

Abstract
Background and Purpose
Amyotrophic lateral sclerosis (ALS), a neurodegenerative disease characterized by the degeneration of upper and lower motor neurons, progressive wasting and paralysis of voluntary muscles and is currently incurable. Although considered to be a pure motor neuron disease, increasing evidence indicates that the sole protection of motor neurons by a single targeted drug is not sufficient to improve the pathological phenotype. We therefore evaluated the therapeutic potential of the multi‐target drug used to treatment of coronary artery disease, trimetazidine, in SOD1G93A mice.
Experimental Approach
As a metabolic modulator, trimetazidine improves glucose metabolism. Furthermore, trimetazidine enhances mitochondrial metabolism and promotes nerve regeneration, exerting an anti‐inflammatory and antioxidant effect. We orally treated SOD1G93A mice with trimetazidine, solubilized in drinking water at a dose of 20 mg kg−1, from disease onset. We assessed the impact of trimetazidine on disease progression by studying metabolic parameters, grip strength and histological alterations in skeletal muscle, peripheral nerves and the spinal cord.
Key Results
Trimetazidine administration delays motor function decline, improves muscle performance and metabolism, and significantly extends overall survival of SOD1G93A mice (increased median survival of 16 days and 12.5 days for male and female respectively). Moreover, trimetazidine prevents the degeneration of neuromuscular junctions, attenuates motor neuron loss and reduces neuroinflammation in the spinal cord and in peripheral nerves.
Conclusion and Implications
In SOD1G93A mice, therapeutic effect of trimetazidine is underpinned by its action on mitochondrial function in skeletal muscle and spinal cord.
Keywords: amyotrophic lateral sclerosis, hypermetabolism, mitochondria, neurodegeneration, SOD1G93A mice, Trimetazidine
Abbreviations
- ACAA2
3‐ketoacyl‐CoA thiolase, mitochondrial/acetyl‐coenzyme A acyltransferase 2
- ALS
amyotrophic lateral sclerosis
- BMI
body mass index
- EDL
extensor digitorum longus
- ETC
electron transport chain
- NMJ
neuromuscular junction
- OCR
oxygen consumption rate
- SOD1
superoxide dismutase
What is already known?
Amyotrophic lateral sclerosis (ALS) is a multifactorial neurodegenerative disease with currently no effective cure.
The failure of the numerous clinical trials in ALS indicates that protection of motor neurons by a single target drug alone is not sufficient to improve the disease outcomes.
What does this study add?
SOD1G93A mice treated with the multi target drug trimetazidine show an improved survival and locomotor performances and a decreased hypermetabolism.
Trimetazidine treatment protects motor neurons and NMJ, improves energy metabolism in spinal cord and skeletal muscle, and decreases neuroinflammation.
What is the clinical significance?
The ability of trimetazidine to improve energy metabolism provides new evidences for a therapeutic approach in the treatment of ALS.
This preclinical study supports the repurposing of trimetazidine in the clinical practice of ALS.
1. INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder that is clinically characterized by the loss of upper and lower motor neurons. Despite intense research studies and numerous clinical trials, ALS currently remains an incurable disease (Wobst et al., 2020).
Metabolic dysfunction has been linked to ALS pathogenesis since the 1970s (Reyes et al., 1984; Van den Bergh et al., 1978). In humans, a decrease in premorbid body mass index (BMI) increases the risk of ALS (O'reilly et al., 2013), while weight loss and hypermetabolism are associated with poor prognosis (Steyn et al., 2018; Van Mantgem, 2020). Accordingly, both ALS patients and animal models share features such as weight loss, energy store depletion, hypermetabolism and alterations in glucose and lipid metabolism (Ferri & Coccurello, 2017; Peggion et al., 2017; Steyn et al., 2018). Overall, these metabolic disorders are likely to promote the use of fat as the main energy source to protect from protein catabolism (Ngo et al., 2014). However, constant lipid consumption results in the exhaustion of energy stores, loss of body fat mass (Dolezalova et al., 2007; Ngo et al., 2014; Tremblay et al., 1990) and a reduction in BMI (Kirk et al., 2019; Ngo et al., 2014).
Despite a growing body of evidence linking metabolic alterations to disease outcome in ALS, interest in targeting metabolic dysfunction as a therapeutic approach has only grown in recent times (Fayemendy et al., 2021; Ferri & Coccurello, 2017; Ngo & Steyn, 2015). In this context, we aimed to evaluate the therapeutic potential of trimetazidine in the SOD1G93A mouse model of ALS. Trimetazidine is an anti‐ischemic drug used for the treatment of coronary artery disease. This drug is classified as a metabolic modulator that inhibits the long‐chain mitochondrial 3‐ketoacyl coenzyme A thiolase (ACAA2), an enzyme that catalyses the oxidation of long‐chain fatty acids (Kalucka et al., 2018; Kantor et al., 2000). Because of its ability to inhibit β‐oxidation and fatty acid uptake, trimetazidine improves glucose metabolism (Fragasso et al., 2003; Kantor et al., 2000). Additionally, trimetazidine is known to increase mitochondrial protein levels and stimulate myogenesis, muscle strength and oxidative metabolism in aged and cachectic muscles, improving muscle quality and neuromuscular communication (Belli et al., 2019; Molinari et al., 2017).
Here, we show that when administered orally at disease onset, trimetazidine improves muscle performance and energy metabolism, while also extending survival of SOD1G93A mice. Moreover, we show that trimetazidine protects neuromuscular junctions (NMJs), preserves spinal motor neurons and reduces neuroinflammation. In this framework, trimetazidine exerts therapeutic benefit by improving mitochondrial function and, ultimately, the production of adenosine triphosphate (ATP).
2. METHODS
2.1. Antibodies
The antibodies used in this study are summarized in Table 1.
TABLE 1.
Antibodies used for western blot (WB) and immunofluorescence (IF)
| Antibodies | Species | Product | Dilution | |
|---|---|---|---|---|
| Primary | CPT1A | Mouse monoclonal | Abcam (Cat#ab83862; RRID:AB_1924900) | WB 1:1000 |
| SLN | Rabbit polyclonal | Millipore (Cat#ABT13; RRID:AB_11203316) | WB 1:1000 | |
| GAPDH | Mouse monoclonal | Santa Cruz Biotechnology (Cat#sc‐20356; RRID:AB_641103) | WB 1:5000 | |
| AGRIN | Mouse monoclonal | Millipore (Cat#MAB5204; RRID:AB_2225272) | WB 1:1000 | |
| PKCθ | Rabbit polyclonal | Cell Signaling Technology (Cat#13643; RRID:AB_2798282) | WB 1:1000 | |
| pPKCθ (Thr538) | Rabbit polyclonal | Cell Signaling Technology (Cat# 9377; RRID:AB_2172071) | WB 1:1000 | |
| LRP4 | Mouse monoclonal | BioLegend (Cat#832201; RRID:AB_2564954) | WB 1:1000 | |
| CD68 | Rat monoclonal | Bio‐Rad/AbD Serotec (Cat#MCA1957GA; RRID:AB_324217) | IF 1:200 | |
| GFAP | Mouse monoclonal | Novus Biological (Cat#NBP1‐05197; RRID:AB_1555288) | IF 1:500 | |
| NFH | Mouse monoclonal | BioLegend (Cat#836001; RRID:AB_2565356) | IF 1:1000 | |
| P2Y12 | Rabbit polyclonal | AnaSpec (Cat#55043A; RRID:AB_2298886) | IF 1:200 | |
| MBP | Rabbit monoclonal | Cell Signaling Technology (Cat#78896; RRID:AB_2799920) | IF 1:200 | |
| SMI 312 | Mouse monoclonal | Biolegend (Cat#837904; RRID:AB_2566782) | IF 1:200 | |
| Synaptophysin | Rabbit monoclonal | Thermo Fisher Scientific (Cat#MA1‐34660; RRID:AB_1960867) | IF 1:200 | |
| Secondary | HRP Conjugate | Goat anti‐rabbit | Bio‐Rad Laboratories (Cat#1706515; RRID:AB_2617112) | WB 1:2500 |
| HRP Conjugate | Goat anti‐mouse | Bio‐Rad Laboratories (Cat#170‐6516, RRID:AB_11125547) | WB 1:2500 | |
| Alexa Fluor 488 | Donkey anti‐rat | ThermoFisher Scientific (Cat#A‐21208; RRID:AB_2535794) | IF 1:200 | |
| Alexa Fluor 488 | Donkey anti‐mouse | ThermoFisher Scientific (Cat#A‐21202; RRID:AB_141607) | IF 1:200 | |
| Cy™3 | Donkey anti‐mouse | Jackson ImmunoResearch Laboratories (Cat#715‐166‐150; RRID:AB_2340816) | IF 1:100 | |
| Cy™3 | Donkey anti‐rabbit | Jackson ImmunoResearch Laboratories (Cat#711‐165‐152; RRID:AB_2307443) | IF 1:100 | |
| Cy™5 | Donkey anti‐rabbit | Jackson ImmunoResearch Laboratories (Cat#711‐175‐152; RRID:AB_2340607) | IF 1:200 | |
| TRITC | Donkey anti‐mouse | Jackson ImmunoResearch Laboratories (Cat#715‐025‐151; RRID:AB_2340767) | IF 1:400 |
Abbreviations: CD68, Cluster of Differentiation 68; CPT1A, Carnitine palmitoyltransferase I A; GAPDH, Glyceraldehyde‐3‐Phosphate Dehydrogenase; GFAP, Glial fibrillary acidic protein; HRP, horseradish peroxidase; LRP4, Low‐density lipoprotein receptor‐related protein 4; MBP, Myelin Basic Protein; NFH, Neurifilament Heavy; PKCθ, Protein Kinasi C Theta; SLN, Sarcolipin; TRITC, tetramethylrhodamine
2.2. Animals
All procedures in Rome were performed in accordance with European Guidelines for the use of animals in research (2010/63/EU) and the requirements of Italian laws (D.L. 26/2014) and were approved by the Animal welfare office, Department of Public Health and Veterinary, Nutrition and Food Safety, General Management of Animal Care and Veterinary Drugs of the Italian Ministry of Health (protocol number 931/2017/PR). Procedures at the University of Queensland were approved by the University of Queensland Animal Ethics Committee and conducted in accordance with the Queensland Government Animal Care and Protection Act 2001, associated Animal Care and Protection Regulations (2002 and 2008) and the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes (2004). Animal studies were conducted in accordance with guidelines reported by ARRIVE (Pierce du Sert et al., 2020) and following the suggestions of the British Journal of Pharmacology (Lilley et al., 2020).
SOD1G93A mice (B6.Cg‐Tg [SOD1 G93A]1Gur/J) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA; RRID:MGI:4835776) and bred and housed in a virus/antigen‐free facility with a light/dark cycle of 12 h at constant temperature and humidity. Food and water were provided ad libitum. Transgenic hemizygous SOD1G93A males were crossbred with C57BL/6 (RRID:MGI:5657312) females and transgenic progeny were genotyped by PCR through hSOD1 oligos and IL2 as housekeeping (sequences in Table 3). Disease onset was defined at 70 days of age by dynamometric evaluation of grip strength, which provides an index of neuromuscular function expressed as maximal muscle strength (Ge et al., 2016). The peak paw grip strength of SOD1G93A mice and wild‐type (WT) control littermates was assessed starting from 49 days of age. The test consisted of three attempts for each mouse, with intervals of 1 min for rest; the mean of the three examinations was normalized to the weight of each respective mouse. The onset of the disease was defined as the age when the peak paw grip strength of SOD1G93A mice was 20% less than that of their wild‐type littermates. To monitor disease progression, behavioural scores and body weight were recorded from 49 days of age (Apolloni et al., 2019; Migliarini et al., 2021). For SOD1G93A mice, disease end stage was defined by full paralysis of hind limbs and loss of the ability to turn within 30 seconds of being positioned on their back. At this stage, corresponding to the humane endpoint, mice were sacrified with CO2 and dislocation of the neck, in order to prevent further distress, according to guidelines for preclinical testing in ALS (Poppe et al., 2014). We assigned disease onset, early symptomatic, symptomatic and late symptomatic at 70, 90, 120 and 150 days of age, respectively.
TABLE 3.
Target genes and primer sequences
| Target | Forward sequence | Reverse sequence |
|---|---|---|
| MyHCIIa | 5′‐AGTCCCAGGTCAACAAGCTG‐3′ | 3′‐GCATGACCAAAGGTTTCACA‐5′ |
| MyHCIIb | 5′‐AGTCCCAGGTCAACAAGCTG‐3′ | 3′‐TTTCTCCTGTCACCTCTCAACA‐5′ |
| Myoglobin | 5′‐CTGTTTAAGACTCACCCTGAGAC‐3′ | 5′‐GGTGCAACCATGCTTCTTCA‐3′ |
| Atrogin1 | 5′‐TGAGCGACCTCAGCAGTTAC‐3′ | 5′‐GCGCTCCTTCGTACTTCCTT‐3′ |
| Col3a1 | 5′‐CCCAACCCAGAGATCCCATT‐3′ | 5′‐GGTCACCATTTCTCCCAGGA‐3′ |
| SOD1 | 5′‐CATCAGCCCTAATCCATCTGA‐3′ | 5′‐CGCGACTAACAATCAAAGTGA‐3′ |
| Il‐2 | 5′‐TAGGCCACAGAATTGAAAGATCT‐3′ | 5′‐GTAGGTGGAAATTCTAGCATCATC C‐3′ |
| TATA box binding protein | 5′‐CCAATGACTCCTATGACCCCTA‐3′ | 3′‐CAGCCAAGATTCACGGTAGAT‐5′ |
Abbreviations: COL3A1, Collagen Type III Alpha 1 Chain; IL‐2, Interleukin 2;MyHCIIa, Myosin heavy chain IIa; MyHCIIb, Myosin heavy chain IIb; SOD1, Superoxide dismutase 1
Mice were anaesthetized with Rompum (xylazine, 20 mg ml−1, 0.5 ml kg−1 Bayer, Milan, Italy) plus Zoletil (tiletamine and zolazepam,100 mg ml−1, 0.5 ml kg−1; Virbac, Milan, Italy), and following loss of the pedal‐withdrawal reflex, they were killed for tissue dissection.
2.3. Liquid chromatography–mass spectrometry (LC–MS) evaluation of trimetazidine concentration in plasma
Different doses of trimetazidine dihydrochloride (Selleckchem), 0, 5, 10, 20 and 40 mg kg−1, were dissolved in drinking water and administered to WT mice (at least n = 3 for each dose). After 1 week of treatment, mouse blood was collected in EDTA‐treated tubes and the plasma was obtained by a 10 min centrifugation at 1000–2000 × g. 100 μl of sample and calibrators were mixed in 300 μl of internal standard working solution containing 25 ng ml−1 trimetazidine‐d8 dihydrochloride (Santa Cruz Biotechnology, Inc. USA) and methanol (LC–MS grade, Biosolve Chimie, France). After centrifugation at 2000 x g for 5 min, 2 μl of sample was injected into a UPLC–MS/MS, and analysed. Chromatographic separation was performed by ACQUITY H‐Class using a BEH C18 1.7 μm, 2.1 × 50 mm column (Waters Corporation, Milford, MA, USA) eluted at a flow rate of 200 μl min−1 with a linear gradient from 95 to 5 water (LC–MS grade, Merck KGaA, Darmstadt, German) 0.05% formic acid in methanol 0.05% formic acid (98%, LC–MS grade, Mallinckrodt Baker Italia, Milano, Italia). The detection of trimetazidine and trimetazidine‐d8 was conducted over multiple reaction monitoring experiments (MRM conditions in Table 2) upon direct infusion of a standard solution (1 μg ml−1) using a triple quadrupole Xevo TQ‐S micro Mass Spectrometer (Waters Corporation) equipped with an ESI source operating in positive mode, with a capillary voltage of 3.15 kV and a desolvation temperature of 600°C. The source of the gas was set as follows: Desolvation at 500 L h−1 and cone at 1 L h−1. The UPLC‐MS/MS system was controlled by MassLynx Mass Spectrometry Software (Waters Corporation; RRID:SCR_014271). Data analysis was performed by software TargetLynx (Waters Corporation).
TABLE 2.
Conditions for the detection of Trimetazidine and Trimetazidine‐d8
| Analyte | Precursor ion mass (m/z) | Product ion mass (m/z) | Collision (V) | Cone (V) |
|---|---|---|---|---|
| Trimetazidine | 267.16 | 181.00 | 11.0 | 18.0 |
| Trimetazidine‐d8 | 275.10 | 181.00 | 11.0 | 18.0 |
2.4. Trimetazidine treatment
Trimetazidine 20 mg kg−1 was dissolved in drinking water and administered to male and female SOD1G93A and WT littermate mice starting from disease onset. Treated and untreated mice from each experimental group/sex (n ≥ 12) were weighed and assessed for grip strength once a week starting from 70 days of age. Assigning 100% performance to each mouse at 70 days of age, we monitored disease progression and considered the midpoint and endpoint as a decrease in grip strength (performed as decribed in the “2.2. Animals” section) of 25% and 50%, respectively. Groups of 120 (n ≥ 8) and 150 (n ≥ 8) day‐old mice were killed for the evaluation of the effects at mid‐ and long‐term trimetazidine treatment respectively. Disease duration was defined as the number of days elapsed between disease onset, and thus the beginning of the treatment, and disease end stage.
2.5. Energy metabolism by indirect calorimetry system
Total energy expenditure of treated and untreated 120 day‐old SOD1G93A and WT littermate mice (n ≥ 6 for each experimental group) was measured through an indirect calorimetry system (TSE PhenoMaster/LabMaster System, Germany) at constant air flow of 0.35 L min−1. Measures were carried out at intervals of 20 min for each mouse, starting at 7:00 PM and ending automatically after 48 h or 72 h (12 h dark–light phase comparison). Mice were assessed under standard nutritional conditions (i.e. standard diet‐fed) and food/water intake and locomotor activity were continuously monitored (Giacovazzo et al., 2018; Scaricamazza et al., 2020; Steyn et al., 2020).
2.6. Glucose tolerance test (GTT)
GTTs was performed on treated and untreated 120 day‐old SOD1G93A and WT littermate mice (n ≥ 6 for each experimental group). Glucose was measured in tail tip blood samples using a Multicare Test Strip apparatus (Biochemical Systems International, Italy). Plasma glucose was assessed at 0, 20, 40 and 60 min after i.p. glucose administration (2 g kg−1), which was delivered after a 16 h overnight fast. The area under the curve was calculated using the trapezoidal rule.
2.7. Electrophoresis and western blotting
Protein obtained from the transverse abdominal (TA) muscle of treated and untreated 120 day‐old SOD1G93A and WT littermate mice (n ≥ 6 for each experimental group) were separated by SDS–polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes (Perkin Elmer, Cat# NBA085B). After blocking with Tris‐buffered saline solution with 0.1% Tween‐20 (TBS‐T) containing 5% BSA, the membranes were incubated overnight at 4°C with primary antibodies diluted in TBS‐T containing 2% BSA. Primary antibodies were detected using the appropriate peroxidase‐conjugated secondary antibody, incubated at room temperature for 1 h, diluted in TBS‐T containing 1% BSA. Immunoblots were visualized using enhanced chemiluminescence (BIO‐RAD ClarityTM Western ECL substrate Cat# 170–5061). The apparent molecular weight of proteins was determined by calibrating the immunoblots with pre‐stained molecular weight markers (Bio‐Rad Laboratories, Cat# 161‐0394). Densitometric analyses were performed using ImageJ (U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2016; RRID:SCR_003070). The relative expression of proteins of interest was normalized to glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) expression levels.
2.8. RNA isolation and real‐time qPCR
Total RNA was extracted from tibialis anterior muscles of treated and untreated 90, 120 and 150 day‐old SOD1G93A and WT littermate mice (n ≥ 5 for each experimental group) with TRIzol® Reagent (Ambion, Life Technologies) and reverse‐transcribed with the ImProm‐II™ Reverse Transcription System (Promega Cat.# A3800). Levels of mRNA expression were measured by Real‐Time qPCR using a Light Cycler 480 SYBR Green System (Roche ETC). Cp values were calculated using the “second derivative max” algorithm of the Lightcycler software (RRID:SCR_012155). TATA box binding protein was used as the housekeeping gene for normalization. Primers sequences are presented in Table 3.
2.9. Skeletal muscle immunofluorescence and histochemistry
Tibialis anterior muscles were embedded in Tissue‐Tek OCT compound and sectioned at 8 μm using a Leica Cryostat. Muscle cryosections were then used for NADH‐tetrazolium reductase activity staining (NADH‐TR). Briefly, muscle cryosections were hydrated in 0.1 M TRIS–HCl, pH 7.5 for 10 min and then incubated in NADH‐TR solution (0.4 mg ml−1 NADH, 0.8 mg ml−1 NTB and 1 M TRIS–HCl, pH 7.5) for 40 min at 37°C in a humidified chamber. Afterwards, excess reagent was removed and the slides were dehydrated in increasing concentrations of ethanol. Slides were then cleared in Xylene and mounted in Eukitt mounting medium. Whole muscle section images were acquired using an Olympus BX‐41, using a SPOT RT 220‐3 camera (Diagnostic Instrument, Inc.) and SPOT advanced software. The percentage of NADH‐positive area was quantified using ImageJ software (RRID:SCR_003070).
For NMJ analysis, the extensor digitorum longus (EDL) muscle was processed as previously described (De Paola et al., 2020). Briefly muscles were dissected and fixed in 4% PFA at 4°C for 3 h. Small bundles of muscle fibres were isolated under a dissecting microscope and immuno‐stained with anti‐neurofilament and anti‐synaptophysin antibodies. TRITC and Cy5 were used as secondary antibodies to visualize neurofilament and synaptophysin primary antibodies respectively. The α subunits of Nicotinic Acetylcholine receptors (nAChRs) were labelled with Alexa Fluor 488–conjugated α‐bungarotoxin (10 nM; Molecular Probes). Z‐stack images were obtained at sequential focal planes (2 μm apart) using a confocal microscope (Laser Scanning TCS SP2; Leica). NMJs was analysed in terms of postsynaptic primary and secondary gutters, number of fragments, and area of nACh receptor clusters. The z‐stacked fluorescence (n = 15–30 NMJs/genotype and treatment) were analysed using ImageJ software (RRID:SCR_003070). The colocalization of synaptophysin and nACh receptors was evaluated using the Fiji ImageJ1 plug‐in (RRID:SCR_002285). Representative images are flattened projections of Z‐stack images.
2.10. Spinal cord and sciatic nerve immunofluorescence and histochemistry
For immunofluorescence and histochemistry analysis, spinal cords and sciatic nerves of treated and untreated 120 and 150 day‐old SOD1G93A and WT mice (n ≥ 4 for each experimental group: n = 4 for WT, n = 6 for WT TMZ, G93A, G93A TMZ), post‐fixed in 4% paraformaldehyde and cryoprotected in 30% sucrose in PB. Spinal cord (L3‐L5) sections were cut at 30 μm‐thickness using a cryostat. For motor neurons count, Nissl with cresyl violet staining was performed as previously described (Mirra et al., 2017). Briefly the sections were stained with 1% Cresyl Violet and dehydrated in 50%–100% alcohol, cleared in xylene and cover‐slipped with Eukitt mounting medium (Sigma‐Aldrich). The ventral horns were photographed at 10 × magnification with a Zeiss Axioskop 2 microscope and large neurons, with a cell body area ≥200 μm2 and a definable cytoplasm with a nucleus and nucleolus, were counted using Neurolucida software (MBF Bioscience, USA; RRID:SCR_001775). For immunofluorescence, the sections were blocked in PBS, 0.3% TitonX‐100 and 10% normal donkey serum (NDS) for 1 h at room temperature. They were then incubated for 48 h at 4°C with the primary antibody in PBS, 0.3% TitonX‐100 and 2% NDS, followed by the appropriate secondary antibody for 3 h at room temperature in the same solution, as previously described (Amadio et al., 2014). To analyse sciatic nerves, transverse 15‐μm‐thick sections were cut using cryostat, and blocked with PBS, 0.2% TitonX‐100 and 10% NDS for 1 h at room temperature. Sections were incubated for 24 h at 4°C with the primary antibody in PBS, 0.2% TitonX‐100 and 2% NDS, followed by the appropriate secondary antibody for 3 h at room temperature. Immunofluorescence on spinal cord and sciatic nerve sections were visualized at 20 × or 40 × magnification with a Zeiss LSM 800 Confocal Laser Scanning Microscope and processed by ZEN 2.6 (Blue Edition) (Carl Zeiss, Milan, Italy) and Adobe Photoshop software (Adobe, San Jose, CA, USA; RRID:SCR_014199). Densitometric analyses were performed using ImageJ (U. S. National Institutes of Health, Bethesda, Maryland, USA, https://imagej.nih.gov/ij/, 1997–2016); (RRID:SCR_003070).
2.11. Bioenergetic analysis of isolated mitochondria
Mitochondria were isolated from the tibialis anterior muscles and spinal cords of treated and untreated 120 day‐old SOD1G93A and WT littermate mice (n ≥ 6 per group). Tissues were homogenized in 210 mM mannitol, 70 mM sucrose, 1 mM EDTA and 10 mM HEPES KOH (pH 7.5) with a Glass/Teflon Potter Elvehjem homogenizer (Salvatori et al., 2017, 2018). The total concentration of mitochondria was determined using the Bradford assay (Bio‐Rad, Cat# 5000006).
Isolated mitochondria, resuspended in a minimum volume of Respiration Buffer (250 mM Sucrose, 15 mM KCl, 1 mM EGTA, 5 mM MgCl2, 30 mM K2HPO4) were used to study mitochondrial function/dysfunction through the Coupling assay, and the study of the sequential flow of electrons through the complexes of the electron transport chain. Briefly, in the Coupling assay, 4 ug of mitochondria were loaded into a seahorse XFe96 microplate and centrifuged for 20 min at 2000 × g. After centrifugation, 180 μl of Respiration Buffer [containing substrates: pyruvate (5 mM), malate (2.5 mM), glutamate (5 mM)] was added to each well and the microplate was incubated at 37°C for 8 min. During plate incubation, the XFe96 cartridge was loaded with drugs at a final concentration of: ADP 1 mM, oligomycin 3 μM, FCCP (2‐[2‐[4‐(trifluoromethoxy)phenyl]hydrazinylidene]‐propanedinitrile) 12 μM, and antimycin A 2 μM. For the Electron Flow Assay, 6 ug of mitochondria were loaded into a seahorse XFe96 microplate and centrifuged for 20 min at 2000 × g. After centrifugation, 180 μl of Respiration Buffer [containing substrates: pyruvate (10 mM), malate (2 mM), FCCP (4 uM)] was added to each well and the microplate was incubated at 37°C for 8 min. During plate incubation, the XFe96 cartridge was loaded with drugs at a final concentration of: ‐ rotenone 2 μm, succinate 10 mM, antimycin A 4 μm, ascorbate and N,N,N,N‐tetramethyl‐p‐phenylenediamine (TMPD) 10 mM and 100 μm, respectively. The microplates containing mitochondria were then loaded into the Seahorse XFe96 Extracellular Flow Analyser (Seahorse Bioscience‐Agilent) and the test was completed according to the protocol developed by Seahorse Agilent. All data were analysed with XFe Wave software and displayed as either point‐to‐point oxygen consumption rates (pmol/minute/well), or absolute oxygen tension in mm Hg O2 versus time.
2.12. Materials
NADH,NTB, TRIZMA, xylene, eukitt, TitonX‐100,PBS, sucrose, EGTA, MgCl2, K2HPO4, rotenone, succinate, ascorbate, N,N,N,N‐tetramethyl‐p‐phenylenediamine(TMPD), pyruvate, malate, glutamate, ADP, oligomycin, FCCP (2‐[2‐[4‐(trifluoromethoxy)phenyl]hydrazinylidene]‐propanedinitrile)antimycin A were obtained from Sigma‐Aldrich.
2.13. Data analysis
Data and statistical analysis complied with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018).
Data are presented as mean ± SEM. Data were analysed using Kaplan–Meier, Student's t‐test (two group comparison), one‐way or two‐way ANOVA as appropriate. Post‐hoc analysis was carried out using Bonferroni or Tukey tests. Differences between groups were considered significant when the P value was less than 0.05. A group of n > 5 was used for each experimental group. Considering the low SEM and the strong significance, the experimental group of histological analysis was composed as follow:‐ n = 4 for WT, n = 6 for WT TMZ, G93A, G93A TMZ. Statistical analysis was carried out using Graphpad Prism 5 software (RRID:SCR_002798).
2.14. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in https://www.guidetopharmacology.org/ and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander et al., 2021).
3. RESULTS
3.1. Trimetazidine improves physical performance and extends lifespan of SOD1G93A mice
Treatment with trimetazidine in SOD1G93A mice was initiated at symptom onset, which corresponded to 70 days of age and 20% decrease in grip strength (Figure S1). Trimetazidine was administered orally in drinking water at 20 mg kg−1 based on plasma bioavailability (Figure S2). This dose achieves a trimetazidine plasma concentration of 70 ng ml−1 in SOD1G93A mice, which is similar to that obtained in humans (Jiao et al., 2007).
Trimetazidine‐treated SOD1G93A mice have a net improvement in grip strength when compared to untreated SOD1G93A mice (Figure 1a,b). The beneficial effect of trimetazidine treatment on grip strength appears to last longer in males than females. However, after 154 days (for females) and 161 days (for males) the data was not statistically significant due to the low number of remaining animals still able to perform the test. Most importantly, the administration of trimetazidine delays the time to reach disease midpoint and endpoint (Figure 1c,d). This is mirrored by a consistent increase in disease duration (Figure 1e,f) and an overall marked extension in the lifespan of SOD1G93A mice (Figure 1g,h).
FIGURE 1.
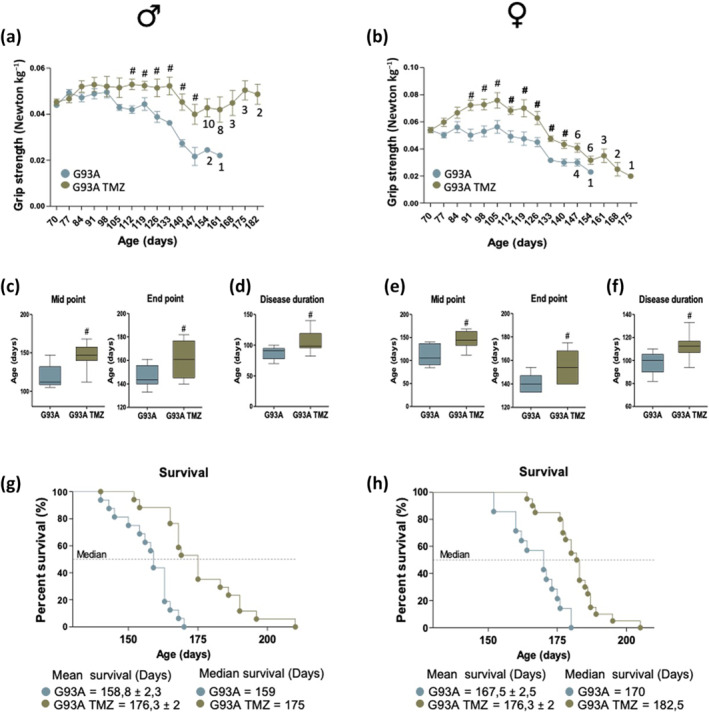
Trimetazidine (TMZ) improves muscle strength and extends lifespan of SOD1G93A mice. (a and b) Maximal grip strength of SOD1G93A mice (males in (a) and females in (b)) treated with (G93A TMZ) or without (G93A) TMZ (20 mg kg−1 in drinking water) from the onset of the disease (70 days of age) through to disease end point (when mice were no longer able to perform the test). Data presented as means ± SEM, # P < 0.05 when compared with untreated littermates SOD1G93A mice, n ≥ 12 per group. Numbers under data points indicate the number of animals remaining in the study from 154 days for males and 147 days for females. (c–e) The midpoint (left panels) and the humane endpoint (middle panels) of disease in TMZ treated (G93A TMZ) and littermates untreated SOD1G93A mice (G93A) are defined by a 25% and 50% decrease in grip strength respectively, starting from the onset of disease; males in (c) and females in (e). Data are presented as means ± SEM, #P < 0.05 when compared with untreated age‐matched SOD1G93A mice, n ≥ 12 per group. (d–f) Disease duration in TMZ treated (G93A TMZ) and untreated (G93A) littermates SOD1G93A mice; males in (d) and females in (f). Data are presented as means ± SEM. # P < 0.05, n ≥ 12 per group. (g and h) Kaplan–Meier survival curves of treated (G93A TMZ) and untreated (G93A) littermates SOD1G93A mice; males in (g) and females in (g), n ≥ 12 per group. P values were obtained using parametric two‐way ANOVA with Bonferroni post hoc test (a, b) or t‐test (e–h)
3.2. Trimetazidine prevents hypermetabolism in symptomatic SOD1G93A mice
We evaluated the impact of trimetazidine treatment on whole‐body metabolism in 120‐day old animals by indirect calorimetry. The increase in energy expenditure (i.e. hypermetabolism), typical of SOD1G93A mice (Scaricamazza et al., 2020; Steyn et al., 2020), is not observed in trimetazidine‐treated male and female mice (Figure 2a–d). Notably, data on locomotor activity were similar among each group (Figure S3), indicating that the observed differences in energy expenditure are not related to alterations of motor behaviour.
FIGURE 2.
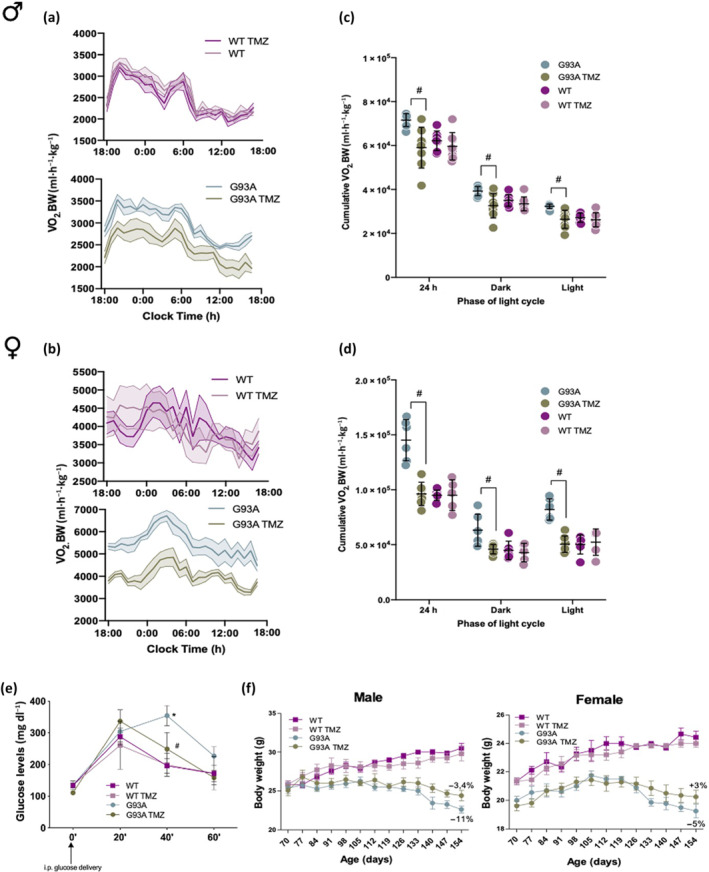
Trimetazidine (TMZ) prevents metabolic alterations in SOD1G93A mice. (a) Data traces representing mean oxygen consumption (a proxy for energy expenditure) over 24 h in 120‐day old male (a) and female (b) wild‐type and SOD1G93A mice untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ) from 70 days of age (disease onset). Dotted lines represent SEM. (c and d) Cumulative oxygen consumption in mice over a 24‐h period and during the light and dark cycle. (e) Glucose tolerance test in 120‐day old wild‐type and SOD1G93A mice untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ) from 70 days of age (disease onset). Blood glucose was assessed before glucose injection (0 min), and 20, 40 and 60 min following glucose injection. (f) Body mass evolution of SOD1G93A and wild‐type littermate mice (males left panel, females right panel) with (G93A TMZ and WT TMZ) or without (G93A and WT) TMZ. Starting body weight at 70 days was assigned as 100%, and the decline in body weight over time was calculated as percentage loss relative to the starting body weight. Data are presented as means ± SEM # P < .05, compared with untreated G93A, n ≥ 6 per group. P values were obtained using one‐way ANOVA with Bonferroni post hoc test
In agreement with previous findings, trimetazidine appears to improve glucose metabolism (Marzilli et al., 2019), as glucose intolerance in 120‐day old SOD1G93A mice was inhibited by trimetazidine (Figure 2e). Despite the beneficial effects induced by trimetazidine on hypermetabolic SOD1G93A mice, trimetazidine does not impact body weight as weight loss in SOD1G93A mice treated with trimetazidine is comparable to non‐treated SOD1G93A mice (Figure 2f).
3.3. Trimetazidine partially reverses the glycolytic‐to‐oxidative switch in muscle from SOD1G93A mice
A progressive switch in skeletal muscle metabolism from a glycolytic phenotype towards an oxidative phenotype has been described in ALS (Ferri & Coccurello, 2017; Scaricamazza et al., 2020; Steyn et al., 2020). One key mechanism by which muscle increases its energy expenditure and promotes lipid oxidation depends on the muscle‐restricted activity of sarcolipin and carnitine palmitoyl‐transferase 1 (Scaricamazza et al., 2020). Sarcolipin, upon binding to the sarcoplasmic reticulum Ca2+‐ATPase (SERCA), inhibits calcium transport to favour ATP hydrolysis (Bal et al., 2012), whereas carnitine palmitoyl‐transferase I allows fatty acids to enter mitochondria. In agreement with this, at 120 days of age, sarcolipin and carnitine palmitoyl‐transferase 1 are significantly upregulated in the tibialis anterior (TA) muscle of SOD1G93A mice when compared to WT mice, while trimetazidine treatment partially attenuates this increase in protein expression (Figure 3a).
FIGURE 3.
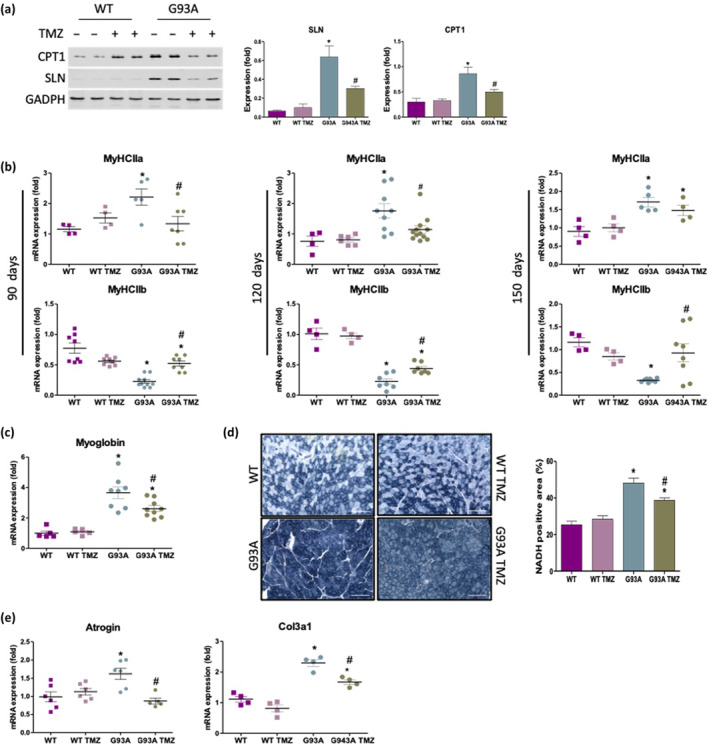
Trimetazidine (TMZ) restores the glycolytic phenotype and reduces atrophic/fibrotic markers in the tibialis anterior (TA) muscle of SOD1G93A mice. (a) Representative western blots (left) and quantification (right) of sarcolipin (SLN) and CPT1 protein expression in the TA of 120‐day old wild‐type (WT) and SOD1G93A (G93A) mice untreated (−) and treated (+) with TMZ. Data are presented as means ± SEM, *P < 0.05 compared with WT, # P < 0.05 compared with G93A, n ≥ 5 per group. (b) Expression level of mRNAs encoding Myosin Heavy Chain isoforms MyHCIIa (top row) and MyHCIIb (bottom row) in TA obtained from 90‐, 120‐ and 150‐day old wild‐type and SOD1G93A and WT mice untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ). Data are presented as means ± SEM, * P < 0.05, compared with WT (arbitrarily set at 1), #P < 0.05, compared with G93A, n ≥ 5 per group. (c) Expression level of mRNAs encoding myoglobin in TA obtained from 120‐day old wild‐type and SOD1G93A mice untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ). Data are presented as means ± SEM, *P < 0.05 compared with WT (arbitrarily set at 1), #P < 0.05 compared with G93A, n ≥ 4 per group. (d) Representative images (left) of in situ NADH‐tetrazolium reductase activity staining in cross sections of TA muscle obtained from 120‐day old wild‐type and SOD1G93A littermates untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ). Scale bar, 200 μm. Graphical representation (right) of the percentage NADH‐positive area of whole transverse sections of TA muscle. Data are presented as means ± SEM, *P < 0.05, compared with WT, # P < 0.05 compared with G93A, n ≥ 5 per group. (e) Expression level of mRNAs encoding atrogin and Col3a1 in TA obtained from 120‐day old wild‐type and SOD1G93A mice untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ). Data are presented as means ± SEM, *P < 0.05 compared with WT, # P < 0.05 compared with G93A, n ≥ 5 per group. P values were obtained using parametric one‐way ANOVA with Bonferroni post hoc test
Next, we evaluated whether trimetazidine treatment affects the mRNA expression levels of myosin heavy chain IIa (MyHCIIa) and IIb (MyCHIIb) isoforms, which are typically expressed by fast‐twitch oxidative and glycolytic myofibers, respectively. We observe a significant reduction in the expression of MyHCIIa (Figure 3b upper panels) and a concomitant upregulation of MyCHIIb (Figure 3b lower panels) in trimetazidine‐treated SOD1G93A mice at 90, 120 and 150 days of age when compared to non‐treated SOD1G93A mice. Accordingly, the oxygen storage protein myoglobin, normally abundant in oxidative myofibers and highly expressed in SOD1G93A mice, is downregulated by trimetazidine treatment (Figure 3c). Assessment of oxidative fibre composition in tibialis anterior muscle via the in situ nicotinamide adenine dinucleotide dehydrogenase‐tetrazolium reductase activity assay (NADH‐TR) reveals a larger oxidative region in SOD1G93A mice vs WT mice (Figure 3d). Treatment with trimetazidine significantly reduces the proportion of oxidative fibres in 90‐ (Figure S4) and 120‐day old (Figure 3d) SOD1G93A mice, without having any effects in WT animals. Finally, trimetazidine reduces the expression of the atrophy marker Atrogin as well as the fibrosis marker Collagen type III alpha 1 chain (Col3a1) in skeletal muscle (Figure 3e).
3.4. Trimetazidine preserves the integrity of neuromuscular junctions in SOD1G93A mice
Since trimetazidine improves grip strength and skeletal muscle metabolic phenotypes in SOD1G93A mice, we next considered if these changes were due to a protective action of trimetazidine on NMJ integrity. Comprehensive morphological analysis of NMJs in the glycolytic extensor digitorum longus (EDL) muscle highlights marked alterations in NMJ integrity and innervation in 120 and 150 day‐old SOD1G93A mice when compared to WT littermates (Figure 4a). In particular, we observe a decrease in the NMJ complexity, as evidenced by the limited level of branching areas (Figure 4b,c), and the tendency, albeit not statistically significant, for greater neuromuscular endplate fragmentation in SOD1G93A mice (Figure 4d). In addition, in 150 day‐old SOD1G93A mice, we detect a significant decrease in NMJ area (Figure 4e). Moreover, quantitative analysis of colocalization between nACh receptor clusters and synaptophysin reveals a significant decrease in the spatial overlap between the two fluorescent signals in SOD1G93A mice at both 120 and 150 days of age (Figure 4f). Crucially, trimetazidine improves NMJ complexity (Figure 4b,c), attenuates NMJ fragmentation (Figure 4d), increases NMJ area (Figure 4e) and enhances the percentage of nACh receptor‐synaptophysin colocalization (Figure 4f) in the EDL muscles of SOD1G93A mice.
FIGURE 4.
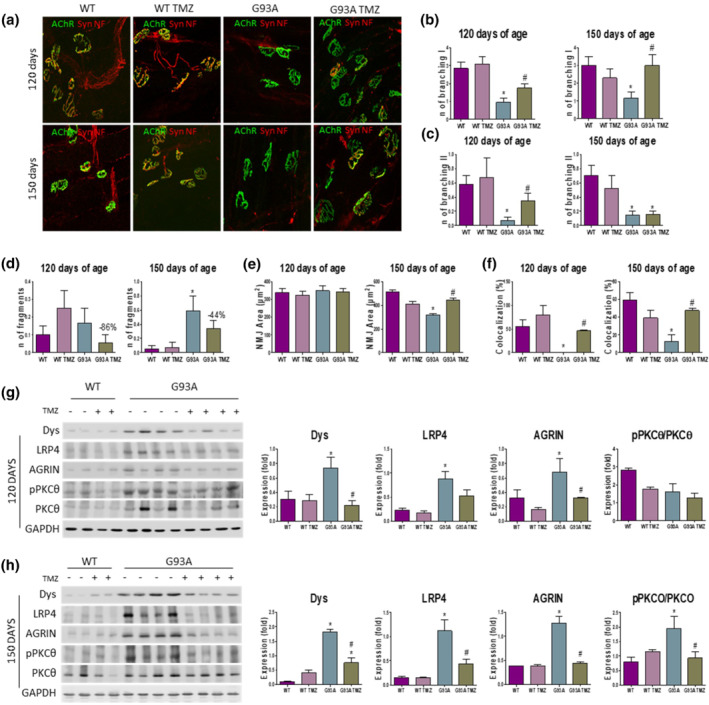
Trimetazidine (TMZ) preserves the integrity of neuromuscular junctions (NMJs). (a) Representative maximum projection images of EDL NMJs from 120‐ and 150‐day old wild‐type and SOD1G93A mice untreated (WT and G93A) and treated with TMZ (WT TMZ and G93A TMZ), stained with synaptophysin (Syn), α‐bungarotoxin (nACh receptor), and Neurofilament (NF). (b–f) Quantitation of NMJ morphometry in EDL muscles of WT, WT TMZ, G93A and G93A TMZ mice at 120 and 150 days of age, highlighting the number of primary (b) and secondary (c) ramifications, the number of fragments per NMJ (d), the area of the nACh receptor clusters (e) and the percentage of colocalized voxels (f). Data are presented as means ± SEM. *P < 0.05, compared with WT mice and #P < 0.05 compared to SOD1G93A mice, n ≥ 4 per group (n = 4 for WT, n = 6 for WT TMZ, G93A, G93A TMZ). The trend towards a reduced neuromuscular endplate fragmentation in TMZ treated SOD1G93A mice (d) is shown as percentage. (g and h) Representative western blots (left) and quantification (right) of Dystrophin (Dys), lipoprotein receptor‐related protein 4 (LRP4), agrin, and phosphorylated Protein Kinase C theta/Protein Kinase C theta (pPKCθ/PKCθ) protein expression in the tibialis anterior of 120‐day old (g) and 150‐day old (h) wild‐type (WT) and SOD1G93A (G93A) mice untreated (−) and treated (+) with TMZ from disease onset (70 days of age). Data are presented as means ± SEM, *P < 0.05, compared with WT mice and # P < 0.05, compared with G93A mice, n ≥ 4 per group (n = 4 for WT, n = 6 for WT TMZ, G93A, G93A TMZ). P value was obtained using parametric one‐way ANOVA with Bonferroni post hoc test
Finally, we analysed selected markers of NMJ stability. Dystrophin, low‐density lipoprotein receptor‐related protein 4 (LRP4) and agrin are critical for maintaining NMJ stability, whereas the phosphorylated form of Protein Kinase C theta (pPKCθ) is typically associated with the destabilization of NMJs (Dobrowolny, Martini, et al., 2018). In line with the reduction in NMJ dismantlement in SOD1G93A mice treated with trimetazidine, we show that pPKCθ levels are reduced in trimetazidine‐treated SOD1G93A mice (Figure 4g,h). While levels of dystrophin, LRP4 and agrin are higher in SOD1G93A mice when compared to WT littermates, trimetazidine treatment normalizes the expression of these proteins in 120‐ and 150‐day old SOD1G93A mice towards WT levels (Figure 4g,h).
3.5. Trimetazidine reduces demyelination and immune cell infiltration in sciatic nerves from SOD1G93A mice
Next, we explored whether trimetazidine treatment might exert a beneficial effect on demyelination, inflammation and axonal loss in the peripheral nervous system of SOD1G93A mice. Cross sections of sciatic nerve from WT and treated and untreated SOD1G93A were assessed for myelin, neurofilament heavy chain and macrophages. As shown in Figure 5, SOD1G93A mice display a Wallerian‐like degeneration characterized by myelin degradation (MBP), axonal cytoskeleton loss (NFH), and immune cell infiltration (CD68). Trimetazidine treatment reduces axonal demyelination, preserves the ordered distribution of axonal fibres, and reduces the infiltration of inflammatory CD68 + cells in 120‐day old SOD1G93A mice (Figure 5a). Comparable trimetazidine effects are also observed in 150‐day old SOD1G93A mice, with the exception of the demyelination process (Figure 5b). Consistent with the removal of myelin debris by an increase in phagocytic GFAP+ Schwan cells (Yang & Wang, 2015) during Wallerian degeneration, we observe a strong decrease in GFAP expression in trimetazidine‐treated SOD1G93A mice when compared with their untreated transgenic littermates at both in 120‐ and 150‐days of age (Figure 5c,d).
FIGURE 5.
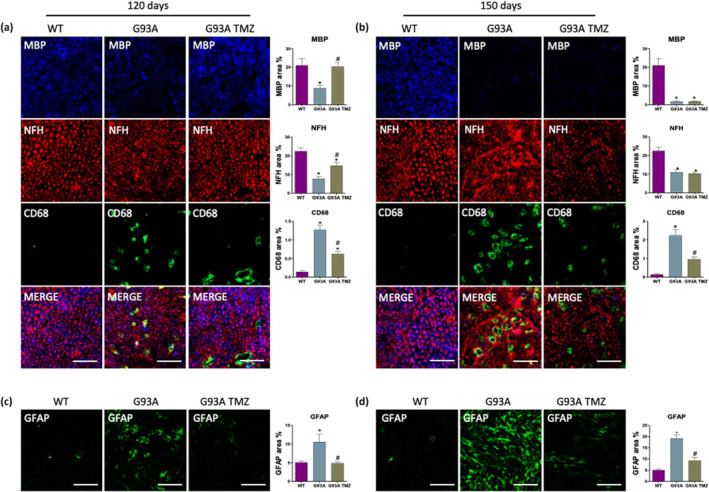
Trimetazidine (TMZ) preserves sciatic nerve integrity. (a and b) Representative images of sciatic nerve sections and quantifications of fluorescence intensity (histograms) from 120‐ and 150‐day old wild‐type mice and SOD1G93A treated without (WT and G93A) or with TMZ (WT TMZ and G93A TMZ) from disease onset (70 days of age). Anti‐MBP stains myelin, anti‐NFH stains the axonal cytoskeleton (neurofilament heavy), anti‐CD68 stains activated macrophages. (c and d) Representative images of sciatic nerve sections and quantifications of fluorescence intensity (histograms) from 120‐ and 150‐day old wild‐type mice and SOD1G93A treated without (WT and G93A) or with TMZ (WT TMZ and G93A TMZ) from disease onset (70 days of age). Anti‐GFAP stains astrocytes. Scale bar 50 μm. Data are presented as means ± SEM, *P < 0.05, compared with WT mice and #P < 0.05, compared with G93A mice, n ≥ 4 per group (n = 4 for WT, n = 6 for WT TMZ, G93A, G93A TMZ). P value was obtained using parametric one‐way ANOVA with Bonferroni post hoc test
3.6. Trimetazidine protects the spinal cord of SOD1G93A mice from motor neurons loss and neuroinflammation
Upstream of NMJ damage, the loss of ventral horn motor neurons is a major pathological feature of ALS and, in particular, of the SOD1G93A mouse. Here we show that the reduction in the number of lower motor neurons in SOD1G93A mice is particularly evident at 120 days of age and that this becomes significantly worse by 150 days of age (43% and 67% of spinal motor neurons loss respectively; Figure 6a,b). Notably, trimetazidine treatment significantly improves motor neuron survival by completely or partially preserving motor neurons from death at 120 days of age (Figure 6a) and 150 days of age (Figure 6b), respectively.
FIGURE 6.
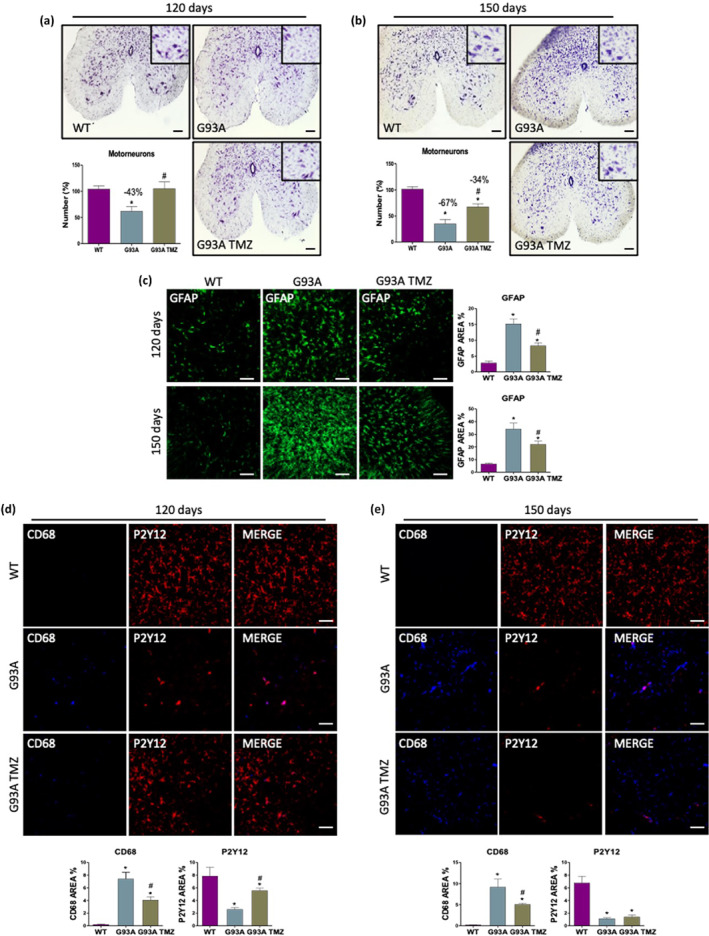
Trimetazidine (TMZ) protects from motor neuron loss and neuroinflammation. (a and b) Representative lumbar spinal cord sections (L3‐L5) stained with cresyl violet (scale bar 100 μm) and quantification of motor neuron number (histograms) in 120‐ (a) and 150‐day old (b) wild‐type mice treated without TMZ (WT) and SOD1G93A mice treated without (G93A) or with TMZ (G93A TMZ) from disease onset (70 days of age). Data are presented as means and ± SEM, *P < 0.05, compared with wild‐type; # P < 0.05, compared with untreated G93A, n ≥ 4 per group (n = 4 for WT, n = 6 for G93A, G93A TMZ). (c) Representative immunofluorescence images and analysis of astrocytes (GFAP) in lumbar spinal cord sections (L3‐L5) of 120‐ and 150‐day old wild‐type mice treated without TMZ (WT) and SOD1G93A mice treated without (G93A) or with TMZ (G93A TMZ) from disease onset (70 days of age). (d and e) Representative immunofluorescence images and analysis of activated microglia (CD68) and resting microglia (P2T12) in lumbar spinal cord sections (L3‐L5) of 120‐ and 150‐day old wild‐type mice treated without TMZ (WT) and SOD1G93A mice treated without (G93A) or with TMZ (G93A TMZ) from disease onset (70 days of age). Scale bar 100 μm. Data are presented as means and ± SEM, *P < 0.05 compared with wild‐type; # P < 0.05, compared with untreated G93A, n ≥ 4 per group (n = 4 for WT, n = 6 for G93A, G93A TMZ). P values were obtained using parametric one‐way ANOVA with Bonferroni post hoc test (a–e)
Motor neuron degeneration is generally accompanied by a strong neuroinflammatory response, which involves, among other factors, the activation of reactive astrocytes and microglia (Volonté et al., 2019). To evaluate the potential effect of the trimetazidine on neuroinflammation, spinal cord sections from untreated and treated mice were probed with antibodies recognizing typical pro‐inflammatory proteins. Reactive GFAP‐positive astrocytes, which are not detectable in WT spinal cords, are evident in SOD1G93A mice at 120 days of age and this becomes more pronounced at 150 days (Figure 6c). Trimetazidine treatment appears to inhibit the activation of reactive astrocytes at both ages, as evidenced by a decrease in GFAP immunoreactivity (Figure 6c). Spinal cord sections from 120‐ and 150‐day old SOD1G93A mice also show increased immunoreactivity for CD68 (a marker of activated microglia) in parallel with a decrease in immunoreactivity for P2Y12 receptor (a marker of resting microglia) when compared to WT mice. Treatment with trimetazidine reduces CD68 immunoreactivity in 120‐ and 150‐day old SOD1G93A mice, and increases P2Y12 immunoreactivity in 120‐day old SOD1G93A mice (Figure 6d,e).
3.7. Trimetazidine improves mitochondrial function in skeletal muscle and spinal cord of SOD1G93A mice
To uncover the potential mechanism(s) through which trimetazidine exerts beneficial effects in SOD1G93A mice, we performed in‐depth analysis of the bioenergetic profiles of mitochondria isolated from the skeletal muscle and spinal cord of trimetazidine‐treated and trimetazidine‐untreated SOD1G93A and WT littermate mice at 120 days of age.
Analysis of Oxygen Consumption Rate (OCR) coupled to ATP production performed through the “coupling assay” shows that trimetazidine improves the functional state of mitochondria (Figure 7a,b). Indeed, basal respiration (Basal), maximal ATP‐coupled respiration obtained by the addition of ADP (State 3), basal ATP synthesis‐uncoupled respiration obtained through the addition of oligomycin (State 4o) and maximal ATP synthesis‐uncoupled respiration obtained by the addition of FCCP (State 3u) are all significantly increased in mitochondria purified from skeletal muscle (Figure 7c) and spinal cord (Figure 7d) of trimetazidine treated SOD1G93A mice when compared to untreated mice. Notably, rescue of State 3 OCR by trimetazidine indirectly highlights an improved ability of mitochondria to produce ATP. Consistent with this, analysis of the electron transport chain (ETC) through the “electron flow assay” reveals improved ETC complex activities in response to trimetazidine treatment (Figure 8a,b). The OCR values obtained by the sequential addition of rotenone, succinate, antimycin A and ascorbate/ N,N,N,N‐tetramethyl‐p‐phenylenediamine (TMDP) show that trimetazidine improves Complex I‐, Complex II/III‐ and Complex IV‐dependent respiration in mitochondria isolated from the tibialis anterior muscle (Figure 8c). In isolated spinal cord mitochondria, trimetazidine restores the activity of Complex I and Complex II/III (Figure 8d).
FIGURE 7.
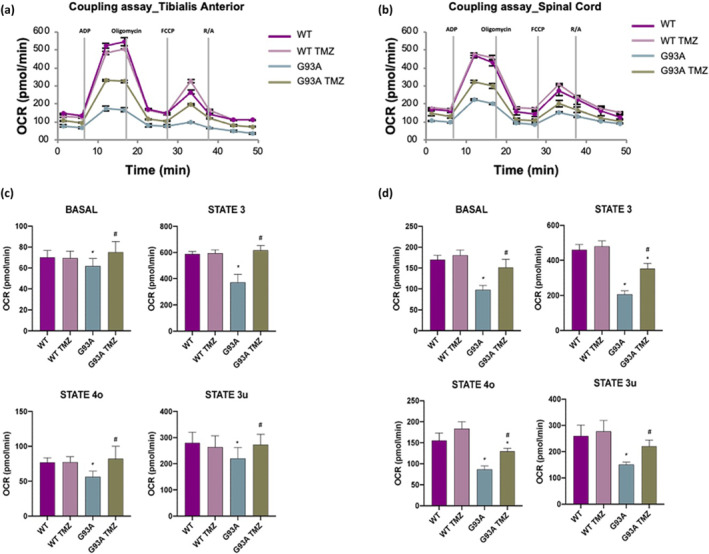
Trimetazidine (TMZ) improves mitochondrial coupling state. (a and b) Representative data traces of oxygen consumption rate (OCR) during a coupling assay on isolated mitochondria purified from the tibialis anterior and spinal cord. (c and d) Mitochondrial respiration states (as OCR) are reported as basal respiration (state 2), maximal coupled respiration (state 3), respiration due to proton leak (state 4o) and maximal uncoupled respiration (state 3u). Data are presented as means ± SEM, *P < 0.05 compared with wild‐type; # P < 0.05 compared with untreated G93A, n ≥ 6 per group. P values were obtained using parametric one‐way ANOVA with Bonferroni post hoc test
FIGURE 8.
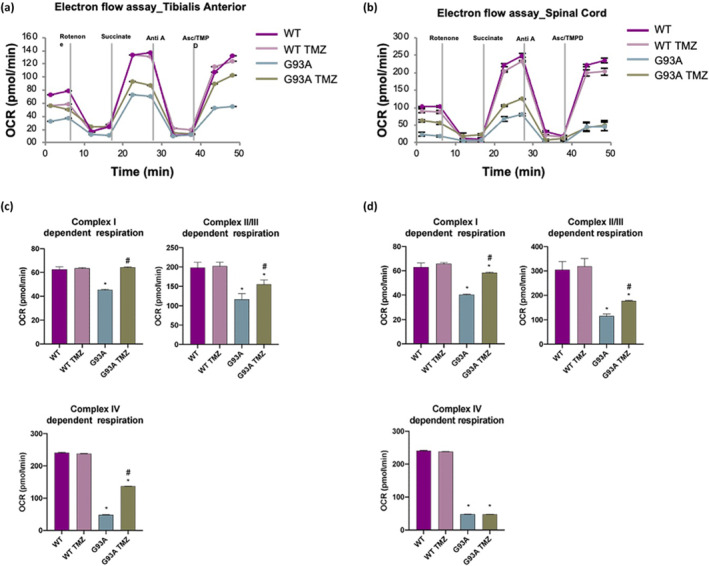
Trimetazidine (TMZ) improves electron transport chain complex activity. (a and b) Representative data traces of oxygen consumption rate (OCR) during an electron flow assay on isolated mitochondria purified from tibialis anterior and spinal cord. (c and d) Quantification of respiration (OCR) activity of complex I, complex II/III and complex IV in the presence of rotenone, succinate, antimycin A and ascorbate/N,N,N,N‐tetramethyl‐p‐phenylenediamine (ASC/TMPD), respectively. Data are presented as means ± SEM, *P < 0.05, compared with wild‐type; # P < 0.05, compared with untreated G93A, n = ≥6 per group. P values were obtained using parametric one‐way ANOVA with Bonferroni post hoc test
4. DISCUSSION
To date, a lack of understanding of the biological processes that trigger the onset and progression of amyotrophic lateral sclerosis (ALS) has impeded efforts to develop effective therapies for the disease. Of the 52 compounds that significantly attenuate disease pathology and improve disease phenotype and survival in mouse models of ALS, none have proven to be effective in clinical trials (Petrov et al., 2017). Due to the multifactorial nature of ALS, previous therapeutic strategies that target a single disease mechanism are likely to be inappropriate for slowing disease progression (Keon et al., 2021). By contrast, a more effective approach could be the use of a compound that acts simultaneously on various disease‐relevant pathways (Gontijo et al., 2019). Trimetazidine is a metabolic modulator that is used to treat cardiomyopathies. It improves metabolic performance of cardiac fibres by targeting a range of molecular pathways (Shu et al., 2021). By inhibiting fatty acid oxidation, trimetazidine is able to enhance glucose metabolism (Marzilli et al., 2019). Moreover, trimetazidine is known to improve mitochondrial function and calcium handling (Argaud et al., 2005; Dedkova et al., 2013; Meng et al., 2006), increase cellular antioxidant capacity (Dhote & Balaraman, 2008) and suppress inflammatory responses (Kuralay et al., 2006; Martins et al., 2012). Given that all of these pathways are affected in ALS, we aimed to assess the therapeutic potential of trimetazidine in the SOD1G93A mouse model of ALS.
Oral administration of trimetazidine from disease onset significantly improved grip strength and the life span of SOD1G93A mice. The beneficial effects of trimetazidine on grip strength, and hence skeletal muscle performance, appear to be due to a partial reversal in the glycolytic‐to‐oxidative switch that has been shown to occur in the skeletal muscle of SOD1G93A mice (Dobrowolny, Lepore, et al., 2018;Scaricamazza et al., 2020; Steyn et al., 2020). In line with this, trimetazidine is known to inhibit fatty acid oxidation by targeting 3‐ketoacyl‐CoA thiolase activity (Amoedo et al., 2021), which would have a net effect of driving muscle towards glucose use and thus more efficient utilization of the available oxygen (Lopaschuk et al., 2003). This notion is further supported by our data demonstrating that trimetazidine was able to downregulate the expression of carnitine palmitoyl‐transferase 1 in skeletal muscle and inhibited glucose intolerance in SOD1G93A mice. Of note, genetic knockout or pharmacological inhibition of carnitine palmitoyl‐transferase 1 has previously been shown to improve disease outcome in SOD1G93A mice (Trabjerg et al., 2021). Thus, the improvement in life span in trimetazidine treated SOD1G93A mice could be due, in part, to the effect of trimetazidine on modulating carnitine palmitoyl‐transferase 1 expression and glucose‐fatty acid‐flux in ALS skeletal muscle.
Since skeletal muscle is the major determinant of whole‐body energy expenditure (Zurlo et al., 1990), it is conceivable that trimetazidine‐induced improvements in skeletal muscle energy metabolism might also decrease the characteristic hypermetabolism (i.e. increased metabolic rate) seen in SOD1G93A mice (Scaricamazza et al., 2020; Steyn et al., 2020). This is of clinical importance as hypermetabolism is associated with poor outcomes in mouse models of ALS and in human ALS (Doshi et al., 2017; Dupuis et al., 2004; Fayemendy et al., 2021; Jésus et al., 2018; Scaricamazza et al., 2020; Steyn et al., 2018). In 2020, we demonstrated that treatment of SOD1G93A mice with the metabolic modulator ranolazine was able to improve locomotor function, decrease metabolic rate and partially rescue the dysfunction of muscle metabolism in SOD1G93A mice (Scaricamazza et al., 2020). A caveat of our 2020 study was that we did not assess the impact of ranolazine on survival. Building on this work, we now show that trimetazidine is able to prevent the emergence of hypermetabolism while also improving survival in SOD1G93A mice. These data highlight that the beneficial effects of trimetazidine are likely to be due to a combination of its metabolic actions of skeletal muscle and whole‐body energy metabolism.
The effect that trimetazidine exerts on skeletal muscle metabolism occurs because of the “metabolic flexibility” of this organ. However, neurons are thought to exhibit a characteristic “metabolic inflexibility” since they use glucose and its metabolite, lactate, as primary energy sources. Therefore, it could be argued that the action of trimetazidine as a metabolic modulator cannot solely explain the beneficial effects on grip strength and survival that we observed in response to the trimetazidine treatment in SOD1G93A mice. Yet, we describe for the first time, a strong positive impact of trimetazidine on the bioenergetics of purified mitochondria from the spinal cords of trimetazidine treated SOD1G93A mice. When considered alongside observations of decreased glucose use (Browne et al., 2006), and increased markers of oxidative stress and lipid peroxidation in brain and spinal cord tissue (Miana‐Mena et al., 2011; Simpson et al., 2004) in ALS, we cannot discount the modulation of lipid metabolism (Tracey et al., 2018) or modulation of cellular metabolism as a whole as a potential therapeutic mechanism of trimetazidine.
In addition to the metabolic effects of trimetazidine, we provide evidence to show that trimetazidine can exert neuroprotection through other mechanisms. We note that trimetazidine protects against the dismantlement of NMJs, an event that typically occurs in the early phase of disease in SOD1G93A mice (Dobrowolny, Martini, et al., 2018; Fischer et al., 2004; Moloney et al., 2014). It has been proposed that NMJ abnormalities predict the degeneration of both distal axons and spinal motor neurons in SOD1G93A mice (Clark et al., 2016; Fischer et al., 2004; Gurney et al., 1994). The beneficial effects of trimetazidine might therefore originate from a delay in NMJ dismantlement, which is considered as the first event of the dying back phenomenon underlying motor neuron degeneration in ALS (Bruneteau et al., 2015; Dupuis et al., 2009; Fischer et al., 2004). Consistent with this, trimetazidine exerted a protective effect on peripheral nerves of SOD1G93A mice, leading to the inhibition of the Wallerian‐like neuronal degeneration, and an attenuation in spinal motor neuron loss. In our experimental paradigm, the neuroprotective effect of trimetazidine on spinal cord motor neurons and peripheral nerves was mirrored by a parallel decrease in neuroinflammation. The state of activation of astrocytes and microglia in the spinal cord and the infiltration of CD68 + macrophages in the sciatic nerve, which increases during the course of disease in SOD1G93A mice (Chiot et al., 2020; Serrano et al., 2019), were mitigated by trimetazidine treatment. Overall, our results are in accordance with reports showing that trimetazidine induces axonal regeneration and myelination in a rodent model of sciatic nerve crush injury (Serarslan et al., 2009) and that it provides anti‐inflammatory effects in various tissues (Wan et al., 2017).
In conclusion, our results describe the efficacy of trimetazidine in counteracting key cellular ALS pathologies and systemic changes in metabolism in SOD1G93A mice. Critically, these changes were observed alongside trimetazidine‐induced improvements in grip strength and survival, providing evidence to progress the repurposing of trimetazidine for ALS. Notably, trimetazidine is currently used for the management of angina pectoris, although its usage has been limited due to the non‐common insurgence of reversible parkinsonian symptoms (Dy et al., 2020; Pintér et al., 2021). In our experimental model, however, no differences were observed in mice locomotor activity upon trimetazidine treatment. Nevertheless, we should stress that the preclinical mouse model employed in our study does not fully recapitulate the pathology. Thus, in‐depth analyses are required to assess the effectiveness and safety of trimetazidine, especially in light of the possible onset of motor deficits, although we envision that the benefits of trimetazidine should outweigh the risks in ALS treatment. However, the ability for trimetazidine to improve grip strength in ALS mice could be rapidly translated to patients to improve skeletal muscle function and quality of life in primis (Scaricamazza et al., 2021). The beneficial effect of trimetazidine on energy metabolism in spinal cord mitochondria also highlights the ability of trimetazidine to target mitochondrial dysfunction as a therapeutic intervention (Mehta et al., 2019) to enhance the chance of success in ALS. Finally, given that trimetazidine prevents hypermetabolism in SOD1G93A mice, our data further substantiate the precedent to target hypermetabolism as a therapeutic approach in ALS.
AUTHOR CONTRIBUTIONS
S.S. and I.S. designed and performed experiments. R.C., F.J.S., S.T.N designed and T.W.T., H.W., F.J.S., S.T.N, G.G. performed the indirect calorimetry experiments. E.F., A.T., V.N., G. D, E.L. and S.A. performed histological analysis. A.P. performed evaluation of trimetazidine concentration in plasma. C.Q. set up the trimetazidine administration method. M.G., E.F., and S.A. expanded and completed the study. J.P.L., F.R., F.J.S., S.T.N, R.C., E.F., A.U., L.P., A.M., N.C. and C.Vo. designed the experiments, analysed and interpreted data, and critically revised the manuscript. A.F. and C.Va. designed the study and wrote the paper.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, Immunoblotting and Immunochemistry and Animal Experimentation, as recommended by funding agencies, publishers and other organizations engaged with supporting research.
Supporting information
Figure S.1. Dinamometric evaluation of grip strength in wild‐type (WT) and SOD1G93A mice littermate mice. Data shows that disease onset occurs at 70 days of age in SOD1G93A mice. At this age, SOD1G93A mice exhibit a 20% decrease in grip strength when compared to their wild‐type (WT) littermates.
Figure S.2 Bio‐availability of Trimetazidine (TMZ) in the plasma of wild‐type mice. Mice were treated for one week with the indicated doses of TMZ (0, 5, 10, 20, 40 mg·kg‐1 ), n ≥ 4/group.
Figure S3. Locomotor activity. The number of horizontal and vertical movements (light‐beam frames broken) was measured over 72 h in SOD1G93A and wild‐type littermate mice (males left panel, females right panel) with (G93A TMZ and WT TMZ) or without (G93A and WT) daily administration of 20 mg·kg‐1 TMZ in drinking water.
Figure S.4. Nicotinamide adenine dinucleotide dehydrogenase‐tetrazolium reductase (NADH‐TR) activity in tibialis anterior (TA) muscle. (A) Representative in situ NADH‐TR activity staining in cross sections of TA muscle obtained from 90‐day old untreated (WT and G93A) and treated (WT TMZ and G93A TMZ) littermate mice. Scale bar, 200μm. (B) Quantification of the percentage of NADH positive areas in cross‐sections of TA muscle from (WT and G93A) and treated (WT TMZ and G93A TMZ) littermate mice. Data are presented as mean +/− SEM, n ≥ 4/group, **P < .05 compared to WT and WT TMZ, # P < .05 compared to G93A, parametric one‐way ANOVA with Bonferroni post‐hoc test.
ACKNOWLEDGEMENTS
This work is dedicated to the memory of our dear Mentor and Friend Maria Teresa Carrì.
This research was funded by Agenzia di Ricerca per la Sclerosi Laterale Amiotrofica (AriSLA) through the HyperALS project (A.F.); AFM‐Telethon project n. 2018; AFM‐Téléthon project n. 21021 (A.F.); Ministero della Salute: RF2019‐12369105 “Validation of a metabolic target as a pharmacodynamic biomarker in ALS patients” (A.F.); Ministero della Salute, Italy–United States of America: n.PGR01040 (C.Va.) (C.Va.); Australian National Health and Medical Research Council: 1185427 (S.T.N. and F.J.S); FightMND: Mid‐Career Research Fellowship (S.T.N); Agenzia Spaziale Italiana ASI_MARS‐PRE 2019‐11‐Udine.0 (A.M.) Fondazione Umberto Veronesi: Grant 2021 (N.C.). Open Access Funding provided by Consiglio Nazionale delle Ricerche within the CRUI‐CARE Agreement. [Correction added on 17 May 2022, after first online publication: CRUI funding statement has been added.]
Scaricamazza, S. , Salvatori, I. , Amadio, S. , Nesci, V. , Torcinaro, A. , Giacovazzo, G. , Primiano, A. , Gloriani, M. , Candelise, N. , Pieroni, L. , Loeffler, J.‐P. , Renè, F. , Quessada, C. , Tefera, T. W. , Wang, H. , Steyn, F. J. , Ngo, S. T. , Dobrowolny, G. , Lepore, E. , … Ferri, A. (2022). Repurposing of Trimetazidine for amyotrophic lateral sclerosis: A study in SOD1G93A mice. British Journal of Pharmacology, 179(8), 1732–1752. 10.1111/bph.15738
Silvia Scaricamazza and Illari Salvatori have equally contributed to this work.
Funding information AFM‐Téléthon, Grant/Award Number: n. 2018 and n. 21021; Agenzia Spaziale Italiana, Grant/Award Number: ASI_MARS‐PRE 2019‐11‐Udine.0; Australian National Health and Medical Research Council, Grant/Award Number: 1185427; FightMND, Grant/Award Number: Mid‐Career Research Fellowship; Fondazione Umberto Veronesi, Grant/Award Number: Grant 2021; Ministero della Salute, Grant/Award Numbers: RF2019‐12369105, n.PGR01040; Ministero della Salute, Italy–United States of America, Grant/Award Number: n.PGR01040 (C.Va.); Agenzia di Ricerca per la Sclerosi Laterale Amiotrofica, Grant/Award Number: HyperALS
Contributor Information
Cristiana Valle, Email: c.valle@hsantalucia.it.
Alberto Ferri, Email: alberto.ferri@cnr.it.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are contained in the body of the manuscript or in the supplemental material. Any additional data used are available from the corresponding authors upon reasonable request.
REFERENCE
- Alexander, S. P. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Aldrich, R. W. , Attali, B. , Baggetta, A. M. , Becirovic, E. , Biel, M. , Bill, R. M. , Catterall, W. A. , … Zhu, M. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Ion channels. British Journal of Pharmacology, 178(S1), S157–S245. 10.1111/bph.15539 [DOI] [PubMed] [Google Scholar]
- Amadio, S. , Parisi, C. , Montilli, C. , Carrubba, A. S. , Apolloni, S. , & Volonté, C. (2014). P2Yreceptor on the verge of a neuroinflammatory breakdown. Mediators of Inflammation, 2014, 1–15. 10.1155/2014/975849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoedo, N. D. , Sarlak, S. , Obre, E. , Esteves, P. , Bégueret, H. , Kieffer, Y. , Rousseau, B. , Dupis, A. , Izotte, J. , Bellance, N. , Dard, L. , Redonnet‐Vernhet, I. , Punzi, G. , Rodrigues, M. F. , Dumon, E. , Mafhouf, W. , Guyonnet‐Dupérat, V. , Gales, L. , Palama, T. , … Rossignol, R. (2021). Targeting the mitochondrial trifunctional protein restrains tumor growth in oxidative lung carcinomas. Journal of Clinical Investigation, 131(1). 10.1172/JCI133081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apolloni, S. , Caputi, F. , Pignataro, A. , Amadio, S. , Fabbrizio, P. , Ammassari‐Teule, M. , & Volonté, C. (2019). Histamine is an inducer of the heat shock response in SOD1‐G93A models of ALS. International Journal of Molecular Sciences, 20(15). 10.3390/ijms20153793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Argaud, L. , Gomez, L. , Gateau‐Roesch, O. , Couture‐Lepetit, E. , Loufouat, J. , Robert, D. , & Ovize, M. (2005). Trimetazidine inhibits mitochondrial permeability transition pore opening and prevents lethal ischemia‐reperfusion injury. Journal of Molecular and Cellular Cardiology, 39(6), 893–899. 10.1016/j.yjmcc.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Bal, N. C. , Maurya, S. K. , Sopariwala, D. H. , Sahoo, S. K. , Gupta, S. C. , Shaikh, S. A. , Pant, M. , Rowland L. A., Bombardier, E. , Goonasekera, S. A. , Tupling, A R. , Molkentin, J. D. , & Periasamy, M. (2012). Sarcolipin is a newly identified regulator of muscle‐based thermogenesis in mammals. Nature Medicine, 18(10), 1575–1579. 10.1038/nm.2897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belli, R. , Bonato, A. , De Angelis, L. , Mirabilii, S. , Ricciardi, M. R. , Tafuri, A. , Molfino, A. , Gorini, S. , Leigheb, M. , Costelli, P. , Caruso, M. , Muscaritoli, M. , & Ferraro, E. (2019). Metabolic reprogramming promotes myogenesis during aging. Frontiers in Physiology, 10, 897. 10.3389/fphys.2019.00897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne, S. E. , Yang, L. , DiMauro, J. P. , Fuller, S. W. , Licata, S. C. , & Beal, M. F. (2006). Bioenergetic abnormalities in discrete cerebral motor pathways presage spinal cord pathology in the G93A SOD1 mouse model of ALS. Neurobiology of Disease, 22(3), 599–610. 10.1016/j.nbd.2006.01.001 [DOI] [PubMed] [Google Scholar]
- Bruneteau, G. , Bauché, S. , Gonzalez de Aguilar, J. L. , Brochier, G. , Mandjee, N. , Tanguy, M.‐L. , Hussain, G. , Behin, A. , Khiami, F. , Sariali, E. , Hell‐Remy, C. , Salachas, F. , Pradat, P. F. , Lacomblez, L. , Nicole, S. , Fontaine, B. , Fardeau, M. , Loeffler, J. P. , Meininger, V. , … Hantaï, D. (2015). Endplate denervation correlates with Nogo‐a muscle expression in amyotrophic lateral sclerosis patients. Annals of Clinical and Translational Neurology, 2(4), 362–372. 10.1002/acn3.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiot, A. , Zaïdi, S. , Iltis, C. , Ribon, M. , Berriat, F. , Schiaffino, L. , Jolly, A. , de la Grange, P. , Mallat, M. , Bohl, D. , Millecamps, S. , Seilhean, D. , Lobsiger, C. S. , & Boillée, S. (2020). Modifying macrophages at the periphery has the capacity to change microglial reactivity and to extend ALS survival. Nature Neuroscience, 23(11), 1339–1351. 10.1038/s41593-020-00718-z [DOI] [PubMed] [Google Scholar]
- Clark, J. A. , Southam, K. A. , Blizzard, C. A. , King, A. E. , & Dickson, T. C. (2016). Axonal degeneration, distal collateral branching and neuromuscular junction architecture alterations occur prior to symptom onset in the SOD1G93A mouse model of amyotrophic lateral sclerosis. Journal of Chemical Neuroanatomy, 76, 35–47. 10.1016/j.jchemneu.2016.03.003 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , Hoyer, D. , Insel, P. A. , Izzo, A. A. , Ji, Y. , MacEwan, D. J. , Sobey, C. G. , Stanford, S. C. , Teixeira, M. M. , Wonnacott, S. , & Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175(7), 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paola, E. , Forcina, L. , Pelosi, L. , Pisu, S. , La Rosa, P. , Cesari, E. , Nicoletti, C. , Madaro, L. , Mercatelli, N. , Biamonte, F. , Nobili, A. , D'Amelio, M. , De Bardi, M. , Volpe, E. , Caporossi, D. , Sette, C. , Musarò, A. , & Paronetto, M. P. (2020). Sam68 splicing regulation contributes to motor unit establishment in the postnatal skeletal muscle. Life Science Alliance, 3(10). 10.26508/lsa.201900637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedkova, E. N. , Seidlmayer, L. K. , & Blatter, L. A. (2013). Mitochondria‐mediated cardioprotection by trimetazidine in rabbit heart failure. Journal of Molecular and Cellular Cardiology, 59, 41–54. 10.1016/j.yjmcc.2013.01.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhote, V. , & Balaraman, R. (2008). Anti‐oxidant activity mediated neuroprotective potential of trimetazidine on focal cerebral ischaemia‐reperfusion injury in rats. Clinical and Experimental Pharmacology and Physiology, 35(5–6), 630–637. 10.1111/j.1440-1681.2008.04845.x [DOI] [PubMed] [Google Scholar]
- Dobrowolny, G. , Lepore, E. , Martini, M. , Barberi, L. , Nunn, A. , Scicchitano, B. M. , & Musarò, A. (2018). Metabolic changes associated with muscle expression of SOD1 G93A. Frontiers in Physiology, 9, 1–9. 10.3389/fphys.2018.00831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolny, G. , Martini, M. , Scicchitano, B. M. , Romanello, V. , Boncompagni, S. , Nicoletti, C. , Pietrangelo, L. , de Panfilis, S. , Catizone, A. , Bouchè, M. , Sandri, M. , Rudolf, R. , Protasi, F. , & Musarò, A. (2018). Muscle expression of SOD1 G93A triggers the dismantlement of neuromuscular junction via PKC‐theta. Antioxidants and Redox Signaling, 28(12), 1105–1119. 10.1089/ars.2017.7054 [DOI] [PubMed] [Google Scholar]
- Dolezalova, R. , Lacinova, Z. , Dolinkova, M. , Kleiblova, P. , Haluzikova, D. , Housa, D. , Papezova, H. , & Haluzik, M. (2007). Changes of endocrine function of adipose tissue in anorexia nervosa: Comparison of circulating levels versus subcutaneous mRNA expression. Clinical Endocrinology, 67(5), 674–678. 10.1111/j.1365-2265.2007.02944.x [DOI] [PubMed] [Google Scholar]
- Doshi, S. , Gupta, P. , & Kalb, R. G. (2017). Genetic induction of hypometabolism by ablation of MC4R does not suppress ALS‐like phenotypes in the G93A mutant SOD1 mouse model. Scientific Reports, 7(1), 13150. 10.1038/s41598-017-13304-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis, L. , Gonzalez de Aguilar, J. L. , Echaniz‐Laguna, A. , Eschbach, J. , Rene, F. , Oudart, H. , Halter, B. , Huze, C. , Schaeffer, L. , Bouillaud, F. , & Loeffler, J. P. (2009). Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE, 4(4), e5390. 10.1371/journal.pone.0005390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuis, L. , Oudart, H. , René, F. , Gonzalez De Aguilar, J. L. , & Loeffler, J. P. (2004). Evidence for defective energy homeostasis in amyotrophic lateral sclerosis: Benefit of a high‐energy diet in a transgenic mouse model. Proceedings of the National Academy of Sciences of the United States of America, 101(30), 11159–11164. 10.1073/pnas.0402026101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dy, A. M. B. , Limjoco, L. L. G. , & Jamora, R. D. G. (2020). Trimetazidine‐induced parkinsonism: A systematic review. Frontiers in Neurology, 11, 44. 10.3389/FNEUR.2020.00044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayemendy, P. , Marin, B. , Labrunie, A. , Boirie, Y. , Walrand, S. , Achamrah, N. , Coëffier, M. , Preux, P. M. , Lautrette, G. , Desport, J. C. , Couratier, P. , & Jésus, P. (2021). Hypermetabolism is a reality in amyotrophic lateral sclerosis compared to healthy subjects. Journal of the Neurological Sciences, 420, 420. 10.1016/j.jns.2020.117257 [DOI] [PubMed] [Google Scholar]
- Ferri, A. , & Coccurello, R. (2017). What is ‘hyper’ in the ALS Hypermetabolism? Mediators of Inflammation. Hindawi Limited, 2017, 1–11. 10.1155/2017/7821672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, L. R. , Culver, D. G. , Tennant, P. , Davis, A. A. , Wang, M. , Castellano‐sanchez, A. , Khan, J. , Polak, M. A. , & Glass, J. D. (2004). Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Experimental Neurology, 185, 232–240. 10.1016/j.expneurol.2003.10.004 [DOI] [PubMed] [Google Scholar]
- Fragasso, G. , Piatti Md, P. M. , Monti, L. , Palloshi, A. , Setola, E. , Puccetti, P. , Calori, G. , Lopaschuk, G. D. , & Margonato, A. (2003). Short‐ and long‐term beneficial effects of trimetazidine in patients with diabetes and ischemic cardiomyopathy. American Heart Journal, 146(5), 854. 10.1016/s0002-8703(03)00415-0 [DOI] [PubMed] [Google Scholar]
- Ge, X. , Cho, A. , Ciol, M. A. , Pettan‐Brewer, C. , Snyder, J. , Rabinovitch, P. , & Ladiges, W. (2016). Grip strength is potentially an early indicator of age‐related decline in mice. Pathobiology of Aging & age‐Related Diseases, 6(1), 32981. 10.3402/pba.v6.32981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacovazzo, G. , Apolloni, S. , & Coccurello, R. (2018). Loss of P2X7 receptor function dampens whole body energy expenditure and fatty acid oxidation. Purinergic Signalling, 14(3), 299–305. 10.1007/s11302-018-9610-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontijo, V. S. , Viegas, F. P. D. , Ortiz, C. J. C. , de Freitas Silva, M. , Damasio, C. M. , Rosa, M. C. , Campos, T. G. , Couto, D. S. , Tranches Dias, K. S. , & Viegas, C. (2019). Molecular hybridization as a tool in the design of multi‐target directed drug candidates for neurodegenerative diseases. Current Neuropharmacology, 18(5), 348–407. 10.2174/1385272823666191021124443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney, M. E. , Pu, H. , Chiu, A. Y. , Canto, M. C. D. , Cynthia, Y. , Alexander, D. D. , Caliendo, J. , Hentati, A. , Kwon, Y. W. , Deng, H. X. , & Siddique, T. (1994). Motor neuron degeneration in mice that express a human cu, Zn superoxide dismutase mutation published by: American Association for the Advancement of Science stable. Science, 264(5166), 1772–1775. http://www.jstor.org/stable/2883932 [DOI] [PubMed] [Google Scholar]
- Jésus, P. , Fayemendy, P. , Nicol, M. , Lautrette, G. , Sourisseau, H. , Preux, P.‐M. , Desport, J. C. , Marin, B. , & Couratier, P. (2018). Hypermetabolism is a deleterious prognostic factor in patients with amyotrophic lateral sclerosis. European Journal of Neurology, 25(1), 97–104. 10.1111/ene.13468 [DOI] [PubMed] [Google Scholar]
- Jiao, Y. , Su, M. , Chen, M. , Jia, W. , Chou, Y. , Huang, Z. , Yang, N. , & Tong, W. (2007). LC/ESI‐MS method for the determination of trimetazidine in human plasma: Application to a bioequivalence study on Chinese volunteers. Journal of Pharmaceutical and Biomedical Analysis, 43(5), 1804–1807. 10.1016/j.jpba.2006.11.040 [DOI] [PubMed] [Google Scholar]
- Kalucka, J. , Bierhansl, L. , Conchinha, N. V. , Missiaen, R. , Elia, I. , Brüning, U. , Scheinok, S. , Treps, L. , Cantelmo, A. R. , Dubois, C. , de Zeeuw, P. , Goveia, J. , Zecchin, A. , Taverna, F. , Morales‐Rodriguez, F. , Brajic, A. , Conradi, L. C. , Schoors, S. , Harjes, U. , … Carmeliet, P. (2018). Quiescent endothelial cells upregulate fatty acid β‐oxidation for Vasculoprotection via redox homeostasis. Cell Metabolism, 28(6), 881–894.e13. 10.1016/j.cmet.2018.07.016 [DOI] [PubMed] [Google Scholar]
- Kantor, P. F. , Lucien, A. , Kozak, R. , & Lopaschuk, G. D. (2000). The antianginal drug trimetazidine shifts cardiac energy metabolism from fatty acid oxidation to glucose oxidation by inhibiting mitochondrial long‐ chain 3‐ketoacyl coenzyme a thiolase. Circulation Research, 86(5), 580–588. 10.1161/01.RES.86.5.580 [DOI] [PubMed] [Google Scholar]
- Keon, M. , Musrie, B. , Dinger, M. , Brennan, S. E. , Santos, J. , & Saksena, N. K. (2021). Destination amyotrophic lateral sclerosis. Frontiers in Neurology, 12. 10.3389/fneur.2021.596006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, S. E. , Tracey, T. J. , Steyn, F. J. , & Ngo, S. T. (2019). Biomarkers of metabolism in amyotrophic lateral sclerosis. Frontiers in Neurology, 10, 191. 10.3389/fneur.2019.00191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuralay, F. , Altekin, E. , Yazlar, A. S. , Onvural, B. , & Goldeli, O. (2006). Suppression of angioplasty‐related inflammation by pre‐procedural treatment with trimetazidine. Tohoku Journal of Experimental Medicine, 208(3), 203–212. 10.1620/tjem.208.203 [DOI] [PubMed] [Google Scholar]
- Lilley, E. , Stanford, S. C. , Kendall, D. E. , Alexander, S. P. H. , Cirino, G. , Docherty, J. R. , George, C. H. , Insel, P. A. , Izzo, A. A. , Ji, Y. , Panettieri, R. A. , Sobey, C. G. , Stefanska, B. , Stephens, G. , Teixeira, M. , & Ahluwalia, A. (2020). ARRIVE 2.0 and the British Journal of Pharmacology: Updated guidance for 2020. British Journal of Pharmacology, 177(16), 3611–3616. 10.1111/BPH.15178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopaschuk, G. D. , Barr, R. , Thomas, P. D. , & Dyck, J. R. B. (2003). Beneficial effects of trimetazidine in ex vivo working ischemic hearts are due to a stimulation of glucose oxidation secondary to inhibition of long‐chain 3‐ketoacyl coenzyme a thiolase. Circulation Research. Circulation Research, 93, e33–e37. 10.1161/01.res.0000086964.07404.a5 [DOI] [PubMed] [Google Scholar]
- Martins, G. F. , de Filho, A. G. S. , de Santos, J. B. F. , Assunção, C. R. C. , Vieira, F. B. , Valência, A. , Carvalho, K. G. , & Jessen, B. (2012). Trimetazidine and inflammatory response in coronary artery bypass grafting. Arquivos Brasileiros de Cardiologia, 99(2), 688–696. 10.1590/S0066-782X2012005000066 [DOI] [PubMed] [Google Scholar]
- Marzilli, M. , Vinereanu, D. , Lopaschuk, G. , Chen, Y. , Dalal, J. J. , Danchin, N. , Etriby, E. , Ferrari, R. , Gowdak, L. H. , Lopatin, Y. , Milicic, D. , Parkhomenko, A. , Pinto, F. , Ponikowski, P. , Seferovic, P. , & Rosano, G. M. C. (2019). Trimetazidine in cardiovascular medicine. International Journal of Cardiology, 293, 39–44. 10.1016/j.ijcard.2019.05.063 [DOI] [PubMed] [Google Scholar]
- Mehta, P. R. , Jones, A. R. , Opie‐Martin, S. , Shatunov, A. , Iacoangeli, A. , Al Khleifat, A. , Smith, B. N. , Topp, S. , Morrison, K. E. , Shaw, P. J. , Shaw, C. E. , Morgan, S. , Pittman, A. , & Al‐Chalabi, A. (2019). Younger age of onset in familial amyotrophic lateral sclerosis is a result of pathogenic gene variants, rather than ascertainment bias. Journal of Neurology, Neurosurgery, and Psychiatry, 90(3), 268–271. 10.1136/jnnp-2018-319089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng, D. , Feng, L. , Chen, X. J. , Yang, D. , & Zhang, J. N. (2006). Trimetazidine improved Ca2+ handling in isoprenaline‐mediated myocardial injury of rats. Experimental Physiology, 91(3), 591–601. 10.1113/expphysiol.2005.032615 [DOI] [PubMed] [Google Scholar]
- Miana‐Mena, F. J. , Piedrafita, E. , González‐Mingot, C. , Larrodé, P. , Muñoz, M. J. , Martínez‐Ballarín, E. , Reiter, R. J. , Osta, R. , & García, J. J. (2011). Levels of membrane fluidity in the spinal cord and the brain in an animal model of amyotrophic lateral sclerosis. Journal of Bioenergetics and Biomembranes, 43(2), 181–186. 10.1007/s10863-011-9348-5 [DOI] [PubMed] [Google Scholar]
- Migliarini, S. , Scaricamazza, S. , Valle, C. , Ferri, A. , Pasqualetti, M. , & Ferraro, E. (2021). Microglia morphological changes in the motor cortex of hSOD1 G93A transgenic ALS mice. Brain Science, 11. 10.3390/brainsci11060807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra, A. , Rossi, S. , Scaricamazza, S. , Di Salvio, M. , Salvatori, I. , Valle, C. , Rusmini, P. , Poletti, A. , Cestra, G. , Carrì, M. T. , & Cozzolino, M. (2017). Functional interaction between FUS and SMN underlies SMA‐like splicing changes in wild‐type hFUS mice. Scientific Reports, 7(1), 2033. 10.1038/s41598-017-02195-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinari, F. , Pin, F. , Gorini, S. , Chiandotto, S. , Pontecorvo, L. , Penna, F. , Rizzuto, E. , Pisu, S. , Musarò, A. , Costelli, P. , Rosano, G. , & Ferraro, E. (2017). The mitochondrial metabolic reprogramming agent trimetazidine as an ‘exercise mimetic’ in cachectic C26‐bearing mice. Journal of Cachexia, Sarcopenia and Muscle, 8(6), 954–973. 10.1002/jcsm.12226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moloney, E. B. , De Winter, F. , Verhaagen, J. , & Kane, C. J. O. (2014). ALS as a distal axonopathy: Molecular mechanisms affecting neuromuscular junction stability in the presymptomatic stages of the disease. Frontiers in Neuroscience, 8(August), 1–18. 10.3389/fnins.2014.00252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo, S. T. , & Steyn, F. J. (2015). The interplay between metabolic homeostasis and neurodegeneration: Insights into the neurometabolic nature of amyotrophic lateral sclerosis. Cell Regeneration, 4, 1–14. 10.1186/s13619-015-0019-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngo, S. T. , Steyn, F. J. , & Mccombe, P. A. (2014). Journal of the neurological sciences body mass index and dietary intervention: Implications for prognosis of amyotrophic lateral sclerosis. Journal of the Neurological Sciences, 340(1–2), 5–12. 10.1016/j.jns.2014.02.035 [DOI] [PubMed] [Google Scholar]
- O'reilly, É. J. , Wang, H. , Weisskopf, M. G. , Fitzgerald, K. C. , Falcone, G. , Mccullough, M. L. , Thun, M. , Park, Y. , Kolonel, L. N. , & Ascherio, A. (2013). Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener, 14(3), 205–211. 10.3109/21678421.2012.735240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peggion, C. , Massimino, M. L. , Biancotto, G. , Angeletti, R. , Reggiani, C. , Sorgato, M. C. , Bertoli, A. , & Stella, R. (2017). Absolute quantification of myosin heavy chain isoforms by selected reaction monitoring can underscore skeletal muscle changes in a mouse model of amyotrophic lateral sclerosis. Analytical and Bioanalytical Chemistry, 409, 2143–2153. 10.1007/s00216-016-0160-2 [DOI] [PubMed] [Google Scholar]
- Percie du Sert, N. , Hurst, V. , Ahluwalia, A. , Alam, S. , Avey, M. T. , Baker, M. , Browne, Wi. J. , Clark, A. , Cuthill, I. C. , Dirnagl, U. , Emerson, M. , Garner, P. , Holgate, S. T. , Howells, D. W. , Karp, N. A. , Lazic, S. E. , Lidster, K. , MacCallum, C. J. , Macleod, M. , … Pearl, E. J. (2020). The ARRIVE guidelines 2.0: Updated guidelines for reporting animal research. PLOS Biology, 18(7), e3000410. 10.1371/journal.pbio.3000410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov, D. , Mansfield, C. , Moussy, A. , & Hermine, O. (2017). ALS clinical trials review: 20 years of failure. Are we any closer to registering a new treatment? Frontiers in Aging Neuroscience, 9(MAR), 1–11. 10.3389/fnagi.2017.00068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pintér, D. , Bereczki, D. , Ajtay, A. , Oberfrank, F. , Janszky, J. , & Kovács, N. (2021). Trimetazidine use in Parkinson's disease: Is it a resolved problem? ENeuro, 8(3), ENEURO.0452–ENEU20.2021. 10.1523/ENEURO.0452-20.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe, L. , Rué, L. , Robberecht, W. , & Van Den Bosch, L. (2014). Translating biological findings into new treatment strategies for amyotrophic lateral sclerosis (ALS). Experimental Neurology. Academic Press Inc, 262, 138–151. 10.1016/j.expneurol.2014.07.001 [DOI] [PubMed] [Google Scholar]
- Reyes, E. T. , Perurena, O. H. , Festoff, B. W. , Jorgensen, R. , & Moore, W. V. (1984). Insulin resistance in amyotrophic lateral sclerosis. Journal of the Neurological Sciences, 63(3), 317–324. 10.1016/0022-510X(84)90154-0 [DOI] [PubMed] [Google Scholar]
- Salvatori, I. , Ferri, A. , Scaricamazza, S. , Giovannelli, I. , Serrano, A. , Rossi, S. , D'Ambrosi, N. , Cozzolino, M. , Giulio, A. D. , Moreno, S. , Valle, C. , & Carrì, M. T. (2018). Differential toxicity of TAR DNA‐binding protein 43 isoforms depends on their submitochondrial localization in neuronal cells. Journal of Neurochemistry, 146(5), 585–597. 10.1111/jnc.14465 [DOI] [PubMed] [Google Scholar]
- Salvatori, I. , Valle, C. , Ferri, A. , & Carrì, M. T. (2017). SIRT3 and mitochondrial metabolism in neurodegenerative diseases. Neurochemistry International, 109, 184–192. 10.1016/j.neuint.2017.04.012 [DOI] [PubMed] [Google Scholar]
- Scaricamazza, S. , Salvatori, I. , Ferri, A. , & Valle, C. (2021). Skeletal muscle in ALS: An unappreciated therapeutic opportunity? Cells. NLM (Medline), 10. 10.3390/cells10030525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaricamazza, S. , Salvatori, I. , Giacovazzo, G. , Loeffler, J. P. , Renè, F. , Rosina, M. , Quessada, C. , Proietti, D. , Heil, C. , Rossi, S. , Battistini, S. , Giannini, F. , Volpi, N. , Steyn, F. J. , Ngo, S. T. , Ferraro, E. , Madaro, L. , Coccurello, R. , Valle, C. , & Ferri, A. (2020). Skeletal‐muscle metabolic reprogramming in ALS‐SOD1G93A mice predates disease onset and is a promising therapeutic target. IScience, 23(5), 101087. 10.1016/j.isci.2020.101087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serarslan, Y. , Bal, R. , Altug, M. E. , Kontaş, T. , Keleş, O. N. , Unal, D. , & Unal, B. (2009). Effects of trimetazidine on crush injury of the sciatic nerve in rats: A biochemical and stereological study. Brain Research, 1247, 11–20. 10.1016/j.brainres.2008.10.007 [DOI] [PubMed] [Google Scholar]
- Serrano, A. , Apolloni, S. , Rossi, S. , Lattante, S. , Sabatelli, M. , Peric, M. , Andjus, P. , Michetti, F. , Carrì, M. T. , Cozzolino, M. , & D'Ambrosi, N. (2019). The S100A4 transcriptional inhibitor Niclosamide reduces pro‐inflammatory and migratory phenotypes of microglia: Implications for amyotrophic lateral sclerosis. Cell, 8(10). 10.3390/cells8101261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu, H. , Peng, Y. , Hang, W. , Zhou, N. , & Wang, D. W. (2021). Trimetazidine in heart failure. Frontiers in Pharmacology. Frontiers Media S.A., 11. 10.3389/fphar.2020.569132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson, E. P. , Henry, Y. K. , Henkel, J. S. , Smith, R. G. , & Appel, S. H. (2004). Increased lipid peroxidation in sera of ALS patients: A potential biomarker of disease burden. Neurology, 62(10), 1758–1765. 10.1212/WNL.62.10.1758 [DOI] [PubMed] [Google Scholar]
- Steyn, F. J. , Ioannides, Z. A. , Van Eijk, R. P. A. , Heggie, S. , Thorpe, K. A. , Ceslis, A. , Heshmat, S. , Henders, A. K. , Wray, N. R. , van den Berg, L. H. , Henderson, R. D. , McCombe, P. A. , & Ngo, S. T. (2018). Hypermetabolism in ALS is associated with greater functional decline and shorter survival. Journal of Neurology, Neurosurgery and Psychiatry, 89(10), 1016–1023. 10.1136/jnnp-2017-317887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyn, F. J. , Li, R. , Kirk, S. E. , Tefera, T. W. , Xie, T. Y. , Tracey, T. J. , Kelk, D. , Wimberger, E. , Garton, F. C. , Roberts, L. , Chapman, S. E. , Coombes, J. S. , Leevy, W. M. , Ferri, A. , Valle, C. , René, F. , Loeffler, J. P. , McCombe, P. A. , Henderson, R. D. , & Ngo, S. T. (2020). Altered skeletal muscle glucose‐fatty acid flux in amyotrophic lateral sclerosis. Brain Communications, 1–17. 10.1093/braincomms/fcaa154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabjerg, M. S. , Andersen, D. C. , Huntjens, P. , Oklinski, K. E. , Bolther, L. , Hald, J. L. , Baisgaard, A. E. , Mørk, K. , Warming, N. , Kullab, U. B. , Kroese, L. J. , Pritchard, C. E. J. , Huijbers, I. J. , & Nieland, J. D. V. (2021). Downregulating carnitine palmitoyl transferase 1 affects disease progression in the SOD1 G93A mouse model of ALS. Communications Biology, 4(1), 509. 10.1038/s42003-021-02034-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey, T. J. , Steyn, F. J. , Wolvetang, E. J. , & Ngo, S. T. (2018). Neuronal lipid metabolism: Multiple pathways driving functional outcomes in health and disease. Frontiers in Molecular Neuroscience, 11(January), 1–25. 10.3389/fnmol.2018.00010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay, A. , Després, J. P. , Leblanc, C. , Craig, C. L. , Ferris, B. , Stephens, T. , & Bouchard, C. (1990). Effect of intensity of physical activity on body fatness and fat distribution. The American Journal of Clinical Nutrition, 51(2), 153–157. 10.1093/ajcn/51.2.153 [DOI] [PubMed] [Google Scholar]
- Van den Bergh, R. , Swerts, L. , Hendrikx, A. , Boni, L. , & Meulepas, E. (1978). Adipose tissue cellularity in patients with amyotrophic lateral sclerosis. Clinical Neurology and Neurosurgery, 80(4), 226–240. 10.1016/S0303-8467(78)80013-4 [DOI] [PubMed] [Google Scholar]
- Van Mantgem, J. (2020). Prognostic value of weight loss in patients with amyotrophic lateral sclerosis: A population‐based study. Journal of Neurology, Neurosurgery, and Psychiatry, 91, 867–875. 10.1136/jnnp-2020-322909 [DOI] [PubMed] [Google Scholar]
- Volonté, C. , Amadio, S. , Fabbrizio, P. , & Apolloni, S. (2019, October 1). Functional microglia neurotransmitters in amyotrophic lateral sclerosis. Seminars in Cell and Developmental Biology. Elsevier Ltd., 94, 121–128. 10.1016/j.semcdb.2019.04.014 [DOI] [PubMed] [Google Scholar]
- Wan, P. , Su, W. , Zhang, Y. , Li, Z. , Deng, C. , & Zhuo, Y. (2017). Trimetazidine protects retinal ganglion cells from acute glaucoma via the Nrf2/ho‐1 pathway. Clinical Science, 131, 2363–2375. 10.1042/CS20171182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wobst, H. J. , Mack, K. L. , Brown, D. G. , Brandon, N. J. , & Shorter, J. (2020). The clinical trial landscape in amyotrophic lateral sclerosis—Past, present, and future. Medicinal Research Reviews. John Wiley and Sons Inc, 40, 1352–1384. 10.1002/med.21661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. , & Wang, K. K. W. (2015). Glial fibrillary acidic protein: From intermediate filament assembly and gliosis to neurobiomarker. Trends in Neurosciences. Elsevier Ltd, 38, 364–374. 10.1016/j.tins.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zurlo, F. , Larson, K. , Bogardus, C. , & Ravussin, E. (1990). Skeletal muscle metabolism is a major determinant of resting energy expenditure. Journal of Clinical Investigation, 86(November), 1423–1427. 10.1172/JCI114857 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S.1. Dinamometric evaluation of grip strength in wild‐type (WT) and SOD1G93A mice littermate mice. Data shows that disease onset occurs at 70 days of age in SOD1G93A mice. At this age, SOD1G93A mice exhibit a 20% decrease in grip strength when compared to their wild‐type (WT) littermates.
Figure S.2 Bio‐availability of Trimetazidine (TMZ) in the plasma of wild‐type mice. Mice were treated for one week with the indicated doses of TMZ (0, 5, 10, 20, 40 mg·kg‐1 ), n ≥ 4/group.
Figure S3. Locomotor activity. The number of horizontal and vertical movements (light‐beam frames broken) was measured over 72 h in SOD1G93A and wild‐type littermate mice (males left panel, females right panel) with (G93A TMZ and WT TMZ) or without (G93A and WT) daily administration of 20 mg·kg‐1 TMZ in drinking water.
Figure S.4. Nicotinamide adenine dinucleotide dehydrogenase‐tetrazolium reductase (NADH‐TR) activity in tibialis anterior (TA) muscle. (A) Representative in situ NADH‐TR activity staining in cross sections of TA muscle obtained from 90‐day old untreated (WT and G93A) and treated (WT TMZ and G93A TMZ) littermate mice. Scale bar, 200μm. (B) Quantification of the percentage of NADH positive areas in cross‐sections of TA muscle from (WT and G93A) and treated (WT TMZ and G93A TMZ) littermate mice. Data are presented as mean +/− SEM, n ≥ 4/group, **P < .05 compared to WT and WT TMZ, # P < .05 compared to G93A, parametric one‐way ANOVA with Bonferroni post‐hoc test.
Data Availability Statement
The data that support the findings of this study are contained in the body of the manuscript or in the supplemental material. Any additional data used are available from the corresponding authors upon reasonable request.