

Abstract
Autophagy is a critical mechanism deployed by eukaryotic cells in response to stress, including viral infection, to boost the innate antimicrobial responses. However, an increasing number of pathogens hijack the autophagic machinery to facilitate their own replication. Influenza A virus (IAV), responsible for several global pandemics, has an intricate dependence on autophagy for successful replication in mammalian cells. To elucidate key chokepoints in the host stress responses facilitating IAV replication, we constructed a meta‐transcriptome of IAV and host gene expression dynamics during early (1–3 hpi), mid (4–6 hpi), and late (8–12 hpi) stages of the viral replication cycle at two multiplicities of infection (MOI): 1 and 5. We supplemented the global transcriptome study with phosphoproteomic analysis of stress‐activated protein kinase (SAPK/JNK) signaling in lung carcinoma (predominantly used as an in vitro model of IAV replication) and normal human bronchial epithelial cells. We report significant differences in the activation profiles of autophagy regulating genes upon IAV infection at the two MOI as well as divergent dependence on ULK1 signaling within the normal and cancer cells. Regardless of the cell model, JNK‐Thr187 signaling was crucial for the production of infectious viral particles.
Keywords: autophagy, infectious particles, Influenza A virus, mechanistic target of rapamycin complex 1 (mTORC1), meta‐transcriptome, RNA‐seq, stress‐activated protein kinase (SAPK/JNK), ULK1 signaling
Genes regulating mTORC1 and ULK1 signaling pathways are simultaneously activated in Influenza A‐infected lung bronchial epithelium cells. The virus load significantly affects the gene expression dynamics of mTORC1. Stress‐activated protein kinase (SAPK/JNK) signaling is triggered upon accumulation of high levels of viral proteins and coincides with the activation of autophagy.
1. INTRODUCTION
Autophagy is a pro‐survival mechanism supporting eukaryotic cell adaptation to various environmental stressors, such as nutrient starvation, UV irradiation, and microbial infections (Hussey et al., 2009; Kim et al., 2011; Kuballa et al., 2012; Qiang et al., 2017; Shang et al., 2011). The current concepts in cellular biology attribute plurality of functions to autophagy, including sustenance of healthy cellular physiology and energy homeostasis; regulation of inflammation; and support of innate and adaptive immunity through pathogen clearance and antigen presentation (Abdulrahman et al., 2011; Deretic et al., 2013; Harris et al., 2011; Kim et al., 2019b; Mintern et al., 2015; Paul & Münz, 2016; Saitoh et al., 2008; Siqueira et al., 2018; Valečka et al., 2018). While the cell signaling pathways regulating autophagy initiation are stress factor specific, the core mechanism of autophagosome biogenesis invariably leads to the formation of double‐membraned vesicles that randomly or selectively engulf cytoplasmic cargo. For the degradation of the captured cargo, autophagosomes fuse with lysosomes forming autolysosomes where low pH and abundance of proteolytic enzymes aid the recycling of damaged organelles into anabolic building blocks and energy source molecules. Additionally, microbes engulfed by the eukaryotic cells via endocytosis are targeted to the autolysosomes for degradation (Levine & Kroemer, 2019; Mizushima et al., 2010).
The role of autophagy as an intrinsic mechanism of eukaryotic cell resistance to pathogens has sparked interest in host‐directed therapeutics development (Kim et al., 2019b; Liang et al., 1998). Due to the great diversity of DNA and RNA virus replication mechanisms, development of broad‐spectrum antiviral therapeutics remains an impossible task. To address this problem, anti‐viral treatment protocols based on augmentation of host cell antimicrobial mechanisms, such as autophagy, can be implemented (Campbell & Spector, 2011; Kobayashi et al., 2020; Shoji‐Kawata et al., 2013). Paradoxically, a growing list of viruses and intracellular bacteria take advantage of host autophagic machinery to promote their own replication (Deretic et al., 2013; Levine et al., 2011; Li et al., 2011; Starr et al., 2012). To further complicate the issue, in discrete cases, autophagy activation has both pro‐ and anti‐viral function (Campbell & Spector, 2011; Kyei et al., 2009). Therefore, identification of pharmacological targets for broad‐spectrum antiviral therapeutics demands in depth understanding of the autophagic machinery components that enhance host defenses and those co‐opted for virus replication.
Influenza A virus (IAV) is an enveloped respiratory pathogen causing seasonal infections with morbidity and mortality among human and animal populations. Historically, influenza viruses have been responsible for several devastating global pandemics (Smith et al., 2009; Sriwilaijaroen & Suzuki, 2020). Periodic cycling of influenza subtypes and strains requires the design of seasonal vaccines that often miss unexpected rearrangements of viral antigens (Viboud et al., 2020). IAVs evolve resistance to antiviral drugs and genomic re‐assortment events further accelerate the global dissemination of resistant viruses (Nelson et al., 2009; Simonsen et al., 2007).
When traditional anti‐viral treatments fail, therapeutics against host regulatory proteins may come to the rescue. It has been previously shown that chemical inhibition of autophagy improved the survival rate of the H5N1‐infected host by reducing inflammation and severe lung damage (Sun et al., 2012). Notwithstanding the evidence, the relationship between IAV and autophagy is quite complicated. The autophagy machinery was found to facilitate influenza virus replication, while at the same time most IAV strains hijack host translational machinery by activating the mechanistic target of rapamycin (mTOR) complex 1 to ensure viral protein synthesis (Kuss‐Duerkop et al., 2017; Zhirnov & Klenk, 2013; Zhou et al., 2009). The IAV dependence on both mTORC1 and autophagy is seemingly paradoxical because activation of mTORC1 through phosphoinositide 3‐kinase (PI3K) and AKT signaling results in mTORC1‐directed phosphorylation and inhibition of autophagy activation kinase 1 (ULK1) complex (Ganley et al., 2009; Jung et al., 2009; Laplante & Sabatini, 2012; Ma & Blenis, 2009).
The IAV causing seasonal epidemics in humans (strains H1N1 and H3N2) utilize their nonstructural protein 1 (NS1) as an anti‐apoptotic agent via direct interaction with the p85β subunit of PI3K and activation of AKT1 signaling pathway (Ehrhardt et al., 2007; Shin et al., 2007; Zhirnov & Klenk, 2007). Thus, the viral NS1 stimulates mTORC1 kinase function to facilitate protein synthesis and cap‐dependent translation initiation through cascade of phosphorylation events, including p70 S6 kinase and 4E‐BP1 (Kuss‐Duerkop et al., 2017; Ma et al., 2011). Activated mTORC1 is supposed to inhibit autophagy through ULK1 signaling; however, it supports the synthesis of viral proteins acting as autophagy stimulation factors, including hemagglutinin (HA) and ion channel matrix 2 (M2) (Zhirnov & Klenk, 2013). Specifically, the influenza virus M2 protein interaction with the microtubule‐associated protein light chain 3 (LC3), a core structural protein of the autophagosomes, was found to inhibit autolysosome formation while redirecting the LC3–M2 complex to the plasma membranes (Beale et al., 2014; Zhirnov & Klenk, 2013). This process is associated with delays in apoptosis initiation and facilitation of virion assembly and budding (Beale et al., 2014; Gannagé et al., 2009).
Current models describe autophagy as a host response mechanism activated during the late stage of IAV replication (Gannagé et al., 2009), while mTORC1 and mTORC2 activation by IAV infection was reported to occur midway through the IAV replication cycle (Kuss‐Duerkop et al., 2017). These models, based on Western blot (WB) phosphoproteomic data analysis, are influenced by two main experimental shortcomings: sensitivity limitations of WB and use of cancer cells as host models for in vitro IAV infection studies. Here, we sought to elucidate the autophagy‐regulating component that is essential for IAV replication in lung epithelial cells. We applied a highly sensitive analytical approach and validated our findings in a noncancer cell model. We performed a RNA‐sequencing‐based meta‐transcriptomic study of IAV and host gene activation dynamics throughout the virus replication cycle at two multiplicities of infection (MOI), 1 and 5. We found that viral load has a strong influence on the mode and dynamics of host cell protective responses and autophagy activation. With a protocol that does not induce autophagy due to serum starvation of the host cells during the virus absorption stage, we detected simultaneous activation of mTOR complex 1 genes and key autophagy regulating genes early upon virus infection (1–3 hr post infection [hpi] at MOI 1). Subsequent phosphoproteomic analysis verified that the classical autophagy activation pathway through ULK1 signaling did not play a significant role in the autophagy activation upon IAV infection, while the activation of c‐jun N‐terminal kinase (JNK) was essential for viral protein accumulation. JNK, a stress‐activated protein kinase (SAPK) activated by various viruses (Clarke et al., 2001; Eliopoulos et al., 1999; McLean & Bachenheimer, 1999), is stimulated by direct interaction with the IAV NS1 protein (Nacken et al., 2014). We show that chemical inhibition of JNK abrogated viral protein accumulation in lung carcinoma A549 cells and significantly inhibited infectious viral titer in noncancer cells. Host‐directed antiviral therapy targeting key regulators of nonclassical autophagy pathways, such as JNK/SAP, could show less toxicity in vivo compared with a pharmacological inhibition of the classical autophagy pathways that are essential for proper host immune defenses.
2. RESULTS
2.1. Serum depletion activates autophagy prior to Influenza A virus replication in vitro
Cleavage of IAV hemagglutinin (HA) is required for fusion of the viral capsid with host‐cell endosomal membranes. Proteases secreted by the respiratory epithelium are responsible for HA cleavage in vivo IAV infection (Klenk & Garten, 1994; Steinhauer, 1999; Zhirnov et al., 2002). The majority of in vitro cell models (MDCK, HEK293, or A549) utilized to study the role of autophagy in IAV replication do not produce proteases that cleave HA necessitating the addition of trypsin to promote virus infectivity. Typically, during the 1‐hr virus absorption stage, the host cells are incubated in serum‐free media containing the infectious virus and TPCK‐trypsin (Beale et al., 2014; Gannagé et al., 2009; Kuss‐Duerkop et al., 2017; Wang et al., 2019; Zhirnov & Klenk, 2013). Applying time‐lapse microscopy, we monitored the cellular localization of transcription factor EB (TFEB) in live cells, as a biomarker of autophagy activation (Napolitano & Ballabio, 2016), and registered TFEB translocation into the nucleus of mock‐treated A549 cells within 40 min of incubation in serum and virus‐free media (Figure S1a). Consistent with TFEB nuclear localization, we observed a significant drop in the levels of phosphorylated ribosomal protein S6 kinase β‐1 (p70S6K1) indicating inhibition of mTORC1 activity (Figure S1b). Thus, serum depravation protocols for in vitro infection of cancer cell lines with IAV generate data that are biased to a condition where mTORC1 function is downregulated and autophagy is induced prior to the initiation of viral replication. To investigate the role of IAV infection on autophagy activation, we adapted a protocol that did not include a serum starvation step. With this protocol, we were able to study the normal course of autophagy activation upon IAV infection (Figure S1c) and found that H3N2 strain, A/Port Chalmers/1/73, showed higher tropism to A549 cells compared with the H1N1 strain, A/PR/8/1934 (Figure S1d). Therefore, in this study we used the H3N2 strain to investigate the role of autophagy in IAV replication in lung epithelial cells.
2.2. Autophagy activation correlates with Influenza virus A load
Employing strand‐specific RNA‐sequencing (RNA‐seq) of infected A549 cells, we simultaneously measured the kinetics of A/PCh/1/73 replication and the expression of host genes affected by IAV replication at 1, 3, 4, 6, 8, and 12 hpi. In order to measure virus replication, we generated RNA‐based growth curves by normalizing the total virus reads to the total host read coverage (Figure 1a). The viral RNA accumulation profile was consistent with the virus titration growth curves (Figure S1d). To reproducibly detect reads from genes with low transcription rates, we performed infections with moderate (1) and high (5) multiplicities of infection (MOI) representing the ratios of host cells to infectious virus particles. At MOI5, we observed a rapid increase in the viral transcript levels that accounted for 10% of the total RNA reads within 2 hr of infection and plateaued between 4 and 8 hpi, accounting for 75% of the total RNA reads (Figure 1a). In contrast, we observed exponential viral growth within 4 hr of infection at MOI1. By 12 hpi, viral RNA levels started to decline in both infection conditions.
FIGURE 1.
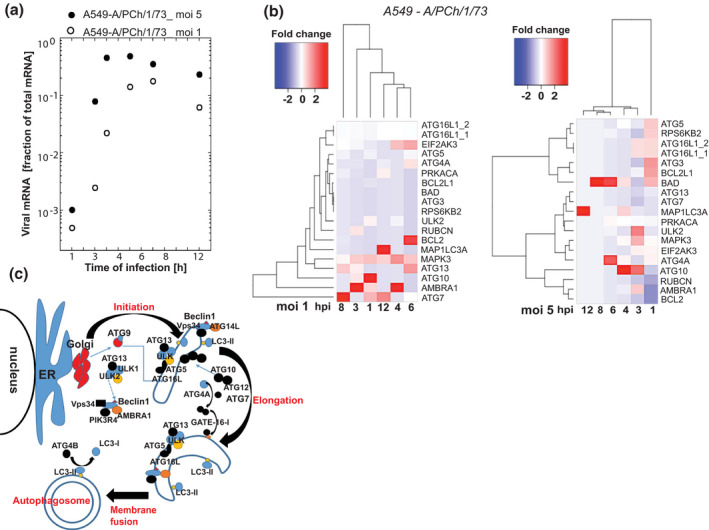
The activation dynamics of genes involved in autophagy regulation are determined by IAV titer. Accumulation of viral transcripts (a) and autophagy regulating gene transcripts (b) in A549 cells at early (1–3 hpi), mid (6–8 hpi), and late stage (12 hpi) of the viral replication cycle at two multiplicities of infection (MOI) as determined by RNA‐seq analysis (FDR‐corrected p < .01). (b) Dendrograms of the hierarchical clustering of DGE show significant difference in gene expression dynamics at the two MOIs. (c) Schematic of the autophagy pathway showing key regulatory proteins involved in each step of autophagosome formation
The objective of our study was to evaluate the effect of IAV replication on the expression profiles of genes involved in the regulation of autophagy, mTOR signaling pathways, and innate immunity. We observed greater dynamics in global host transcriptome (with DGE ≥ twofold change) during early stages of IAV infection compared with later timepoints of virus replication when data were normalized to time‐matched mock‐treated cells (Figures 1 and 2). Virus load played a significant role in the gene expression dynamics.
FIGURE 2.
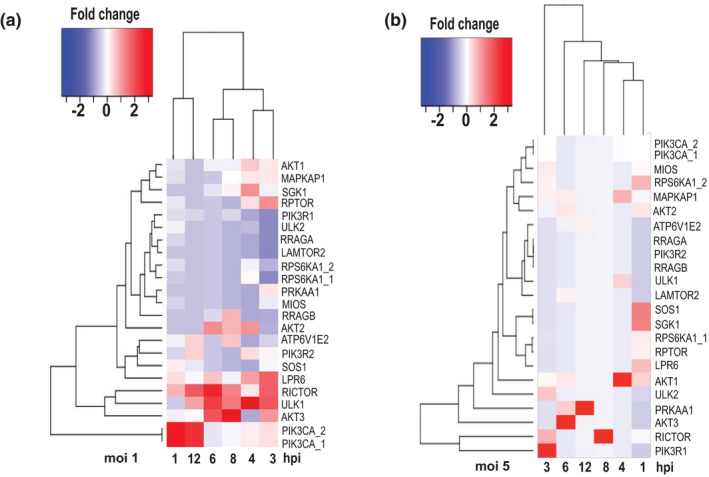
Transcriptional activation of mTORC and ULK1 pathways in IAV‐infected A549 cells. Gene activation dynamics were determined in A549 cells infected with IAV P/Ch/1/73 at MOI1 and MOI5 versus mock treated cells at 1, 3, 4, 6, 8, and 12 hpi. Fold change differences of gene expression levels in IAV‐infected relative to mock‐treated cells are statistically significant between the two groups (FDR‐corrected p < .01)
At a moderate virus load (MOI1), within 4 hr of infection, we observed a sequential activation of genes that regulate the early stages of the autophagy pathway (Figure 1b). We witnessed initiation of autophagy elongation phase by the activation of ULK2 and Autophagy and Beclin‐1 Regulator protein (AMBRA1) (1 hpi) followed by ATG13 and RUBCN (3 hpi) transcript accumulation. Activation of ATG7 and ATG10 gene expression corresponds to the elongation of nascent autophagosomal membrane at 1 hpi. ATG4A, a regulator of the final stage of functional autophagosome formation (phagophore membrane fusion), was differentially upregulated at 6 hpi, while the MAPK1LC3 gene activity, facilitating the phagophore membrane fusion, remained unchanged up to 12 hpi.
During the period of exponential virus replication (1–4 hpi), we observed a gradual accumulation of the AMBRA1 transcripts encoding for a core subunit of the PI3K complex that is essential for the initiation and maturation of autophagosomes. AMBRA1 gene expression declined at later stages of infection (6–12 hpi) when the viral load surpassed 50% of total RNA reads, overwhelming the host transcription machinery.
The gene activation dynamics of autophagy regulators showed oscillatory pattern at high virus load (MOI5). Compared with DGE in cells infected with lower MOI, at MOI 5 gene expression of AMBRA1, ULK2, ATG10, and ATG16L spiked 3 hpi instead of 1 hpi. We could not detect ATG13 activation in the samples infected with IAV at MOI5, while this key regulator of the autophagy initiation was differentially activated for 5 hr (3–8 hpi) in A549 cells infected at MOI1. It is possible that DGE dynamics could have been accelerated at high virus load (MOI5), and therefore, we were only able to detect the “tail” of the gene activation burst at 1 hpi for genes regulating the early stages of autophagosome formation: ATG5, ATG16L1, and ATG3 (Figure 1b). The observed delay of ATG10 activation at MOI5 (3–4 hpi) compared with infection at MOI1 (1–3 hpi) may be indicative of a secondary oscillatory burst in the activation of autophagy gene regulators due to increased virus burden.
A steady accumulation of BAD gene transcripts was the characteristic of the A549 cells infected at MOI5 (Figure 1b). BAD protein positively regulates apoptosis by forming dimers with cell death‐reversing BCL‐2 protein. Consequently, the transcript levels of BCL2L1 were upregulated within 1 hr of infection at MOI5. Another DGE profile specific to lung carcinoma cells infected with high IAV load was the upregulation of RPS6KB2, a mTOR substrate responsible for stimulation of protein synthesis through phosphorylation of S6 ribosomal protein and eukaryotic translation initiation factor 4B (eIF4B). In contrast, RPS6KB2 was not differentially expressed in A549 cells infected at MOI1, while the transcript levels of global protein synthesis inhibitor, EIF2AK3, increased between 4 and 6 hpi. The cause for EIF2AK3 gene activation could be energy depletion in the lung cells hosting rapidly replicating virus. Interestingly, EIF2AK3 gene transcription exceeded background levels within the first 3 hr of infection at a high viral load. These data suggest that host cells attempted to counteract viral replication by shutting down protein synthesis, while virus co‐opted host cell protein synthesis machinery for replication (Figure 1b).
MAPK3/ERK controls various biological functions, including cell growth, survival, and differentiation. Depending on interaction with a pathway‐specific substrate, MAPK3 can activate translation through EIF4EBP1 and apoptosis through phosphorylation of BAD. Additionally, MAPK3 activates mTORC1 by phosphorylation of RAPTOR and TSC complex (Carriere et al., 2011; Zoncu et al., 2011). Activated mTORC1 positively regulates protein synthesis and inhibits activation of autophagy pathway through ULK1/2 complex. Surprisingly, MAPK3 and autophagy regulating genes were simultaneously activated during the exponential phase of IAV replication (Figure 1b, MOI1). At higher MOI (5), MAPK3 gene expression spiked at 3 hpi and returned to base levels thereafter. These results suggest that cell stress induced by the high burden of viral protein synthesis could initiate negative feedback mechanisms to activate MAP3K transcription.
Collectively, our unbiased transcriptome study of lung cancer cells infected with IAV demonstrated that the autophagy pathway was activated during the exponential stage of virus replication. High virus load (MOI5) stimulated both pro‐apoptotic gene groups (BAD and BCL2L1) and genes essential for enhanced protein synthesis (RPS6KB2) within an hour of infection. At MOI1, genes regulating initiation of autophagy were activated during the exponential phase of IAV replication. Autophagy gene expression showed oscillatory behavior both at high (MOI5) and low (MOI1) virus load.
2.3. IAV replication simultaneously activated genes regulating mTORC1 and ULK1 pathways
mTOR regulatory complex is activated at nutrient‐rich environment to positively regulate protein synthesis, cell growth, and differentiation. A common set of protein kinase regulatory proteins associate with the proteins Raptor and Rictor to form two complexes: mTORC1 and mTORC2 respectively, each complex carrying a distinct function. Activation of mTOR1 facilitates protein synthesis through phosphorylation of ribosomal protein kinase S6K (RPS6K), while mTORC2 regulates cytoskeleton dynamics and cell survival (Jung et al., 2009; Laplante & Sabatini, 2012). To promote cell survival, mTORC2 phosphorylates and activates SGK1 and AKT kinases. Growth factors activate mTORC1 through the phosphoinositide‐3 kinase (PI3K)‐AKT or SOS‐MAPK3/ERK signaling axis (Mendoza et al., 2011; Xu et al., 2020). Additionally, the LRP‐GSK axis neutralizes the inhibitory action of the TSC1/2 complex (Inoki et al., 2006).
Previous studies have shown that H1N1 IAV ramps up the production of viral proteins through direct activation of PI3K (Kuss‐Duerkop et al., 2017; Zhirnov & Klenk, 2013). Here, we observed transcription activation of PI3K catalytic subunit alpha (PI3KCA) within an hour of H3N2 IAV infection at MOI1. Thereafter, PI3KCA transcript levels gradually decreased during the exponential accumulation of viral RNA to spike again at 12 hpi (Figure 2a, MOI1). In contrast, we detected no activation of PI3KCA gene expression upon infection with high MOI (Figure 2a, MOI5). Similarly, gene transcription of Raptor (RPTOR) and its activator AKT1 was stimulated early upon IAV infection at MOI1 and was inhibited at later stage of the virus replication cycle (Figure 2, MOI1). Both genes were slightly activated within an hour of IAV infection at high virus load (Figures 1a and 2, MOI5).
Interestingly, the AKT isoforms were activated upon IAV infection in a sequential order: AKT1 at 3–4 hpi, AKT2 at 4–8 hpi with maximum expression levels at 6 hpi, and AKT3 at 3–8 hpi (Figure 2, MOI1). At high virus load, the AKT3 levels peaked at 6 hpi (Figure 2, MOI5). AKT kinases regulate many processes including cell metabolism and survival via mTOR dependent and independent signaling pathways. The timing of AKT1/2/3 isoform, RPTOR, and RICTOR gene activation suggested that AKT1 and AKT2 regulate protein synthesis through mTORC1 signaling, while AKT3 gene function can be linked to cell survival through mTORC2 activation. Exponential IAV growth coincided with activation of RICTOR gene transcription (Figures 1a and 2, MOI1). RICTOR gene activation followed oscillatory pattern similar to DGE of the major autophagy regulators both at lower and high virus load. In comparison, SOS1 and SGK1 genes regulating cell survival and stress responses were upregulated only at the early stages of virus replication (Figure 2).
The ULK1 gene was upregulated upon IAV infection at MOI 1 and sustained transcription activity until the later stage of viral replication (12 hpi). In contrast, at high virus infection load (MOI5) we observed a less pronounced ULK1 upregulation at 3 hpi followed by gene inhibition at later stages of virus replication (Figure 2, MOI5). Notably, ULK2 gene transcription was inhibited in host cells infected with the lower viral load (MOI1). In contrast, ULK2 gene was activated at the early stage (3 hpi) of IAV infection at MOI5. The ULK2 expression profile was consistent with that of AMPK subunit alpha (PRKAA1) and showed inverse DGE pattern to the ULK1 gene expression (Figure 2). In summary, our transcriptome data indicate that upon IAV infection at MOI1 nutrient depletion may not contribute to the stimulation of the autophagy pathway.
Collectively, we observed steady accumulation of ULK1, MAPK3/ERK (Figure 1b, MOI1), and LRP6 (Figure 2, MOI1) gene transcripts throughout the virus replication cycle indicating that genes regulating autophagy and mTORC1 activation were simultaneously activated. The oscillatory pattern of key autophagy (ATG7, Figure 1b, MOI1) and mTORC2 (RICTOR, Figure 2, MOI1) gene expression profiles suggests that viral replication activates two major cell survival pathways: autophagy and mTORC2.
2.4. IAV replication activates autophagy in lung epithelium cells independently of the ULK1 signaling pathway
Our global transcriptome analysis revealed previously uncharacterized correlation between IAV load and activation of ULK gene transcription. While ULK1 gene was consistently transcribed in A549 cells throughout the IAV virus replication cycle at MOI1, its expression dynamics remained mostly unchanged at MOI5 (Figure 2, Figure S2). Therefore, we further investigated the role ULK1 plays in autophagy activation and IAV replication at a post‐transcriptional level.
Applying Western blot (WB) analysis, we could barely detect viral protein accumulation 12 hpi at MOI1 (Figure S3), while the transcriptome analysis showed that viral RNA had reached maximum levels at 4 hpi (Figure 1a). We could detect accumulation of viral proteins (M2 and NP) with WB as early as 6 hpi at MOI5 (Figure 3a). At this high virus load, total IAV transcript levels reached their maximum levels between 3 and 4 hpi and declined at 6 hpi (Figure 1). The observed time gap between transcript and protein accumulation is largely due to the substantial difference in target detection sensitivity between the two analytical methods. Quantitative analysis of percentage infected cells via flow cytometry indicated that at MOI1 close to 10% of the total cell population harbored the virus, while at MOI5 half of the population become infected (Figure S3). Since both methods analyze bulk transcript or protein levels, the WB data could only reveal the biomarkers with the most prominent changes elicited by IAV infection.
FIGURE 3.
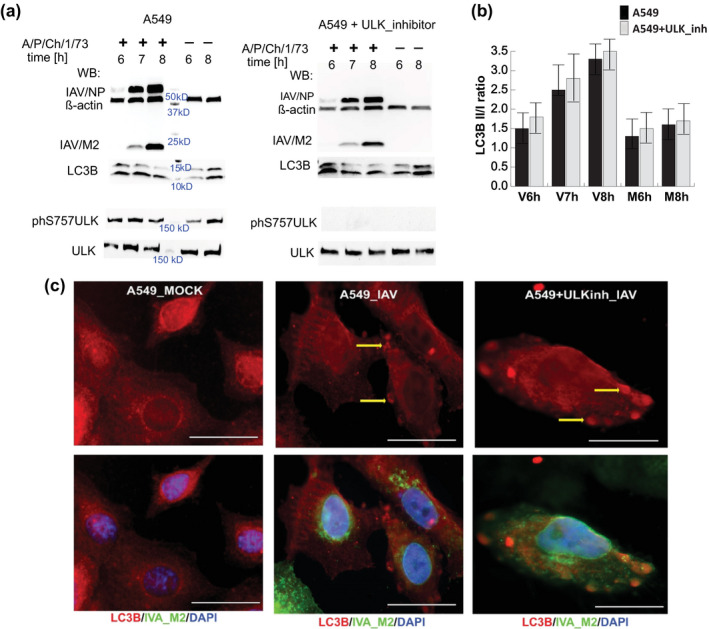
Activation of autophagy in IAV P/Ch/1/73 infected A549 cells is not dependent on ULK‐S757 phosphorylation by mTORC1. (a) Western blot analysis was performed on total protein isolated from IAV‐infected (+) and mock‐treated (−) A549 cells at mid (6 hpi) and later stage (7–8 hpi) of virus replication cycle. The ratio between lipidated (LC3B‐II) and unprocessed (LC3B‐I) forms of MAP1LC3B protein was used to determine the autophagy activation event. Untreated and SBI0206965 [10 μM] treated A549 cells were infected with IAV P/Ch/1/73 at MOI5 and total protein was collected at the indicated hours post infection (hpi) together with the time‐matched mock treated samples. Accumulation of the viral nucleoprotein (NP) and matrix 2 (M2) protein was analyzed relative to beta‐Actin protein levels. (b) Quantitative analysis of LC3B protein band densities was performed on five independent experiments with Image Lab 6.0 (BioRad). Shown are the mean and standard deviations. (c) Fluorescent microscopy of A549 cells mock‐treated or infected with IAV P/Ch/1/73 at MOI5. A549 cells treated with the ULK inhibitor, SBI0206965 [10 μM], were equally infected and processed. Cells were fixed 8 hpi and were treated with primary antibodies against LC3B protein (red) and IAV matrix 2 (green). The cell nuclei were stained with DAPI (blue). The yellow arrows indicate at autophagosome formations. The white scale bar corresponds to 10 μm. Shown is a representative of three independent biological replicas of IAV infected A549 cells
Regardless of WB sensitivity limitations, we could observe activation of autophagy through accumulation of lipidated LC3B‐II form of the microtubule‐associated light chain protein that coincided with IAV protein accumulation (Figure 1a) (Mizushima et al., 2010). When autophagy is activated via the classical pathway, the levels of ULK1 phosphorylation at Ser757 subside due to inhibition of mTORC1 activity (Kim et al., 2011). Since the global transcriptome analysis showed continuous activation of ULK1 gene expression at MOI1 and no significant changes in gene activity at MOI5, we further investigated the role of ULK1 in infected cells enduring metabolic stress due to high virus protein levels at MOI5. Inhibition of ULK1 phosphorylation at S757 with small molecule SBI0206965 did not change the course of autophagy activation upon IAV infection as we observed continuous accumulation of lipidated LC3‐II form in virus‐infected cells (Figure 3a,b). At the same time, we observed no reduction of viral protein levels in A549 cells treated with the ULK1 inhibitor (Figure 3a).
We further validated our WB results by applying immunocytofluorescence with anti‐LC3B antibodies. We observed autophagosome vesicle formations in IAV‐infected A549 cells when ULK1 signaling was inhibited with a small molecule (Figure 3c) thus confirming the WB‐based proteome data. Collectively our data indicated that autophagy was activated through an alternative to ULK1 signaling pathway during IAV infection.
2.5. IAV infection upregulates AKT and Stress Response Kinase JNK activity in A549 cells midway through the virus replication cycle
Previous in vitro studies of IAV infection have demonstrated that viral proteins stimulate mTORC1 activity to ensure virus protein expression and promote successful replication (Zhirnov & Klenk, 2007, 2013). Additionally, mTORC2 function supports host survival in the later stages of IAV replication when cells undergo significant stress (Kuss‐Duerkop et al., 2017).
To validate our findings from the global transcriptome analysis of mTOR‐regulatory complex differential gene expression, we measured mTORC1 and mTORC2 activity in IAV infected A549 cells. We observed that at MOI1 the phosphorylation levels of p70 S6 kinase 1 (S6K), a mTORC1 substrate, remained unchanged, while AKT1‐Ser473 phosphorylation, a mTORC2 substrate, increased throughout the virus replication cycle (Figure 4a). Notably, the exponential increase in phosphorylated AKT‐Ser473, an indicator of cell stress, coincided with the accumulation of viral transcripts (Figure 1a). Our results also demonstrated that the Thre389 phosphorylation on the S6K increased proportionally to the virus load at the later stage of virus replication (Figure 4b).
FIGURE 4.
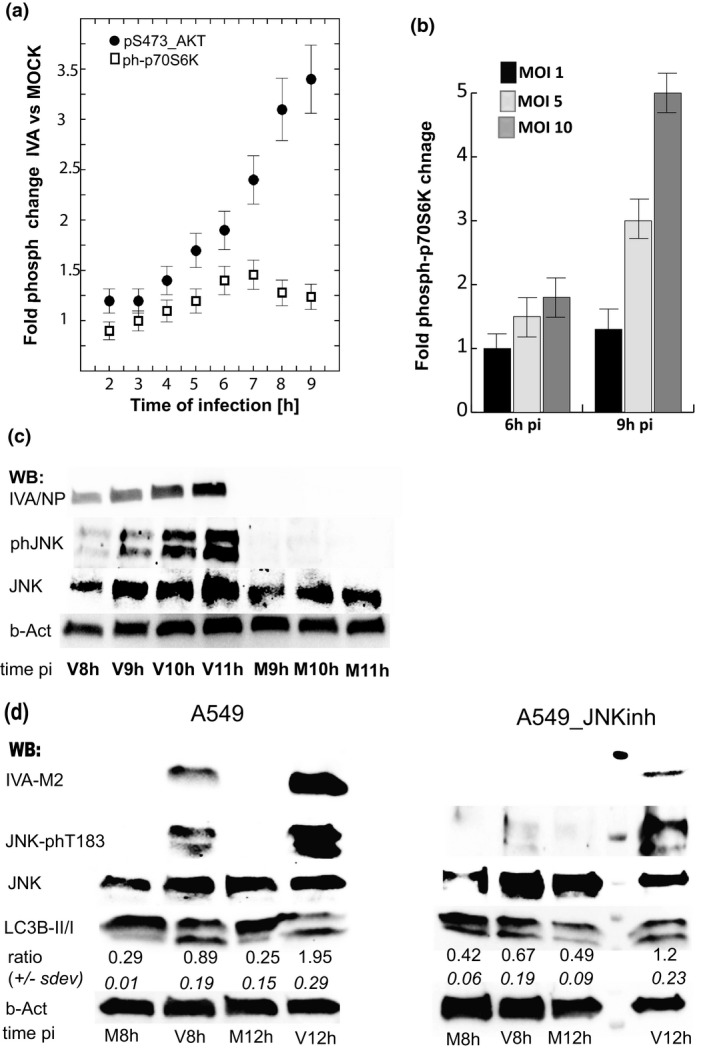
Phosphorylation of SAPK/JNK‐Thr183/Tyr185 coincides with AKT‐pSer473 anti‐apoptotic signaling and is essential for viral protein accumulation in IAV P/Ch/1/73‐infected A549 cells. (a) The mTORC1 and mTORC2 activity was deduced from the phosphorylation status of their corresponding substrates, p70S6K‐Thr389 and AKT‐S473, in IAV‐infected (MOI1) cells. (b) mTORC1 activity was measured at later stage of IAV infection (6 and 8 hpi) at MOI 1, 5, and 10. (c) Western blot analysis of phosphorylated JNK‐Thr183/Tyr185, total JNK, and IAV nucleocapsid protein (IAV‐NP) in samples isolated from virus‐infected (V) A549 cells at MOI5 and mock‐treated (M) cells. JNK signaling was consequently activated with accumulation of viral protein at mid‐to‐late stage of the virus replication cycle. (d) Western blot analysis shows that chemical inhibition of SAPK/JNK signaling with SP600125 [10 μM] resulted in inhibition of virus protein (IAV‐M2) accumulation in A549 cells. In the same samples, increase in the LC3B‐II/LC3B‐I ratio indicates that high viral protein levels are responsible for the SAPK/JNK and autophagy activation. Shown is a representative of four independent virus infection (MOI5) experiments
The phosphoproteomic data presented here demonstrated that pro‐survival mechanisms dependent on AKT‐Ser473 phosphorylation were activated at mid throughout the late stage of the virus replication cycle, while mTORC1 activity was not affected in cells infected with IAV at MOI1. We detected mTORC1 activation in the later stages of IAV replication specifically at very high MOI, when mTORC1 function was required to satiate the high demand for protein synthesis (Figure 4b: 9 hpi vs. 6 hpi).
Together with the activation of AKT anti‐apoptotic signaling, we investigated the JNK stress‐response regulator upon virus infection. JNK activation is a common response to many stress stimuli, including endoplasmic reticulum (ER) excessive stress, causing apoptotic cell death through regulation of the BCL2 family proteins (Raciti et al., 2012; Wei et al., 2008; Yamamoto et al., 1999). Our Western blot results showed consistent accumulation of Thr183‐phosphorylated JNK in IAV‐infected cells. This was proportional to the synthesis of viral proteins (Figure 4c). Activation of JNK signaling occurred midway through the IAV (H3N2) replication cycle and coincided with the AKT‐pSer473 anti‐apoptotic signaling (Figure 4a,c). For comparison, we detected accumulation of phosphorylated JNK‐Thr185 at later time upon infection with similar MOI of IAV (H1N1) subtype (Figure S3c). These findings underline strong correlation between the accumulation of high virus protein amount and stress‐activated JNK signaling. Importantly, pharmacological inhibition of JNK signaling through Thr183 phosphorylation resulted in a significant decrease in IAV protein synthesis and a weaker autophagy pathway stimulation, measured as reduced levels of the lipidated LC3B‐II (Figure 4d). We observed that the LC3‐II/LC3‐I ratio increased eightfold within 12 hr of virus infection compared to the time matched mock‐treated A549 cells with functional JNK signaling and only twofold in the IAV infected cells exposed to JNK inhibitor (Figure 4d).
Taken together, the phosphoproteomic analysis of AKT‐Ser473 and JNK‐Thr187 signaling upon infection of A549 cells with IAV indicate that viral proteins stimulated host anti‐apoptotic adaptations through activation of AKT and JNK stress response mechanisms. Pharmacological inhibition of JNK signaling dramatically reduced viral protein synthesis in IAV‐infected lung carcinoma A549 cells.
2.6. Pharmacological inhibition of JNK‐Thr187 signaling decreased IAV titer in normal human bronchial epithelial cells (NHBE) in vitro
Since the cancer cells, including the A549 line, used as models to study IAV replication may regulate stress response and survival mechanisms differently in comparison to normal human lung cells, we further investigated the role of JNK signaling in NHBE cells in vitro infected with the H3N2 A/PCh/1/73 strain.
Western blot analysis of total protein extracted from mock‐treated and IAV‐infected NHBE cells showed different responses to ULK1 and JNK signaling inhibition when compared with the A549 cancer cell line. The NHBEC were more sensitive to the small molecule compounds SBI0206965 and SP600125 targeting ULK1 and JNK signaling, respectively, therefore we applied lower drug concentrations. Unexpectedly, ULK1 inhibition caused a decline of JNK‐T183 phosphorylation in IAV‐infected NHBEC, although the viral protein (M2) accumulation and activation of LC3B synthesis remained unaffected (Figure 5a). In contrast to the A549 cancer cells, we detected LC3B accumulation only in IAV‐infected NHBEC, either untreated or treated with SBI0206965 to inhibit ULK1 signaling. LC3B in NHBEC treated with the JNK inhibitor SP600125 was barely detectable even in IAV‐infected populations (Figure 5a). Immunofluorescence analysis, a more sensitive protein detection method, showed that IAV‐infected NHBEC produced LC3B. Furthermore, the percentage of NHBEC harboring IAV_M2 protein dropped significantly upon pharmacological inhibition of JNK signaling (Figure 5b) indicating at its significance for the IAV (H3N2) replication.
FIGURE 5.
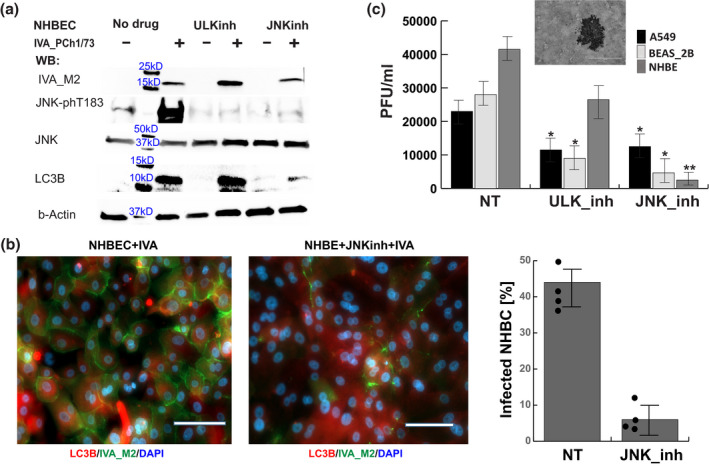
Noncancer lung bronchial epithelial cells exhibit higher dependence on JNK signaling for production of infectious IAV particles. (a) Chemical inhibition of ULK (SBI0206965 [1 μM]) and JNK (SP600125 [5 μM]) signaling showed divergent effects on the production of viral protein and LC3B in normal human bronchial epithelial cells (NHBE). Shown is a representative of three independent Western blot experiments of later stage (24 hpi) IAV infection at MOI5. (b) Immunofluorescence with antibodies targeting LC3B and IAV protein M2 demonstrated the effect of JNK‐phT183 inhibition on the reduction of virus‐infected NHBEC. Images were acquired 24 hpi at MOI5. The white scale bar corresponds to 60 µm. The graph presents the percent of cells harboring viral M2 protein. Image quantification of IAV‐M2‐labeled cells was performed on >300 cells in random fields for four independent infection experiments. (c) Inhibition of virus protein synthesis was consistent with reduction of infectious virus load, measured as plaque forming units per ml conditioned media (PFU/ml), generated by A549, BEAS‐2B, and NHBE treated with inhibitors of ULK or JNK signaling compared with the not treated (NT) controls. The PFU/ml were determined 40 hpi with IAV‐A/PCh/1/73 at MOI5. The statistical significance of the results was determined from four independent infection experiments with Student t‐test (*p < .01 and **p < .001)
We further validated the role of JNK and ULK1 signaling in IAV replication analyzing the infectious virus titers generated by NHBE, adenovirus transformed NHBE (BEAS‐2B) and lung epithelium carcinoma cell line (A549). We found that NHBE from three genetically unrelated healthy donors produced infectious virus titers comparable to those released by the immortalized cell lines. Pharmacological inhibition of JNK function reduced the levels of infectious IAV particles by twofold in the transformed cell lines and by 20‐fold in the NHBE cells (Figure 5c). These results complemented our Western blot and immunocytochemistry studies and demonstrated that JNK‐signaling inhibition in NHBEC has stronger effect on IAV replication in comparison to cell models based on immortalized cell lines.
Collectively, our data indicated that IAV (H3N2) activates autophagy independently of the classical ULK1 signaling pathway in normal and cancer (A549) lung epithelial cells. Although, IAV infection universally activated JNK signaling in all tested cell models, the immortalized cell lines were less dependent on this regulatory response for production of infectious virus compared with NHBEC. The JNK‐Thr187 signaling appeared as a stress regulatory mechanism activated by the rampant synthesis of viral proteins. Therefore, inhibition of JNK‐Thr187 signaling decreased the host cell capacity as a virus “factory” and resulted in a significant reduction of infectious IAV titer in vitro.
2.7. IAV matrix protein M2 interaction with the host LC3B protein activates autophagosome formation
Our Western blot results have consistently indicated that IAV infection activates autophagy in strict correlation to accumulation of viral proteins (Figures 3a, 4d, and 5a). Since detection of the lipidated LC3B‐II form was contingent to high viral protein load, we designed experiments to test whether direct interaction between the viral M2 protein and the autophagosome core protein occurs in situ and activates autophagy independently of the classical (ULK1) or alternative (JNK) signaling cascades. Previous studies have inferred interaction between the two proteins from in vitro binding studies and infections with M2‐deficient H1N1 virus (Beale et al., 2014). Here, we utilized split green fluorescent protein (GFP) methodology (Cabantous et al., 2005) to directly test M2‐LC3 interaction in live cells. We tagged each LC3B and M2 proteins with GFP fragments 10 (G10) and 11 (G11). Plasmids expressing proteins tagged with the GFP fragments were co‐transfected in HEK293T cells. No fluorescence was detected in transfected cells prior to addition of the GFP1‐9 scaffold domain (data not shown). Since the GFP1‐9 utilized in this study was not optimized to fold properly in mammalian cells, we supplied purified and folded GFP1‐9 peptide in trans similar to the antibody staining techniques. Thus, we indirectly registered protein–protein interactions between G10 and G11 fragments that occurred in live cells after we visualized this interaction in fixed cells supplemented with the GFP1‐9 scaffold.
Guided by previous knowledge that IAV M2 protein forms homo‐dimer and tetramer complexes (Holsinger & Lamb, 1991), we co‐transfected plasmids expressing M2 protein tagged with G10 and G11 as a positive control for our assay. As expected, complementation of the split‐GFP fragments in HEK293T cells expressing M2‐tagged proteins resulted in fluorescence that highlighted the extracellular membrane and intracellular membranous structures where M2 resides (Figure 6a, upper left panel). Self‐oligomerization of LC3B protein has not been previously reported; therefore, we applied G10 and G11‐tagged at the N‐terminus of LC3B‐expresing plasmids as a negative control. Unexpectedly, split‐GFP complementation of the LC3B‐tagged proteins recovered the GFP fluorescence activity (Figure 6a, upper right panel). The fluorescent signal was consistent with that of native LC3B‐I protein cellular localization (Figure 3c). Finally, co‐transfection of plasmids expressing tagged LC3B and M2 proteins produced fluorescently labeled cell membrane and phagosomes consistent with the localization of LC3B‐II and M2 (Figure 6a, lower panel).
FIGURE 6.
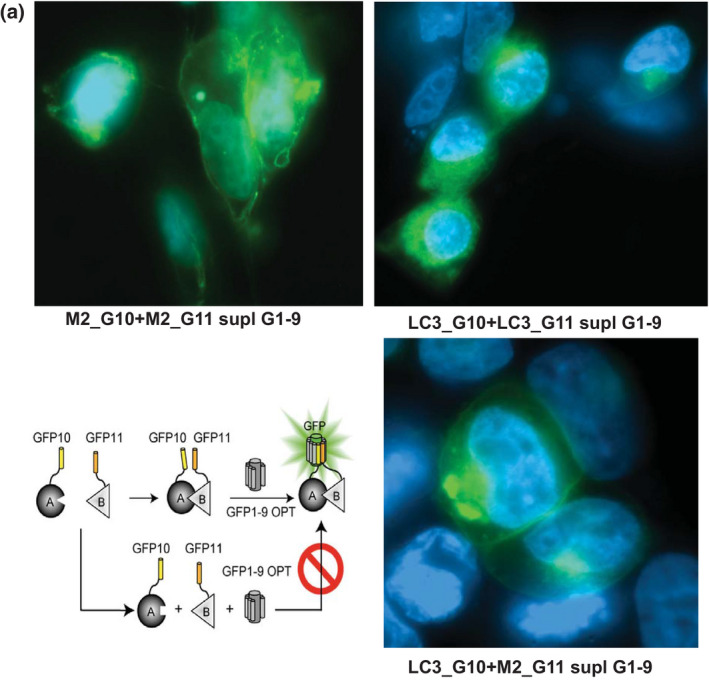
IAV‐M2 recruits LC3B in the cell membranes. Fluorescent microscopy of HEK293T cells transfected with plasmids expressing G10‐ and G11‐tagged IAV‐M2 and LC3B proteins demonstrate that direct interaction between the two proteins occurs on the cell plasma membrane or intracellular membranous vesicles. Shown are images of split‐GFP complementation assay 48 hr after transfection of HE293T cells with the corresponding plasmids. The GFP1‐9 scaffold was supplied as purified and folded protein in fixed and permeabilized cells
Altogether, data presented here demonstrate direct interaction between the viral M2 protein and LC3B that occurs on the cell membranes. The split‐GFP assay shows that the IAV matrix protein recruited LC3B to the extracellular membrane and vice versa, the M2 protein localized at the autophagosome membranes where LC3B‐II naturally resides. The viral M2 protein stimulated the LC3B lipidation required for the autophagosome maturation.
3. DISCUSSION
Autophagy has been shown to significantly impact the virus life cycle, particularly the fledgling phase of viral RNA synthesis (Wang et al., 2019) and presents an appealing target for host‐directed antiviral therapeutics. However, the role of autophagy during viral infection varies by cell type and viral strain—making the critical investigation of autophagy during IAV infection, of which there are an estimated 198 subtypes, uniquely challenging. Here, we evaluated the effect of IAV replication on the expression profiles of genes involved in autophagy regulation and mTOR signaling. The majority of in vitro IAV infection studies are based on cell models in which autophagy activation predated virus infection. By omitting cell serum starvation during the virus absorption step, we were able to capture autophagy activation during IAV infection that is more representative of the events occurring in vivo.
We observed global host transcriptome changes (LFC ≥2) that varied between the early stages of IAV replication and later timepoints depending on the ratio between infectious virus and host cells, indicating that virus load played a significant role in host gene expression dynamics (Figures 1 and 2). Despite host cells' efforts to shut down protein synthesis, IAV replication activated mTORC1 signaling, positively regulating protein synthesis in nutrient‐rich conditions. Both mTORC1 and mTORC2 carry out distinct functions: activation of mTOR1 facilitates protein synthesis and inhibits autophagy via repression of ULK1 signaling, while mTORC2 regulates cytoskeleton dynamics and cell survival (Oh & Jacinto, 2011; Sabatini, 2017). Despite the activation of mTORC1, IAV replication also simultaneously activated autophagy independent of ULK1 signaling. The ULK1 gene executes a feedback loop between mTORC1 signaling and the autophagy pathway, inhibiting mTORC1 via interaction with RPTOR during stress (Dunlop et al., 2011; Jung et al., 2011; Luo et al., 2018). RPTOR gene activation was limited to the early stages of IAV infection and may not have had the capacity to inhibit ULK1 function, particularly given the continuous ULK1 transcript accumulation throughout the virus replication cycle (Figure 2 and Figure S2). At the same time, RICTOR gene activation occurred throughout the viral replication cycle playing a major role for mTORC2 signaling in IAV infected cells (Luo et al., 2018; Sarbassov et al., 2004). Our global transcriptome analysis of IAV (H3N2)‐infected cells revealed remarkable dependence of ULK1 and ULK2 gene activation on the viral load. At lower MOI1, the ULK1 gene was continuously activated throughout the IAV replication cycle. At MOI5, ULK1 gene expression was mostly suppressed and ULK2 was activated briefly at the early stage of IAV replication. We further examined the role of ULK1 signaling on post‐transcriptional level and found that pharmacological inhibition of ULK1 had no significant effect on IAV replication and activation of autophagy in the lung cancer A549 cell line (Figure 3).
In response to increased viral burden, we observed secondary oscillatory burst of autophagy gene regulator activation as infection progressed (3–4 hpi), particularly related to protein synthesis (EIF2AK3 and MAPK3). However, at a higher viral load (MOI5), MAPK3 gene expression spiked at 3 hpi and subsequently returned to base levels. This suggests that cell stress burdened by viral protein synthesis could initiate a feedback mechanism activating MAP3K transcription. These findings implicate host cell protein synthesis downregulation as a mechanism for impeding viral replication. In turn, activation of genes regulating mTORC1 activity at early stage of IAV replication signified viral hijacking of host protein synthesis machinery (Figure 1b and Zhirnov & Klenk, 2007; Ehrhardt et al., 2007; Shin et al., 2007). However, the steady accumulation of MAPK3/ERK (Figure 1b, MOI1) and LRP6 (Figure 2, MOI1) gene transcripts throughout IAV infection indicates that mTORC1 activation could be sustained on a post‐transcriptional level (Figure 4b: 9 hpi vs. 6 hpi).
The oscillatory pattern of key autophagy (ATG7, Figure 1b, MOI1) and mTORC2 (RICTOR, Figure 2, MOI1) gene expression profiles demonstrates that viral replication activates two major cell survival pathways: mTORC1 and autophagy that is independent of ULK1 signaling, also found in Poliovirus infection (Corona Velazquez et al., 2018). This observation and the known relationship between LC3B‐II and viral protein (M2) accumulation leading to autophagosome formation (Beale et al., 2014; Gannagé et al., 2009) could explain an autophagy activation alternative to the classical ULK1 signaling pathways. Furthermore, our data show that pharmacological inhibition of ULK1 function prior to and upon IAV infection did not change the course of autophagy activation. Together, these results demonstrate that ULK1‐independent simultaneous activation of mTORC1 and autophagy is possible when orchestrated by IAV proteins boosting their own amplification (Kuss‐Duerkop et al., 2017; Shin et al., 2007; Zhirnov & Klenk, 2007).
Previous studies have shown that direct interaction between IAV Matrix 2 (M2) ion‐channel protein with LC3 blocks autophagosome fusion with lysosomes (Gannagé et al., 2009) and redirects LC3 to the plasma membrane to facilitate virus budding (Beale et al., 2014). High levels of M2 responsible for accumulation of LC3II and formation of autophagosomes were linked to retardation of viral growth and decreased shedding of infectious virus particles (Calderon et al., 2019). Applying split‐GFP technology, we provided direct evidence for IAV/M2‐LC3 protein interaction causing localization of the LC3 to the cell wall membranes (Figure 6a). This method is superior to the derivation of protein–protein interactions from colocalization studies of LC3 and M2 fused to full‐length fluorescent proteins since the fluorescent proteins tend to dimerize and produce false‐positive puncta formations. We observed increased formation of autophagosomes in stable cell lines expressing LC3–GFP or LC3–Cherry fusion proteins and ruled out their application for autophagy activation studies. Thus, our study concurs with the hypothesis that the viral protein M2 is responsible for the activation of autophagy independently of the classical or alternative signaling pathway.
Additionally, we found that cell survival responses regulated by AKT‐Ser473 and JNK‐Thr187 became activated midway throughout the virus replication cycle. Pharmacological inhibition of JNK signaling dramatically reduced viral protein synthesis in IAV‐infected NHBE cells, while there was a modest effect in the cancer cell line A549. Our findings are in agreement with previous study reporting a key role of JNK signaling in the host cell permissiveness to Influenza virus replication. Avian IAV subtypes, H5N1 and H9N2, activated JNK signaling in chicken fibroblasts. In the same host cells, infection with the H1N1 subtype, known as swine flu causing seasonal epidemics in humans, did not activate JNK phosphorylation (Zhang et al., 2019). In our studies, we observed that both H1N1 and H3N2 subtypes activated JNK signaling in human lung cell line and this correlated with the replication efficiency of the virus (Figures 4 and 5 and Figures S1d and S3c). Additionally, we observed unexpected decrease in JNK‐Thr187 levels in normal human lung epithelium cultures infected with IAV (H2N3) when ULK1 signaling was targeted with a selective inhibitor SBI0206965. This could be the cause of off‐target inhibition of kinases regulating JNK function in this cell type, including FAK (Egan et al., 2015; Liu et al., 2007; Martin et al., 2018). Regardless, the IAV replication and production of infectious virus particles remained unchallenged when compared with the experimental group treated with JNK‐specific inhibitor SP600125 (Figure 5) indicating that even at reduced activity JNK signaling could support IAV infection. Only upon complete inhibition of the JNK function, the IAV titer was reduced. Collectively, our data indicate that lung carcinoma A549 show reduced sensitivity to inhibition of JNK signaling during IAV replication compared with NHBEC, supporting the idea that cell models utilized to study host–pathogen interaction would produce subjective results. Our study solidified the link between IAV infection and JNK‐Thr187 signaling as a stress regulatory mechanism accompanying the rampant synthesis of viral proteins and activating alternative autophagy pathways (Clarke et al., 2001; Eliopoulos et al., 1999; McLean & Bachenheimer, 1999; Nacken et al., 2014; Zhang et al., 2019). Therefore, therapies that decrease the host cell capacity as a viral particle “factory” through inhibition of JNK‐Thr187 signaling can result in a significant reduction of infectious IAV titer.
In conclusion, our findings indicate that the IAV load is responsible for differential gene expression dynamics within the cell host. We demonstrate that IAV replication simultaneously activated transcription of key genes regulating mTORC1 and autophagy in human lung epithelial cells. Host stress response mechanisms regulated by the c‐jun N‐terminal kinase are co‐opted by the IAV replication machinery to boost viral titer and their inhibition has the propensity to serve as host‐directed antiviral therapy. We also observed differences between NHBE cells and cancer cell lines in their response to virus invasion. In the search of universal, effective and safe antiviral therapies, we call for comprehensive comparison of data originating from diverse and relevant cell models of virus infection.
4. EXPERIMENTAL PROCEDURES
4.1. Cell culture, media, and chemical reagents
A549 carcinoma human alveolar epithelial cells (ATCC, CCL‐185) and Madin‐Darby canine kidney (MDCK) cells (ATCC, CCL‐34) were maintained in high‐glucose DMEM (Gibco‐Thermo Fisher, Grand Island, NY) and EMEM (Gibco‐Thermo Fisher, Grand Island, NY), respectively, supplemented with 10% (v/v) fetal bovine serum (Gibco‐Thermo Fisher, Grand Island, NY). BEAS‐2B (ATCC, CRL‐9609), a polyoma virus transformed normal human lung bronchial epithelial cells, and Normal Human Bronchial Epithelial (NHBE, Lonza‐CC2541) were maintained in Bronchial Epithelial Cell Growth medium obtained from Lonza/Clonetics Co as a BEGM kit (CC‐3170). All cells were cultured at 37°C and 5% CO2.
To inhibit ULK1 signaling, A549 cells were cultured in 10%‐DMEM media supplemented with 10 μM SBI0206965 (Selleckchem, Pittsburgh, PA) for 6 hr prior to IAV infection, and drug concentration was maintained in the media post‐virus infection. Since BEAS‐2B and NHBE have lower expression levels of ULK1, SBI0206965 was added at 1 μM to the BEGM growth media. We observed high toxicity at 10 μM SBI0206965 for both noncarcinoma cell types, which could be caused by an off‐target drug effect at this concentration. To inhibit JNK‐Thr187 phosphorylation, we added 10 μM SP600125 (Selleckchem, Pittsburgh, PA) to A549 growth media 6 hr prior to IAV infection, while 5 and 1 μM were added to the culture media of NHBE and BEAS‐2B, respectively, due to sensitivity issues resulting from JNK inhibition.
4.2. Viruses and virus infection procedures
The following IAV strains were used in this study: A/PR/8/1934 (H1N1) and A/Port Chalmers/1/73 (H3N2). The virus work was performed in compliance with CDC guidelines for biosafety level 2 agents. Virus stocks were prepared from MDCK cells infected at MOI of 0.01 in the following media: EMEM, 10 mM HEPES (Gibco), 0.125% BSA (Gibco), 1 µg/ml TPCK trypsin (Worthington Biomedical Corporation). MDCK cells were incubated for 1 hr at 37°C in virus media, washed with PBS, and overlaid with serum‐free EMEM containing 1 µg/ml TPCK trypsin. Conditioned media was harvested when a cytopathic effect was observed (usually 24 and 48 hpi for A/PCh/1/73 and, A/PR/8/34 respectively), centrifuged at 1,000 g for 10 min, passed through 0.45 μm Nalgene filter units, and aliquoted in 50% BSA media for storage at −80°C. The virus titer was determined in MDCK cells by plaque assays, as described previously (Szretter et al., 2006), and immunoplaque analysis was performed with anti‐NP antibody (ThermoFisher, PA5‐32242) and alkaline phosphatase‐conjugated anti‐rabbit antibody.
To study the dynamics of viral RNA replication, A549 cells were plated 18 hr prior to infection in high glucose DMEM supplemented with 5% or 10% fetal clone III serum at 1 × 106 per well in six‐well format dishes. In order to standardize the multiplicity of virus infection (MOI) between independent virus infection experiments, the cell number was calculated from a control sample immediately before IAV infection. A549 cells were mock treated in six‐well plates with virus free media or were infected with 250 µl virus containing media. To compare the effect of serum starvation on autophagy activation we utilized two protocols of virus activation for absorption.
The serum‐starved protocol has been broadly used in in vitro IAV infections and is based on dilution of viral stocks in serum‐free media supplemented with 1 µg/ml TPCK trypsin followed by incubation of virus media and host cells for 1 hr at 37°C. After 1 hr, 1 ml of EMEM supplemented with 5% Fetal clone III serum (5% DMEM) was added and cells were incubated at 37°C prior to harvest.
In the serum replete protocol, 20 µg TPCK‐treated trypsin (Warthington, Lakewood, NJ) was added to 1 ml virus stock (harvested from MDCK producer cells in serum free media) and was incubated for 10 min at 37°C to activate cleavage of viral HA glycoprotein to HA1 and HA2. Equal volume (1 v/v) of EMEM supplemented with 10% Fetal clone III serum (10% EMEM) was added to the TPCK trypsin‐treated virus to neutralize the enzyme. To determine the titer of infectious IAV particles, 250 µl of the trypsin pre‐activated virus diluted to 2 × 105 (approximated from the pfu/ml of the original virus stock) in 10%‐EMEM were layered over 2 × 106 MDCK cells in a 6‐well plate and incubated for 1 hr at 37°C. In parallel, 250 µl of the original virus stock diluted to MOI 0.1 in serum‐free EMEM supplemented with 1 µg/ml TPCK‐treated trypsin was layered over 2 × 106 MDCK cells in a 6‐well plate and incubated for 1 hr at 37°C. Following the 1 hr virus absorption step, 2 ml 10%‐EMEM were added to the infected cells and the incubation at 37°C continued for 48 hr. Conditioned media from IAV‐infected MDCK cells was serially diluted (log10) in serum‐free EMEM media supplemented with 1 µg/ml TPCK‐treated trypsin to determine the virus titer in MDCK cells by plaque assays, as described previously (Szretter et al., 2006). On average, the titer (pfu/ml) produced by cells infected with trypsin pre‐activated IAV in serum replete media was tenfold lower compared with the infections in serum‐free media. To normalize the MOI between the two protocols, A549 cells were infected at tenfold higher virus titer for the serum replete protocol compared with the serum‐free protocol. Cells were collected at 1, 3, 6, and 8 hpi and the levels of viral RNA were determined via quantitative RT‐PCR with Taqman probe (5′‐/56‐FAM/AGCTCTCGG/ZEN/ACGAARGGCARCGA/3IABkFQ/‐3′) and primers (5′‐TCCTCTGCATTGTCTCCGAA‐3′ and 5′‐TGTCYTTYCAGGGGCGGG‐3′) targeting the IAV NP6 transcript. With this quantitative method, we observed comparable levels of viral transcripts (ϪCt normalized to GAPDH transcripts) between the two infection protocols when tenfold higher virus titer was applied in the serum‐replete protocol. For the transcriptome and WB studies, A549 cells were plated 18 hr prior to infection in 10% DMEM at 1 × 106 per well in six‐well format dishes. Pre‐activated virus stock was adjusted to the required MOI in 10% DMEM and was layered on target cells at 250 µl. After 1 hr of incubation at 37°C, A549 mock and virus infected cells were topped with fresh 10%‐DMEM to a total volume of 2 ml and were incubated at 37°C prior to harvest.
4.3. RNA isolation and sample preparation for Illumina RNA Mi‐seq analysis
At indicated time points, post initiation of infection, cells were washed with cold phosphate buffered saline (PBS) and lysed in the well with 350 µl 1% beta‐mercaptoethanol supplemented RLT buffer from the RNeasy Plus Mini kit (QIAGEN, Maryland). Total RNA was isolated according to the manufacturer's protocol, including on‐column removal of gDNA. After elution, total RNA was treated with Turbo DNA‐free Kit (AM1907, Thermo Fisher Scientific) for complete elimination of gDNA. RNA quality was determined by both direct observation of rRNA on 1% denaturing agarose gel and by Agilent bioanalyzer scan. For the preparation of Illumina libraries, 5 µg total RNA per sample was processed with the TruSeq Stranded Total RNA Ribo‐Zero H/M/R kit (RS‐122‐2201, Illumina). Following rRNA removal, polyA‐mRNA species were separated from the nonpolyA RNA pool with the polyA Spin mRNA Isolation Kit (S1560S, NEB). The two RNA sample pools were further quantified by Qubit (Invitrogen) and 15 ng was subjected to size fragmentation followed by cDNA synthesis and the addition of a terminal tagged oligonucleotide. Tagged cDNA was purified with Agencourt AMpure XP beads and amplified with 15 cycles of PCR. Fragment length distribution of all libraries was analyzed on a BioAnalyzer high‐sensitivity LabChip. Diluted libraries (5 pmol) were multiplexed and sequenced on the Illumina MiSeq (2‐ by 250‐bp paired‐end run with 3–4 million reads/sample).
4.4. Analysis of RNAseq data and differential gene expression assay
RNAseq data were analyzed on the EDGE Bioinformatics platform (Li et al., 2017). Using the PiReT application (https://github.com/mshakya/piret), we performed quality control on the RNA‐seq data using FaQC v2.09 (Lo & Chain, 2014). The reads that passed the QC were mapped using HISAT2 v2.1.0 against its graph FM index (GFM) human reference genome (Kim et al., 2019a). Htseq‐count v0.11.1 was used to calculate the number of reads mapped to coding sequences (Anders et al., 2015). Applying count data, we calculated transcripts per kilobase per million (TPM) and found differentially expressed genes using the R package DESeq2 v1.18.1. Heatmaps showing fold change (FC) were made using the R package heatmap v1.0.10 which clustered both rows and columns based on FC values.
4.5. Data availability
All transcriptomics data have been submitted to GEO under accession number GSE162295 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162295) and NCBI tracking system #21497852.
4.6. Western blot analysis and fluorescent microscopy
Uninfected cells and cells infected with A/Port Chalmers/1/73 (H3N2) at MOI5 were collected after the treatment of interest and were solubilized in RIPA Lysis and Extraction Buffer (Thermo Scientific, Grand Island, NY) supplemented with a cocktail of protease and phosphatase inhibitor mixture (Thermo Scientific). Lysates were cleared at 14,000 rpm for 15 min, and the total protein amount in the supernatants was quantified with the BCA kit (Pierce). Equal amounts of protein (5 μg) were resolved by Tris–Glycine–SDS PAGE electrophoresis (4%–15% Mini‐PROTEAN, BioRad, Hercules, CA). After the SDS–PAGE the proteins were transferred onto PVDF membranes (Millipore, Billerica, MA), blocked with 5% nonfat dried milk in TBST (25 mM Tris [pH 7.6], 137 mM NaCl, and 0.2% Tween 20) and incubated overnight at 4°C with primary antibodies in 1% bovine serum albumin in phosphate‐buffered saline. Membranes washed from the primary antibodies were incubated for 2 hr in HRP‐conjugated secondary antibody and were analyzed for immunoreactive proteins with luminol reagent (Santa Cruz Biotechnology) by BioRad Molecular Imager ChemiDoc XRS+. Image Lab 6.0 software was applied for quantitative analysis of the protein band intensity. The following primary antibodies were used in this study: anti‐actin (sc‐58673, 2Q1055); anti‐Influenza A Virus M2 Protein [14C2] (ab5416); anti‐Influenza A Virus Nucleoprotein [9C11] (SAB5300169); anti‐LC3B (CST‐2775); anti‐mTOR [7C10] (CST‐2983); anti‐phospho‐mTOR(S2448) [D9C2] (CST‐5536); anti‐ULK1 [D8H5] (CST‐8054); anti‐phospho‐ULK1(S757) [D706U] (CST‐14202); anti‐SAPK/JNK (CST‐9252); anti‐phospho‐SAPK/JNK(Thr183/Tyr185) (CST‐9251).
For the fluorescent microscopy studies, cells were plated on borosilicate chamber slides pre‐coated with fibronectin. IAV‐infected and mock‐treated samples were fixed with 4% paraformaldehyde at various time points, incubated in blocking solution (PBS+3% bovine serum albumin) and labeled with mouse anti‐Influenza A Virus M2 Protein [14C2] (ab5416). Following overnight incubation with the anti‐IVA/M2 primary antibody, cells were washed in PBS and permeabilized (PBS+0.25% TritonX) prior to incubation with rabbit anti‐LC3B antibody (Sigma, L7543). Alexa Fluor488 goat anti‐mouse (ab150113) and Alexa Fluor555 goat anti‐rabbit IgGs were used to fluorescently label IVA/M2 and LC3B proteins, respectively. Images were obtained from Zeiss Axio Observer.Z1 using ZenPro software. To determine the percentage of infected cells, images with fluorescently labeled anti‐IVA/M2 antibody were obtained with 20× objective choosing fields with >300 cells. For each culture condition, five images were taken from nonoverlapping fields and were analyzed with ImageJ. A cell counter plugin was created with the following parameters: (a) manual threshold, (b) fill holes, (c) enhance contrast and saturated pixels at 0.3%, (d) watershed separation, and (e) particle analysis at size 100‐infinity and circularity at 0.2–0.1. The fraction of cells labeled with anti‐viral antibody was calculated as percent of total cell population labeled with DAPI.
4.7. ELISA
Endogenous levels of phosphorylated AKT at Ser473 were determined with FastScan™ Phospho‐Akt (Ser473) ELISA Kit (Cell Signaling technologies, CST#80895) in cleared cell lysates of mock treated and virus infected 106 A549 cells. Absolute values of phosphorylated Ser473‐AKT were determined with a standard curve correlating the positive control phospho‐Akt(Ser473) concentration (μg/ml) to absorbance at 450 nM. We utilized PathScan Phospho‐p70 S6 Kinase (Thr389) Sandwich ELISA kit (Cell Signaling technologies, CST#7063) to determine mTORC1 activity by measuring the phosphorylated levels of its primary substrate p70S6K. In this assay, the levels of phosphorylated Thr389 (detection antibody) were normalized to the total levels of p70S6K protein (capture antibody). A Bioteck Synergy 2 plate reader was used to collect absorbance at 450 nm.
4.8. Plasmids and reagents for split‐GFP in situ protein‐protein interactions
To study direct interaction of IAV M2 and host LC3B proteins, we used a split green fluorescent protein (GFP) assay, as previously described (Cabantous et al., 2005). gBLOCK gene fragments, encoding for GFP11‐ and GFP10‐tagged M2 and LC3B, were chemically synthesized by Twist Biosciences Inc. The synthetic DNA contained a set of primer sequences for amplification and Hind III and EcoR V restriction enzyme sites. The genes also contained methionine required for the start and a stop codon. The 5′ end of all gene constructs contained CGATTAACGGTCTCTGGGGAAGCTTACCATG the 3′end of the sequences contained TAAGATATCAAAAGGAGACCGTTAATCTAAAATCATTATTTG. The primers used for amplification of the constructs were forward primer CGATTAACGGTCTCTGGGG and reverse primer CAAATAATGATTTTAGATTAACGG. All genes were PCR amplified using Q5 proof reading polymerase (New England Biolabs, M0491). Purified PCR products encoding the GFP Tag were cloned into a pcDNA 5.1 vector plasmid encoding influenza A M2 protein (Gabbard et al., 2009). Restriction enzymes Hind III (NEB, #R0104) and EcoRV (NEB #R0195) were used to create sticky ends on the PCR and plasmid vector, followed by gel extraction and purification (Qiagen #28706) prior to T4 DNA (NEB, #B0202) ligation. The plasmid vectors encoding tagged‐M2 and LC3B proteins were verified via Sanger sequencing, and the amino acid sequence of the various constructs are presented in Supplementary Material (S3). For the Split GFP assay, the plasmid vectors encoding for tagged M2 and LC3B proteins were transfected in HEK293T cells with Lipofectamine 3000 (Thermo Fisher/Invitrogen). At day 3 post‐transfection, the cells were fixed with 4% paraformaldehyde (15 min at room temperature) and permeabilized with 0.3% Triton X100 (10 min at room temperature). After blocking with 2% BSA in phosphate buffer saline (PBS), the cells were incubated overnight at 4°C with purified protein fraction of GFP1‐9 (10 μg/ml). Only when in very close proximity, the GFP11 and GFP10 peptides can be complimented with the larger fraction GFP1‐9 to produce folded and fluorescent GFP (Cabantous et al., 2013). Imaging was performed on Zeiss Axio Observer.Z1 and processed with ZenPro software.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
S.M.‐V. and T.A.S.B was involved in conceptualization; S.M.‐V., T.A.S.B, N.V., C.D.G., and S.B. performed methodology; S.M.‐V., T.A.S.B, S.R.S., and G.X. did formal analysis; S.M.‐V., T.A.S.B., S.R.S., and N.V. performed investigation; S.M.‐V. and T.A.S.B. wrote – original draft preparation; S.M.‐V., T.A.S.B., and N.V. wrote – review and editing; S.M.‐V., S.R.S., and G.W. supervised; S.M.‐V., G.W., and S.R.S did funding acquisition.
Supporting information
Fig S1‐3
Table S1
Supplementary Material
Bell, T.A.S. , Velappan, N. , Gleasner, C.D. , Xie, G. , Starkenburg, S.R. , Waldo, G. et al. (2022) Nonclassical autophagy activation pathways are essential for production of infectious Influenza A virus in vitro. Molecular Microbiology, 117, 508–524. 10.1111/mmi.14865
Funding information
Funding for this work was provided by Los Alamos National Laboratory R&D grants 202000696ER to S.M‐V., 20160054DR to G.W., and 20210082DR to S.R.S.
DATA AVAILABILITY STATEMENT
All transcriptomics data have been submitted to GEO under accession number GSE162295 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162295) and NCBI tracking system #21497852.
REFERENCES
- Abdulrahman, B.A. , Khweek, A.A. , Akhter, A. , Caution, K. , Kotrange, S. , Abdelaziz, D.H.A. et al. (2011) Autophagy stimulation by rapamycin suppresses lung inflammation and infection by Burkholderia cenocepacia in a model of cystic fibrosis. Autophagy, 7(11), 1359–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P.T. & Huber, W. (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31(2), 166–169. 10.1093/bioinformatics/btu638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beale, R. , Wise, H. , Stuart, A. , Ravenhill, B.J. , Digard, P. & Randow, F. (2014) A LC3‐interacting motif in the influenza A virus M2 protein is required to subvert autophagy and maintain virion stability. Cell Host and Microbe, 15(2), 239–247. 10.1016/j.chom.2014.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous, S. , Nguyen, H.B. , Pedelacq, J.‐D. , Koraïchi, F. , Chaudhary, A. , Ganguly, K. et al. (2013) A new protein‐protein interaction sensor based on tripartite split‐GFP association. Scientific Reports, 3(1), 2854. 10.1038/srep02854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabantous, S. , Terwilliger, T.C. & Waldo, G.S. (2005) Protein tagging and detection with engineered self‐assembling fragments of green fluorescent protein. Nature Biotechnology, 23(1), 102–107. 10.1038/nbt1044 [DOI] [PubMed] [Google Scholar]
- Calderon, B.M. , Danzy, S. , Delima, G.K. , Jacobs, N.T. , Ganti, K. , Hockman, M.R. et al. (2019) Dysregulation of M segment gene expression contributes to influenza A virus host restriction. PLoS Pathogens, 15(8), e1007892. 10.1371/journal.ppat.1007892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, G.R. & Spector, S.A. (2011) Hormonally active vitamin D3 (1alpha,25‐dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV‐1 infection. Journal of Biological Chemistry, 286(21), 18890–18902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriere, A. , Romeo, Y. , Acosta‐Jaquez, H.A. , Moreau, J. , Bonneil, E. , Thibault, P. et al. (2011) ERK1/2 phosphorylate Raptor to promote Ras‐dependent activation of mTOR complex 1 (mTORC1). The Journal of Biological Chemistry, 286(1), 567–577. 10.1074/jbc.M110.159046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, P. , Meintzer, S.M. , Widmann, C. , Johnson, G.L. & Tyler, K.L. (2001) Reovirus infection activates JNK and the JNK‐dependent transcription factor c‐Jun. Journal of Virology, 75(23), 11275–11283. 10.1128/JVI.75.23.11275-11283.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corona Velazquez, A. , Corona, A.K. , Klein, K.A. & Jackson, W.T. (2018) Poliovirus induces autophagic signaling independent of the ULK1 complex. Autophagy, 14(7), 1201–1213. 10.1080/15548627.2018.1458805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic, V. , Saitoh, T. & Akira, S. (2013) Autophagy in infection, inflammation and immunity. Nature Reviews Immunology, 13(10), 722–737. 10.1038/nri3532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop, E.A. , Hunt, D.K. , Acosta‐Jaquez, H.A. , Fingar, D.C. & Tee, A.R. (2011) ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy, 7(7), 737–747. 10.4161/auto.7.7.15491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan, D.F. , Chun, M.G. , Vamos, M. , Zou, H. , Rong, J. , Miller, C.J. et al. (2015) Small molecule inhibition of the autophagy kinase ULK1 and identification of ULK1 substrates. Molecular Cell, 59(2), 285–297. 10.1016/j.molcel.2015.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt, C. , Wolff, T. , Pleschka, S. , Planz, O. , Beermann, W. , Bode, J.G. et al. (2007) Influenza A virus NS1 protein activates the PI3K/Akt pathway to mediate antiapoptotic signaling responses. Journal of Virology, 81(7), 3058–3067. 10.1128/JVI.02082-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos, A.G. , Blake, S.M. , Floettmann, J.E. , Rowe, M. & Young, L.S. (1999) Epstein‐Barr virus‐encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2. Journal of Virology, 73(2), 1023–1035. 10.1128/JVI.73.2.1023-1035.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbard, J. , Velappan, N. , Di Niro, R. , Schmidt, J. , Jones, C.A. , Tompkins, S.M. et al. (2009) A humanized anti‐M2 scFv shows protective in vitro activity against influenza. Protein Engineering, Design and Selection, 22(3), 189–198. 10.1093/protein/gzn070 [DOI] [PubMed] [Google Scholar]
- Ganley, I.G. , du Lam, H. , Wang, J. , Ding, X. , Chen, S. & Jiang, X. (2009) ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. Journal of Biological Chemistry, 284(18), 12297–12305. 10.1074/jbc.M900573200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannagé, M. , Dormann, D. , Albrecht, R. , Dengjel, J. , Torossi, T. , Rämer, P.C. et al. (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host and Microbe, 6(4), 367–380. 10.1016/j.chom.2009.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris, J. , Hartman, M. , Roche, C. , Zeng, S.G. , O'Shea, A. , Sharp, F.A. et al. (2011) Autophagy controls IL‐1beta secretion by targeting pro‐IL‐1beta for degradation. Journal of Biological Chemistry, 286(11), 9587–9597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holsinger, L.J. & Lamb, R.A. (1991) Influenza virus M2 integral membrane protein is a homotetramer stabilized by formation of disulfide bonds. Virology, 183(1), 32–43. 10.1016/0042-6822(91)90115-R [DOI] [PubMed] [Google Scholar]
- Hussey, S. , Travassos, L.H. & Jones, N.L. (2009) Autophagy as an emerging dimension to adaptive and innate immunity. Seminars in Immunology, 21(4), 233–241. 10.1016/j.smim.2009.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki, K. , Ouyang, H. , Zhu, T. , Lindvall, C. , Wang, Y. , Zhang, X. et al. (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell, 126(5), 955–968. 10.1016/j.cell.2006.06.055 [DOI] [PubMed] [Google Scholar]
- Jung, C.H. , Jun, C.B. , Ro, S.H. , Kim, Y.M. , Otto, N.M. , Cao, J. et al. (2009) ULK‐Atg13‐FIP200 complexes mediate mTOR signaling to the autophagy machinery. Molecular Biology of the Cell, 20(7), 1992–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung, C.H. , Seo, M. , Otto, N.M. & Kim, D.H. (2011) ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy, 7(10), 1212–1221. 10.4161/auto.7.10.16660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Paggi, J.M. , Park, C. , Bennett, C. & Salzberg, S.L. (2019a) Graph‐based genome alignment and genotyping with HISAT2 and HISAT‐genotype. Nature Biotechnology, 37(8), 907–915. 10.1038/s41587-019-0201-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, J. , Kundu, M. , Viollet, B. & Guan, K.L. (2011) AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology, 13(2), 132–141. 10.1038/ncb2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y.S. , Silwal, P. , Kim, S.Y. , Yoshimori, T. & Jo, E.‐K. (2019b) Autophagy‐activating strategies to promote innate defense against mycobacteria. Experimental and Molecular Medicine, 51(12), 1–10. 10.1038/s12276-019-0290-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenk, H.‐D. & Garten, W. (1994) Host cell proteases controlling virus pathogenicity. Trends in Microbiology, 2(2), 39–43. 10.1016/0966-842X(94)90123-6 [DOI] [PubMed] [Google Scholar]
- Kobayashi, S. , Yoshii, K. , Phongphaew, W. , Muto, M. , Hirano, M. , Orba, Y. et al. (2020) West Nile virus capsid protein inhibits autophagy by AMP‐activated protein kinase degradation in neurological disease development. PLoS Pathogens, 16(1), e1008238. 10.1371/journal.ppat.1008238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuballa, P. , Nolte, W.M. , Castoreno, A.B. & Xavier, R.J. (2012) Autophagy and the immune system. Annual Review of Immunology, 30(1), 611–646. 10.1146/annurev-immunol-020711-074948 [DOI] [PubMed] [Google Scholar]
- Kuss‐Duerkop, S.K. , Wang, J. , Mena, I. , White, K. , Metreveli, G. , Sakthivel, R. et al. (2017) Influenza virus differentially activates mTORC1 and mTORC2 signaling to maximize late stage replication. PLoS Pathogens, 13(9), e1006635. 10.1371/journal.ppat.1006635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyei, G.B. , Dinkins, C. , Davis, A.S. , Roberts, E. , Singh, S.B. , Dong, C. et al. (2009) Autophagy pathway intersects with HIV‐1 biosynthesis and regulates viral yields in macrophages. The Journal of Cell Biology, 186(2), 255–268. 10.1083/jcb.200903070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante, M. & Sabatini, D.M. (2012) mTOR signaling in growth control and disease. Cell, 149(2), 274–293. 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, B. & Kroemer, G. (2019) Biological functions of autophagy genes: a disease perspective. Cell, 176(1–2), 11–42. 10.1016/j.cell.2018.09.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine, B. , Mizushima, N. & Virgin, H.W. (2011) Autophagy in immunity and inflammation. Nature, 469(7330), 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. , Liu, Y. , Wang, Z. , Liu, K. , Wang, Y. , Liu, J. et al. (2011) Subversion of cellular autophagy machinery by hepatitis B virus for viral envelopment. Journal of Virology, 85(13), 6319–6333. 10.1128/JVI.02627-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P.E. , Lo, C.C. , Anderson, J.J. , Davenport, K.W. , Bishop‐Lilly, K.A. , Xu, Y. et al. (2017) Enabling the democratization of the genomics revolution with a fully integrated web‐based bioinformatics platform. Nucleic Acids Research, 45(1), 67–80. 10.1093/nar/gkw1027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang, X.H. , Kleeman, L.K. , Jiang, H.H. , Gordon, G. , Goldman, J.E. , Berry, G. et al. (1998) Protection against fatal Sindbis virus encephalitis by beclin, a novel Bcl‐2‐interacting protein. Journal of Virology, 72(11), 8586–8596. 10.1128/JVI.72.11.8586-8596.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Xu, S.W. , Kennedy, L. , Pala, D. , Chen, Y. , Eastwood, M. et al. (2007) FAK is required for TGFbeta‐induced JNK phosphorylation in fibroblasts: implications for acquisition of a matrix‐remodeling phenotype. Molecular Biology of the Cell, 18(6), 2169–2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo, C.‐C. & Chain, P.S.G. (2014) Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinformatics, 15(1), 366. 10.1186/s12859-014-0366-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. , Xu, W. , Li, G. & Cui, W. (2018) Weighing in on mTOR complex 2 signaling: the expanding role in cell metabolism. Oxidative Medicine and Cellular Longevity, 2018, 7838647. 10.1155/2018/7838647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. , Sun, Q. , Mi, R. & Zhang, H. (2011) Avian influenza A virus H5N1 causes autophagy‐mediated cell death through suppression of mTOR signaling. Journal of Genetics and Genomics, 38(11), 533–537. 10.1016/j.jgg.2011.10.002 [DOI] [PubMed] [Google Scholar]
- Ma, X.M. & Blenis, J. (2009) Molecular mechanisms of mTOR‐mediated translational control. Nature Reviews Molecular Cell Biology, 10(5), 307–318. 10.1038/nrm2672 [DOI] [PubMed] [Google Scholar]
- Martin, K.R. , Celano, S.L. , Solitro, A.R. , Gunaydin, H. , Scott, M. , O'Hagan, R.C. et al. (2018) A potent and selective ULK1 inhibitor suppresses autophagy and sensitizes cancer cells to nutrient stress. iScience, 8, 74–84. 10.1016/j.isci.2018.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean, T.I. & Bachenheimer, S.L. (1999) Activation of cJUN N‐terminal kinase by herpes simplex virus type 1 enhances viral replication. Journal of Virology, 73(10), 8415–8426. 10.1128/JVI.73.10.8415-8426.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza, M.C. , Er, E.E. & Blenis, J. (2011) The Ras‐ERK and PI3K‐mTOR pathways: cross‐talk and compensation. Trends in Biochemical Sciences, 36(6), 320–328. 10.1016/j.tibs.2011.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mintern, J.D. , Macri, C. , Chin, W.J. , Panozza, S.E. , Segura, E. , Patterson, N.L. et al. (2015) Differential use of autophagy by primary dendritic cells specialized in cross‐presentation. Autophagy, 11(6), 906–917. 10.1080/15548627.2015.1045178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N. , Yoshimori, T. & Levine, B. (2010) Methods in mammalian autophagy research. Cell, 140(3), 313–326. 10.1016/j.cell.2010.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacken, W. , Anhlan, D. , Hrincius, E.R. , Mostafa, A. , Wolff, T. , Sadewasser, A. et al. (2014) Activation of c‐jun N‐terminal kinase upon influenza A virus (IAV) infection is independent of pathogen‐related receptors but dependent on amino acid sequence variations of IAV NS1. Journal of Virology, 88(16), 8843–8852. 10.1128/JVI.00424-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napolitano, G. & Ballabio, A. (2016) TFEB at a glance. Journal of Cell Science, 129(13), 2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, M.I. , Simonsen, L. , Viboud, C. , Miller, M.A. & Holmes, E.C. (2009) The origin and global emergence of adamantane resistant A/H3N2 influenza viruses. Virology, 388(2), 270–278. 10.1016/j.virol.2009.03.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, W.J. & Jacinto, E. (2011) mTOR complex 2 signaling and functions. Cell Cycle, 10(14), 2305–2316. 10.4161/cc.10.14.16586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, P. & Münz, C. (2016) Autophagy and mammalian viruses: roles in immune response, viral replication, and beyond. Advances in Virus Research, 95, 149–195. [DOI] [PubMed] [Google Scholar]
- Qiang, L. , Sample, A. , Shea, C.R. , Soltani, K. , Macleod, K.F. & He, Y.‐Y. (2017) Autophagy gene ATG7 regulates ultraviolet radiation‐induced inflammation and skin tumorigenesis. Autophagy, 13(12), 2086–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raciti, M. , Lotti, L.V. , Valia, S. , Pulcinelli, F.M. & Di Renzo, L. (2012) JNK2 is activated during ER stress and promotes cell survival. Cell Death and Disease, 3(11), e429. 10.1038/cddis.2012.167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini, D.M. (2017) Twenty‐five years of mTOR: uncovering the link from nutrients to growth. Proceedings of the National Academy of Sciences of the United States of America, 114(45), 11818–11825. 10.1073/pnas.1716173114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh, T. , Fujita, N. , Jang, M.H. , Uematsu, S. , Yang, B.G. , Satoh, T. et al. (2008) Loss of the autophagy protein Atg16L1 enhances endotoxin‐induced IL‐1beta production. Nature, 456(7219), 264–268. [DOI] [PubMed] [Google Scholar]
- Sarbassov, D.D. , Ali, S.M. , Kim, D.H. , Guertin, D.A. , Latek, R.R. , Erdjument‐Bromage, H. et al. (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin‐insensitive and raptor‐independent pathway that regulates the cytoskeleton. Current Biology, 14(14), 1296–1302. 10.1016/j.cub.2004.06.054 [DOI] [PubMed] [Google Scholar]
- Shang, L. , Chen, S. , Du, F. , Li, S. , Zhao, L. & Wang, X. (2011) Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proceedings of the National Academy of Sciences of the United States of America, 108(12), 4788–4793. 10.1073/pnas.1100844108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, Y.K. , Li, Y. , Liu, Q. , Anderson, D.H. , Babiuk, L.A. & Zhou, Y. (2007) SH3 binding motif 1 in influenza A virus NS1 protein is essential for PI3K/Akt signaling pathway activation. Journal of Virology, 81(23), 12730–12739. 10.1128/JVI.01427-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoji‐Kawata, S. , Sumpter, R. , Leveno, M. , Campbell, G.R. , Zou, Z. , Kinch, L. et al. (2013) Identification of a candidate therapeutic autophagy‐inducing peptide. Nature, 494(7436), 201–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen, L. , Viboud, C. , Grenfell, B.T. , Dushoff, J. , Jennings, L. , Smit, M. et al. (2007) The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Molecular Biology and Evolution, 24(8), 1811–1820. 10.1093/molbev/msm103 [DOI] [PubMed] [Google Scholar]
- Siqueira, M.D.S. , Ribeiro, R.D.M. & Travassos, L.H. (2018) Autophagy and its interaction with intracellular bacterial pathogens. Frontiers in Immunology, 9, 935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, G.J. , Vijaykrishna, D. , Bahl, J. , Lycett, S.J. , Worobey, M. , Pybus, O.G. et al. (2009) Origins and evolutionary genomics of the 2009 swine‐origin H1N1 influenza A epidemic. Nature, 459(7250), 1122–1125. [DOI] [PubMed] [Google Scholar]
- Sriwilaijaroen, N. & Suzuki, Y. (2020) Host receptors of influenza viruses and coronaviruses‐molecular mechanisms of recognition. Vaccines, 8(4), 587. 10.3390/vaccines8040587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starr, T. , Child, R. , Wehrly, T.D. , Hansen, B. , Hwang, S. , López‐Otin, C. et al. (2012) Selective subversion of autophagy complexes facilitates completion of the Brucella intracellular cycle. Cell Host and Microbe, 11(1), 33–45. 10.1016/j.chom.2011.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhauer, D.A. (1999) Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology, 258(1), 1–20. 10.1006/viro.1999.9716 [DOI] [PubMed] [Google Scholar]
- Sun, Y. , Li, C. , Shu, Y. , Ju, X. , Zou, Z. , Wang, H. et al. (2012) Inhibition of autophagy ameliorates acute lung injury caused by avian influenza A H5N1 infection. Science Signaling, 5(212), ra16. 10.1126/scisignal.2001931 [DOI] [PubMed] [Google Scholar]
- Szretter, K.J. , Balish, A.L. & Katz, J.M. (2006) Influenza: propagation, quantification, and storage. Current Protocols in Microbiology, 3(1), 15G.1.1–15G.1.22. 10.1002/0471729256.mc15g01s3 [DOI] [PubMed] [Google Scholar]
- Valečka, J. , Almeida, C.R. , Su, B. , Pierre, P. & Gatti, E. (2018) Autophagy and MHC‐restricted antigen presentation. Molecular Immunology, 99, 163–170. [DOI] [PubMed] [Google Scholar]
- Viboud, C. , Gostic, K. , Nelson, M.I. , Price, G.E. , Perofsky, A. , Sun, K. et al. (2020) Beyond clinical trials: evolutionary and epidemiological considerations for development of a universal influenza vaccine. PLoS Pathogens, 16(9), e1008583. 10.1371/journal.ppat.1008583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R. , Zhu, Y. , Zhao, J. , Ren, C. , Li, P. , Chen, H. et al. (2019) Autophagy promotes replication of influenza A virus in vitro. Journal of Virology, 93(4), e01984‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, Y. , Pattingre, S. , Sinha, S. , Bassik, M. & Levine, B. (2008) JNK1‐mediated phosphorylation of Bcl‐2 regulates starvation‐induced autophagy. Molecular Cell, 30(6), 678–688. 10.1016/j.molcel.2008.06.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, F. , Na, L. , Li, Y. & Chen, L. (2020) Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell and Bioscience, 10(1), 54. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yamamoto, K. , Ichijo, H. & Korsmeyer, S. J. (1999) BCL‐2 is phosphorylated and inactivated by an ASK1/Jun N‐Terminal protein kinase pathway normally activated at G 2 /M. Molecular and Cellular Biology, 19(12), 8469–8478. 10.1128/mcb.19.12.8469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Ruan, T. , Sheng, T. , Wang, J. , Sun, J. , Wang, J. et al. (2019) Role of c‐Jun terminal kinase (JNK) activation in influenza A virus‐induced autophagy and replication. Virology, 2(526), 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhirnov, O.P. , Ikizler, M.R. & Wright, P.F. (2002) Cleavage of influenza A virus hemagglutinin in human respiratory epithelium is cell associated and sensitive to exogenous antiproteases. Journal of Virology, 76(17), 8682–8689. 10.1128/JVI.76.17.8682-8689.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhirnov, O.P. & Klenk, H.D. (2007) Control of apoptosis in influenza virus‐infected cells by up‐regulation of Akt and p53 signaling. Apoptosis, 12(8), 1419–1432. 10.1007/s10495-007-0071-y [DOI] [PubMed] [Google Scholar]
- Zhirnov, O.P. & Klenk, H.D. (2013) Influenza A virus proteins NS1 and hemagglutinin along with M2 are involved in stimulation of autophagy in infected cells. Journal of Virology, 87(24), 13107–13114. 10.1128/JVI.02148-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Z. , Jiang, X. , Liu, D. , Fan, Z. , Hu, X. , Yan, J. et al. (2009) Autophagy is involved in influenza A virus replication. Autophagy, 5(3), 321–328. 10.4161/auto.5.3.7406 [DOI] [PubMed] [Google Scholar]
- Zoncu, R. , Efeyan, A. & Sabatini, D.M. (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology, 12(1), 21–35. 10.1038/nrm3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐3
Table S1
Supplementary Material
Data Availability Statement
All transcriptomics data have been submitted to GEO under accession number GSE162295 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162295) and NCBI tracking system #21497852.
All transcriptomics data have been submitted to GEO under accession number GSE162295 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE162295) and NCBI tracking system #21497852.