

Abstract
Azoreductases require NAD(P)H to reduce azo dyes but the high cost of NAD(P)H limits its application. Formate dehydrogenase (FDH) allows NAD(P)+ recycling and therefore, the fusion of these two biocatalysts seems promising. This study investigated the changes to the fusion protein involving azoreductase (AzoRo) of Rhodococcus opacus 1CP and FDH (FDHC23S and FDHC23SD195QY196H) of Candida boidinii in different positions with His‐tag as the linker. The position affected enzyme activities as AzoRo activity decreased by 20‐fold when it is in the N‐terminus of the fusion protein. FDHC23S+AzoRo was the most active construct and was further characterized. Enzymatic activities of FDHC23S+AzoRo decreased compared to parental enzymes but showed improved substrate scope – accepting bulkier dyes. Moreover, pH has an influence on the stability and activity of the fusion protein because at pH 6 (pH that is suboptimal for FDH), the dye reduction decreased to more than 50 % and this could be attributed to the impaired NADH supply for the AzoRo part.
Keywords: azoreductases, Brilliant Black BN, diazo dyes, fusion proteins, NADH regeneration systems
A fusion protein comprised of formate dehydrogenase and azoreductase was constructed to allow the simultaneous activity of dye reduction and NAD+ regeneration. The fusion protein showed improved substrate scope, but it only showed partial degradation for the bulkier azo dyes.
Introduction
The emergence of textile industries and other relevant industries, has paved the way to the synthesis of azo dyes. [1] Azo dyes have become a staple to the industry because of the variety of colors produced and high stability in light and washing. [2] However, the use of such xenobiotics such as azo dyes has significant tradeoffs to human health and the environment. [3] As a result, different methods such as the use of physicochemical methods like coagulation‐flocculation and even the utilization of biological materials have been developed to mitigate these problems.[ 4 , 5 ]
As part of the biological method, enzymes such as azoreductases have been thoroughly investigated. Several papers have already reported the degradation of azo dyes by different microorganisms.[ 6 , 7 , 8 ] But decades later, the application of azoreductases to an industrial scale have faced so many hurdles. To begin with, there is still no trend as to what kind of azo dyes can an azoreductase accept even if the enzymes belong to the same clade. [9] In addition, various studies have shown that there are azoreductases that can only accept one or two substrates.[ 10 , 11 ]
Flavin‐containing azoreductases (mainly FMN‐dependent) degrade dyes via a ping‐pong mechanism, reducing the flavin co‐factor by NAD(P)H and the reduced equivalents are transferred from flavin to the azo bond.[ 3 , 9 , 11 ] This reaction happens twice until the products are obtained. This co‐substrate requirement of NAD(P)H limits the application for these enzymes since the practical cost outweighs the benefits, especially when it will be applied for wastewater treatments.[ 12 , 13 , 14 ]
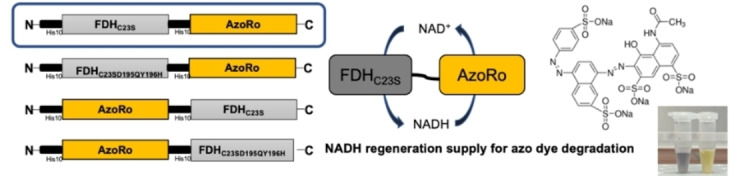
To mitigate this limitation, several NAD(P)H regeneration systems such as formate dehydrogenase and glucose dehydrogenase have now been designed and investigated to help circumvent the problem.[ 15 , 16 , 17 , 18 ] Formate dehydrogenase uses a cheaper substrate, formate (COOH−), and oxidizes it to carbon dioxide, donating the electrons to a second substrate, NAD+. [16] The compatibility of the mechanisms and the requirements for enzymes such as formate dehydrogenase and azoreductases make them interesting candidates for a fusion protein (Figure 1).
Figure 1.

Schematic view on the reaction mechanism of an azoreductase fused with a formate dehydrogenase to produce a self‐sufficient system for the reduction of azo dyes. The formate dehydrogenase oxidizes formate to carbon dioxide and donates the electron to the second substrate, NAD+, to produce NADH. NADH is then used by the azoreductase to reduce the azo bond, and thus cycled between the two fused proteins. The reaction to reduce the azo dyes completely is a two‐step process until the final products are obtained. The hydrazo‐intermediate does not accumulate.
Fusion proteins are constructed by putting two genes as one open reading frame (ORF) and placing a peptide linker in‐between. [19] This allows co‐purification of two biocatalysts, thereby saving time and resources. Other studies have also shown enhanced expression, improved stability, and better catalytic activity. [20] However, studies on fusion proteins are still in their early phase. Fusing two biocatalysts can be delicate as there are no proper methods to predictably design linkers. There is still a lot of work that is needed to be done since the outcome for the two biocatalysts can either have beneficial or detrimental effects.[ 20 , 21 ]
Most studies have focused on the use of flexible linkers composed of Gly or Ser or rigid linkers composed of Pro.[ 21 , 22 ] His, on the other hand, is often used to design (mostly terminal) tags for the affinity purification of recombinant proteins since those tags (typically composed of 6 to 10 His) have a high affinity towards nickel comprising resins. After binding such tagged proteins to Ni‐loaded resins, other proteins can be separated by washing the material with buffer containing a low imidazole concentration and finally the tagged protein can be eluted via the increase of imidazole in the elution buffer. Since most recombinant proteins often have His tags on either the N‐ or C‐terminal side, it would be an interesting way to see the effect of combining two proteins through a His tag.
Therefore, this study focuses on the construction of a fusion protein comprised of the formate dehydrogenase (FDH) from Candida boidinii and azoreductase (AzoRo) from Rhodococcus opacus 1CP with the His10‐tag as the linker. We aim to characterize the purified fusion protein – providing insights on the changes that may have happened to these enzymes and allowing a closer look and understanding of this bifunctional biocatalyst.
Results and Discussion
Fusion protein construction, expression optimization, and protein purification
Few papers have now shown and proved the possibility of combining NAD(P)H regeneration systems together with enzymes that use NAD(P)H for enzymatic reactions such as azoreductases.[ 23 , 24 , 25 ] The FDH used from Candida boidinii has a point mutation at the 23rd residue, changing from Cys to Ser. [26] This C23S variant was described to be more stable compared to the wild‐type enzyme. In addition, the wild‐type FDH of C. boidinii does not accept NADP+. Therefore, the double mutant D195QY196H was used as it showed overall better catalytic efficiency with NADP+. [27] Meanwhile, the azoreductase AzoRo used from Rhodococcus opacus 1CP shows a limited substrate scope, only accepting (1) (Figure 2) as a substrate but can use both NADH and NADPH as electron donor (co‐substrate) to reduce (1). This AzoRo operates better on acidic pH and the enzyme activity drops from neutral to basic pH.[ 11 , 28 , 29 ]
Figure 2.

Structure of the azo dyes used in the study: (1) methyl red (2) methyl orange (3) Sudan orange G (4) Allura red AC (5) Brilliant Black BN (6) Ponceau S. Samples (1–4) represent the dyes with single azo bond while samples (5) and (6) represent the dyes with two azo bonds.
Both enzymes were individually produced as N‐terminal His10‐tagged proteins demonstrating the functionality of this small fusion tag.[ 11 , 28 , 29 ] Several studies now demonstrate the importance of finding the right linkers when fusing two proteins.[ 21 , 30 , 31 ] The linkers used can influence protein solubility, protein interactions, substrate channeling, solvent accessibility, protein flexibility, and a lot more. [21] Most commonly used residues for linking two proteins are Gly, Ser, and Pro – depending on the aims and goals. Gly and Ser are described as flexible linkers, allowing flexibility for two proteins and improving the protein folding.[ 21 , 30 , 32 ] Meanwhile, Pro linkers reduce interactions of two proteins, increase stiffness and structural independence of the proteins. [33] This will maintain a certain distance between two proteins and keep their independent functions. [21] So far, no study has ever used His tags as linkers of two proteins. Since the His‐tag is beneficial for protein purification and had no negative effect on both of our target enzymes, we hypothesized that it might work also as a linker for the construction of bifunctional fusion proteins. This might support protein purification even further by adding more His residues to the target protein.
To understand the effects of fusing these two proteins together, four constructs were made (Figure 3). The order of the protein sequence can influence structure and function when combining proteins together. [20] Therefore, the position of the proteins at the N‐ and C‐terminal sides was varied. The constructs, AzoRo+FDHC23S and AzoRo+FDHC23SD195QY196H, signify that the AzoRo was placed at the N‐terminus while the FDH is at the C‐terminus (Figure 3). The same concept applies to FDHC23S+AzoRo and FDHC23SD195QY196H+AzoRo, respectively.
Figure 3.
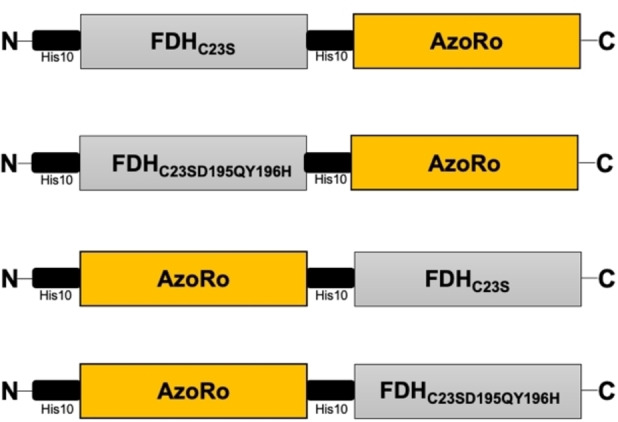
Schematic diagram of the four fusion protein constructs.
Different strategies were used for gene expression since the formation of inclusion bodies was apparent with these fusion proteins. These inclusion bodies in Escherichia coli can be attributed to improper protein folding, aggregation, high rate of gene expression, and degradation. [34] To prevent the possible formation of inclusion bodies, some studies have shown that the components/additives of the growth medium used for gene expression can play a role. LBNB, which has 3‐times more salts than normal LB, exerts high osmotic pressure and therefore leads to accumulation of osmolytes. [35] Osmolytes such as betaine, glycerol, and DMSO can act as a chemical chaperone by increasing protein stability of the native protein and by assisting on the refolding of the unfolded protein. [36] This is because the backbone of a protein in denatured state gets highly exposed to the osmolyte leading to the destabilization of the denatured state due to unfavorable interactions rather than the destabilization of the native state.[ 37 , 38 ] Osmolytes also lead to preferential hydration of the native state of the protein since they mainly act on the denatured state. Meanwhile, presence of ethanol allows the E. coli to mimic a heat‐shock response. [35] Synthesis of heat shock proteins happens, and these heat shock proteins act as molecular chaperones which also help prevent protein aggregation, rearrange disulfide bonds, and enhance solubility of proteins. To address the problem of inclusion bodies, different media containing either high salt concentrations (LBNB) with betaine, glycerol or ethanol were used.
The use of different E. coli strains also plays an important role for protein solubility. The parental strain BL21(DE3) was originally used as an expression strain. However, several trials to purify the fusion proteins from this E. coli strain remained futile – forming inclusion bodies even with the combination of different media to slow down protein production.
Therefore, another approach was made by using different E. coli strains such as SHuffle and C41(DE3).[ 39 , 40 ] The E. coli SHuffle is a derivative of the K‐12 strain that was engineered to facilitate post‐translational modifications such as disulfide bond formation. [39] The C41(DE3) strain is a mutant derivative of BL21(DE3). [40] It was observed that several genes poorly expressed by means of BL21(DE3), likely as a the corresponding proteins were toxic to these cells, were better expressed with C41(DE3). [40] This might be due to the fact that T7 RNA polymerase is faster than the E. coli RNA polymerase in strain BL21(DE3) and therefore proper protein production can be hampered.[ 41 , 42 ] In C41(DE3), a plausible mutation in the T7 RNA polymerase could prevent the uncoupling of transcription and translation and thus lead to improved expression yields.
Large and modified proteins generally need tailored conditions for protein production. The mix and match of different media and different E. coli strains for gene expression was done in this study wherein the combination of C41(DE3) and LBNB led to the soluble production of these His‐linked fusion proteins (Table S1). The study of Oganesyan et al. also showed that the target proteins used for the study showed better solubility with LBNB and betaine. [35] Moreover, using His as linkers might have also affected the solubility of the fusion protein. Therefore, screening for other linkers with varying properties and length can be recommended. FDH of C. boidinii has a size of 44 kDa and AzoRo from R. opacus 1CP has a size of 25 kDa. Together, the fusion protein constructs yield a size of about 70 kDa. The SDS‐PAGE gels showed a band in the 70 kDa range (Figure S1).
Screening of the fusion protein constructs
FDHC23S, regardless of its position in the construct, showed the best activity of NADH production from NAD+. The fusion protein constructs with the FDHC23SD195QY196H, whether it is in the N‐ or C‐terminus, have shown a 4‐fold loss of enzymatic activity for NADH production when comparing to the fusion protein construct with FDHC23S (Figure 4A). Meanwhile, AzoRo activity was greatly impaired when placed on the N‐terminus part of the fusion construct, whether it was paired with FDHC23S or FDHC23SD195QY196H.
Figure 4.

Thermal stability of the (A) FDH part of the fusion protein with 50 mM phosphate buffer, pH 7 containing 125 mM formate and 1 mM NAD+ and the (B) azoreductase part of the fusion protein using 50 mM phosphate buffer, pH 7 containing 150 μM NADH, 50 μM FMN, and 25 μM (1). The protein was incubated in different temperatures and the activity was measured from different time points of incubation up to 2 h. Results for 50 °C was not shown since the enzymes were rendered inactive after 1 h.
For NAD(P)H production, FDHC23SD195QY196H+AzoRo and AzoRo+FDHC23SD195QY196H showed subpar activities compared to the fusion protein constructs with FDHC23S (Figure S2). For checking AzoRo activity using NADPH as a co‐substrate, the constructs that contained AzoRo in the N‐terminal side barely had conversion, as expected (Figure S3).
This just goes to show that when constructing fusion proteins, several outcomes are possible. Some proteins do not get affected at all when placed in different positions in the ORF such as the case of the combination of a Baeyer‐Villiger monooxygenase (BVMO) and a phosphite dehydrogenase (PTDH) where the placement did not play any role to the activities of both proteins. [45] Meanwhile, a different effect was seen in the fusion of an alcohol dehydrogenase (ADH) and a cyclohexanone monooxygenase (CHMO) where the placement of CHMO did not have any influence to its activities but for ADH, the placement showed varying activities. [46]
Since FDHC23S+AzoRo exhibited the best activity from all tested variants for both FDH and AzoRo part, further characterization was done with this construct.
Influence of pH, temperature, and metal ions to FDHC23S+AzoRo
FDHC23S+AzoRo was tested on different pH values to see if the behavior for each part would be different. For the FDH part, the highest activity was achieved with phosphate buffer and Tris‐HCl buffer at pH 8 with an activity of 0.86 U/mgprotein and 0.8 U/mgprotein, respectively (Figure S4). When pH 7 was used, a 50 % drop on its activity was observed for Tris‐HCl while only a 10 % decrease was seen for phosphate buffer. Significant decrease of activity was observed at pH 6. As for the AzoRo from R. opacus 1CP, it was reported that its optimum pH is at pH 6 (Figure S4). [11] It showed about 2 U/mgprotein for both Tris‐HCl and phosphate buffer at pH 6. This showed that the activity of AzoRo in terms of pH is inversely proportional to that of FDH. The activity of AzoRo decreased dramatically by about 7‐fold when pH 7 was used and further decreased at pH 8 (Figure S4). Finding the right pH for the fusion protein to operate on is very crucial as the activities can be affected. As a result, phosphate buffer at pH 7 was used for the subsequent assays.
The effect of temperature to FDHC23S+AzoRo was also investigated. The stability of both parts of the fusion protein was tested wherein the FDH and AzoRo activities were measured from differing time points of incubations for up to 2 h. The enzyme was incubated at different temperatures. As seen in Figure 4, the stability of FDH to 25 °C, 30 °C, and 40 °C was very apparent as the activities were sustained after 2 h. Meanwhile, the activity of AzoRo was more erratic when incubated at different temperatures. At 25 °C, the activity was consistent for 2 h. Increased activities were also observed with 30 °C and 40 °C but prolonged incubation with both temperatures showed a gradual decrease in the activity of AzoRo. After 2 h incubation at 40 °C, a total loss of 50 % can be seen (Figure 4).
At 50 °C, the FDH part already lost 50 % of its activity after 1 h of incubation while AzoRo part had only 33 % of activity after 30 min of incubation (data not shown). After 1 h of incubation, no activity can be detected anymore for AzoRo. This shows that the fusion protein does not act as a single unit as the FDH part was more stable than the AzoRo part. This has not always been the case as the fusion protein containing the phenylacetone monooxygenase (PAMO) and PTDH had similar thermostability as compared to separate enzymes. [45]
It was observed that the addition of metal ions has an effect to the activity of AzoRo wherein the addition of manganese ions led to an increase of activity by 119 %. [11] Different metal ions were also used in this study. No metal ions have shown improved properties to the activity of FDH and AzoRo. As a matter of fact, all metal ions inhibited enzymatic activity (Figure S5), showing more than 50 % decrease in activity especially for the activity of FDH. This was also observed from Lu et al. where the addition of metal ions led to the changes in the AzoRo activity coupled with glucose‐1‐dehydrogenase. [23] Moreover, it can also be speculated that the two His‐tags might interact with the metal ions and thus, affect enzymatic properties.
Insights to the FDH and AzoRo activity of FDHC23S+AzoRo
The kinetic parameters were determined to get a deeper picture on the effects of fusing FDH and AzoRo. The FDHC23S from C. boidinii was also measured to compare the differences between them. As seen in Table 1, the parental FDH still showed better affinity to formate and NAD+. The K M value of the FDH part of the fusion protein FDHC23S+AzoRo worsened by at least 10‐fold for formate and almost 2‐fold for NAD+ (Table 1). Although turnover numbers (k cat) were relatively similar for the free FDH and FDHC23S+AzoRo, the catalytic efficiency of FDHC23S+AzoRo was evidently worse than that of the single enzyme. Some studies have shown how enzyme activity was greatly affected after fusing two proteins. The study of Jiang and Fang showed that the combination of phenylalanine dehydrogenase (PheDH) and FDH also caused severe effects on their kinetic parameters. [19]
Table 1.
Kinetic parameters of FDHC23S activity from Candida boidinii and the FDH activity of FDHC23S+AzoRo at 25 °C. The reaction mixture (1 mL) contained 2.6 μg of enzyme in a 50 mM phosphate buffer, pH 7 with varying mixture of either NAD+ or formate.
|
|
Substrate |
K M [mM] |
k cat [s−1] |
k cat/K M [mM/s] |
|---|---|---|---|---|
|
FDHC23S |
Formate[a] NAD+[b] |
4.23±0.07 0.05±0.02 |
1.78±0.04 1.93±0.12 |
0.42±0.06 35.8±5.32 |
|
FDHC23S + AzoRo |
Formate[a] NAD+[b] |
48.9±4.88 0.08±0.02 |
1.80±0.06 1.37±0.04 |
0.04±0.01 16.3±2.34 |
[a] This indicates that the formate was varied (5 mM to 200 mM) with a constant concentration of 1 mM NAD+ in the reaction. [b] This indicates the NAD+ was varied (0.1 mM to 1 mM) with a constant concentration of 125 mM formate in the reaction.
The parental AzoRo was previously described to only accept (1) as a substrate. Studies have shown that it was more active at acidic pH (pH 4) but the most stable at pH 6. At pH 6, the reduction of (1) with 150 μM NADH was about 20 U/mgprotein. Following NADH consumption when (1) is present, AzoRo showed about a K M of 10.07 μM and a Vmax of 51.48 U/mgprotein. [11] At pH 7, the activity of parental AzoRo decreased, and data fitting was not possible to obtain a proper Michaelis‐Menten kinetics measurement. [28] For reduction of (1), the parental AzoRo from the study showed an activity of about 5.49±0.14 U/mg when 150 μM NADH was used as a co‐substrate and have a k cat of 2.3 s−1 (Table 2). [28] When the artificial electron donor, BNAH (1‐benzyl‐1,4‐dihydronicotinamide), was used, it had an activity of 6.76±0.13 U/mg at 150 μM concentration. [28] With BNAH, it was possible to obtain the kinetic properties of the parental AzoRo at neutral pH, following the reduction of (1). The kinetic parameters were as follows: (K M=12.45±0.47 μM, k cat=3.02 s−1, k cat /K M=0.24 μM/s). Considering that BNAH was an NADH mimic, it could still be observed that the parental AzoRo accepted (1) better. Moreover, it barely accepted (5) (Figure 5).
Table 2.
Kinetic parameters of the AzoRo part from Rhodococcus opacus 1CP and the AzoRo activity of FDHC23S+AzoRo at 25 °C. The reaction mixture (1 mL) contained 2.6 μg of enzyme in a 50 mM phosphate buffer, pH 7 with 150 μM NADH, 50 μM FMN, and varying concentrations of the azo dye (10–60 μM for (1) at 430 nm, 5–40 μM for (5) at 570 nm).
|
|
Substrate |
K M [μM] |
k cat [s−1] |
k cat/K M [μM/s] |
|---|---|---|---|---|
|
AzoRo |
(1) (5) |
n. d.[a] n. d.[b] |
2.32±0.06 n. d.[b] |
n. d.[a] n. d.[b] |
|
FDHC23S+AzoRo |
(1) (5) |
73.9±47.5 6.01±1.39 |
1.58±0.66 0.23±0.02 |
0.021±0.014 0.037±0.011 |
n. d.=not determined. [a] The K M cannot be determined for (1) because no proper Michaelis‐Menten kinetic was possible at pH 7. [28] [b] The substrate (5) was not accepted by the parental AzoRo from Rhodococcus opacus 1CP.
Figure 5.

Specific activity of the individual FDH and AzoRo part of the fusion protein constructs. (A) Production of NADH from NAD+, detecting the activity at 340 nm. The assay was done using 50 mM phosphate buffer, pH 7 containing 125 mM formate and 1 mM NAD+ in a reaction volume of 1 mL. (B) Degradation of (1), detecting the activity at 430 nm. The assay was done using 50 mM phosphate buffer, pH 7 containing 150 μM NADH, 50 μM FMN, and 25 μM (1) in a reaction volume of 1 mL. A total of 2.6 μg protein was used for each reaction.
AzoR from E. coli, an azoreductase with high structural similarity to AzoRo, showed that the prosthetic groups of FMN binds on the C‐terminus end of the enzyme. [43] Given the similarity of these two azoreductases, it can then be speculated that the addition of His‐tag linker and FDH at the C‐terminus of AzoRo may have altered properties of the azoreductase part such as its redox properties. It could be that the placement of the AzoRo also hindered proper folding of the protein resulting to a degraded or a less active enzyme. This detrimental effect was also seen when an elastin‐like polypeptide (ELP) was fused with different proteins wherein the activity of the protein of interest showed differences on its activity when the pentapeptide tag was placed at the N‐terminal part. [44]
With the fusion of FDHC23S to AzoRo, the property of the azoreductase part changed. When looking at the K M values (Table 2), the fusion protein had poorer substrate binding affinity values for (1) than of (5). This meant that (1), which was supposed to be the preferred substrate of AzoRo, cannot bind properly anymore to the catalytic pocket. Meanwhile, (5) showed a lower K M value, suggesting that it was binding better to the catalytic pocket of AzoRo part of the fusion protein. It can be surmised that the catalytic pocket was altered with the addition of the His‐tag linker and FDH for the fusion protein. Although the turnover number for (1) was higher than (5), FDHC23S+AzoRo is more efficient in catalyzing (5) than (1). The low turnover numbers might be because (1) has only one azo bridge and therefore, the catalysis to final products is faster as opposed to (5) with two azo bridges.
In vitro degradation of azo dyes by FDHC23S+AzoRo without NADH supply and the effect of pH
To further prove the activity of FDHC23S+AzoRo as one unit of a bifunctional biocatalyst, degradation of dyes was done without NADH supply. The azo dyes (1), (2), and (5) (Figure 2) were initially tested at pH 6 with a concentration of 25 μM and 100 μM (data not shown). After 1 h, 25 μM of (1) was completely degraded while about 80 % decrease was achieved with (2). (5), on the other hand, was only degraded for about 20 %. For 100 μM concentration, the construct did not perform any better. (1) was not completely degraded. This was unexpected as AzoRo from R. opacus 1CP operates the best at pH 6 on this substrate. [11] Moreover, (5) and (2) were only degraded by 10 % and 40 % at this pH, respectively. This result could be attributed to the impaired supply of NADH as the activity of FDH drops at acidic pH.
As the construct did not operate well at pH 6, degradation of dyes was then tested at pH 7 and pH 8. Additional azo dyes namely, (3), (4), and (6) (Figure 2), were also investigated. These dyes have never been reported to be accepted by the original AzoRo from R. opacus 1CP. All dyes were tested only at 100 μM concentrations at this point.
As shown in Figure 6A and 6B, the construct performed better at pH 7 and pH 8 as (1) and (5) were completely degraded after 20 h reaction. This was a significant change from the previous result with pH 6. (3) was also completely degraded at both pH 7 and pH 8 after 40 h. On the other hand, for the dyes such as (2), (6), and (4), the pH seemed to have influenced the dye degradation process. (2) was degraded up to 75 % at pH 7 while only by 40 % at pH 6 and pH 8. This contrasted with (6) and (4) wherein both dyes were only degraded by 25 % at pH 7 even after 40 h of reaction while (6) was degraded by 60 % and (4) up to 40 % at pH 8. The improved activity of the fusion protein construct, FDHC23S+AzoRo, at pH 7 and pH 8 suggested that the optimal conditions for the FDH part to function properly might be a crucial factor to dye degradation.
Figure 6.
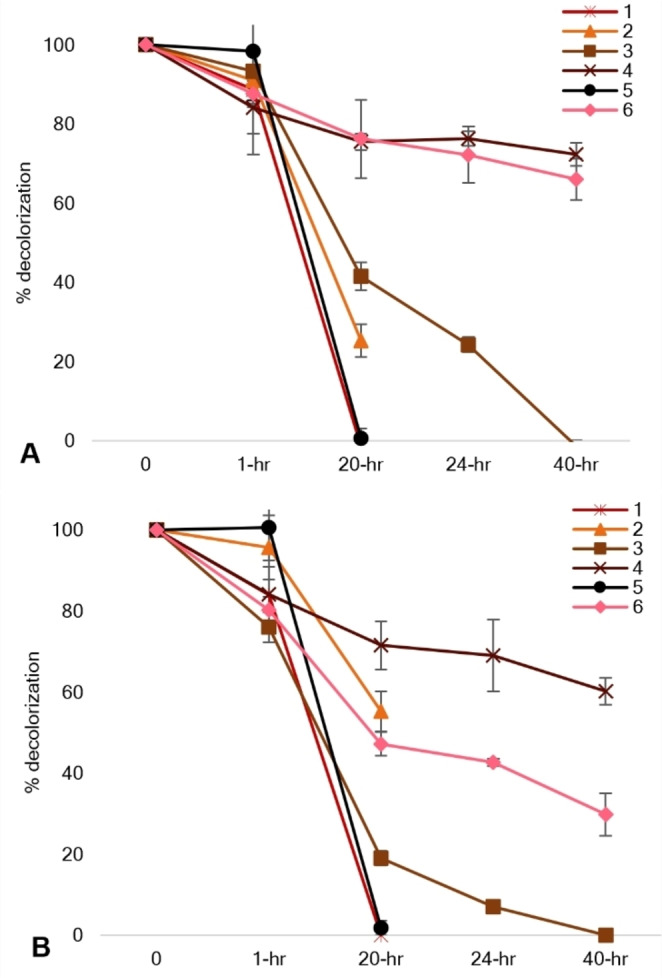
In vitro degradation of the azo dyes by FDHC23S+AzoRo. No NADH was supplied to the reaction. The reaction was done in 50 mM phosphate buffer with 125 mM formate, 1 mM NAD+, 50 μM FMN, 100 μM dye (A) at pH 7 and (B) at pH 8, both setups at 30 °C. A total of 2.6 μg protein was used for each reaction.
As suggested earlier, the activity of FDH to generate NADH from NAD+ was compromised when pH 6 was used. Even though the original AzoRo works optimally at pH 6, the degradation of (1) was also not that efficient. It could be surmised that substrate channeling of NADH for the reduction of azo dyes might have been affected or halted.
Proposed mechanism of degradation by FDHC23S+AzoRo using (5) as a model substrate
Degradation of dyes has been widely documented for whole cell cultures and purified enzymes such as azoreductases. Several studies have already shown how azo dyes with single bridges are being attacked by azoreductases. For example, products of (1) degradation from an azoreductase treatment yield 2‐aminobenzoic acid and N’‐N’‐dimethyl‐p‐phenylenediamine.[ 11 , 47 ] However, only few studies have elucidated how a diazo dye is reduced. For the diazo dye Navitan Fast Blue S5R, the azoreductase from Pseudomonas aeruginosa reduces the dye to three products namely, metanilic acid, 1’4‐diaminonaphthalene, and peri acid. [48] Franconibacter sp., on the other hand, was shown to degrade (6) as confirmed by GC‐MS determination of the products: sulfonamine and 2’5‐diaminobenzenesulfonic acid. [49] On the other hand, 8‐aminonaphthol‐2,5‐disulfonic acid, was not observed but rather, 1‐amino‐2‐naphtol was found – suggesting that the supposed product undergoes further desulfonation by the organism. This demonstrates that the route how azo bonds are attacked by either the organism or the enzyme can differ.
For FDHC23S+AzoRo, the degradation mechanism for (5) was investigated because the native AzoRo was unable to effectively convert (5) [11] but the fusion protein did reduce it (Figure 4). Hence, the fusion character might have changed the structure of AzoRo active site and thus the mechanism or capability to become a more promiscuous enzyme. During this degradation of (5) by FDHC23S+AzoRo, we observed the formation of an orange color. This is in line with earlier reports as Lang et al demonstrated that the azoreductase originating of a Dermacoccus sp. reduced (5). [49] There the substrate underwent a two‐step reduction process with the first reduction happening with the azo linkage between the naphthalene rings, producing an orange by‐product. [49] Hence, this by‐product was also observed after treatment of (5) by FDHC23S+AzoRo. Several trials and attempts were made to verify the process. The reactions were also further incubated to see if the orange by‐product will be further reduced but the result was futile.
To further investigate mechanism and orange by‐product observed, HPLC analysis was first used to compare the changes between the control and the sample. A peak from the control was eluting at 5.4 min with a wavelength of 574 nm, corresponding to (5). After treatment of the substrate (5) by the fusion protein, this peak decreased and another peak appeared with a wavelength of 450 nm, corresponding to the orange by‐product also detected from the study of Lang et al. [50] This orange intermediate of (5) degradation was further reduced by Dermacoccus sp. and therefore produced a colorless solution and completed the two‐step degradation process. [50] In contrast in our investigation, the constructed fusion FDHC23S+AzoRo did not produce this colorless solution. Hence, only one of the two azo bonds was cleaved.
LC‐MS was then used to detect the products from the reaction. The signal spectra from the MS showed three fragment ions of m/z 202.5 [M−2H]2−, 405.9 [M−H]−, and 427.9 [M−2H+Na]− and correspond to the compound 8‐amino‐5‐((4‐sulfonatophenyl)diazenyl) naphthalene‐2‐sulfonic acid (Compound 4), the orange by‐product (Figure 7). Meanwhile, the other naphthol‐based compound (Compound 3) from the first reduction cannot be detected. The absence of (E)‐4‐acetamido‐6‐((4‐amino‐7‐sulfonaphthalen‐1‐yl)diazenyl)‐5‐hydroxynaphthalene‐1,7‐disulfonic acid (Compound 2) and only a slight signal of fragment ions with m/z 172 [M−H]−, corresponding to sulfanilic acid (Compound 1) indicate a clear preference for the 1st‐cleavage site (Figure 7). Additional slight signal of fragment ions with m/z 237 [M−H]− corresponding to 5,8‐diaminonaphthalene‐2‐sulfonic acid (Compound 5), imply that the orange intermediate was further cleaved at the 2nd‐cleavage site to some minor extent, which is in agreement with the small amounts of Compound 6 detected (Figure 7). It is therefore hypothesized that the dye underwent only a partial degradation process with clear preference for the 1st‐cleavage site and only minor cleavage at the 2nd‐cleavage site (5).
Figure 7.

Proposed degradation mechanism of (5) by the fusion protein construct, FDHC23S+AzoRo.
Conclusions
The study shows that the fusion of an NADH regeneration system such as formate dehydrogenase and an azoreductase through His‐tag linkers is possible. However, careful construction of the position and design of a bifunctional catalyst affects the separate activities of the enzymes. Further research can be done by trying different linkers such as flexible linkers, rigid linkers, or helical linkers to investigate the influence of linkers to the fusion proteins. It is important to check factors such as pH as it greatly influenced the dyes that were degraded by FDHC23S+AzoRo. Moreover, it was observed that the structure of the azoreductase might have also been affected as the substrate promiscuity improved, allowing bulkier substrates to be accepted.
Experimental Section
General experimental methods: Chemicals and reagents were purchased from TCI and Sigma‐Aldrich. The expression vector, pET16bP, was used for the cloning of formate dehydrogenase (FDH) and azoreductase (AzoRo).[ 11 , 51 , 52 , 53 ] All constructs were initially transformed to Escherichia coli DH5α then the plasmids were isolated using NucleoSpin plasmid mini prep kit for plasmid DNA (Macherey‐Nagel). Preculture for expression strains were done using lysogeny broth (LB) with 10 g/L of tryptone, 10 g/L of NaCl, 5 g/L of yeast extract. [53] A final concentration of 100 μg/mL ampicillin was used for LB broth and agar plates. Cultures were incubated at 37 °C for 24 h, 140 rpm.
Plasmid construction and cloning of the fusion protein: The cloning of the codon‐optimized azoRo (KT923288) was described in the study of Qi et al. [11] Meanwhile, the gene fdh was also codon optimized for protein expression. The codon optimized fdh (OL449253) was synthesized (Eurofins) in a pEX_K4 plasmid. The gene was obtained via restriction digestion of NdeI and NotI sites. Both the azoRo and fdh fragments were ligated into a pET16bP vector using the same restriction sites. E. coli DH5α was transformed with the plasmids and the plasmids were then isolated using the GeneJET Plasmid Mini Prep Kit (ThermoFischer). The inserts were checked via PCR using pETcheck primers (Table 3). The construction of the FDHC23SD195QY196H was done by using the pET16bP_FDHC23S as a template for a PCR reaction, amplifying the gene of interest with the overlapping primers C and D (Table 3) to change Asp195 to Glu and Tyr196 to His, allowing the possibility to accept NADP+. [27]
Table 3.
Primers used in the study. Each primer is designated with a letter.
|
Primer name |
Primer sequence |
|
|---|---|---|
|
PET_check fw |
5’‐CATCACAGCAGCGGCCATATCGAAG‐3’ |
A |
|
PET_check rv |
5’‐CAGCTTCTTTTCGGGCTTTGTTAG‐3’ |
B |
|
FDH_ D195QY196H_ fw |
5’‐TTAACCCGAAAGAACTGCTGTATTACCAG CATCAGGCATTACCGAAAGAAGCCG‐3’ |
C |
|
FDH_ D195QY196H_rv |
5’‐CGGCTTCTTTCGGTAATGCCTGATGC TGGTAATACAGCAGTTCTTTCGGGTTAA‐3’ |
D |
|
PET‐fw‐Gibson |
5’‐TGAGCGGCCGCACTTAAGTTACG CGTGGATCC‐3’ |
E |
|
PET‐Azorev‐Gibson |
5’‐ACGTACAAGAGAAGTCACCCAACGTTGGTC‐3’ |
F |
|
PET‐FDHrev‐Gibson |
5’‐TTTCTTATCGTGTTTCCCATACGCTTTCG‐3’ |
G |
|
Overlap‐AzoFDH‐Gibson |
5’‐GGGTGACTTCTCTTGTACGTGGCCATCATCAT CATCATCATC‐3’ |
H |
|
Overlap‐FDHAzo‐Gibson |
5’‐ATGGGAAACACGATAAGAAAGGCCATCAT CATCATCATCATC‐3’ |
I |
|
Overlap‐insert‐PET |
5’‐GTAACTTAAGTGCGGCCGCTCA‐3’ |
J |
|
AzoRo_ internalcheck |
5’‐GAGCACCTGGTTCCATTAGC‐3’ |
K |
|
FDH_ internalcheck |
5’‐GGCAAATTTGATTATCGCCCAC‐3’ |
L |
The plasmids, pET16bP_AzoRo, pET16bP_FDHC23S and pET16bP_FDHC23SD195QY196H, were used as a parental template for the construction of the fusion proteins. A total of four genes encoding the fusion proteins were cloned via Gibson assembly. [55] As linker, a sequence encoding for a His10‐tag was placed between the genes of azoRo and fdh.
For pET16bP_AzoRo+FDHC23S and pET16bP_AzoRo+FDHD195QY196H, pET16bP_AzoRo was linearized via PCR (Eppendorf) using primers E and F (Table 3) with 30 cycles and used as the vector. Each cycle consists of denaturation for 15 s at 95 °C, annealing for 15 s at 60 °C, and elongation for 1 min 45 s at 72 °C. The inserts FDHC23S and FDHD195QY196H was amplified via PCR from pET16bP_FDH using primers H+J (Table 3) with the following conditions: denaturation for 15 s at 95 °C, annealing for 15 s at 58 °C, and elongation for 45 s at 72 °C. The two PCR products were digested overnight with DpnI and ligated together using NEBuilder Assembly Mastermix (NEB).
For pET16bP_FDHC23S+AzoRo and pET16bP_FDHD195QY196H+AzoRo, the PCR conditions were the same. However, pET16bP_FDH was used as a template and primers E+G were used for vector amplification while for the insert AzoRo, the pET16bP_AzoRo was used as a template and primers I+J were used for the insert amplification.
E. coli DH5α was transformed with the constructs and the plasmids were purified (ThermoFisher Genejet Plasmid Miniprep Kit). The plasmids were controlled via Sanger sequencing (Microsynth Seqlab) using the primer K or L to check if the proper inserts were incorporated.
Gene expression and protein purification: Plasmids of the fusion protein constructs were transformed to different E. coli T7 expression systems such as BL21(DE3), SHuffle, and C41(DE3). Transformation was done through heat‐shock method by subjecting the cells at 42 °C for 1 min. The cells were placed on ice for 3 min and a 950 μL antibiotic‐free LB broth was pipetted into the tube. The regeneration was done at 37 °C for 1 h. The cells were then spun down at 4,700×g for 7 min (ThermoScientific Heraeus Fresco 17). The supernatant was discarded, and the pellets were resuspended with LB medium. A 50 μL aliquot was plated onto an LB agar plate with ampicillin (100 μg/mL) and the plates were incubated at 37 °C for 24 h.
Isolated colony from the LB agar plate was taken using a sterile inoculating loop and transferred to an LB medium to serve as a preculture. After 24 h, the precultures were transferred to a fresh expression medium. Different expression media such as LB, LBNB, LB with 10 % glycerol, and LB with 1 % ethanol were used to optimize the best conditions for the fusion proteins production. The cultures were grown at 37 °C until an OD of 0.5–0.8 was obtained. The temperature was lowered to 20 °C and a final concentration of 0.1 mM isopropyl‐β‐D‐1‐thiogalactopyranoside (IPTG) was added to the culture for induction. The cells were incubated overnight and were harvested the next day at 5,000×g for 30 min (Sorvall RC 5C Plus). Cell pellets were stored at −20 °C for further use.
For protein purification, the cell pellets were thawed and resuspended with 25 mM Tris‐HCl buffer, pH 7.5. DNAse I was added to the solution. The cells were then lysed using an ultrasonic cell disruptor with an MS72 tip (Sonopuls, Bandelin, VWR). The conditions for the cell lysis were as follows: 30 s with 1 min rest, repeating the process 10 times. The disrupted cell cultures were centrifuged for 30 min at 17,000×g at 4 °C (ThermoScientific Heraeus Fresco 17) to obtain the crude extract.
The crude cell extracts were then subjected for protein purification through fast protein liquid chromatography (FPLC, Äkta Start, GE Healthcare) using a nickel affinity column (HisTrap, GE Healthcare). Tris‐HCl buffer (25 mM Tris, 500 mM NaCl, pH 7.5) was used as the binding buffer. The bounded fusion protein was eluted from the nickel column by employing a gradient of 20 to 100 % Tris‐HCl buffer containing imidazole (25 mM Tris, 500 mM NaCl, 500 mM imidazole, pH 7.5). The purified proteins were precipitated using a saturated ammonium sulfate solution. Pellets obtained were then re‐dissolved with 100 mM phosphate buffer, pH 7.
Different samples were taken during protein expression and purification such as after induction, crude extract, cell pellet, flowthrough, washing, and elution for sodium dodecyl sulfate‐polyacrylamide gel electrophoresis (SDS‐PAGE) analysis. [55] This was to verify the fusion protein according to its size. The protein samples were mixed with a 2× SDS sample buffer (300 mM Tris‐HCl pH 6.8, 4 % SDS, 30 % glycerol, 5 % β‐mercaptoethanol, 0.2 % dithiothreitol, and 0.01 % bromophenol blue) and were denatured at 90 °C for 10 mins. The protein samples were then pipetted into the wells of the SDS gel and the Thermo Scientific PageRuler prestained protein ladder was used as the marker. Proteins were quantified using the Bradford Assay (Rotiquant; Carl Roth, Germany) using bovine serum albumin as the standard.[ 56 , 57 ]
Biochemical characterization of the fusion protein: The activities of the fusion protein were measured using a UV‐vis spectrophotometer (Cary 60 UV‐vis Agilent) at 25 °C. FDH part of FDHC23S+AzoRo was measured in a quartz cuvette containing 1 mM NAD+, 125 mM formate, and the enzyme. NADH generation at 340 nm was followed.
Meanwhile, azoreductase part of FDHC23S+AzoRo was also carried out in a quartz cuvette containing 150 μM NADH, 50 μM FMN, 25 μM of the azo dye substrate, and the enzyme. Degradation of azo dyes at their respective wavelengths were followed ((1) 430 nm and (5) 571 nm).
The optimum pH for each enzyme of the fusion protein was determined by checking activities for each segment at pH 6, 7, and 8. For thermal stability, the proteins were incubated at different time points and the enzyme activity was tested for each time point. For metal ions, the components for a cuvette reaction were the same but just supplemented with additional metal ions (Cu2+, Mg2+, Ca2+, Mn2+, Zn2+) with a final concentration of 50 μM.
Kinetic analysis of the fusion protein: Steady‐state kinetic measurements were done as described above. Initial velocities of the enzymatic reaction for FDH and AzoRo part were measured. Apparent kinetic parameters were obtained through a non‐linear Michael‐Menten assumption. The specific activity was determined as described previously wherein 1 U represents the conversion of 1 μmol NAD+, 1 μmol NADH or substrate per min. For all enzymatic reactions, a total of 2.6 μg of proteins was used.
In vitro degradation of azo dyes and assessment of substrate scope via HPLC: To further prove that the fusion protein was functional, in vitro degradation of dyes was conduction. The components of the biocatalytic reactions comprised of 125 mM formate, 1 mM NAD+, 50 μM FMN, 100 μM of the substrate, and 2.6 μg enzyme. The reactions were done on a 1.5 mL microcentrifuge tube at 30 °C with a shaking of 600 rpm (LLG‐uniTHERMIX 2 pro). A 50 μL volume was obtained from the samples and the reaction was stopped with equal parts of methanol (MeOH). The mixture was then centrifuged for 30 min and the supernatant was carefully taken. The reaction sampling time points were done after 1 h, 20 h, 24 h, 40 h. Additional setup without the enzymes served as a negative control. All measurements were done in triplicates.
The samples were subjected to reverse phase – high pressure liquid chromatography (RP‐HPLC) analysis (ThermoScientific Dionex Ultimate 3000) to determine if the azo dyes were really degraded and that the results observed were not just because of pH change.
A C18 reversed‐phase column (Knauer Eurospher 100‐5 C18, 125×4 mm) was used. Different instrument methods were employed to elute the azo dyes properly. For (1), the dye was eluted using a 40–95 % gradient of MeOH with 0.1 % TFA H2O at a flow rate of 0.7 mL/min, detecting it at 490 nm. For (5), an isocratic method of 50 % MeOH and 50 % H2O with 50 mM ammonium acetate was used at a flow rate of 1.0 mL/min, via 575 nm. (2) was also eluted using an isocratic method composed of 60 % MeOH and 40 % H2O with 0.1 % TFA at a flow rate of 0.7 mL/min, detecting it at 440 nm. For (6) and (4), 520 nm and 508 nm were used to detect the dyes, respectively, with a buffer composition of 70 % MeOH and 30 % ammonium acetate at 0.7 mL/min. For (3), 80 % MeOH and 20 % H2O with 0.1 %TFA was used at a flow rate of 0.7 mL/min, detecting it at 375 nm.
Extraction and analysis of dye degradation product: LC‐MS analysis of the decolorization products were performed with a LC‐MS 8030 (Shimadzu). Separation was conducted through a reverse phase C18 column (Kinetex®, 2.6 μm, 100 Å, 100×2.1 mm) and a guard column at 40 °C with mobile phase A, consisting of water with 10 mM ammonium acetate (LiChropur™, Merck, Germany) and mobile phase B, consisting of methanol. Elution program was 10 % B for the first 2 min, followed by a linear gradient up to 90 % B for 6 min, at a flow rate of 0.4 mL/min. Products were detected by a diode array detector (DAD) at 254 nm. Mass spectrometry was performed with an electron spray ionization (ESI) unit in negative mode.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
A. C. N. was supported by a pre‐doctoral scholarship from Katholischer Akademischer Ausländer‐Dienst (KAAD). F. S. was supported by Federal Ministry of Education and Research in the frame of the BioConversion project (BMBF; 03INT513BF). D. T. acknowledges the DFG Research Training Group GRK 2341 “Microbial Substrate Conversion (MiCon)” for support and training of our graduate students. We thank for the Open Access funding which was provided by Projekt DEAL and Ruhr‐Universität Bochum, Germany. Open Access funding enabled and organized by Projekt DEAL.
A. C. R. Ngo, F. P. J. Schultes, A. Maier, S. N. H. Hadewig, D. Tischler, ChemBioChem 2022, 23, e202100643.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1. Saratale R. G., Saratale G. D., Chang J. S., Govindwar S. P., J. Inst. Chem. 2011, 42, 138–157. [Google Scholar]
- 2. Husain Q., Crit. Rev. Biotechnol. 2006, 26, 201–221. [DOI] [PubMed] [Google Scholar]
- 3. Pandey A., Singh P., Iyengar L., Int. Biodeterior. Biodegrad. 2007, 59, 73–84. [Google Scholar]
- 4. Forgacs E., Cserhati T., Oros G., Environ. Int. 2004, 30, 953–971. [DOI] [PubMed] [Google Scholar]
- 5. Misal S. A., Gawai K. R., Biores. Bioproc. 2018, 5, 1–9. [Google Scholar]
- 6. Kalyani D. C., Telke A. A., Dhanve R. S., Jadhav J. P., J. Hazard. Mater. 2009, 163, 735–742. [DOI] [PubMed] [Google Scholar]
- 7. Lade H. S., Waghmode T. R., Kadam A. A., Govindwar S. P., Int. Biodeterior. Biodegrad. 2012, 72, 94–107. [Google Scholar]
- 8. Goud B. S., Cha H. L., Koyyada G., Kim J. H., Curr. Microbiol. 2020, 77, 3240–3255. [DOI] [PubMed] [Google Scholar]
- 9. Suzuki H., Appl. Microbiol. Biotechnol. 2019, 103, 3965–3978. [DOI] [PubMed] [Google Scholar]
- 10. Chen H., Feng J., Kweon O., Xu H., Cerniglia C. E., BMC Biochem. 2010, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qi J., Schlömann M., Tischler D., J. Mol. Catal. B 2016, 130, 9–17. [Google Scholar]
- 12. Liu W., Wang P., Biotechnol. Adv. 2007, 25, 369–384. [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Saba T., Yiu H. H., Howe R. F., Anderson J. A., Shi J., Chem 2017, 2, 621–654. [Google Scholar]
- 14. Saba T., Burnett J. W., Li J., Wang X., Anderson J. A., Kechagiopoulos P. N., Wang X., Catal. Today 2020, 339, 281–288. [Google Scholar]
- 15. Ferry J. G., FEMS Microbiol. Rev. 1990, 7, 377–382. [DOI] [PubMed] [Google Scholar]
- 16. Tishkov V. I., Popov V. O., Biochemistry 2004, 69, 1252. [DOI] [PubMed] [Google Scholar]
- 17. Weckbecker A., Hummel W., in Microbial Enzymes and Biotransformations, Methods in Biotechnology, Vol. 17 (Ed.: Barredo J. L.), Humana, 2005, pp. 225–238. [Google Scholar]
- 18. Xu Z., Jing K., Liu Y., Cen P., J. Ind. Microbiol. Biotechnol. 2007, 34, 83–90. [DOI] [PubMed] [Google Scholar]
- 19. Jiang W., Fang B. S., J. Ind. Microbiol. Biotechnol. 2016, 43, 577–584. [DOI] [PubMed] [Google Scholar]
- 20. Aalbers F. S., Fraaije M. W., ChemBioChem 2019, 20, 20–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chen X., Zaro J. L., Shen W. C., Adv. Drug Delivery Rev. 2013, 65, 1357–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klein J. S., Jiang S., Galimidi R. P., Keeffe J. R., Bjorkman P. J., Protein Eng. Des. Sel. 2014, 27, 325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu L. L., Yang Y. Y., Lin H., Gao F., Zhao Y. H., Int. Biodeterior. Biodegrad. 2014, 87, 81–86. [Google Scholar]
- 24. Rathod J., Dhebar S., Archana G., Int. Biodeterior. Biodegrad. 2017, 124, 91–100. [Google Scholar]
- 25. Dong H., Guo T., Zhang W., Ying H., Wang P., Wang Y., Chen Y., Int. J. Biol. Macromol. 2019, 140, 1037–1046. [DOI] [PubMed] [Google Scholar]
- 26. Slusarczyk H., Felber S., Kula M. R., Pohl M., Eur. J. Biochem. 2000, 267, 1280–1289. [DOI] [PubMed] [Google Scholar]
- 27. Andreadeli A., Platis D., Tishkov V. I., Popov V. O., Labrou N. E., FEBS J. 2008, 275, 3859–3869. [DOI] [PubMed] [Google Scholar]
- 28. Qi J., Paul C. E., Hollmann F., Tischler D., Enzyme Microb. Technol. 2017, 100, 17–19. [DOI] [PubMed] [Google Scholar]
- 29. Ngo A. C. R., Qi J., Juric C., Bento I., Tischler D., Arch. Biochem. Biophys. 2022, 717, 109123. [DOI] [PubMed] [Google Scholar]
- 30. Argos P., J. Mol. Biol. 1990, 211, 943–958. [DOI] [PubMed] [Google Scholar]
- 31. George R., Heringa J., Protein Eng. 2002,15, 871–879. [DOI] [PubMed] [Google Scholar]
- 32. Waldo G., Standish B., Berendzen J., Terwilliger T., Nat. Biotechnol. 1999, 17, 691–695. [DOI] [PubMed] [Google Scholar]
- 33. Williamson M. P., Biochem. J. 1994, 297, 249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bhatwa A., Wang W., Hassan Y. I., Abraham N., Li X. Z., Zhou T., Front. Bioeng. Biotechnol. 2021, 9, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Oganesyan N., Ankoudinova I., Kim S. H., Kim R., Protein Expression Purif. 2007, 52, 280–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aioanei D., Lv S., Tessari I., Rampioni A., Bubacco L., Li H., Samori B., Brucale M., Angew. Chem. Int. Ed. 2011, 50, 4394–4397; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4486–4489. [Google Scholar]
- 37. Gekko K., Timasheff S. N., Biochemistry 1981, 20, 4677–4686. [DOI] [PubMed] [Google Scholar]
- 38. Bolen D. W., Baskakov I. V., J. Mol. Biol. 2001, 310, 955–963. [DOI] [PubMed] [Google Scholar]
- 39. Lobstein J., Emrich C. A., Jeans C., Faulkner M., Riggs P., Berkmen M., Microb. Cell Fact. 2012, 11, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miroux B., Walker J. E., J. Mol. Biol. 1996, 260, 289–298. [DOI] [PubMed] [Google Scholar]
- 41. Iost I., Dreyfus M., EMBO J. 1995, 14, 3252–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Makarova O. V., Makarov E. M., Sousa R., Dreyfus M., Proc. Natl. Acad. Sci. USA 1995, 92, 12250–12254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ito K., Nakanishi M., Lee W. C., Sasaki H., Zenno S., Saigo K., Kitade Y., Tanokura M., J. Biol. Chem. 2006, 281, 20567–20576. [DOI] [PubMed] [Google Scholar]
- 44. Christensen T., Amiram M., Dagher S., Trabbic-Carlson K., Shamji M. F., Setton L. A., Chilkoti A., Protein Sci. 2009, 18, 1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Torres Pazmiño D. E., Snajdrova R., Baas B. J., Ghobrial M., Mihovilovic M. D., Fraaije M. W., Angew. Chem. 2008, 120, 2307–2310; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2008, 47, 2275–2278. [DOI] [PubMed] [Google Scholar]
- 46. Aalbers F. S., Fraaije M. W., Appl. Microbiol. Biotechnol. 2017, 101, 7557–7565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vatandoostarani S., Lotfabad T. B., Heidarinasab A., Yaghmaei S., Int. Biodeterior. Biodegrad. 2017, 125, 62–72. [Google Scholar]
- 48. Nachiyar C. V., Rajakumar G. S., Enzyme Microb. Technol. 2005, 36, 503–509. [Google Scholar]
- 49. Baena-Baldiris D., Montes-Robledo A., Baldiris-Avila R., ACS Omega 2020, 5, 28146–28157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lang W., Sirisansaneeyakul S., Martins L. O., Ngiwsara L., Sakairi N., Pathom-aree W., Okuyama M., Mori H., Kimura A., J. Environ. Manage. 2014, 132, 155–164. [DOI] [PubMed] [Google Scholar]
- 51. Riedel A., Heine T., Westphal A. H., Conrad C., Rathsack P., van Berkel W. J. H., Tischler D., AMB Expr. 2015, 5, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tischler D., Kermer R., Gröning J. A. D., Kaschabek S. R., van Berkel W. J. H., Schlömann M., J. Bacteriol. 2010, 192, 5220–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bertani G., J. Bacteriol. 1951, 62, 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gibson D. G., Young L., Chuang R. Y., Venter J. C., Hutchison C. A., Smith H. O., Nat. Methods 2009, 6, 343–345. [DOI] [PubMed] [Google Scholar]
- 55. Tischler D., Eulberg D., Lakner S., Kaschabek S. R., van Berkel W. J., Schlömann M., J. Bacteriol. 2009, 191, 4996–5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bradford M. M., Anal. Biochem. 1976, 72, 248–254. [DOI] [PubMed] [Google Scholar]
- 57. Oelschlägel M., Zimmerling J., Schlömann M., Tischler D., Microbiology 2014, 160, 2481–2491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.