09 | 0.23 | 0.9 | 2.3 | NA | 100 mM | ||||||
| GSH‐PEG liposomal (HSPC phospholipid) | 0.10 | 0.09 | 0.82 | 0.9 | 8.2 | ||||||||
DAMGO, [D‐Ala2, N‐MePhe4, Gly‐ol]‐enkephalin; DHP, Diphenhydramine; EYPC, egg yolk phosphatidyl choline; HSPC, hydrogen soy phosphatidyl choline; MTX, Methotrexate; NA, not applicable.
HOW DOES DISEASE INFLUENCE CNS DRUG DELIVERY?
The general paradigm is that BBB functionality may change in disease conditions and therefore will affect CNS drug distribution and CNS target site concentrations that drive the CNS effects. Many qualitatively obtained changes in BBB properties have been reported for neurodegenerative CNS diseases that all imply that CNS drug distribution would change as a result. 87 , 88 , 89 , 90 However, the number of studies that have quantitatively measured BBB transport and intra‐brain distribution of drugs in the context of neurodegenerative CNS diseases are surprisingly low.
What happens with small molecules?
Parkinson’s disease
No changes in BBB transport of L‐DOPA was found using microdialysis in the unilateral rotenone rat model of Parkinson’s disease, despite Parkinson’s disease‐like pathology, indicated by a huge reduction of tyrosine hydroxylase as well as by substantially reduced levels and higher elimination rates of DOPAC and HVA. 91 Likewise, in α‐synuclein transgenic mice, no changes were found in the extent of CNS distribution (Kp ,uu,brain) of digoxin, levofloxacin, paliperidone, oxycodone, and diazepam, compared with wild‐type mice. 92
Epilepsy
P‐gp expression is often taken as a biomarker of transporter functionality and changes in P‐gp expression due to epilepsy have been interpreted as a reason for pharmacoresistance (transporter hypothesis), without assessing (long term) changes in BBB P‐gp functionality. In a kainate rat model of epilepsy, where status epilepticus was induced, brain capillary protein expression of P‐gp as well as BBB P‐gp functionality was assessed in individual rats. Although substantial changes in P‐gp expression were found that turned back to baseline after 30 days following status epilepticus, no changes were observed in BBB transport of the strong P‐gp substrate quinidine. 93
Alzheimer’s disease
In aged AβPP‐transgenic mice and wild type mice, Kp ,uu,brain was assessed for digoxin, levofloxacin, paliperidone, oxycodone, and diazepam. Despite Aβ pathology, no differences in Kp ,uu,brain were found. 92 In an in vitro study, the effect of Alzheimer’s disease and inflammatory insult on the function of LAT1 at the mouse BBB (for [14C]‐L‐leucine) as well as LAT function and expression in mouse primary astrocytes was studied and the results showed that the presence of APP/PS1 mutations did not change. 94
Lipopolysaccaride induces inflammation
In the in vitro study above, also the effect of an inflammatory insult using lipopolysaccharide on LAT1 function at the mouse BBB and expression in mouse primary astrocytes was assessed. 94 In addition, here, no changes in LAT1 function at the BBB, and LAT1 function and expression in mouse primary astrocytes were found.
Pericyte deficiency
Research implicates that pericytes are key components of the neurovascular unit, which induce BBB properties by regulating transcytosis in the BBB. 95 Using the brain uptake technique in pericyte‐deficient (Pdgfbret/ret) mice, CNS distribution of diazepam, oxycodone, and paliperidone was studied. No changes in the unidirectional transfer coefficients (K in) were found between the pericyte‐deficient and wild type littermates. 96 The combined use of in vivo and in vitro data on diazepam, digoxin, levofloxacin, oxycodone, and paliperidone neither show differences in BBB transport (Kp ,uu,brain) in pericyte‐deficiency. 97
Traumatic brain injury
Patients with human traumatic brain injury were studied regarding morphine brain delivery, by comparing the brain injury site and the “better” brain site using microdialysis. 40 Kp ,uu,brain was 0.64 in the “better” human brain tissue indicating active BBB efflux of morphine, and was substantially changed to 1.0 at the injury site, indicating lack of efflux function due to the traumatic injury. In addition, a corresponding increased brain exposure of morphine was found in experimental meningitis with lipopolysaccharide injections in pigs, studied with microdialysis. 98
What happens with large molecules?
Large molecules cross the BBB to an extremely low extent and BBB targeting vectors have to be used for transport systems that rely on vesicular trafficking. For example, receptor‐specific monoclonal antibodies cross the BBB via receptor‐mediated transport. 7 , 8 , 9 Of interest is that pericyte deficiency increases the permeability of the BBB to tracers related to endothelial transcytosis. 99 In addition, many studies reporting dysfunctional CNS barriers due to increased transcytosis have implicated caveolae as the main contributors of barrier leakage. 100 Although much has been learned on the mechanisms that regulate transcytosis at the BBB, it is still to a large extent a black box, and the basic cell biology of transcytosis at the BBB is still elusive. An overview of specific contributions of potentially multiple pathways to overall transcytosis at the BBB in physiological and pathophysiological states would deserve a review on its own. 7 , 8 , 9 We can conclude that further studies providing quantitative (and generic) information on BBB transport and intra‐brain distribution of large molecules are strongly needed.
DISCUSSION AND CONCLUSIONS
The anatomy and physiology of the CNS is complex and transport of drugs into, within, and out of the CNS is driven by the BBB, the brain parenchyma and fluid spaces, and drug‐specific properties. In combination, they determine the drug concentration‐time profiles in specific CNS compartments.
Three fundamental PK properties underlying CNS drug delivery are rate, extent, and binding. Different methodologies can be used to measure these properties for individual drugs, also making use of the pH partition theory. To this end, the brain can be looked upon “as a whole” or further subdivided into physiological compartments (brain regions, cells, lysosomes, and/or CSF compartments). The focus of such quantitative CNS drug delivery studies so far has been on small molecule drugs.
As indicated, the CNS PK processes are interdependent, which creates a need for integrative approaches to generate smart data to understand rate and extent of BBB transport and intra‐brain and CNS target site distribution, something that can be accomplished with the Mastermind Research Approach. With the CMA, steady‐state relationship between plasma PK and CNS PK in different animal brain compartments can be obtained in a more high‐throughput mode, which makes it very useful in drug discovery. Then, the rat and human validated physiologically based LEICNSPK3.0 models can be used to predict CNS drug concentration‐time profiles in multiple brain and CSF compartments in animals and humans in health and disease, based on drug properties and CNS (patho)physiology for which quantitative data are available, but also as “what‐if” scenarios. This can be used in any stage of CNS drug development as well as in the clinical setting.
With regard to CNS diseases, such as Alzheimer’s and Parkinson’s disease, many studies have indicated a “leaky” barrier and changes in active transporter expressions on the basis of morphologic techniques molecular biological and/or marker compound /tracer investigations. However, quantitative animal studies on BBB transport and intra‐brain distribution of small molecules indicate that their CNS distribution remains unchanged. All together, these studies indicate that for small molecules, the concept of a leaky barrier within neurodegenerative conditions has to be interpreted with caution when estimating CNS drug distribution. Thus, it seems that the capability of the highly dynamic BBB to regulate brain drug exposure still seems to be intact despite the presence of pathology, whereas also changes in BBB expression of active transporters do not necessarily reflect changes in BBB functionality of these transporters. Predictions with the LEICNSPK3.0 model indicate the same situation in humans. There is, however, an exception where the BBB transport of morphine is affected by traumatic brain injury conditions.
For large molecules that have a high potential to treat CNS diseases, a lot has been learned on the mechanisms that regulate transcytosis at the BBB, but it is still to a large extent a black box, and the basic cell biology of transcytosis at the BBB is still elusive. Large efforts are still needed to provide more knowledge on BBB transport of the very important new biologics, to increase their use in CNS diseases.
Here, quantitative integrative data on BBB transport and intra‐brain distribution for large molecules are needed in healthy as well as CNS disease conditions. Fast, real‐time dynamics of transcytosis has been demonstrated in the lungs using intravital microscopy. This technique has not yet been used for brain transcytosis, but would be very helpful for investigating rate, direction, and extent of vesicular BBB trafficking. 101 Knowledge gathered so far indicates that vector‐guided BBB transcytosis may be far more sensitive to disease conditions, which is another level of complexity in developing large molecule drug treatment of CNS diseases. Overall, it seems that small drug molecules have the advantage of less complex BBB transport and intra‐brain distribution processes, whereas also their CNS delivery is less influenced by changes induced by CNS diseases.
FUNDING
No funding was received for this work.
CONFLICT OF INTEREST
The authors declared no competing interests for this work.
- 1. Abbott, N.J. , Patabendige, A.A. , Dolman, D.E. , Yusof, S.R. & Begley, D.J. Structure and function of the blood‐brain barrier. Neurobiol. Dis. 37, 13–25 (2010). [DOI] [PubMed] [Google Scholar]
- 2. Ronaldson, P.T. & Davis, T.P. Regulation of blood‐brain barrier integrity by microglia in health and disease: a therapeutic opportunity. J. Cereb. Blood Flow Metab. 40, S6–S24 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nag, S. Morphology and molecular properties of cellular components of normal cerebral vessels. Methods Mol. Med. 89, 3–36 (2003). [DOI] [PubMed] [Google Scholar]
- 4. Boström, E. , Simonsson, U.S.H. & Hammarlund‐Udenaes, M. In vivo blood‐brain barrier transport of oxycodone in the rat: indications for active influx and implications for pharmacokinetics/pharmacodynamics. Drug Metab. Dispos Biol. Fate Chem. 34, 1624–1631 (2006). [DOI] [PubMed] [Google Scholar]
- 5. Sadiq, M.W. et al. Diphenhydramine active uptake at the blood‐brain barrier and its interaction with oxycodone in vitro and in vivo. J. Pharm. Sci. 100, 3912–3923 (2011). [DOI] [PubMed] [Google Scholar]
- 6. Stanimirovic, D.B. , Bani‐Yaghoub, M. , Perkins, M. & Haqqani, A.S. Blood‐brain barrier models: in vitro to in vivo translation in preclinical development of CNS‐targeting biotherapeutics. Expert Opin. Drug Discov. 10, 141–155 (2015). [DOI] [PubMed] [Google Scholar]
- 7. Pardridge, W.M. Delivery of biologics across the blood‐brain barrier with molecular trojan horse technology. BioDrugs 31, 503–519 (2017). [DOI] [PubMed] [Google Scholar]
- 8. Yu, Y.J. & Watts, R.J. Developing therapeutic antibodies for neurodegenerative disease. Neurotherapeutics 10, 459–472 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Haqqani, A.S. et al. Endosomal trafficking regulates receptor‐mediated transcytosis of antibodies across the blood brain barrier. J. Cereb. Blood Flow Metab. 38, 727–740 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hammarlund‐Udenaes, M. , Fridén, M. , Syvänen, S. & Gupta, A. On the rate and extent of drug delivery to the brain. Pharm. Res. 25, 1737–1750 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gupta, A. , Chatelain, P. , Massingham, R. , Jonsson, E.N. & Hammarlund‐Udenaes, M. Brain distribution of cetirizine enantiomers: comparison of three different tissue‐to‐plasma partition coefficients: K(p), K(p, u), and K(p, uu). Drug Metab. Dispos. Biol. Fate Chem. 34, 318–323 (2006). [DOI] [PubMed] [Google Scholar]
- 12. Hellinger, E. et al. Comparison of brain capillary endothelial cell‐based and epithelial (MDCK‐MDR1, Caco‐2, and VB‐Caco‐2) cell‐based surrogate blood‐brain barrier penetration models. Eur. J. Pharm. Biopharm. 82, 340–351 (2012). [DOI] [PubMed] [Google Scholar]
- 13. Veszelka, S. et al. Comparison of a rat primary cell‐based blood‐brain barrier model with epithelial and brain endothelial cell lines: gene expression and drug transport. Front. Mol. Neurosci. 11, 166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Culot, M. et al. A simple method for assessing free brain/free plasma ratios using an in vitro model of the blood brain barrier. PLoS One 8, e80634 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cecchelli, R. et al. Modelling of the blood‐brain barrier in drug discovery and development. Nat. Rev. Drug Discov. 6, 650–661 (2007). [DOI] [PubMed] [Google Scholar]
- 16. Booth, R. & Kim, H. Permeability analysis of neuroactive drugs through a dynamic microfluidic in vitro blood‐brain barrier model. Ann. Biomed. Eng. 42, 2379–2391 (2014). [DOI] [PubMed] [Google Scholar]
- 17. Hakkarainen, J.J. et al. Comparison of in vitro cell models in predicting in vivo brain entry of drugs. Int. J. Pharm. 402, 27–36 (2010). [DOI] [PubMed] [Google Scholar]
- 18. Hakkarainen, J.J. , Pajander, J. , Laitinen, R. , Suhonen, M. & Forsberg, M.M. Similar molecular descriptors determine the in vitro drug permeability in endothelial and epithelial cells. Int. J. Pharm. 436, 426–443 (2012). [DOI] [PubMed] [Google Scholar]
- 19. Lippmann, E.S. et al. Derivation of blood‐brain barrier endothelial cells from human pluripotent stem cells. Nat. Biotechnol. 30, 783–791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lippmann, E.S. , Azarin, S.M. , Palecek, S.P. & Shusta, E.V. Commentary on human pluripotent stem cell‐based blood‐brain barrier models. Fluids Barriers CNS 17, 64 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Weksler, B. , Romero, I.A. & Couraud, P.O. The hCMEC/D3 cell line as a model of the human blood brain barrier. Fluids Barriers CNS 10, 16 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cecchelli, R. et al. A stable and reproducible human blood‐brain barrier model derived from hematopoietic stem cells. PLoS One 9, e99733 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Urich, E. , Patsch, C. , Aigner, S. , Graf, M. , Iacone, R. & Freskgård, P.‐O. Multicellular self‐assembled spheroidal model of the blood brain barrier. Sci. Rep. 3, 1500 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cho, C.F. et al. Blood‐brain‐barrier spheroids as an in vitro screening platform for brain‐penetrating agents. Nat. Commun. 8, 15623 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bagchi, S. , Chhibber, T. , Lahooti, B. , Verma, A. , Borse, V. & Jayant, R.D. In‐vitro blood‐brain barrier models for drug screening and permeation studies: an overview. Drug Des. Devel. Ther. 13, 3591–3605 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Radan, M. , Djikic, T. , Obradovic, D. & Nikolic, K. Application of in vitro PAMPA technique and in silico computational methods for blood‐brain barrier permeability prediction of novel CNS drug candidates. Eur. J. Pharm. Sci. 168, 106056 (2022). [DOI] [PubMed] [Google Scholar]
- 27. Cory Kalvass, J. & Maurer, T.S. Influence of nonspecific brain and plasma binding on CNS exposure: implications for rational drug discovery. Biopharm. Drug Dispos. 23, 327–338 (2002). [DOI] [PubMed] [Google Scholar]
- 28. Suzuki, T. , Oshimi, M. , Tomono, K. , Hanano, M. & Watanabe, J. Investigation of transport mechanism of pentazocine across the blood‐brain barrier using the in situ rat brain perfusion technique. J. Pharm. Sci. 91, 2346–2353 (2002). [DOI] [PubMed] [Google Scholar]
- 29. Smith, Q.R. & Allen, D.D. In situ brain perfusion technique. Methods Mol. Med. 89, 209–218 (2003). [DOI] [PubMed] [Google Scholar]
- 30. Zhao, R. , Kalvass, J.C. & Pollack, G.M. Assessment of blood‐brain barrier permeability using the in situ mouse brain perfusion technique. Pharm. Res. 26, 1657–1664 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Fridén, M. , Ducrozet, F. , Middleton, B. , Antonsson, M. , Bredberg, U. & Hammarlund‐Udenaes, M. Development of a high‐throughput brain slice method for studying drug distribution in the central nervous system. Drug Metab. Dispos. Biol. Fate Chem. 37, 1226–1233 (2009). [DOI] [PubMed] [Google Scholar]
- 32. Loryan, I. , Fridén, M. & Hammarlund‐Udenaes, M. The brain slice method for studying drug distribution in the CNS. Fluids Barriers CNS 10, 6 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ito, K. et al. Quantitative membrane protein expression at the blood‐brain barrier of adult and younger cynomolgus monkeys. J. Pharm. Sci. 100, 3939–3950 (2011). [DOI] [PubMed] [Google Scholar]
- 34. Uchida, Y. Quantitative proteomics‐based blood‐brain barrier study. Biol. Pharm. Bull. 44, 465–473 (2021). [DOI] [PubMed] [Google Scholar]
- 35. Uchida, Y. et al. Abundant Expression of OCT2, MATE1, OAT1, OAT3, PEPT2, BCRP, MDR1, and xCT transporters in blood‐arachnoid barrier of pig and polarized localizations at CSF‐ and blood‐facing plasma membranes. Drug Metab. Dispos. Biol. Fate Chem. 48, 135–145 (2020). [DOI] [PubMed] [Google Scholar]
- 36. De Lange, E.C.M. Utility of cerebrospinal fluid in translational neuroscience. J. Pharmacokinet. Pharmacodyn. 40, 315–326 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Fridén, M. et al. Structure‐brain exposure relationships in rat and human using a novel data set of unbound drug concentrations in brain interstitial and cerebrospinal fluids. J. Med. Chem. 52, 6233–6243 (2009). [DOI] [PubMed] [Google Scholar]
- 38. Saleh, M.A.A. , Loo, C.F. , Elassaiss‐Schaap, J. & De Lange, E.C.M. Lumbar cerebrospinal fluid‐to‐brain extracellular fluid surrogacy is context‐specific: insights from LeiCNS‐PK3.0 simulations. J. Pharmacokinet. Pharmacodyn. 48, 725–741 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. de Lange, E.C. The mastermind approach to CNS drug therapy: translational prediction of human brain distribution, target site kinetics, and therapeutic effects. Fluids Barriers CNS 10, 12 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ederoth, P. et al. Blood‐brain barrier transport of morphine in patients with severe brain trauma. Br. J. Clin. Pharmacol. 57, 427–435 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ketharanathan, N. et al. Analgosedation in paediatric severe traumatic brain injury (TBI): practice, pitfalls and possibilities. Childs Nerv. Syst. 33, 1703–1710 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Custers, M.L. et al. Applicability of cerebral open flow microperfusion and microdialysis to quantify a brain‐penetrating nanobody in mice. Anal. Chim. Acta. 1178, 338803 (2021). [DOI] [PubMed] [Google Scholar]
- 43. Chang, H.Y. , Morrow, K. , Bonacquisti, E. , Zhang, W. & Shah, D.K. Antibody pharmacokinetics in rat brain determined using microdialysis. mAbs 10, 843–853 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chang, H.Y. et al. Brain pharmacokinetics of anti‐transferrin receptor antibody affinity variants in rats determined using microdialysis. mAbs 13, 1874121 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Müllauer, J. et al. Pharmacokinetic modeling of P‐glycoprotein function at the rat and human blood‐brain barriers studied with (R)‐[11C]verapamil positron emission tomography. EJNMMI Res. 2, 58 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gustafsson, S. , Eriksson, J. , Syvänen, S. , Eriksson, O. , Hammarlund‐Udenaes, M. & Antoni, G. Combined PET and microdialysis for in vivo estimation of drug blood‐brain barrier transport and brain unbound concentrations. NeuroImage 155, 177–186 (2017). [DOI] [PubMed] [Google Scholar]
- 47. Sehlin, D. , Fang, X.T. , Meier, S.R. , Jansson, M. & Syvanen, S. Pharmacokinetics, biodistribution and brain retention of a bispecific antibody‐based PET radioligand for imaging of amyloid‐beta. Sci. Rep. 7, 17254 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Storelli, F. , Anoshchenko, O. & Unadkat, J.D. Successful prediction of human steady‐state unbound brain‐to‐plasma concentration ratio of P‐gp substrates using the proteomics‐informed relative expression factor approach. Clin. Pharmacol. Ther. 110, 432–442 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bendels, S. , Kansy, M. , Wagner, B. & Huwyler, J. In silico prediction of brain and CSF permeation of small molecules using PLS regression models. Eur. J. Med. Chem. 43, 1581–1592 (2008). [DOI] [PubMed] [Google Scholar]
- 50. Grumetto, L. , Russo, G. & Barbato, F. Immobilized artificial membrane HPLC derived parameters vs PAMPA‐BBB data in estimating in situ measured blood‐brain barrier permeation of drugs. Mol. Pharm. 13, 2808–2816 (2016). [DOI] [PubMed] [Google Scholar]
- 51. Russo, G. , Grumetto, L. , Szucs, R. , Barbato, F. & Lynen, F. Determination of in vitro and in silico indexes for the modeling of blood‐brain barrier partitioning of drugs via micellar and immobilized artificial membrane liquid chromatography. J. Med. Chem. 60, 3739–3754 (2017). [DOI] [PubMed] [Google Scholar]
- 52. Loryan, I. et al. Molecular properties determining unbound intracellular and extracellular brain exposure of CNS drug candidates. Mol. Pharm. 12, 520–532 (2015). [DOI] [PubMed] [Google Scholar]
- 53. Varadharajan, S. et al. Exploring in silico prediction of the unbound brain‐to‐plasma drug concentration ratio: model validation, renewal, and interpretation. J. Pharm. Sci. 104, 1197–1206 (2015). [DOI] [PubMed] [Google Scholar]
- 54. Chen, H. , Winiwarter, S. , Fridén, M. , Antonsson, M. & Engkvist, O. In silico prediction of unbound brain‐to‐plasma concentration ratio using machine learning algorithms. J. Mol. Graph. Model. 29, 985–995 (2011). [DOI] [PubMed] [Google Scholar]
- 55. Liu, H. , Dong, K. , Zhang, W. , Summerfield, S.G. & Terstappen, G.C. Prediction of brain: blood unbound concentration ratios in CNS drug discovery employing in silico and in vitro model systems. Drug Discov. Today 23, 1357–1372 (2018). [DOI] [PubMed] [Google Scholar]
- 56. Dolgikh, E. et al. QSAR model of unbound brain‐to‐plasma partition coefficient, Kp, uu, brain: incorporating P‐glycoprotein efflux as a variable. J Chem. Inf. Model 56, 2225–2233 (2016). [DOI] [PubMed] [Google Scholar]
- 57. Zhang, Y.Y. , Liu, H. , Summerfield, S.G. , Luscombe, C.N. & Sahi, J. Integrating in silico and in vitro approaches to predict drug accessibility to the central nervous system. Mol. Pharm. 13, 1540–1550 (2016). [DOI] [PubMed] [Google Scholar]
- 58. Kosugi, Y. , Mizuno, K. , Santos, C. , Sato, S. , Hosea, N. & Zientek, M. Direct comparison of the prediction of the unbound brain‐to‐plasma partitioning utilizing machine learning approach and mechanistic neuropharmacokinetic model. AAPS J. 23, 72 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gupta, M. , Bogdanowicz, T. , Reed, M.A. , Barden, C.J. & Weaver, D.F. The Brain Exposure Efficiency (BEE) score. ACS Chem. Neurosci. 11, 205–224 (2020). [DOI] [PubMed] [Google Scholar]
- 60. Ball, K. , Bouzom, F. , Scherrmann, J.‐M. , Walther, B. & Declèves, X. A physiologically based modeling strategy during preclinical CNS drug development. Mol. Pharm. 11, 836–848 (2014). [DOI] [PubMed] [Google Scholar]
- 61. Gaohua, L. , Neuhoff, S. , Johnson, T.N. , Rostami‐Hodjegan, A. & Jamei, M. Development of a permeability‐limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: Estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab. Pharmacokinet. 31, 224–233 (2016). [DOI] [PubMed] [Google Scholar]
- 62. Yamamoto, Y. , Danhof, M. & de Lange, E.C.M. Erratum to: microdialysis: the key to physiologically based model prediction of human CNS target site concentrations. AAPS J. 19, 1249–1252 (2017). [DOI] [PubMed] [Google Scholar]
- 63. Yamamoto, Y. , Danhof, M. & de Lange, E.C.M. Microdialysis: the key to physiologically based model prediction of human CNS target site concentrations. AAPS J. 19, 891–909 (2017). [DOI] [PubMed] [Google Scholar]
- 64. Yamamoto, Y. et al. Predicting drug concentration‐time profiles in multiple CNS compartments using a comprehensive physiologically‐based pharmacokinetic model. CPT Pharmacometrics Syst. Pharmacol. 6, 765–777 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yamamoto, Y. et al. Prediction of human CNS pharmacokinetics using a physiologically‐based pharmacokinetic modeling approach. Eur. J. Pharm. Sci. 112, 168–179 (2018). [DOI] [PubMed] [Google Scholar]
- 66. Saleh, M.A.A. & de Lange, E.C.M. Impact of CNS diseases on drug delivery to brain extracellular and intracellular target sites in human: a "WHAT‐IF" simulation study. Pharmaceutics 13, 95 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Loryan, I. et al. Mechanistic understanding of brain drug disposition to optimize the selection of potential neurotherapeutics in drug discovery. Pharm. Res. 31, 2203–2219 (2014). [DOI] [PubMed] [Google Scholar]
- 68. Fridén, M. et al. Measurement of unbound drug exposure in brain: modeling of pH partitioning explains diverging results between the brain slice and brain homogenate methods. Drug Metab. Dispos. Biol. Fate Chem. 39, 353–362 (2011). [DOI] [PubMed] [Google Scholar]
- 69. Chang, H.Y. , Wu, S. , Meno‐Tetang, G. & Shah, D.K. A translational platform PBPK model for antibody disposition in the brain. J. Pharmacokinet. Pharmacodyn. 46, 319–338 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Syvänen, S. et al. Species differences in blood‐brain barrier transport of three positron emission tomography radioligands with emphasis on P‐glycoprotein transport. Drug Metab. Dispos. Biol. Fate Chem. 37, 635–643 (2009). [DOI] [PubMed] [Google Scholar]
- 71. Loryan, I. et al. In‐depth neuropharmacokinetic analysis of antipsychotics based on a novel approach to estimate unbound target‐site concentration in CNS regions: link to spatial receptor occupancy. Mol. Psychiatry 21, 1527–1536 (2016). [DOI] [PubMed] [Google Scholar]
- 72. Groenendaal, D. et al. Pharmacokinetic/pharmacodynamic modelling of the EEG effects of opioids: the role of complex biophase distribution kinetics. Eur. J. Pharm. Sci. 34, 149–163 (2008). [DOI] [PubMed] [Google Scholar]
- 73. Shaffer, C.L. , Osgood, S.M. , Mancuso, J.Y. & Doran, A.C. Diphenhydramine has similar interspecies net active influx at the blood‐brain barrier. J. Pharm. Sci. 103, 1557–1562 (2014). [DOI] [PubMed] [Google Scholar]
- 74. Suzuki, T. et al. Involvement of an influx transporter in the blood‐brain barrier transport of naloxone. Biopharm. Drug Dispos. 31, 243–252 (2010). [DOI] [PubMed] [Google Scholar]
- 75. Puris, E. et al. Mechanistic study on the use of the l‐type amino acid transporter 1 for brain intracellular delivery of Ketoprofen via prodrug: a novel approach supporting the development of prodrugs for intracellular targets. Mol. Pharm. 16, 3261–3274 (2019). [DOI] [PubMed] [Google Scholar]
- 76. Puris, E. , Gynther, M. , Auriola, S. & Huttunen, K.M. L‐Type amino acid transporter 1 as a target for drug delivery. Pharm. Res. 37, 88 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Terstappen, G.C. , Meyer, A.H. , Bell, R.D. & Zhang, W. Strategies for delivering therapeutics across the blood‐brain barrier. Nat. Rev. Drug Discov. 20, 362–383 (2021). [DOI] [PubMed] [Google Scholar]
- 78. Juhairiyah, F. & de Lange, E.C.M. Understanding drug delivery to the brain using liposome‐based strategies: studies that provide mechanistic insights are essential. AAPS J. 23, 114 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Carreno, F. , Paese, K. , Silva, C.M. , Guterres, S.S. & Dalla Costa, T. Pharmacokinetic investigation of quetiapine transport across blood‐brain barrier mediated by lipid core nanocapsules using brain microdialysis in rats. Mol. Pharm. 13, 1289–1297 (2016). [DOI] [PubMed] [Google Scholar]
- 80. Lindqvist, A. , Jonsson, S. & Hammarlund‐Udenaes, M. Exploring factors causing low brain penetration of the opioid peptide DAMGO through experimental methods and modeling. Mol. Pharm. 13, 1258–1266 (2016). [DOI] [PubMed] [Google Scholar]
- 81. Hu, Y. , Gaillard, P.J. , de Lange, E.C.M. & Hammarlund‐Udenaes, M. Targeted brain delivery of methotrexate by glutathione PEGylated liposomes: How can the formulation make a difference? Eur. J. Pharm. Biopharm. 139, 197–204 (2019). [DOI] [PubMed] [Google Scholar]
- 82. Hu, Y. , Gaillard, P.J. , Rip, J. , de Lange, E.C.M. & Hammarlund‐Udenaes, M. In vivo quantitative understanding of pegylated liposome's influence on brain delivery of diphenhydramine. Mol. Pharm. 15, 5493–5500 (2018). [DOI] [PubMed] [Google Scholar]
- 83. Hu, Y. & Hammarlund‐Udenaes, M. Perspectives on nanodelivery to the brain: prerequisites for successful brain treatment. Mol. Pharm. 17, 4029–4039 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hu, Y. , Hammarlund‐Udenaes, M. & Friden, M. Understanding the influence of nanocarrier‐mediated brain delivery on therapeutic performance through pharmacokinetic‐pharmacodynamic modeling. J. Pharm. Sci. 108, 3425–3433 (2019). [DOI] [PubMed] [Google Scholar]
- 85. Lindqvist, A. , Rip, J. , van Kregten, J. , Gaillard, P.J. & Hammarlund‐Udenaes, M. In vivo functional evaluation of increased brain delivery of the opioid peptide DAMGO by glutathione‐PEGylated liposomes. Pharm. Res. 33, 177–185 (2016). [DOI] [PubMed] [Google Scholar]
- 86. Lindqvist, A. , Fridén, M. & Hammarlund‐Udenaes, M. Pharmacokinetic considerations of nanodelivery to the brain: using modeling and simulations to predict the outcome of liposomal formulations. Eur. J. Pharm. Sci. 92, 173–182 (2016). [DOI] [PubMed] [Google Scholar]
- 87. Lossinsky, A.S. & Shivers, R.R. Structural pathways for macromolecular and cellular transport across the blood‐brain barrier during inflammatory conditions. Review. Histol. Histopathol. 19, 535–564 (2004). [DOI] [PubMed] [Google Scholar]
- 88. Atilano‐Roque, A. , Roda, G. , Fogueri, U. , Kiser, J.J. & Joy, M.S. Effect of disease pathologies on transporter expression and function. J. Clin. Pharmacol. 56(Suppl 7), S205–S221 (2016). [DOI] [PubMed] [Google Scholar]
- 89. McInerney, M.P. , Short, J.L. & Nicolazzo, J.A. Neurovascular alterations in Alzheimer's disease: transporter expression profiles and CNS drug access. AAPS J. 19, 940–956 (2017). [DOI] [PubMed] [Google Scholar]
- 90. Ahishali, B. & Kaya, M. Evaluation of blood‐brain barrier integrity using vascular permeability markers: evans blue, sodium fluorescein, albumin‐alexa fluor conjugates, and horseradish peroxidase. Methods Mol. Biol. 2367, 87–103 (2021). [DOI] [PubMed] [Google Scholar]
- 91. Ravenstijn, P.G. , Drenth, H.J. , O'Neill, M.J. , Danhof, M. & de Lange, E.C. Evaluation of blood‐brain barrier transport and CNS drug metabolism in diseased and control brain after intravenous L‐DOPA in a unilateral rat model of Parkinson's disease. Fluids Barriers CNS 9, 4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Gustafsson, S. , Lindström, V. , Ingelsson, M. , Hammarlund‐Udenaes, M. & Syvänen, S. Intact blood‐brain barrier transport of small molecular drugs in animal models of amyloid beta and alpha‐synuclein pathology. Neuropharmacology 128, 482–491 (2018). [DOI] [PubMed] [Google Scholar]
- 93. De Lange, E.C.M. , Vd Berg, D.J. , Bellanti, F. , Voskuyl, R.A. & Syvanen, S. P‐glycoprotein protein expression versus functionality at the blood‐brain barrier using immunohistochemistry, microdialysis and mathematical modeling. Eur. J. Pharm. Sci. 124, 61–70 (2018). [DOI] [PubMed] [Google Scholar]
- 94. Gynther, M. et al. Alzheimer's disease phenotype or inflammatory insult does not alter function of L‐type amino acid transporter 1 in mouse blood‐brain barrier and primary astrocytes. Pharm. Res. 36, 17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Ben‐Zvi, A. et al. Mfsd2a is critical for the formation and function of the blood‐brain barrier. Nature 509, 507–511 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Mihajlica, N. , Betsholtz, C. & Hammarlund‐Udenaes, M. Rate of small‐molecular drug transport across the blood‐brain barrier in a pericyte‐deficient state. Eur. J. Pharm. Sci. 124, 182–187 (2018). [DOI] [PubMed] [Google Scholar]
- 97. Mihajlica, N. , Betsholtz, C. & Hammarlund‐Udenaes, M. Pharmacokinetics of pericyte involvement in small‐molecular drug transport across the blood‐brain barrier. Eur. J. Pharm. Sci. 122, 77–84 (2018). [DOI] [PubMed] [Google Scholar]
- 98. Tunblad, K. , Ederoth, P. , Gardenfors, A. , Hammarlund‐Udenaes, M. & Nordstrom, C.H. Altered brain exposure of morphine in experimental meningitis studied with microdialysis. Acta Anaesthesiol. Scand. 48, 294–301 (2004). [DOI] [PubMed] [Google Scholar]
- 99. Armulik, A. et al. Pericytes regulate the blood‐brain barrier. Nature 468, 557–561 (2010). [DOI] [PubMed] [Google Scholar]
- 100. Goulatis, L.I. & Shusta, E.V. Protein engineering approaches for regulating blood‐brain barrier transcytosis. Curr. Opin. Struct. Biol. 45, 109–115 (2017). [DOI] [PubMed] [Google Scholar]
- 101. Ayloo, S. & Gu, C. Transcytosis at the blood‐brain barrier. Curr. Opin. Neurobiol. 57, 32–38 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Xie, R. & Hammarlund‐Udenaes, M. Blood‐brain barrier equilibration of codeine in rats studied with microdialysis. Pharm. Res. 15, 570–575 (1998). [DOI] [PubMed] [Google Scholar]
- 103. Tunblad, K. , Jonsson, E.N. & Hammarlund‐Udenaes, M. Morphine blood‐brain barrier transport is influenced by probenecid co‐administration. Pharm. Res. 20, 618–623 (2003). [DOI] [PubMed] [Google Scholar]
- 104. Bouw, M.R. , Gardmark, M. & Hammarlund‐Udenaes, M. Pharmacokinetic‐pharmacodynamic modelling of morphine transport across the blood‐brain barrier as a cause of the antinociceptive effect delay in rats–a microdialysis study. Pharm. Res. 17, 1220–1227 (2000). [DOI] [PubMed] [Google Scholar]
- 105. Tunblad, K. , Hammarlund‐Udenaes, M. & Jonsson, E.N. Influence of probenecid on the delivery of morphine‐6‐glucuronide to the brain. Eur. J. Pharm. Sci. 24, 49–57 (2005). [DOI] [PubMed] [Google Scholar]
- 106. Xie, R. , Bouw, M.R. & Hammarlund‐Udenaes, M. Modelling of the blood‐brain barrier transport of morphine‐3‐glucuronide studied using microdialysis in the rat: involvement of probenecid‐sensitive transport. Br. J. Pharmacol. 131, 1784–1792 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Friden, M. , Ljungqvist, H. , Middleton, B. , Bredberg, U. & Hammarlund‐Udenaes, M. Improved measurement of drug exposure in the brain using drug‐specific correction for residual blood. J. Cereb. Blood Flow Metab. 30, 150–161 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Dagenais, C. , Graff, C.L. & Pollack, G.M. Variable modulation of opioid brain uptake by P‐glycoprotein in mice. Biochem. Pharmacol. 67, 269–276 (2004). [DOI] [PubMed] [Google Scholar]
- 109. Lindqvist, A. , Rip, J. , Gaillard, P.J. , Björkman, S. & Hammarlund‐Udenaes, M. Enhanced brain delivery of the opioid peptide DAMGO in glutathione pegylated liposomes: a microdialysis study. Mol. Pharm. 10, 1533–1541 (2013). [DOI] [PubMed] [Google Scholar]
- 110. Hu, Y. , Rip, J. , Gaillard, P.J. , de Lange, E.C.M. & Hammarlund‐Udenaes, M. The impact of liposomal formulations on the release and brain delivery of methotrexate: an in vivo microdialysis study. J. Pharm. Sci. 106, 2606–2613 (2017). [DOI] [PubMed] [Google Scholar]