

Summary
High‐throughput single‐cell RNA sequencing (scRNA‐Seq) identifies distinct cell populations based on cell‐to‐cell heterogeneity in gene expression. By examining the distribution of the density of gene expression profiles, we can observe the metabolic features of each cell population.
Here, we employ the scRNA‐Seq technique to reveal the entire biosynthetic pathway of a flower volatile.
The corolla of the wild tobacco Nicotiana attenuata emits a bouquet of scents that are composed mainly of benzylacetone (BA). Protoplasts from the N. attenuata corolla limbs and throat cups were isolated at three different time points, and the transcript levels of > 16 000 genes were analyzed in 3756 single cells.
We performed unsupervised clustering analysis to determine which cell clusters were involved in BA biosynthesis. The biosynthetic pathway of BA was uncovered by analyzing gene co‐expression in scRNA‐Seq datasets and by silencing candidate genes in the corolla.
In conclusion, the high‐resolution spatiotemporal atlas of gene expression provided by scRNA‐Seq reveals the molecular features underlying cell‐type‐specific metabolism in a plant.
Keywords: 10x genomics, benzylacetone, corolla, floral scents, Nicotiana attenuata, scRNA‐Seq
Introduction
Flowers produce diverse volatiles for various ecological interactions. More than 1700 different floral scents have been identified (Knudsen et al., 2006): these scents are classified as terpenoids, fatty acid‐derivatives, and phenylpropanoid/benzenoids, according to their biosynthetic pathways (Muhlemann et al., 2014; Knudsen & Gershenzon, 2020). One molecular mechanism underlying floral scent diversity is the duplication followed by subsequent mutation (neofunctionalization) of biosynthetic genes (Baudino et al., 2020). A mutation in a coding region can generate new volatile compounds by binding to the original substrate or to similar substrates. For example, a single amino‐acid substitution in Oryza sativa terpene synthase 1 (TPS1) changes the ratio between two products: (E)‐β‐caryophyllene and germacrene A (Chen et al., 2014). In addition, a single amino‐acid substitution in salicylic acid methyltransferase isolated from Datura wrightii (Barkman et al., 2007) and in O‐methyltransferase isolated from Thalictrum tuberosum (Frick & Kutchan, 1999) alters a binding affinity to substrates, which results in different products. Mutations in a cis‐regulatory element may trigger changes in the spatiotemporal expression of biosynthetic enzymes, resulting in the generation of a new metabolic pathway. Because small changes in enzymes can produce new compounds, identifying the genes responsible for floral scent biosynthesis requires a multidisciplinary approach that includes functional prediction based on sequence homology to characterized enzymes, biochemical assay, and in vivo gene manipulation.
To identify the genes responsible for floral scent production, DNA/RNA sequencing techniques, microarrays, and several statistical methods have been widely used. Candidate genes have been prioritized by investigating correlations among transcript abundances and target compounds (Verdonk et al., 2005; Magnard et al., 2015; Xu et al., 2018). By analyzing differential gene expression among the rose cultivars that vary widely in floral scent emission, researchers identified a sesquiterpene synthase (Guterman et al., 2002) and a nudix hydrolase (Magnard et al., 2015), a new type of monoterpene synthase. In addition, when a biosynthetic gene in a multi‐step pathway is available, this gene can be used as ‘bait’ to calculate its degree of association to the entire transcriptome (gene co‐expression network analysis) (Wisecaver et al., 2017), and a gene cluster that regulates the production of metabolites can be constructed (Li et al., 2020). For instance, TPS11 enzyme in Arabidopsis thaliana flowers synthesizes (+)‐α‐barbatene and (+)‐thujopsene. These volatiles are further oxidized by CYP706A3, which was discovered through co‐expression analysis with TPS11 (Boachon et al., 2019). These successful identification of biosynthetic genes of specialized volatiles through gene‐to‐metabolite or gene‐to‐gene correlation requires genetic, environmental, tissue‐specific diversity in the material analyzed (Wong et al., 2020).
Flower scents are mainly synthesized in the petals. In particular, several volatile biosynthetic genes are detected in the epidermal cells of petals, for example, (S)‐linalool synthase (Dudareva et al., 1996) and isoeugenol O‐methyltransferase (Dudareva & Pichersky, 2000) in Clarkia breweri flowers; benzoic acid carboxyl methyltransferase in Antirrhinum majus flowers (Kolosova et al., 2001); and orcinol O‐methyltransferase in rose cultivars (Scalliet et al., 2006). Separating scent‐producing cells from the rest of the cells in the entire petals can increase the accuracy of a co‐expression analysis: laser microdissection procedures and marker‐based cell sorting methods have been used to isolate cells (Libault et al., 2017; Wong et al., 2020). Recently, several high‐throughput single‐cell RNA sequencing (scRNA‐Seq) techniques have emerged for analyzing transcriptomes expressed in single cells isolated from tissues or organs. The scRNA‐Seq database shows cellular heterogeneity and molecular processes specific to cell types in plant tissues, such as roots (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Ryu et al., 2019; Shulse et al., 2019), leaves (Lopez‐anido et al., 2020; Kim et al., 2021), and shoot apical meristems (Satterlee et al., 2020; Zhang et al., 2021). Assuming that all biosynthetic genes of a given floral scent are expressed in a single cell and that the regulation of those genes is coordinated, the scRNA‐Seq database of petals can be useful in prioritizing candidate genes, as it allows researchers to calculate correlations among genes expressed in several thousands of petal cells (Wong et al., 2020).
Flowers in the wild tobacco Nicotiana attenuata emit benzylacetone (BA, 4‐phenyl‐2‐butanone) at night to attract pollinators, such as Manduca sexta hawkmoths (Euler & Baldwin, 1996; Baldwin, 1997; Haverkamp et al., 2016), and to prevent floral damage from cucumber beetles (Kessler et al., 2019). Most BA is produced from corolla limbs (Euler & Baldwin, 1996; Kessler & Baldwin, 2007), and is regulated by the circadian clock (Yon et al., 2016). l‐Phenylalanine (Phe) is a precursor of BA, and N. attenuata phenylalanine ammonia‐lyase 4 (NaPAL4) is known to be the first enzyme in the BA biosynthetic pathway (Guo et al., 2020). In addition, the co‐silencing of polyketide synthase 2/3 (NaPKS2/3, previously NaCHAL2/3) reduced the level of BA emission (Kessler et al., 2008; Guo et al., 2020), which suggested that one or more NaPKSs catalyze a condensation reaction with malonyl‐Coenzyme A (CoA) to produce BA intermediates, as shown in an ornamental rhubarb (Rheum palmatum, Abe et al., 2001). In the plant kingdom, the major Phe‐derived floral volatiles are phenylpropanoid (a phenyl ring with a three‐carbon side chain, C6–C3) and benzenoid (C6–C1) (Widhalm & Dudareva, 2015), whose biosyntheses are generally initiated by PAL and two types of CoA ligases (4‐coumarate‐CoA ligase, 4CL; cinnamate‐CoA ligase, CNL) (Klempien et al., 2012; Qualley et al., 2012). However, BA possesses a C6–C4 structure (phenylbutanoids). To identify the entire biosynthetic pathway of this rare floral scent (Knudsen et al., 2006), we performed the scRNA‐Seq of protoplasts isolated from the N. attenuata corolla limbs and throat cups, and defined distinct cell clusters based on the cell‐to‐cell variation in gene expression. Gene co‐expression analysis with scRNA‐Seq datasets and in vivo and in vitro characterization of enzyme activity enable us to identify the entire set of BA biosynthetic genes.
Materials and Methods
Plant materials and protoplasting
Seeds of N. attenuata Utah 39th inbred line originating from a natural population in Utah were germinated on Petri dishes with Gamborg’s B5 medium including vitamins (Duchefa Biochemie, Haarlem, the Netherlands), as described previously (Krügel et al., 2002). The original collection of the seeds was done on private lands and no specific permissions for seed collections were necessary. Plants were grown under 16 h : 8 h, light : dark conditions at 26°C with ±2°C variation. Protoplasts were isolated from approximately 40 corolla limbs and throat cups of N. attenuata at Zeitgeber Time 8 (ZT 8, 8 h after the onset of light), ZT 12, and ZT 16 of the first day of flower opening. Corolla limbs and throat cups were excised by scalpel and immediately immersed in a plant culture dish containing 25 ml of cell‐wall‐degrading enzyme mixtures (1.5% (w/v) cellulase R‐10; Yakult, Tokyo, Japan; 0.4% (w/v) macerozyme R‐10, Yakult; 9% (w/v) mannitol, Duchefa Biochemie; 0.1% (w/v) 2‐(N‐morpholino)ethanesulfonic acid (MES), Duchefa Biochemie; 0.1% bovine serum albumin (BSA), Sigma‐Aldrich, St Louis, MO, USA) (Yoo et al., 2007). After 5 min of vacuum treatment, tissues were shaken at 20 rpm on an orbital shaker for 30 min in the dark, and the cell suspension was filtered onto a Petri dish using a 40 μm cell strainer (Corning, NY, USA). The protoplast suspensions were centrifuged at 100 g for 5 min, and the resulting pellets were resuspended and washed with 10% (w/v) mannitol solution. Cells were washed, centrifuged again, and finally filtered with 40 μm Flowmi tip strainer (Bel‐Art SP Scienceware; Wayne, NJ, USA). Cell concentration and viability were calculated using 0.2% trypan blue on Cellometer Auto 2000 (Nexcelom Bioscience, Lawrence, MA, USA). All samples showed a viability over 70%. The entire protoplasting procedure was completed in 2 h.
Generation of single‐cell RNA sequencing library and feature‐barcode matrices
Cell suspensions were loaded into the 10x Genomics Chromium single‐cell microfluidics device with the Single Cell 3′ Library & Gel Bead Kit v.3 (10x Genomics, leasanton, CA, USA) according to the manufacturer’s instructions. Twelve PCR cycles were used for complementary DNA (cDNA) amplification, and 14 to 15 PCR cycles were performed for ligating adaptors. The library quality and size were checked using DNA high‐sensitivity D5000 ScreenTape System (Agilent Technologies, Santa Clara, CA, USA). The single‐cell RNA sequencing (scRNA‐Seq) libraries were sequenced on the HiSeq X Ten platform (Illumina, San Diego, CA, USA) at Macrogen (Seoul, Republic of Korea). Approximately 400 000 reads and 1300 genes were detected per cell. Total 19 553, 19 842, and 18 917 genes were detected from ZT 8, ZT 12, and ZT 16 datasets, respectively.
Sequencing data were demultiplexed with mkfastq command in Cell Ranger (v.3.1.0) (10x Genomics) to generate fastq files. Expression matrices were also generated through the Cell Ranger pipeline, by aligning reads to the N. attenuata genome (NIATTr2). The Cellranger mkref command was used after filtering the GTF file to include only protein‐coding genes in the N. attenuata genome. Feature‐barcode matrix was generated after preprocessing, aligning, counting UMI, and filtering cells using Cell Ranger count command with default options.
Single‐cell expression matrices were row‐summed to generate pseudo‐bulk datasets from scRNA‐Seq datasets. Conventional bulk RNA sequencing (bulk RNA‐Seq) was performed with corolla limbs at three different time points: ZT 8, ZT 12, and ZT 16. Pearson's correlation coefficient was calculated between these pseudo‐bulk datasets and conventional bulk RNA‐Seq datasets. Some of the stress‐related genes were highly upregulated in three scRNA‐Seq datasets (log2 fold change (FC) > 5); these genes (described in Supporting Information Table S1) were excluded from downstream analysis.
Uniform manifold approximation and projection visualization and marker gene identification
After filtering matrices, the seurat R package (v.3.2.0) was used for dimension reduction and visualization (Satija et al., 2015; Butler et al., 2018). Cells that expressed < 200 genes were discarded. To integrate the single‐cell libraries generated from three different time points, the datasets were normalized separately by sctransform (SCT) (Hafemeister & Satija, 2019), and all features were selected for downstream integration. Pearson residuals were calculated by running PrepSCTIntegration, and anchors were identified by SCT normalization. Principal component analysis was conducted on the integrated dataset. Based on the top 50 principal components, distinct cell populations were defined by unsupervised clustering analysis (0.4 resolution) and visualized by uniform manifold approximation and projection (UMAP).
Marker genes of each cluster through all time points were selected by Wilcoxon rank sum test in Seurat's FindConservedMarkers function. Genes over 0.25 log‐FC relative to other clusters and Bonferroni‐corrected P‐value < 0.05 were chosen as upregulated genes in individual clusters. Average levels of transcripts and percentages of cells expressing the marker per cluster were visualized by color and size in dot plots or in violin plots. Based on the predicted functions of marker genes, we characterized clusters as epidermis, parenchyma, chlorenchyma, pollen, and vasculatures.
Gene co‐expression analysis
Twenty bulk RNA‐Seq datasets containing 14 tissue types from N. attenuata were downloaded from the sequence read archive database (Li et al., 2016). TPM values of each gene were used for calculating Spearman’s correlation coefficients between genes, and coefficients of each gene with N. attenuata PAL4 (A4A49_25502) were extracted and visualized as histograms. For scRNA‐Seq co‐expression analysis, unique molecular identifier (UMI) values of genes were divided by the sum of UMI in each cell and multiplied by 100 000 to normalize the values. The normalized UMIs of each gene were used for calculating Spearman’s correlation coefficients between genes.
Genes with at least one UMI were assigned a value of 1, and others with no UMIs were assigned a value of 0. Cells with the value 1 of target gene were regarded as the cells expressing the gene. This binary matrix was then used as an input for an UpSet plot. The UpSet plot was drawn in complexupset package (v.0.7.3) (Lex et al., 2014; Krassowski, 2020). For visualizing the cells that express four BA biosynthetic genes, the FeaturePlot function in the seurat package was used with the parameters: min.cutoff =‘q10’, max.cutoff = ‘q90’, and blend = T.
Virus‐induced gene silencing
To silence a target gene in N. attenuata corolla, virus‐induced gene silencing (VIGS) using tobacco rattle virus (TRV)‐based vectors was performed (Saedler & Baldwin, 2004). A 250 bp fragment of the gene of interest was designed using the SGN VIGS Tool (Fernandez‐Pozo et al., 2015) and cloned into the TRV2 vector. The Agrobacterium tumefaciens AGL1 strain harboring the TRV2 target gene was mixed with Agrobacterium containing TRV1 just before infiltration. About 1 to 2 ml of Agrobacterium solution was injected into N. attenuata rosette leaves (three leaves per plant). The infiltrated plants were incubated in the dark at 22°C for 2 d and then acclimated under weak light conditions in a growth chamber for 2 d. Later, the infiltrated plants were grown under 16 h : 8 h, light : dark conditions at 26°C with ±2°C variation. TRV2‐eGFP was used as a negative control, and phytoene desaturase (PDS) gene was silenced (TRV2‐PDS) in parallel to check whether VIGS works in flowers for each experimental condition. We measured the level of BA and BA‐biosynthetic genes in flowers from virus‐infected plants once TRV2‐PDS flowers produced white flowers. The silencing efficiency of individual corolla was measured by quantitative reverse transcription polymerase chain reaction (qRT‐PCR). Total RNA was extracted using RNeasy Plant Mini Kit (Qiagen, Hilden, Germany), and genomic DNA was removed by treating RNase‐free DNase I (Qiagen). Extracted RNA was reverse‐transcribed using oligo dT and SuperScript™ IV reverse transcriptase (Thermo Fisher Scientific, Waltham, MA, USA) following manufacturer’s protocols. Quantitative RT‐PCR was performed on AriaMx real‐time PCR System (Agilent Technologies) with TB Green Premix EX Taq (Takara, Shiga, Japan). Transcript levels were quantified by the comparative CT method (Schmittgen & Livak, 2008). The C q values were normalized to the value of the internal control gene, NaEF1a (elongation factor 1a, ∆C q). To calculate individual FC values, each biological replicate sample (∆C q) was normalized by one calibrator sample in the TRV2‐GFP control group (∆∆C q) and presented as points in graph. Primers used in this study are listed in Table S2.
Headspace and internal volatile sampling and gas chromatography–mass spectrometry (GC–MS) analysis
Flower headspace volatiles were trapped in silicone tubing (ST) and analyzed as previously described (Kallenbach et al., 2014). Briefly, before flowers start to open, a flower was excised and enclosed in a clean 8 ml glass vial with 300 μl of ultrapure water. Clean ST, 1 cm in length, was placed directly over the corolla limb for 3 h (ZT 14–ZT 17), when the BA emission peaked (Yon et al., 2016) and our last sampling for scRNA‐Seq (ZT 16) was done. GCMS‐QP2020 (Shimadzu, Kyoto, Japan) connected with a TD‐30 thermal desorption unit (Shimadzu) was used for gas analysis. All standard compounds used in this study were purchased from Sigma‐Aldrich.
For internal volatile sampling, 50 mg of corolla limbs and throat cups was finely ground in liquid nitrogen and resuspended in 1 ml of saturated calcium chloride solution spiked with tetralin as an internal standard (Joo et al., 2019). A 1 cm length of clean ST was incubated in the resuspension solution overnight and shaken at 80 rpm. Incubated STs were rinsed with ultrapure water and dried under a gentle nitrogen flow. Room temperature‐equilibrated STs were analyzed in GCMS‐QP2020 connected with a TD‐30 thermal desorption unit (Shimadzu).
Functional evaluation of benzylacetone biosynthetic enzymes
Codon‐optimized BA biosynthetic gene sequences (Bioneer, Daejeon, Korea) or native coding sequences (CDSs) were cloned into a pET50 vector. Escherichia coli Rosetta (DE3) cells were transformed with pET50‐a target gene and grown in LB broth media containing 50 µg ml−1 of kanamycin and 30 µg ml−1 of chloramphenicol at 37°C. When the optical density measured at 600 nm (OD600) reached 0.5–0.6, protein expression was induced by adding 0.35 mM of isopropyl β‐d‐1‐thiogalactopyranoside (IPTG). After incubating an additional 18–20 h at 18°C, cells were harvested and lysed in cell resuspension buffer supplemented with 0.5 mM phenylmethylsulfonyl fluoride (PMSF) by sonication. Cell resuspension buffers for each protein were prepared as previously described (Abe et al., 2001; Koeduka et al., 2011; Klempien et al., 2012); for purifying recombinant proteins, soluble protein fractions were obtained by centrifugation at 16 200 g for 20 min at 4°C and loaded onto Ni‐NTA agarose resin (Thermo Fisher Scientific), following manufacturer’s instructions. The size and purity of eluted proteins were confirmed by Coomassie staining. To get rid of excess imidazole, proteins were dialyzed against the final dissolving buffers and then stored at −80°C.
The CoA ligase activity of Na4CL1 with t‐cinnamic acid (Sigma‐Aldrich) and its derivatives (4‐coumaric acid, caffeic acid and ferulic acid; Sigma‐Aldrich) was determined spectrophotometrically, as previously described (Klempien et al., 2012). Briefly, the spectrophotometric assay was performed by monitoring the increase in absorption maximum on a microplate reader (311, 333, 346, and 345 nm for cinnamoyl‐, coumaroyl‐, caffeoyl‐, and feruloyl‐CoA, respectively). The assay reaction was performed with 250 µl solution containing 1.76 µg of purified Na4CL1 protein in 50 mM Tris/K phosphate/Na citrate pH 8.5, 2.5 mM adenosine triphosphate (ATP), 5 mM magnesium chloride (MgCl2), 0.4 mM CoA, and various concentration of t‐cinnamic acid and its derivatives. Reactions were started by adding substrates to each well. The extinction coefficient used for calculating cinnamoyl‐CoA formation was 22 000 ml mmol−1 cm−1 (Lee et al., 1997); likewise, 21 000 ml mmol−1 cm−1 for coumaroyl‐CoA, 18 000 ml mmol−1 cm−1 for caffeoyl‐CoA, and 19 000 ml mmol−1 cm−1 for feruloyl‐CoA were used as extinction coefficients.
We purified NaPKS2 and NaPKS3 proteins as described previously (Abe et al., 2001) and mixed NaPKS2 or NaPKS3 with the products formed during Na4CL1 spectrophotometric assays and malonyl‐CoA. The reaction mixture contained 50 µl of Na4CL1 products (intended to contain final 40 µM of cinnamoyl‐CoA in 500 µl reaction mixture), 40 µM malonyl‐CoA, and 10 µg of NaPKS2 or NaPKS3 in a final volume of 500 µl of 100 mM potassium phosphate buffer containing 1 mM ethylenediaminetetraacetic acid (EDTA, pH 8.0). Mixtures were incubated at 30°C for 2 h, and reactions were stopped by adding 50 µl of glacial acetic acid. The products were immediately extracted with 1 ml of ethyl acetate via vigorous vortexing, and the ethyl acetate layer was concentrated using a freeze‐dryer. All products were injected for gas chromatography–mass spectrometry (GC‐MS) via a thermal desorption unit (Shimadzu).
Reducing activity of NaAER1 was determined spectrophotometrically, with modified reaction conditions as previously described (Koeduka et al., 2011). The assay mixture contained 5 µg of purified NaAER1 proteins, 0.5 mM NADPH, and putative substrates (benzalacetone and vanillylidenacetone from Sigma‐Aldrich; 4‐hydroxy‐benzalacetone from AA Blocks, San Diego, CA, USA) in different concentrations in final 200 µl of 125 mM MES‐KOH (pH 7.5) buffer. Reactions were started by adding substrates. Decreasing absorbance at 340 nm for benzalacetone and 370 nm for 4‐hydroxy‐benzalacetone and vanillylidenacetone was monitored with the plate reader (Infinite M Nano; Tecan, Männedorf, Switzerland). For GC–MS analysis, the reaction mixture (3 µg NaAER1–0.5 mM benzalacetone) was incubated for 20 min at room temperature, and the product was extracted with 1 ml of ethyl acetate. Standard compound injections verified that the product of the NaAER1–benzalacetone reaction was benzylacetone.
Generation of promoter::eGFP lines and confocal imaging
Reporter lines were generated by transforming enhanced green fluorescent protein (eGFP) reporter plasmids to N. attenuata hypocotyl explants. Promoter sequences (2 kb upstream region from the NaPAL4 and NaPKS2 genes) were cloned into eGFP expressing binary plasmid derived from pH2GW7 (Karimi et al., 2002). Agrobacterium‐mediated transformation and tissue culture were performed as previously described, and three independent transformants for each line were investigated (Kang et al., 2020). Transgenic flowers were fixed in 4% paraformaldehyde fixative containing 0.25% of glutaraldehyde, 0.1% of Triton X‐100, and 0.1% of Tween 20 for overnight at 4°C. Samples were dehydrated and embedded in paraffin with a Leica EG1150H (Leica, Wetzlar, Germany). Cross‐sections were cut at 10 μm using a microtome (Leica) and placed on slides, dried at 42°C. Sections were treated with xylene substitute to remove paraffin. Fluorescence signals were measured in flowers of regenerated reporter lines using an LSM780 confocal laser‐scanning microscope (Carl Zeiss, Oberkochen, Germany). Enhanced GFP was excited at 488 nm and emission signals were captured from 493 nm to 540 nm.
Results
Single‐cell RNA sequencing defines distinct cell types in the corolla
To generate a single‐cell transcriptome atlas of genes involved in BA biosynthesis, we isolated protoplasts from N. attenuata corolla limbs and throat cups at three time points (ZT 8, ZT 12, and ZT 16, one biological replicate) when BA emission increases (Fig. 1a). The protocol of protoplast isolation was optimized to retain cell integrity and the temporal transcriptome; the whole process, from flower excision to generating single‐cell gel bead‐in‐emulsion, took c. 2 h. The 10x Genomics Chromium and the Illumina sequencing platforms were used to generate scRNA‐Seq libraries. To evaluate the processes involved in scRNA‐Seq, we generated a pseudo‐bulk dataset for each time point by pooling scRNA‐Seq UMIs and performed conventional bulk RNA‐Seq with corolla limbs and throat cups collected at the same time points (ZT 8, ZT 12, and ZT 16, two biological replicates); these pseudo‐bulk datasets were compared with the bulk RNA‐Seq datasets. The global transcriptome profiles of two datasets were highly correlated (Pearson’s correlation, r > 0.8, Supporting Information Fig. S1). We found several hypothetical proteins, ethylene‐responsive factors, and zinc finger proteins that were highly induced in scRNA‐Seq libraries compared to in bulk RNA‐Seq results (Table S1, log2 FC > 5). Because the protoplast isolation process is known to induce several ethylene‐responsive factors and zinc‐finger proteins (Birnbaum et al., 2003), we excluded genes that were consistently induced in three scRNA‐Seq libraries from further analysis (Table S1). In single cell population, we detected 82–87% of genes that were detected in bulk RNA‐Seq analysis (Fig. S2). A total of 16 465 genes detected in all time points was used for further analysis.
Fig. 1.
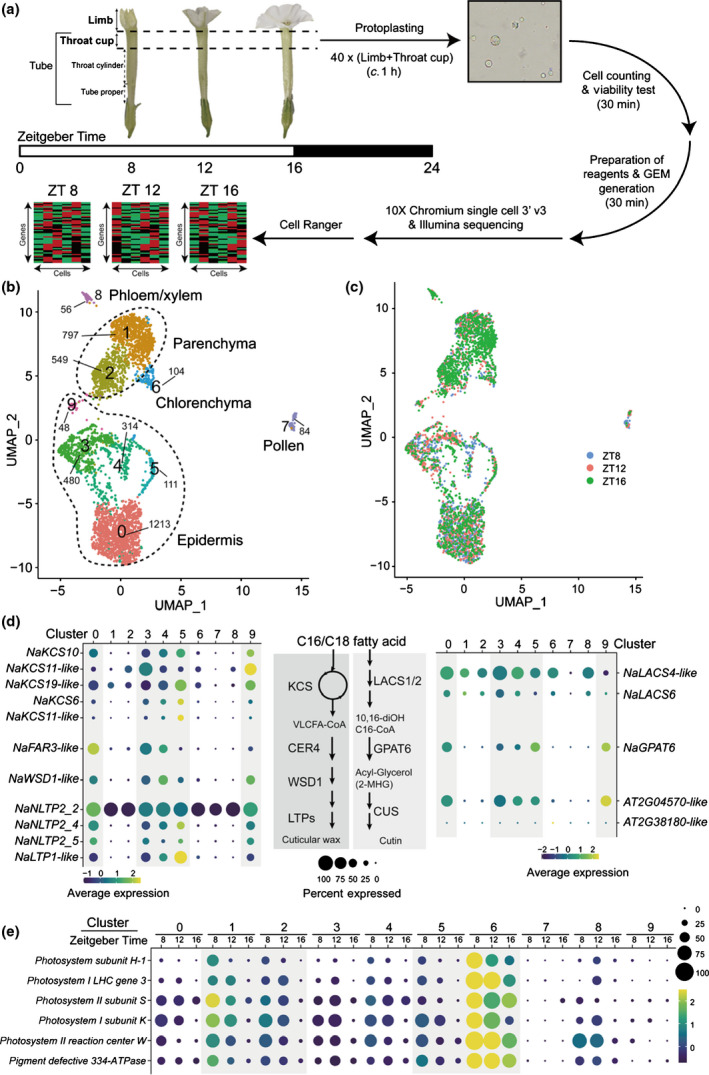
Single‐cell RNA sequencing (scRNA‐Seq) characterizes distinct Nicotiana attenuata corolla cell types. (a) Corolla limbs and throat cups were sampled from 40 N. attenuata flowers at each Zeitgeber Time (ZT), and scRNA‐Seq libraries were generated. The total experimental procedures were finished within 2 h to reduce the effect of protoplasting. (b) A uniform manifold approximation and projection (UMAP) plot of the 3756 cells classified into five major cell types, composed of a total of 10 distinct clusters. The number of cells of each cluster is given next to clusters. (c) UMAP plot showing dimensional reduction of the distribution of 3756 corolla cells from three scRNA‐Seq libraries. (d) Most biosynthetic genes of cuticular wax and cutin are highly enriched in clusters 0, 3, 4, 5, and 9. These genes are known to be expressed in the epidermis. CER4/FAR3, ECERIFERUM4/fatty acid reductase 3; CUS, cutin synthase‐like; GPAT6, glycerol‐3‐phosphate acyltransferase 6; KCS, 3‐ketoacyl‐CoA synthase; LACS1/2, long‐chain acyl‐CoA synthase 1/2; LTP, lipid‐transfer proteins; NLTP, nonspecific lipid‐transfer proteins; VLCFA, very‐long‐chain fatty acids; WSD1, wax ester synthase/diacylglycerol acyltransferase 1. (e) Genes associated with photosynthesis are highly expressed in cluster 6 at all time points and in clusters 1, 2, and 6 at ZT 8. Photosynthetic genes have been reported to be expressed specifically in the inner layer cells, like mesophyll or parenchymal cells (Langdale & Nelson, 1991; Stockhaus et al., 1997).
We next categorized single cells into clusters based on their shared transcriptomic profiles using the Seurat package (Satija et al., 2015; Butler et al., 2018). Cells from three time points were projected and visualized on a UMAP plot, which showed 10 distinct clusters in two‐dimensional space (Figs 1b,c, S3). Because there were no well‐known marker genes for flower cells, we assigned cell identities based on the putative biochemical functions of cluster‐specific genes. The N. attenuata transcriptome was annotated based on the homology of protein‐coding sequences to previously investigated genes in several model plants (Brockmöller et al., 2017). For instance, in cluster 7, several pectin esterase genes, extension families, and homologs of pollen allergens were specifically detected; these cells were defined as pollen that had attached to the corolla’s surface (Figs S4, S5). In cluster 8, we detected several orthologs of AtSWEET11/12 and AtSULTR2;1 genes, which are known to be specifically expressed in phloem and xylem (Figs S4, S7 see later) (Chen et al., 2012; Maruyama‐Nakashita et al., 2015; Kim et al., 2021).
We assigned the remaining major clusters to three groups of cells: epidermis, chlorenchyma, and parenchyma. Epidermal cells are known to express wax/cutin biosynthetic genes for synthesizing cuticular wax and cutin (Suh et al., 2005). Several genes involved in cuticular wax biosynthesis were specifically enriched in clusters 0, 3, 4, 5, and 9: ketoacyl‐CoA synthase, ECERIFERUM4, wax ester synthase/diacylglycerol acyltransferase 1, and several lipid‐transfer proteins, which may participate in wax synthesis or transport, were highly expressed (Figs 1d, S6). We also found that clusters 0, 3, 4, 5, and 9 contained higher levels of cutin biosynthetic genes (glycerol‐3‐phosphate acyltransferase 6 and cutin synthase‐like) compared to other clusters; their protein sequences are similar to the well‐studied orthologs of Arabidopsis thaliana (Fig. S7). These results suggest that clusters 0, 3, 4, 5, and 9 are epidermal cells. Cluster 6 was putatively defined as a chlorenchyma cell, because it contained highly expressed complex subunits of photosystems I and II (Figs 1e, S8). As shown in Fig. 1(a), the throat cup of the corolla retained chloroplasts. Photosynthetic genes are well‐known to have a clear diurnal rhythm, with high levels during the day and low levels during the night (Kim et al., 2011). Our scRNA‐Seq data showed that the transcript levels of several photosynthetic genes gradually decreased from ZT 8 (in the middle of the day) to ZT 16 (reflecting the transition time from light to dark) (Fig. 1e). Clusters 1 and 2 had low levels of cuticle biosynthetic genes and relatively high levels of photosynthetic genes. These results suggest that in the corolla, clusters 1 and 2 are parenchyma cells.
Correlation analysis at single‐cell resolution prioritizes candidate genes for benzylacetone biosynthesis
In the corolla, the initial step of BA biosynthesis is the nonoxidative deamination of Phe by the action of NaPAL4 (Fig. 2a) (Guo et al., 2020). The product of NaPAL4 enzyme, t‐cinnamic acid, is likely converted to cinnamoyl‐CoA by a homologous protein of 4‐coumarate:CoA ligase (4CL) or cinnamate:CoA ligase (CNL) (Klempien et al., 2012) without further hydroxylation, because BA has no hydroxyl group on the aromatic ring. Next, we hypothesized that the cinnamoyl‐CoA is likely condensed with one molecule of malonyl‐CoA, which yields benzalacetone (4‐phenyl‐3‐buten‐2‐one), a C6–C4 moiety of phenylbutanoid. Abe et al. (2001) found that a novel polyketide synthase named benzalacetone synthase produces 4‐hydroxybenzalacetone (4‐(4‐hydroxyphenyl)‐3‐buten‐2‐one) from 4‐coumaroyl‐CoA and one molecule of malonyl‐CoA in R. palmatum (Abe et al., 2001; Morita et al., 2010; Shimokawa et al., 2012). Nicotiana attenuata contains four polyketide synthase enzymes (NaPKS1/2/3/4, renamed from NaCHAL1/2/3/4, respectively) homologous to chalcone synthase or benzalacetone synthase (Fig. S9) (Abe et al., 2001); silencing NaPKSs in N. attenuata flowers dramatically reduces the level of BA emission (Kessler et al., 2008; Guo et al., 2020). In the target region of the previous RNA‐interference (RNAi) construct, the NaPKS3 (A4A49_39367) sequence differed from the NaPKS1 (A4A49_08280), NaPKS2 (A4A49_34074), and NaPKS4 (A4A49_65921) sequences by only 34, 10, and 39 nucleotides, respectively (Kessler et al., 2008) (Fig. S10). Therefore, we were not sure whether one or some NaPKSs help convert cinnamoyl‐CoA to benzalacetone. Finally, benzalacetone appears to be reduced by an unknown reductase, which leads to the production of BA.
Fig. 2.
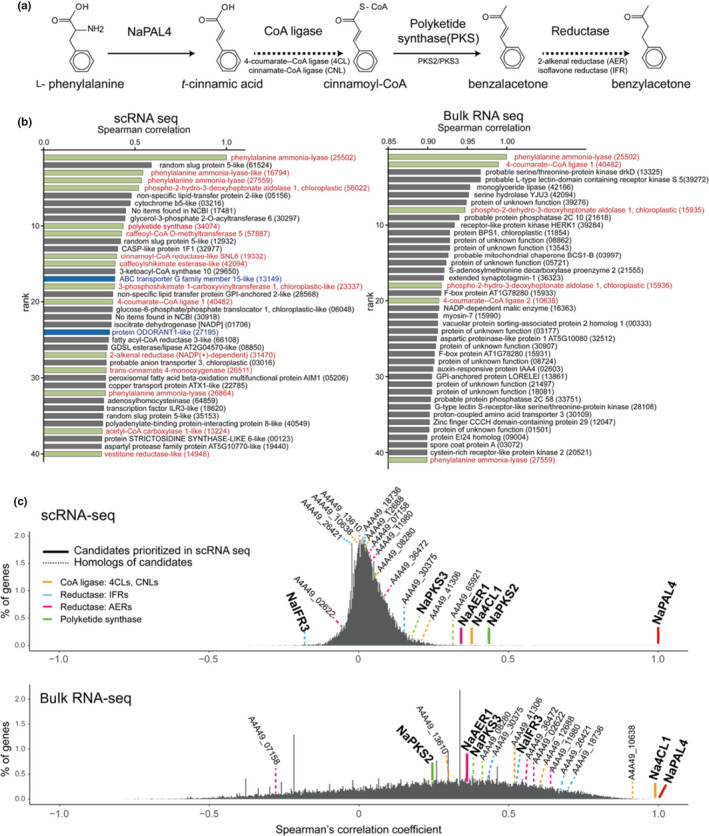
Identification of benzylacetone (BA) biosynthetic genes via correlation analysis at single‐cell resolution. (a) BA biosynthetic pathway proposed based on the biochemical function of NaPAL4 and NaPKS1/2/3/4 (solid lines), which has been characterized previously. Dashed lines indicate a putative reaction mediated by unknown enzymes: Coenzyme A (CoA) ligases and reductases; NaPAL4, Nicotiana attenuata phenylalanine ammonia‐lyase 4; NaPKS, N. attenuata polyketide synthase. Genes tested in this study are annotated below the arrows. (b) Two correlation analyses display different top‐ranked gene profiles. Genes are ranked by their correlation coefficients with NaPAL4. Single‐cell RNA sequencing (scRNA‐Seq) based analysis (left panel) generated more relevant profiles where several phenylpropanoid pathway associated genes were included; green bars: putative phenylpropanoid pathway genes, blue bars: homologs of transporter (ABC transporter) and transcription factor (ODORANT1) studied in petunia. (c) Percentage of genes (y‐axis) that have a given Spearman’s rank correlation coefficient (x‐axis) with NaPAL4 expression in scRNA‐Seq and bulk RNA‐Seq. Genes encoding CoA ligase, reductases, and polyketide synthases appear in different colors on the histograms. Thick lines highlight the CoA ligase, reductase, and polyketide synthase most correlated with NaPAL4 in scRNA‐Seq. NaAER1, N. attenuata 2‐alkenal reductases 1; NaCNL, N. attenuata cinnamate:CoA ligase; NaIFR, N. attenuata isoflavone reductase; Na4CL1, N. attenuata 4‐coumarate:CoA ligase.
To identify BA biosynthetic genes, we hypothesized that all BA biosynthetic genes would be co‐regulated over time at the single‐cell level. Because NaPAL4 is known to be the first enzyme for BA biosynthesis, we calculated the Spearman’s rank correlation coefficient of NaPAL4 expression with the whole gene expression detected in our scRNA‐Seq data. For comparison, we also calculated the same gene‐to‐gene correlation in 20 previously published bulk RNA‐Seq datasets from tissues including those of seed, leaf, root, stem, anther, style, stigma, ovary, nectary, pedicel, and corolla (Li et al., 2016; Brockmöller et al., 2017). We compared functional relationships among the top 40 ranked genes with NaPAL4 in both datasets (Fig. 2b): genes involved in phenylpropanoid metabolism were detected more in scRNA‐Seq (13 among 40 genes) than in bulk RNA‐Seq (six among 40 genes).
Next, we prioritized candidate genes: CoA ligases, PKSs, and reductases in scRNA‐Seq data based on the correlation with NaPAL4. The top 40 ranked genes in scRNA‐Seq and bulk RNA‐Seq data contained one common 4CL/CNL‐like gene (Na4CL1, A4A49_40482), suggesting that Na4CL1 was the strongest candidate enzyme for producing BA. Among four NaPKSs, NaPKS2 was the gene that ranked highest in scRNA‐Seq datasets, followed by NaPKS3 (Fig. 2c). However, in bulk RNA‐Seq datasets, NaPKS3 was correlated more with NaPAL4 than was NaPKS2 (Fig. 2c). Five homologs of 2‐alkenal reductase (AER) and four homologues of isoflavone reductase (IFR), another family of reductases, were found in the scRNA‐Seq and bulk RNA‐Seq datasets. Interestingly, one putative AER (NaAER1, A4A49_31470) was highly ranked in scRNA‐Seq (Fig. 2b,c). In the bulk RNA‐Seq, all IFR genes and three of five AER homologs were more highly correlated with NaPAL4 expression than was NaAER1 (Fig. 2c). The distribution of the correlation coefficient in scRNA‐Seq differed from its distribution in bulk RNA‐Seq. A large number of genes in scRNA‐Seq appeared to have a low correlation with NaPAL4: 99.46% of genes had a correlation coefficient between −0.25 and 0.25. The histogram from bulk RNA‐Seq showed a broad and skewed distribution of correlation coefficients.
Na4CL1, NaPKS2, and NaAER1 are involved in benzylacetone biosynthesis
To test the model for BA biosynthesis that emerged from the co‐expression analysis in scRNA‐Seq datasets (Fig. 2a), we first silenced the expression of Na4CL1 by VIGS and measured the level of floral volatiles in the headspace and in endogenous cells. Silencing Na4CL1 reduced levels of BA both in the flower headspace (Fig. 3a) and in endogenous cells (Fig. 3b). It is noteworthy that other Na4CL1 homologs (A4A49_41306 and A4A49_10638) and NaCNL homologs (A4A49_13610 and A4A49_12688), which are expressed in N. attenuata flowers, were not silenced in Na4CL1‐silenced corolla (Figs S11, S12). To evaluate the catalytic ability of Na4CL1, purified Na4CL1 proteins were tested with t‐cinnamic acid and several C6–C3 compounds (4‐coumaric acid, caffeic acid, and ferulic acid) under the same conditions (Fig. 3c). Best‐fit kinetic values were calculated by fitting with the Michaelis–Menten curve. Na4CL1 showed the ability to catalyze the formation of CoA‐thioesters of t‐cinnamic acid and similar C6–C3 compounds. Determined apparent Michaelis constant (K M) values showed that Na4CL1 has a higher affinity to t‐cinnamic acid and 4‐coumaric acid than to caffeic acid and ferulic acid. Considering the catalytic rate constant (k cat) values of t‐cinnamic acid and derivatives, it seems that Na4CL1 can activate carboxylate groups in several C6–C3 compounds in N. attenuata, mediating reactions on carbon chains of phenylpropanoids. We next tried to clarify which NaPKSs were involved in BA biosynthesis. Although we designed the VIGS construct to be as specific as possible, the high sequence similarity observed among NaPKSs likely led to NaPKS1/2/3/4 being silenced (Figs 4a, S10, S13). As previously reported, silencing NaPKSs reduced levels of BA in the headspace (Fig. 4a) and corolla limb (Fig. 4b). Although both NaPKS2 and NaPKS3 were strongly expressed in flower tissues (Fig. S14), scRNA‐Seq datasets indicated that NaPAL4 was more correlated with NaPKS2 than with NaPKS3 (Fig. 2c); NaPAL4 and NaPKS2 were mainly expressed in clusters 0, 3, and 4, but NaPKS3 was expressed in the entire clusters (Fig. 6, see later). Interestingly, when we incubated the same amount of purified NaPKS2 or NaPKS3 proteins with Na4CL1 products (cinnamoyl‐CoA) and malonyl‐CoA (as carbon donor), we detected benzalacetone in the NaPKS2‐containing mixture but not in the NaPKS3‐containing mixture (Fig. 4c). The result suggests that NaPKS2 may play a major role for BA biosynthesis, as predicted by scRNA‐Seq correlation analysis (Fig. 2c).
Fig. 3.
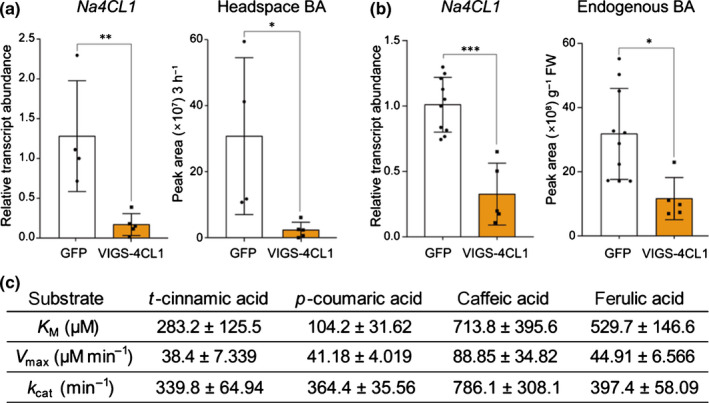
Nicotiana attenuata 4‐coumarate:CoA ligase (Na4CL1) participates in benzylacetone (BA) biosynthesis in N. attenuata flowers. (a, b) Mean (± SD) levels of headspace BA and endogenous BA in corollas were measured in Na4CL1‐silenced flowers (mean ± SD; *, P < 0.05; **, P < 0.01; ***, P < 0.001; two‐tailed Student’s t‐test). Green fluorescent protein (GFP), plant infected TRV2:GFP (a truncated form), which was used as a negative control; VIGS‐4CL1, a Na4CL1‐silenced plant, which was generated by virus‐induced gene silencing (VIGS). (c) Kinetic parameters (best‐fit value ±SE) of Na4CL1 determined from in vitro enzyme assays of heterologously purified Na4CL1 protein. Best‐fit catalytic values were calculated by fitting with the Michaelis–Menten curve, showing that Na4CL1 can catalyze the formation of Coenzyme A (CoA)‐thioesters of t‐cinnamic acid and similar C6–C3 compounds.
Fig. 4.
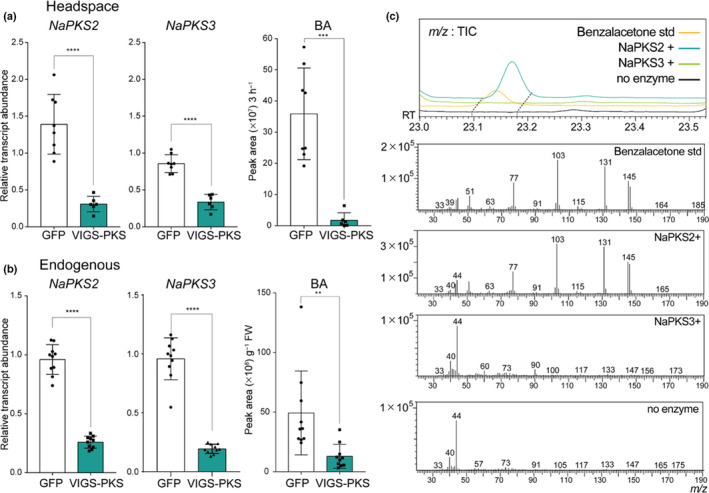
Nicotiana attenuata polyketide synthase 2 (NaPKS2) is the enzyme that participates in benzylacetone (BA) biosynthesis in N. attenuata flowers. (a) NaPKS2 and NaPKS3 are co‐silenced in NaPKS2/3‐silenced corollas. Green fluorescent protein (GFP), plant infected TRV2‐GFP (a truncated form), which was used as a negative control; VIGS‐PKS, a NaPKSs co‐silenced plant, which was generated by virus‐induced gene silencing (VIGS) (mean ± SD; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; two‐tailed Student’s t‐test). Mean (±SD) levels of headspace BA show that NaPKS2/3‐silenced flowers emit less BA. (b) NaPKS2/3‐silenced flowers harbor less endogenous BA in corolla tissue. (mean ± SD; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001; two‐tailed Student’s t‐test). (c) In vitro enzyme assay reveals that NaPKS2, not NaPKS3, can synthesize benzalacetone, a putative intermediate of BA biosynthesis. A novel peak detected in NaPKS2‐contained reaction mixtures represents benzalacetone, based on its fragmentation pattern and standard compound injections.
Fig. 6.
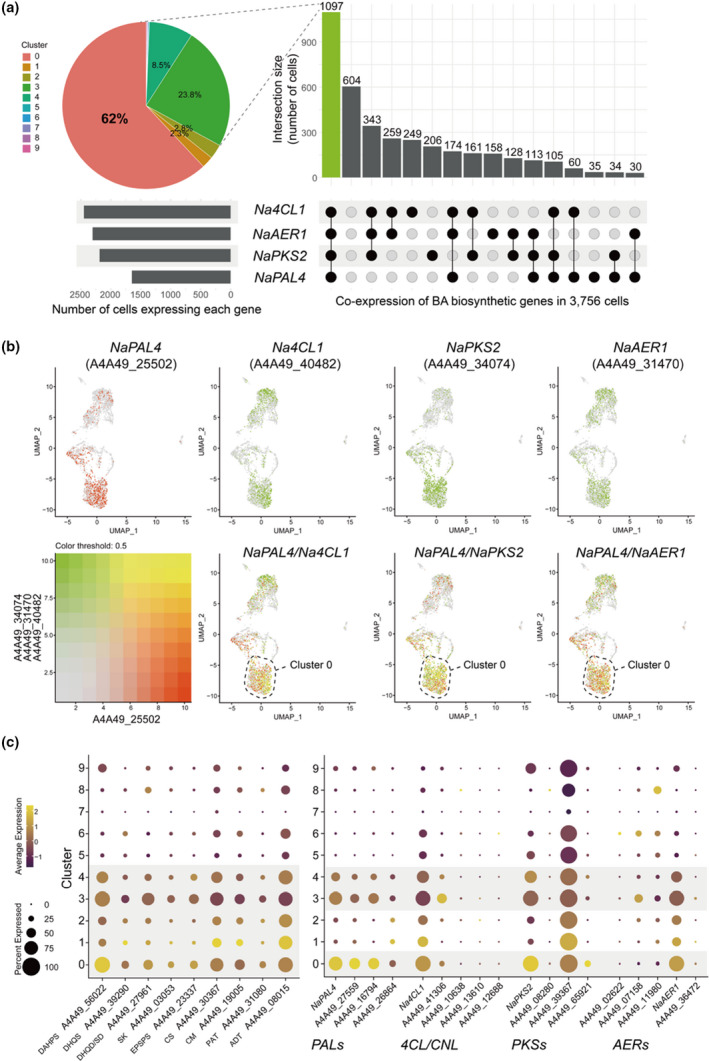
Characterization of benzylacetone (BA) biosynthetic cell clusters. (a) An UpSet plot shows numbers of cells harboring individual gene or combination of NaPAL4, Na4CL1, NaPKS2, and NaAER1 genes. A pie chart represents the distribution of clusters of cells expressing all four genes. Clusters 0, 3, and 4 (epidermis) contain 94.3% of cells co‐expressing all four genes. (b) Featureplots visualize co‐expression between NaPAL4 and Na4CL1, NaPKS2, and NaAER1, respectively. Cells that co‐express genes mostly locate in cluster 0. (c) Dotplots of l‐phenylalanine (Phe) biosynthetic genes (primary metabolism) and BA biosynthetic genes (secondary metabolism). Phe is mainly synthesized in clusters 0 to 4, and BA synthesis occurs in clusters 0, 3, and 4. NaPAL4, Nicotiana attenuata phenylalanine ammonia‐lyase 4; Na4CL1, N. attenuata 4‐coumarate:CoA ligase; NaPKS2, N. attenuata polyketide synthase 2; NaAER1, N. attenuata 2‐alkenal reductase 1; DAHPS, phospho‐2‐dehydro‐3‐deoxyheptonate aldolase; DHQS, 3‐dehydroquinate synthase; DHQD/SD, bifunctional 3‐dehydroquinate dehydratase/shikimate dehydrogenase; SK, shikimate kinase; EPSPS, 3‐phosphoshikimate 1‐carboxyvinyltransferase 1; CS, chorismate synthase; CM, chorismate mutase; PAT, bifunctional aspartate aminotransferase and glutamate/aspartate‐prephenate aminotransferase; ADT, arogenate dehydratase/prephenate dehydratase.
To identify the last step enzyme of BA biosynthesis, we silenced the candidate reductase NaAER1 by VIGS and measured the level of floral volatiles in the headspace and in endogenous cells. Silencing NaAER1 impaired BA biosynthesis in corollas without silencing other homologs; the levels of BA emission and endogenous BA were reduced in NaAER1‐silenced flowers (Figs 5a,b, S15). We detected a novel compound emitted from NaAER1‐silenced flowers, which was absent in headspace of control flowers (Fig. 5c). Metabolite fragmentation patterns and standard compound injections verified that this compound is benzalacetone, the nonreduced form of BA. The kinetics analysis of NaAER1 proteins showed that benzalacetone had a lower K M value than did other derivatives with hydroxyl or methoxy groups (Fig. 5d). Extinction coefficients used in NaAER1 kinetics were calculated experimentally: 6.314 l mmol−1 cm−1 for NADPH (340 nm), 0.6843 l mmol−1 cm−1 for benzalacetone (340 nm), 2.638 l mmol−1 cm−1 for NADPH (370 nm), 3.903 l mmol−1 cm−1 for 4‐hydroxy‐benzalacetone (370 nm), and 7.565 l mmol−1 cm−1 for vanillylidenacetone (370 nm) (Fig. S16). Generalized Beer–Lambert laws for complex mixtures were adopted for calculating decreasing amounts of substrates (Wypych, 2015). In addition, GC–MS confirmed that purified NaAER1 proteins with NADPH, a biological reducing agent, can convert benzalacetone to BA (Fig. S17). The accumulation of benzalacetone in NaAER1‐silenced corollas and the in vitro assay showed that NaAER1 is the final enzyme involved in BA biosynthesis.
Fig. 5.
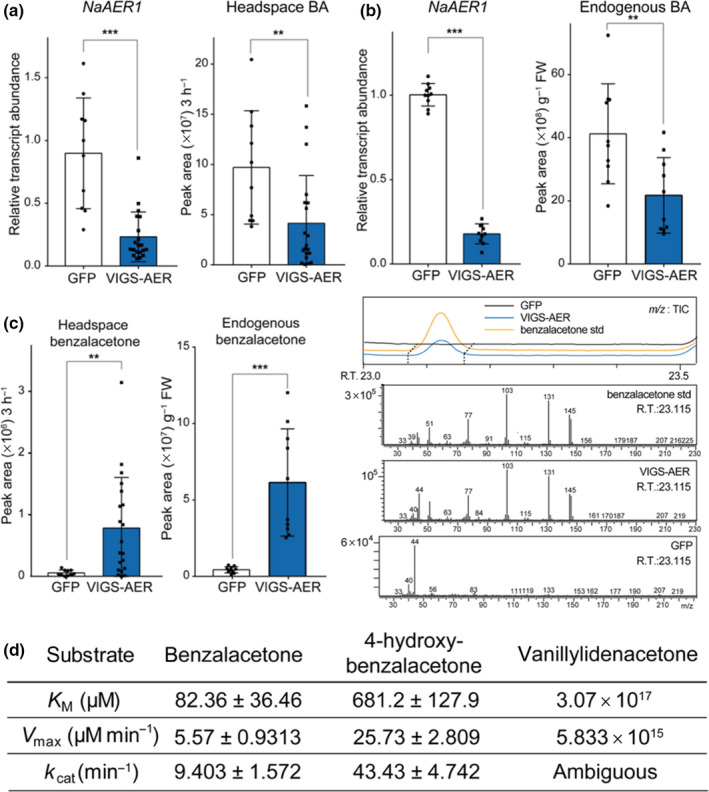
Nicotiana attenuata 2‐alkenal reductase 1 (NaAER1) catalyzes the final step in benzylacetone (BA) biosynthesis. (a, b) Mean (± SD) levels of headspace BA and endogenous BA measured in NaAER1‐silenced flowers (mean ± SD; **, P < 0.01; ***, P < 0.001; two‐tailed Student’s t‐test). BA levels were reduced in NaAER1‐silenced flowers. Green fluorescent protein (GFP), plant infected TRV2‐GFP (a truncated form), which was used as a negative control; VIGS‐AER, a NaAER1‐silenced plant, which was generated by virus‐induced gene silencing (VIGS). (c) Benzalacetone, a metabolic intermediate of BA, was emitted from NaAER1‐silenced flowers and not from control flowers (mean ± SD; **, P < 0.01; ***, P < 0.001; two‐tailed Student’s t‐test). Right panel shows a novel gas chromatography–mass spectrometry (GC–MS) peak in headspace of a NaAER1‐silenced flower, which has the same retention time and mass fragmentation pattern as the benzalacetone standard. (d) Kinetic parameters (best‐fit value ± SE) of NaAER1 determined from in vitro enzyme assays of heterologously purified NaAER1 protein. Best‐fit catalytic values were calculated by fitting with the Michaelis–Menten curve.
Recently, Guo et al. (2020) reported that NaIFR3 correlated strongly with NaPAL4 in the same bulk RNA‐Seq dataset used in this study. Although we also observed the positive correlation between NaIFR3 and NaPAL4 in bulk RNA‐Seq data, there was no positive correlation between these two genes in the scRNA‐Seq dataset (Fig. 2c). Furthermore, we found that levels of BA emission in NaIFR3‐silenced corollas did not differ significantly from levels in control corollas (Fig. S18a), even though the silencing efficacy of NaIFR3 in our study was approximately three‐fold higher than in the previous study (Guo et al., 2020). Because there was a large variation in BA emission from NaIFR3‐silenced corollas, we measured the endogenous levels of BA in control and NaIFR3‐silenced corollas. No difference in endogenous BA levels with a low variation was found between control and NaIFR3‐silenced corollas (Fig. S18b).
Benzylacetone is synthesized in the epidermal layer
Among the 3756 cells, 1648 contained at least one molecule of NaPAL4 messenger RNA (mRNA) (Fig. S19). Given that 80% of NaPAL4‐expressing cells belonged to epidermal cells (clusters 0, 3, and 4), we hypothesize that BA is synthesized in the epidermal layer of the corolla. We calculated the number of cells harboring individual gene or combination of NaPAL4, Na4CL1, NaPKS2, and NaAER1 genes. Of 3756 cells, 1097 contained all four genes; 94% of these cells were found in clusters 0, 3, and 4 (epidermal cells) (Fig. 6a). Most NaPAL4‐expressing cells (67%) contained all three genes: Na4CL1, NaPKS2, and NaAER1. To visualize the spatial distribution of cells in which two target genes were co‐expressed, we merged two featureplots of NaPAL4 and one of Na4CL1, NaPKS2, and NaAER1 expression (Fig. 6b). The results showed that a high level of co‐expression (yellowish cells) was mainly found in cluster 0, implicating the scent‐biosynthesis step that occurs in the epidermis. To validate the location of BA‐biosynthetic cells, we generated NaPAL4 promoter‐GFP lines and NaPKS2 promoter‐GFP lines in N. attenuata. In both lines, GFP expression was strongly detected in the corolla epidermis, especially in the adaxial region (Fig. S20), which suggests that clusters 0, 3, and 4 are the epidermal cells. Phe biosynthetic genes were detected mainly in clusters 0, 3, and 4, but these genes were also expressed in other clusters (clusters 1 and 2) (Fig. 6c). Dotplots also showed that NaPAL2/3/4 with Na4CL1, NaPKS2, and NaAER1 were highly expressed in the epidermal cells (Fig. 6c).
Diurnal rhythms of benzylacetone (BA) biosynthetic genes and BA emissions
Because N. attenuata flowers emit BA rhythmically (Yon et al., 2016), we asked whether all BA biosynthetic genes share this diurnal expression pattern. In our experiments, BA emission began to increase near ZT 12 and peaked at ZT 16 (Fig. 7a). Quantitative PCR analysis showed that the transcript levels of NaPAL4, Na4CL1, NaPKS2, and NaAER1 peaked c. 4 h before BA emission levels peaked, suggesting that BA emission is regulated mainly at the transcriptional level. The scRNA‐Seq data also showed similar expression patterns of BA biosynthetic genes. For instance, the violin plots illustrated a similar time‐dependent expression of NaPAL4 transcripts in clusters 0, 3, and 4 (Fig. 7b). The transcript levels of the other BA biosynthetic genes in each cluster also depended on time with slight differences: Na4CL1 and NaAER1 expression peaked at ZT 12 in all clusters, but NaPKS2 expression increased from ZT 8 to ZT 16 in the epidermal clusters only (Fig. 7b).
Fig. 7.
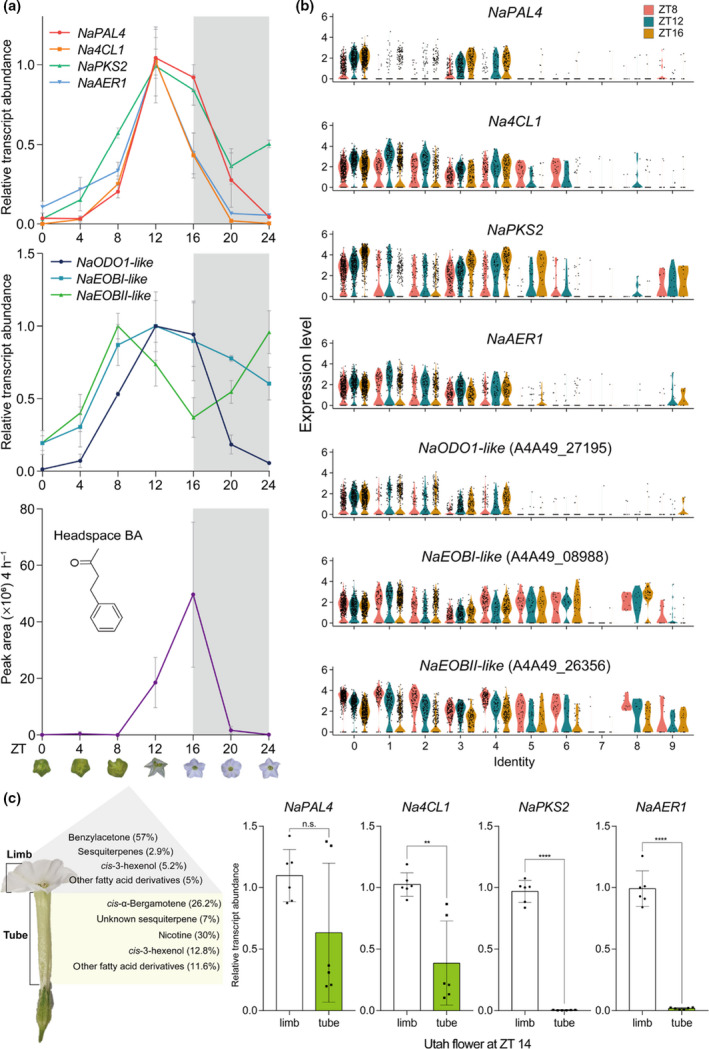
Diurnal rhythm of benzylacetone (BA) biosynthesis genes and its expression pattern in flower tissues. (a) Rhythmic emission of BA and expression of related genes. Relative transcript levels (mean ± SD) of NaPAL4, Na4CL1, NaPKS2, and NaAER1 peak at Zeitgeber Time (ZT) 12, 4 h before the peak of BA emission. NaODO1‐like and NaEOBI‐like also peak at ZT 12 when the BA synthesis genes peak, and NaEOBII‐like peaks 4 h earlier than other MYP transcription factors. NaEOBI/II‐like, Nicotiana attenuata EMISSION OF BENZENOID I/II‐like; NaODO1‐like, N. attenuata ODORANT1‐like. (b) Violin plots show the expression levels of BA biosynthetic genes and MYB transcription factors across all clusters at three different time points. The levels of Na4CL1 and NaAER1 transcript peak at ZT 12 (widest sections of violin plots are higher than ZT 8 and ZT 16), as shown in (a). NaPAL4 and NaPKS2 transcript levels at ZT 16 are higher than those at ZT 8 and 12 in clusters 0, 3, and 4. Violin plots of MYB transcription factors also reflect transcriptional dynamics shown in (a). (c) Kessler & Baldwin (2007) reported the percentage of N. attenuata floral scents in the floral headspace. BA is synthesized mainly in the corolla limb, and cis‐α‐bergamotene is synthesized in the corolla tube. Quantitative transcript analysis of BA biosynthetic genes in dissected flower tissues of limb and tube reveals that the lack of BA emission in the corolla tube resulted from the limited expression of NaPKS2 and NaAER1 (mean ± SD; n.s., not significant, **, P < 0.01; ****, P < 0.0001; two‐tailed Student’s t‐test). NaPAL4, N. attenuata phenylalanine ammonia‐lyase 4; Na4CL1, N. attenuata 4‐coumarate:CoA ligase; NaPKS2, N. attenuata polyketide synthase 2; NaAER1, N. attenuata 2‐alkenal reductase 1.
In petunia flowers, rhythmic scent emission is regulated by several transcriptional activators: Petuniaxhybrida ODORANT1 (PhODO1) and EMISSION OF BENZENOID I/II (PhEOBI/II) (Dexter et al., 2007; Klempien et al., 2012; Fenske et al., 2015; Amrad et al., 2016). PhODO1 peaks in the evening, right before scent emission peaks (Verdonk et al., 2005; Spitzer‐Rimon et al., 2013). PhEOBII peaks in the morning (Spitzer‐Rimon et al., 2010), and directly activates PhODO1 and PhEOBI. (Van Moerkercke et al., 2011). In the N. attenuata genome database, we identified several homologs of three petunia MYB transcription factors (Brockmöller et al., 2017). Among these homologs, we found that one homolog of PhODO1 and two homologs of PhEOBI/II were expressed in N. attenuata flowers. The amino acid sequence alignment between N. attenuata and petunia is shown in Fig. S21. Quantitative PCR analysis showed that NaODO1‐like and NaEOBI‐like peaked concurrently with BA biosynthetic genes (ZT 12), and NaEOBII‐like peaked during the daytime (Fig. 7a); these transcript dynamics were also observed in the violin plots (Fig. 7b). Single‐cell RNA‐Seq data showed that NaODO1‐like expression rather than NaEOBI/II had a high correlation with NaPAL4 (Fig. 2b) and was confined to clusters 0, 1, 2, 3, and 4, which suggests that NaODO1‐like regulates BA biosynthetic genes.
Next, we measured transcript levels of BA biosynthetic genes in the corolla limb and tube (Fig. 7c). Although transcript levels of NaPAL4 in the corolla limb did not differ with the level in the corolla tube, Na4CL1 expression was lower in the corolla tube than in the corolla limb. The NaPKS2 and NaAER1 transcripts were barely detected in the corolla tube, which is consistent with the known location of BA biosynthesis (Euler & Baldwin, 1996; Kessler & Baldwin, 2007).
Discussion
BA is synthesized mainly in epidermal cells, as revealed from how cell populations clustered and from the spatial distribution of BA biosynthetic genes. NaPAL4, the first enzyme required for BA biosynthesis, was found in the epidermis (Fig. S20). Most cells (67%) expressing NaPAL4, the gene required for the initial step in BA biosynthesis, harbored all other BA biosynthetic genes, indicating that the single‐cell transcriptome map of NaPAL4 largely concurred with the map of BA‐producing cells in the corolla. Only 45% of Na4CL1‐, 50% of NaPKS2‐, and 48% of NaAER1‐expressing cells contained the entire set of BA biosynthetic genes. High levels of NaPAL4 and Na4CL1 transcripts were found in the corolla limb and in the corolla tube (Fig. 7c), although no BA is produced in the tube. In contrast, NaPKS2 and NaAER1 transcripts were detected in the corolla limb and not in the tube, suggesting that the expression of NaPKS2 and NaAER1 determines BA production at the sub‐organ level. Phe biosynthetic genes were mainly found in the epidermal clusters (Fig. 6c), which suggests that primary metabolism in corollas is specialized to support the high demands of secondary metabolite production.
Transcriptional heterogeneity, both in different plant tissues and amongst accessions with distinctive floral volatile productions, has been used to identify key enzymes for volatile biosynthesis. Bulk RNA‐Seq analysis is a powerful method with which to examine gene‐to‐gene correlation in several samples (Boachon et al., 2015, 2019; Xu et al., 2018). In addition, bulk RNA‐Seq analysis is useful for predicting a target gene among several homologous genes in plant tissues (Wong et al., 2020): Petunia CNL gene was identified by sequence comparison and bulk RNA‐Seq analysis (Klempien et al., 2012). We analyzed publicly available bulk RNA‐Seq datasets containing 14 different tissues from N. attenuata, because BA is synthesized in the corolla but not in other tissues. The metabolic heterogeneity among these tissues was used to identify a CoA ligase, Na4CL1, in BA biosynthesis (Fig. 2). However, the bulk RNA‐Seq datasets used in this study were insufficient for exploring the entire BA biosynthetic pathway, perhaps because NaPAL4 expressed in other plant tissues may be involved in other phenylpropanoid pathway.
A previous study (Guo et al., 2020) has suggested a BA biosynthetic pathway that differs from our biosynthetic pathway. Guo et al. (2020) suggested that t‐cinnamic acid is condensed with one malonyl‐CoA by an unknown CoA ligase and malonyl transferase; the malonylated compound is then reduced by NaIFR3, and the acyl side chain of the reduced compound is removed by NaPKS3. Here, we propose that NaPKS2 condenses cinnamoyl‐CoA with one malonyl‐CoA to form benzalacetone; no additional malonyl transferase is necessary for BA biosynthesis. Although the protein sequence of NaPKS2 shared a high similarity with NaPKS3, scRNA‐Seq analysis predicts that NaPKS2 is the key enzyme for BA biosynthesis. Purified NaPKS2 proteins produced benzalacetone from the mixture of cinnamoyl‐CoA and malonyl‐CoA (Fig. 4c). Although purified NaPKS3 proteins produced no benzalacetone in vitro, more genetic evidence is needed to examine the biochemical function of NaPKS3 in vivo. We also propose that NaAER1 is the major reductase for BA biosynthesis; the levels of BA in the headspace and in endogenous corolla cells were lower in NaAER1‐silenced corollas than in control corollas. The nonreduced form of BA was found in the headspace and in endogenous cells of NaAER1‐silenced corollas. Although Guo et al. (2020) showed that NaIFR3‐silenced flowers emitted less BA than did control flowers, we found that silencing NaIFR3 did not alter the headspace and endogenous levels of BA in the corollas. NaIFR3 expression was shown to be highly correlated with NaPAL4 expression in the bulk RNA‐Seq dataset, but not in the scRNA‐Seq dataset (Figs 2c, S22). NaAER1 was more correlated with NaPAL4 than was NaIFR3 in the scRNA‐Seq dataset (Fig. 2). This discrepancy may have originated from the large variation in the headspace levels of BA emitted from NaIFR3‐silenced corollas (Fig. S18) and the unintended sample selection bias in the previous study. Members of an IFR family are known to bind a flavonoid, which has a C6–C3–C6 carbon skeleton (Wang et al., 2006; Cheng et al., 2015). Silencing NaIFR3, which may alter the level of flavonoids, affects BA emission but not BA biosynthesis.
We analyzed the transcriptomic profiles of corolla at the single cell level, which reveals the existence of distinctive cell identities in N. attenuata corolla cells. This cell‐to‐cell heterogeneity in the corolla helps us to identify unknown BA biosynthetic genes. To increase cell heterogeneity, we performed more time‐point sampling rather than biological replicate sampling. Our major aim was to identify floral scent (BA) biosynthetic genes using gene‐to‐gene correlation. We thought that time‐series sampling is useful to increase the power of correlation analysis, because N. attenuata flowers synthesize BA diurnally (Fig. 7). That is why we performed scRNA‐Seq experiments at three different time points instead of using three biological replicates. Rather than retrieving the average transcript levels of genes in tissue, we can examine gene‐to‐gene correlation using the transcript levels of genes expressed in a single cell. However, the lack of replicates may weaken the correlation analysis because we were not able to differentiate between technical noise and biological variability.
Single‐cell RNA‐Seq generates high‐dimensional data, which may provide more reliable correlation among genes (Shinozaki et al., 2018). Bulk RNA‐Seq data contain tens of thousands of variables (gene number), but the number of samples is much smaller than the number of genes. However, the scRNA‐Seq data have thousands of variables (gene numbers) with large numbers of cells. It is noteworthy to mention that scRNA‐Seq, which can only detect a thousand genes in a single cell, might contain numerous false zeros in a matrix. Bulk RNA‐Seq, in contrast, can capture over 10 000 genes in a sample; with proper sampling design, bulk RNA‐Seq can improve the identification of biosynthetic genes. In addition, our protoplasting method may provoke cell‐type‐dependent dissociation, which can be solved by single‐nucleus RNA‐Seq (Wu et al., 2019). Although gene co‐expression analysis of scRNA‐Seq data is limited by low sequencing depth per cell (dropout events) and a high level of technical noise, scRNA‐Seq analysis can be used to investigate biosynthetic genes of a target metabolite.
Recent technological advances, such as live single‐cell MS enable us to examine metabolic profiling in single cells (Fujii et al., 2015). Apparent cell types like trichomes or guard cells have been extensively studied for their single‐cell‐type metabolism (Misra et al., 2014). However, it is hard to measure levels of a metabolite of low abundance within a single cell. The detection of intermediates is also difficult due to their rapid turnover rates. For instance, we were not able to detect benzalacetone (BA intermediate) in N. attenuata headspace. In this study, without the measurement of BA intermediate, we were able to identify benzalacetone synthase (NaPKS2) and reductase (NaAER1) in the corolla. This result suggests that scRNA‐Seq analysis coupled with single‐cell metabolite analysis will enhance our understanding of metabolic heterogeneity and pathways in a single cell.
Author contributions
S‐GK conceived and supervised the project. MK generated scRNA‐Seq libraries and performed bioinformatic analysis. YC generated cDNA libraries for bulk RNA‐ Seq. MK and HK analyzed plant volatiles. MK and HK performed VIGS experiments. MK conducted quantitative PCR analysis and in vitro enzyme assay. S‐GK and MK wrote the manuscript.
Supporting information
Fig. S1 Correlation analysis between bulk‐RNA‐Seq and scRNA‐Seq datasets.
Fig. S2 QC metrics of scRNA‐Seq datasets.
Fig. S3. Cells are displayed by an integrated uniform manifold approximation and projection (UMAP) plot in two dimensions.
Fig. S4 Pollen and vasculature‐specific marker genes are exclusively expressed in clusters 7 and 8, respectively.
Fig. S5 Validation of pollen‐specific marker genes.
Fig. S6 Putative wax and cutin biosynthesis genes are more expressed in clusters 0, 3, 4, 5, and 9.
Fig. S7 Protein sequences of marker genes are similar to their Arabidopsis thaliana orthologs.
Fig. S8 Subunits of photosystems I and II complexes are enriched in clusters 1, 2, and 6.
Fig. S9 NaPKS1/2/3/4 share high sequence similarity with polyketide synthase proteins of other species.
Fig. S10 Target sequences for NaPKS RNAi experiments (used in previous studies, NaCHAL) and VIGS experiments (used in this study).
Fig. S11 Cladogram of Nicotiana attenuata 4CL, CNL homologs within acyl‐activating enzyme (AAE) superfamily.
Fig. S12 4CL and CNL homologs are not co‐silenced in Na4CL1‐silenced flowers.
Fig. S13 Co‐silencing of NaPKS1/2/3/4 in NaPKS3‐VIGS flowers.
Fig. S14 Tissue‐specific expression of NaPKS2 and NaPKS3 in Nicotiana attenuata.
Fig. S15 AER homologs are not co‐silenced in NaAER1‐silenced flowers.
Fig. S16 Extinction coefficients used for NaAER1 kinetics.
Fig. S17 NaAER1 can reduce benzalacetone to benzylacetone.
Fig. S18 Silencing NaIFR transcription does not alter BA biosynthesis in corolla.
Fig. S19 NaPAL4 transcripts are highly expressed in clusters 0, 3, and 4.
Fig. S20 Expression of NaPAL4 and NaPKS2 in Nicotiana attenuata flowers.
Fig. S21 Nicotiana attenuata homologs of Petunia×hybrida MYB transcription factors.
Fig. S22 NaIFR3 expression patterns in scRNA‐Seq dataset.
Table S1 One hundred and twenty‐one genes excluded for analysis.
Table S2 Primers used in this study.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
The authors appreciate Dr Inkyung Jung, Seongwan Park, and Mooyoung Kim for helping them to generate a scRNA‐Seq library; Dr Gisuk Lee and Dr Yongsung Joo for help with metabolite analysis; San‐Hae Lim with protein purification; Guo Han, Shuqing Xu, and Ian T. Baldwin for comments on BA biosynthetic pathway; Emily Wheeler for editorial assistance. This work is supported by grants from the Samsung Science & Technology Foundation (SSTF‐BA1901‐10, S‐GK) and TJ Park Doctoral Science Fellowship (MK). The authors declare no competing interests.
Data availability
Single‐cell RNA‐Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession no. GSE193464. Other data generated during this study are included in this published article and Supporting Information files.
References
- Abe I, Takahashi Y, Morita H, Noguchi H. 2001. Benzalacetone synthase. A novel polyketide synthase that plays a crucial role in the biosynthesis of phenylbutanones in Rheum palmatum . European Journal of Biochemistry 268: 3354–3359. [DOI] [PubMed] [Google Scholar]
- Amrad A, Moser M, Mandel T, de Vries M, Schuurink RC, Freitas L, Kuhlemeier C. 2016. Gain and loss of floral scent production through changes in structural genes during pollinator‐mediated speciation. Current Biology 26: 3303–3312. [DOI] [PubMed] [Google Scholar]
- Baldwin IT, Preston C, Euler M, Gorham D. 1997. Patterns and consequences of benzyl acetone floral emissions from Nicotiana attenuata plants. Journal of Chemical Ecology 23: 2327–2343. [Google Scholar]
- Barkman TJ, Martins TR, Sutton E, Stout JT. 2007. Positive selection for single amino acid change promotes substrate discrimination of a plant volatile‐producing enzyme. Molecular Biology and Evolution 24: 1320–1329. [DOI] [PubMed] [Google Scholar]
- Baudino S, Hugueney P, Caissard J‐C. 2020. Chapter 12: evolution of scent genes. In: Pichersky E, Dudareva N, eds. Biology of plant volatiles. Boca Raton, FL, USA: CRC Press. [Google Scholar]
- Birnbaum K, Shasha DE, Wang JY, Jung JW, Lambert GM, Galbraith DW, Benfey PN. 2003. A gene expression map of the Arabidopsis root. Science 302: 1956–1960. [DOI] [PubMed] [Google Scholar]
- Boachon B, Burdloff Y, Ruan JX, Rojo R, Junker RR, Vincent B, Nicolè F, Bringel F, Lesot A, Henry L et al. 2019. A promiscuous CYP706A3 reduces terpene volatile emission from Arabidopsis flowers, affecting florivores and the floral microbiome. Plant Cell 31: 2947–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boachon B, Junker RR, Miesch L, Bassard JE, Höfer R, Caillieaudeaux R, Seidel DE, Lesot A, Heinrich C, Ginglinger JF et al. 2015. CYP76C1 (Cytochrome P450)‐mediated linalool metabolism and the formation of volatile and soluble linalool oxides in Arabidopsis flowers: a strategy for defense against floral antagonists. Plant Cell 27: 2972–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmöller T, Ling Z, Li D, Gaquerel E, Baldwin IT, Xu S. 2017. Nicotiana attenuata Data Hub (NaDH): an integrative platform for exploring genomic, transcriptomic and metabolomic data in wild tobacco. BMC Genomics 18: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. 2018. Integrating single‐cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36: 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Li G, Köllner TG, Jia Q, Gershenzon J, Chen F. 2014. Positive Darwinian selection is a driving force for the diversification of terpenoid biosynthesis in the genus Oryza . BMC Plant Biology 14: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LQ, Qu XQ, Hou BH, Sosso D, Osorio S, Fernie AR, Frommer WB. 2012. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335: 207–211. [DOI] [PubMed] [Google Scholar]
- Cheng Q, Li N, Dong L, Zhang D, Fan S, Jiang L, Wang X, Xu P, Zhang S. 2015. Overexpression of soybean isoflavone reductase (GmIFR) enhances resistance to phytophthora sojae in soybean. Frontiers in Plant Science 6: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denyer T, Ma X, Klesen S, Scacchi E, Nieselt K, Timmermans MCP. 2019. Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high‐throughput single‐cell RNA sequencing. Developmental Cell 48: 840–852.e5. [DOI] [PubMed] [Google Scholar]
- Dexter R, Qualley A, Kish CM, Ma CJ, Koeduka T, Nagegowda DA, Dudareva N, Pichersky E, Clark D. 2007. Characterization of a petunia acetyltransferase involved in the biosynthesis of the floral volatile isoeugenol. The Plant Journal 49: 265–275. [DOI] [PubMed] [Google Scholar]
- Dudareva N, Cseke L, Blanc VM, Pichersky E. 1996. Evolution of floral scent in Clarkia: novel patterns of S‐linalool synthase gene expression in the C. breweri flower. Plant Cell 8: 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudareva N, Pichersky E. 2000. Biochemical and molecular genetic aspects of floral scents. Plant Physiology 122: 627–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euler M, Baldwin IT. 1996. The chemistry of defense and apparency in the corollas of Nicotiana attenuata . Oecologia 107: 102–112. [DOI] [PubMed] [Google Scholar]
- Fenske MP, Hewett Hazelton KD, Hempton AK, Shim JS, Yamamoto BM, Riffell JA, Imaizumi T. 2015. Circadian clock gene LATE ELONGATED HYPOCOTYL directly regulates the timing of floral scent emission in Petunia . Proceedings of the National Academy of Sciences, USA 112: 9775–9780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Pozo N, Rosli HG, Martin GB, Mueller LA. 2015. The SGN VIGS tool: user‐friendly software to design virus‐induced gene silencing (VIGS) Constructs for functional genomics. Molecular Plant 8: 486–488. [DOI] [PubMed] [Google Scholar]
- Frick S, Kutchan TM. 1999. Molecular cloning and functional expression of O‐methyltransferases common to isoquinoline alkaloid and phenylpropanoid biosynthesis. The Plant Journal 17: 329–339. [DOI] [PubMed] [Google Scholar]
- Fujii T, Matsuda S, Tejedor ML, Esaki T, Sakane I, Mizuno H, Tsuyama N, Masujima T. 2015. Direct metabolomics for plant cells by live single‐cell mass spectrometry. Nature Protocols 10: 1445–1456. [DOI] [PubMed] [Google Scholar]
- Guo H, Lackus ND, Köllner TG, Li R, Bing J, Wang Y, Baldwin IT, Xu S. 2020. Evolution of a novel and adaptive floral scent in wild tobacco. Molecular Biology and Evolution 37: 1090–1099. [DOI] [PubMed] [Google Scholar]
- Guterman I, Shalit M, Menda N, Piestun D, Dafny‐Yelin M, Shalev G, Bar E, Davydov O, Ovadis M, Emanuel M et al. 2002. Rose scent: genomics approach to discovering novel floral fragrance‐related genes. Plant Cell 14: 2325–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister C, Satija R. 2019. Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biology 20: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp A, Yon F, Keesey IW, Mißbach C, Koenig C, Hansson BS, Baldwin IT, Knaden M, Kessler D. 2016. Hawkmoths evaluate scenting flowers with the tip of their proboscis. eLife 5: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean‐Baptiste K, McFaline‐Figueroa JL, Alexandre CM, Dorrity MW, Saunders L, Bubb KL, Trapnell C, Fields S, Queitsch C, Cuperusa JT. 2019. Dynamics of gene expression in single root cells of Arabidopsis thaliana . Plant Cell 31: 993–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo Y, Schuman MC, Goldberg JK, Wissgott A, Kim SG, Baldwin IT. 2019. Herbivory elicits changes in green leaf volatile production via jasmonate signaling and the circadian clock. Plant, Cell & Environment 42: 972–982. [DOI] [PubMed] [Google Scholar]
- Kallenbach M, Oh Y, Eilers EJ, Veit D, Baldwin IT, Schuman MC. 2014. A robust, simple, high‐throughput technique for time‐resolved plant volatile analysis in field experiments. The Plant Journal 78: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Ahn H, Rothe E, Baldwin IT, Kim SG. 2020. A robust genome‐editing method for wild plant species Nicotiana attenuata . Plant Biotechnology Reports 14: 585–598. [Google Scholar]
- Karimi M, Inzé D, Depicker A. 2002. GATEWAY vectors for Agrobacterium‐mediated plant. Trends in Plant Science 7: 193–195. [DOI] [PubMed] [Google Scholar]
- Kessler D, Baldwin IT. 2007. Making sense of nectar scents: the effects of nectar secondary metabolites on floral visitors of Nicotiana attenuata . The Plant Journal 49: 840–854. [DOI] [PubMed] [Google Scholar]
- Kessler D, Bing J, Haverkamp A, Baldwin IT. 2019. The defensive function of a pollinator‐attracting floral volatile. Functional Ecology 33: 1223–1232. [Google Scholar]
- Kessler D, Gase K, Baldwin IT. 2008. Field experiments with transformed plants reveal the sense of floral scents. Science 321: 1200–1202. [DOI] [PubMed] [Google Scholar]
- Kim J‐Y, Symeonidi E, Pang TY, Denyer T, Weidauer D, Bezrutczyk M, Miras M, Zöllner N, Hartwig T, Wudick MM et al. 2021. Distinct identities of leaf phloem cells revealed by single cell transcriptomics. Plant Cell 33: 511–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SG, Yon F, Gaquerel E, Gulati J, Baldwin IT. 2011. Tissue specific diurnal rhythms of metabolites and their regulation during herbivore attack in a native tobacco, Nicotiana attenuata . PLoS ONE 6: e26214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klempien A, Kaminaga Y, Qualley A, Nagegowda DA, Widhalm JR, Orlova I, Shasany AK, Taguchi G, Kish CM, Cooper BR et al. 2012. Contribution of CoA ligases to benzenoid biosynthesis in petunia flowers. Plant Cell 24: 2015–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen JT, Eriksson R, Gershenzon J, Ståhl B. 2006. Diversity and distribution of floral scent. Botanical Review 72: 1–120. [Google Scholar]
- Knudsen JT, Gershenzon J. 2020. The chemical diversity of floral scent. In: Pichersky E, Dudareva N, eds. Biology of plant volatiles. Boca Raton, FL, USA: CRC Press, 22. [Google Scholar]
- Koeduka T, Watanabe B, Suzuki S, Hiratake J, Mano J, Yazaki K. 2011. Characterization of raspberry ketone/zingerone synthase, catalyzing the alpha, beta‐hydrogenation of phenylbutenones in raspberry fruits. Biochemical and Biophysical Research Communications 412: 104–108. [DOI] [PubMed] [Google Scholar]
- Kolosova N, Sherman D, Karlson D, Dudareva N. 2001. Cellular and subcellular localization of S‐adenosyl‐L‐methionine : benzoic acid carboxyl methyltransferase, the enzyme responsible for biosynthesis of the volatile ester methylbenzoate in snapdragon flowers. Plant Physiology 126: 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassowski M. 2020. complexupset . doi: 10.5281/zenodo.3700590. [DOI] [Google Scholar]
- Krügel T, Lim M, Gase K, Halitschke R, Baldwin IT. 2002. Agrobacterium‐mediated transformation of Nicotiana attenuata, a model ecological expression system. Chemoecology 12: 177–183. [Google Scholar]
- Langdale JA, Nelson T. 1991. Spatial regulation of photosynthetic development in C4 plants. Trends in Genetics 7: 191–196. [DOI] [PubMed] [Google Scholar]
- Lee D, Meyer K, Chapple C, Douglas CJ. 1997. Antisense suppression of 4‐coumarate : coenzyme A ligase activity in Arabidopsis leads to altered lignin subunit composition. Plant Cell 9: 1985–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lex A, Gehlenborg N, Strobelt H, Vuillemot R, Pfister H. 2014. UpSet: visualization of intersecting sets. IEEE Transactions on Visualization and Computer Graphics 20: 1983–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Heiling S, Baldwin IT, Gaquerel E. 2016. Illuminating a plant’s tissue‐specific metabolic diversity using computational metabolomics and information theory. Proceedings of the National Academy of Sciences, USA 113: E7610–E7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Wang Z, Wang K, Liu Y, Hong Y, Chen C, Guan X, Chen Q. 2020. Co‐expression network analysis uncovers key candidate genes related to the regulation of volatile esters accumulation in Woodland strawberry. Planta 252: 1–15. [DOI] [PubMed] [Google Scholar]
- Libault M, Pingault L, Zogli P, Schiefelbein J. 2017. Plant systems biology at the single‐cell level. Trends in Plant Science 22: 949–960. [DOI] [PubMed] [Google Scholar]
- Lopez‐anido CB, Vatén A, Smoot NK, Sharma N, Guo V, Gong Y, Gil XA, Weimer AK, Bergmann DC. 2020. Single‐cell resolution of lineage trajectories in the Arabidopsis stomatal lineage and developing leaf. Developmental Cell 56: 1043–1055.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnard JL, Roccia A, Caissard JC, Vergne P, Sun P, Hecquet R, Dubois A, Saint OLH, Jullien F, Nicolè F et al. 2015. Biosynthesis of monoterpene scent compounds in roses. Science 349: 81–83. [DOI] [PubMed] [Google Scholar]
- Maruyama‐Nakashita A, Watanabe‐Takahashi A, Inoue E, Yamaya T, Saito K, Takahashi H. 2015. Sulfur‐responsive elements in the 3ʹ‐nontranscribed intergenic region are essential for the induction of SULFATE TRANSPORTER 2;1 gene expression in Arabidopsis roots under sulfur deficiency. Plant Cell 27: 1279–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra BB, Assmann SM, Chen S. 2014. Plant single‐cell and single‐cell‐type metabolomics. Trends in Plant Science 19: 637–646. [DOI] [PubMed] [Google Scholar]
- Morita H, Shimokawa Y, Tanio M, Kato R, Noguchi H, Sugio S, Kohno T, Abe I. 2010. A structure‐based mechanism for benzalacetone synthase from Rheum palmatum . Proceedings of the National Academy of Sciences, USA 107: 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlemann JK, Klempien A, Dudareva N. 2014. Floral volatiles: from biosynthesis to function. Plant, Cell & Environment 37: 1936–1949. [DOI] [PubMed] [Google Scholar]
- Qualley AV, Widhalm JR, Adebesin F, Kish CM, Dudareva N. 2012. Completion of the core β‐oxidative pathway of benzoic acid biosynthesis in plants. Proceedings of the National Academy of Sciences, USA 109: 16383–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu KH, Huang L, Kang HM, Schiefelbein J. 2019. Single‐cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiology 179: 1444–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saedler R, Baldwin IT. 2004. Virus‐induced gene silencing of jasmonate‐induced direct defences, nicotine and trypsin proteinase‐inhibitors in Nicotiana attenuata . Journal of Experimental Botany 55: 151–157. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, Regev A. 2015. Spatial reconstruction of single‐cell gene expression data. Nature Biotechnology 33: 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterlee JW, Strable J, Scanlon MJ. 2020. Plant stem‐cell organization and differentiation at single‐cell resolution. Proceedings of the National Academy of Sciences, USA 117: 33689–33699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scalliet G, Lionnet C, Le BM, Dutron L, Magnard J‐L, Baudino S, Bergougnoux V, Jullien F, Chambrier P, Vergne P et al. 2006. Role of petal‐specific orcinol O‐methyltransferases in the evolution of rose scent. Plant Physiology 140: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmittgen TD, Livak KJ. 2008. Analyzing real‐time PCR data by the comparative CT method. Nature Protocols 3: 1101. [DOI] [PubMed] [Google Scholar]
- Shimokawa Y, Morita H, Abe I. 2012. Benzalacetone synthase. Frontiers in Plant Science 3: 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki Y, Nicolas P, Fernandez‐Pozo N, Ma Q, Evanich DJ, Shi Y, Xu Y, Zheng Y, Snyder SI, Martin LBB et al. 2018. High‐resolution spatiotemporal transcriptome mapping of tomato fruit development and ripening. Nature Communications 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shulse CN, Cole BJ, Ciobanu D, Lin J, Yoshinaga Y, Gouran M, Turco GM, Zhu Y, O’Malley RC, Brady SM et al. 2019. High‐throughput single‐cell transcriptome profiling of plant cell types. Cell Reports 27: 2241–2247.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer‐Rimon B, Farhi M, Albo B, Cna'ani A, Ben Zvi MM, Masci T, Edelbaum O, Yu Y, Shklarman E, Ovadis M et al. 2013. The R2R3‐MYB‐like regulatory factor EOBI, acting downstream of EOBII, regulates scent production by activating ODO1 and structural scent‐related genes in petunia. Plant Cell 24: 5089–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitzer‐Rimon B, Marhevka E, Barkai O, Marton I, Edelbaum O, Masci T, Prathapani NK, Shklarman E, Ovadis M, Vainstein A. 2010. EOBII, a gene encoding a flower‐specific regulator of phenylpropanoid volatiles’ biosynthesis in petunia. Plant Cell 22: 1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockhaus J, Schlue U, Koczor M, Chitty JA, Taylor WC, Westhoff P. 1997. The promoter of the gene encoding the C4 form of phosphoenolpyruvate carboxylase directs mesophyll‐specific expression in transgenic C4 Flaveria spp. Plant Cell 9: 479–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh MC, Samuels AL, Jetter R, Kunst L, Pollard M, Ohlrogge J, Beisson F. 2005. Cuticular lipid composition, surface structure, and gene expression in Arabidopsis stem epidermis. Plant Physiology 139: 1649–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Moerkercke A, Haring MA, Schuurink RC. 2011. The transcription factor EMISSION of BENZENOIDS II activates the MYB ODORANT1 promoter at a MYB binding site specific for fragrant petunias. The Plant Journal 67: 917–928. [DOI] [PubMed] [Google Scholar]
- Verdonk JC, Haring MA, Van Tunen AJ, Schuurink RC. 2005. ODORANT1 regulates fragrance biosynthesis in petunia flowers. Plant Cell 17: 1612–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, He X, Lin J, Shao H, Chang Z, Dixon RA. 2006. Crystal structure of isoflavone reductase from Alfalfa (Medicago sativa L.). Journal of Molecular Biology 358: 1341–1352. [DOI] [PubMed] [Google Scholar]
- Widhalm JR, Dudareva N. 2015. A familiar ring to it: biosynthesis of plant benzoic acids. Molecular Plant 8: 83–97. [DOI] [PubMed] [Google Scholar]
- Wisecaver JH, Borowsky AT, Tzin V, Jander G, Kliebenstein DJ, Rokas A. 2017. A global coexpression network approach for connecting genes to specialized metabolic pathways in plants. Plant Cell 29: 944–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DCJ, Peakall R, Pichersky E. 2020. The role of transcriptome analysis in shaping the discovery of plant volatile genes. In: Pichersky E, Dudareva N, eds. Biology of plant volatiles. Boca Raton, FL, USA: CRC Press. [Google Scholar]
- Wu H, Kirita Y, Donnelly EL, Humphreys BD. 2019. Advantages of single‐nucleus over single‐cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. Journal of the American Society of Nephrology 30: 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wypych G. 2015. 3 – Mechanisms of UV stabilization. In: Wypych G, ed. Handbook of UV degradation and stabilization, 2 nd edn. Toronto, ON, Canada: ChemTec Publishing, 37–65. [Google Scholar]
- Xu H, Moghe GD, Wiegert‐Rininger K, Schilmiller AL, Barry CS, Last RL, Pichersky E. 2018. Coexpression analysis identifies two oxidoreductases involved in the biosynthesis of the monoterpene acid moiety of natural pyrethrin insecticides in Tanacetum cinerariifolium . Plant Physiology 176: 524–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yon F, Joo Y, Cortés Llorca L, Rothe E, Baldwin IT, Kim SG. 2016. Silencing Nicotiana attenuata LHY and ZTL alters circadian rhythms in flowers. New Phytologist 209: 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SD, Cho YH, Sheen J. 2007. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nature Protocols 2: 1565–1572. [DOI] [PubMed] [Google Scholar]
- Zhang T, Chen Y, Wang J, Zhang T, Chen Y, Wang J. 2021. A single‐cell analysis of the Arabidopsis vegetative shoot apex. Developmental Cell 56: 1–19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Correlation analysis between bulk‐RNA‐Seq and scRNA‐Seq datasets.
Fig. S2 QC metrics of scRNA‐Seq datasets.
Fig. S3. Cells are displayed by an integrated uniform manifold approximation and projection (UMAP) plot in two dimensions.
Fig. S4 Pollen and vasculature‐specific marker genes are exclusively expressed in clusters 7 and 8, respectively.
Fig. S5 Validation of pollen‐specific marker genes.
Fig. S6 Putative wax and cutin biosynthesis genes are more expressed in clusters 0, 3, 4, 5, and 9.
Fig. S7 Protein sequences of marker genes are similar to their Arabidopsis thaliana orthologs.
Fig. S8 Subunits of photosystems I and II complexes are enriched in clusters 1, 2, and 6.
Fig. S9 NaPKS1/2/3/4 share high sequence similarity with polyketide synthase proteins of other species.
Fig. S10 Target sequences for NaPKS RNAi experiments (used in previous studies, NaCHAL) and VIGS experiments (used in this study).
Fig. S11 Cladogram of Nicotiana attenuata 4CL, CNL homologs within acyl‐activating enzyme (AAE) superfamily.
Fig. S12 4CL and CNL homologs are not co‐silenced in Na4CL1‐silenced flowers.
Fig. S13 Co‐silencing of NaPKS1/2/3/4 in NaPKS3‐VIGS flowers.
Fig. S14 Tissue‐specific expression of NaPKS2 and NaPKS3 in Nicotiana attenuata.
Fig. S15 AER homologs are not co‐silenced in NaAER1‐silenced flowers.
Fig. S16 Extinction coefficients used for NaAER1 kinetics.
Fig. S17 NaAER1 can reduce benzalacetone to benzylacetone.
Fig. S18 Silencing NaIFR transcription does not alter BA biosynthesis in corolla.
Fig. S19 NaPAL4 transcripts are highly expressed in clusters 0, 3, and 4.
Fig. S20 Expression of NaPAL4 and NaPKS2 in Nicotiana attenuata flowers.
Fig. S21 Nicotiana attenuata homologs of Petunia×hybrida MYB transcription factors.
Fig. S22 NaIFR3 expression patterns in scRNA‐Seq dataset.
Table S1 One hundred and twenty‐one genes excluded for analysis.
Table S2 Primers used in this study.
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
Single‐cell RNA‐Seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO series accession no. GSE193464. Other data generated during this study are included in this published article and Supporting Information files.