

Abstract
H2O2 is a bulk chemical used as “green” alternative in a variety of applications, but has an energy and waste intensive production method. The electrochemical O2 reduction to H2O2 is viable alternative with examples of the direct production of up to 20% H2O2 solutions. In that respect, we found that the dinuclear complex Cu2(btmpa) (6,6’‐bis[[bis(2‐pyridylmethyl)amino]methyl]‐2,2’‐bipyridine) reduces O2 to H2O2 with a selectivity up to 90 % according to single linear sweep rotating ring disk electrode measurements. Microbalance experiments showed that complex reduction leads to surface adsorption thereby increasing the catalytic current. More importantly, we kept a high Faradaic efficiency for H2O2 between 60 and 70 % over the course of 2 h of amperometry by introducing high potential intervals to strip deposited copper (depCu). This is the first example of extensive studies into the long term electrochemical O2 to H2O2 reduction by a molecular complex which allowed to retain the high intrinsic selectivity of Cu2(btmpa) towards H2O2 production leading to relevant levels of H2O2.
Keywords: copper complexes, electrocatalysis, homogeneous catalysis, hydrogen peroxide, oxygen reduction
Optimised catalytic activity: Electrochemical H2O2 production is a promising sustainable alternative to the anthraquinone method. In that perspective, we have studied the dinuclear copper complex Cu2(btmpa) that has a high intrinsic H2O2 selectivity. By extensive, long term studies, we found that complex accumulation on the electrode increases the catalytic activity and that high potential intervals strip away deposited copper. Through this procedure we were able to reach a 60 to 70 % Faradaic efficiency during long term amperometry experiments.

1. Introduction
H2O2 is a bulk chemical that is produced on a 4.5 million ton scale [1] and used in many applications [2] such as bleaching (largest single use), [3] waste water treatment, [4] disinfecting, and industrial organic synthesis. [5] It is considered as an environmentally friendly chemical oxidant because the decomposition products are water and/or O2. However, its current production method is far from environmentally friendly. Over 90 % of the worldwide H2O2 production proceeds via the anthraquinone process.[ 2 , 6 ] Here, anthraquinones are used as redox mediators that first undergo reduction with H2, followed by a separate re‐oxidation in the presence of air (O2) which produces H2O2 selectively. Liquid‐liquid extractions are required to extract H2O2 given that these reactions take place in organic solvent. Consequently, the obtained H2O2 is contaminated with organic impurities. As a result, most of the cost and energy of producing H2O2 result from the purification of this extract.
The electrochemical reduction of O2 to H2O2 is a viable alternative and was first reported in 1939 by Berl. [7] In fact, it has been industrialized in the Huron‐Dow process which is mostly used for on‐site production of alkaline peroxide mixtures for the paper bleaching industry. Nevertheless, this only covers a negligible fraction of the total H2O2 production.[ 2 , 6 ] To overcome the problem of separating the H2O2 from the aqueous electrolyte, solid electrolyte cells in combination with flow cell chemistry have recently been proposed as a feasible option. [8] The cathode, where O2 reduction takes place, can be made of several materials. Noble metals usually catalyze the full 4 electron reduction to H2O or they interact weakly with O2 resulting in low rates and a high overpotential. Alloys combine these characteristics and result in better catalysts. [9] Another interesting approach is the use of carbon based electrodes because these have an intrinsic selectivity towards the formation of H2O2 when performing O2 reduction. [10] Nevertheless, their reactivity is quite poor, and application of such materials therefore requires large overpotentials. [9a] Improvements can be made by increasing the defect [11] and/or oxygen content, [12] doping with heteroatoms, [13] or doping with metals as single‐site catalysts. The latter approach is challenging since metal–support interactions for carbon are relatively weak. [9a] In those cases, molecular complexes can improve adsorption through the ligand‐carbon interactions. Most molecular catalysts, that have been reported to perform the reduction of O2 to H2O2, have only been studied in non‐aqueous solvents. [14] Mechanisms and selectivity depend significantly on the acid type and acid strength and cannot be directly translated to aqueous solutions. Until now, high selectivity for electrocatalytic H2O2 production in aqueous solutions is observed only for a few manganese, [15] iron, [16] copper,[ 15b , 17 ] and cobalt complexes.[ 15b , 16d , 18 ] The initial high selectivity for H2O2 is generally restricted to a small potential window and only observed for a few minutes. Longer measurements are rarely performed and if so, they typically result in an overall 4 electron selectivity either due to over‐reduction of H2O2 or due to the disproportionation of H2O2, also catalyzed by the same molecular catalysts. [16b] Thus far, there is only the exception of a cobalt tetrakis(N‐methyl‐4‐pyridyl)porphyrin complex that was reported with high selectivity (>90 %) for H2O2 after 2 h of electrolysis, but no further details were provided to support this claim. [18a]
Our group reported [Cu(tmpa)(L)]2+ (Cu(tmpa) (Scheme 1), tmpa=tris(2‐pyridylmethyl)amine, L=solvent) for the electrochemical 4 electron reduction of O2 to H2O that proceeds in a stepwise mechanism with H2O2 as detectable intermediate. [17] At pH 7, two separate catalytic cycles for O2 to H2O2 and H2O2 to H2O reduction take place with onsets of 0.50 and 0.45 V versus the reversible hydrogen electrode (RHE), respectively. Cu(tmpa) is an intrinsic very fast catalyst for the O2 to H2O2 reduction, but over‐reduction of H2O2 to H2O is fast as well. For that reason, we set out to investigate the dicopper complex [Cu2(btmpa)(L)4]4+ (Cu2(btmpa), btmpa=6,6’‐bis[[bis(2‐pyridylmethyl)amino]methyl]‐2,2’‐bipyridine) which is consisting of two Cu(tmpa) moieties fused via a covalent bond between one of the three pyridines on each moiety resulting in a bipyridine backbone (Scheme 1). [19] An earlier report suggested that the CuI complex [Cu2(btmpa)(MeCN)2(ClO4)2]2+ has a CuI geometry that was different from CuI(tmpa), consequently a positively shifted redox potential of the CuII/I redox couple compared to Cu(tmpa). As a result isolated and well‐characterized [CuI 2(btmpa)](ClO4)2 shows a diminished reactivity towards O2 with respect to Cu(tmpa) in acetonitrile. [19c] We found that these changes in geometric properties in particular affected the electrochemical H2O2 reduction resulting in a high selectivity up to 100 % for H2O2. In addition, we performed the first systematic study of long bulk electrosynthesis of H2O2 by a molecular catalyst. By performing long term amperometry measurements, we were able to identify factors that limit the Faradaic efficiency; were able to improve the overall electrosynthesis process, and thereby achieved a record breaking Faradaic efficiency of 65 % for H2O2 over the course of 2 h.
Scheme 1.

Structures of Cu(tmpa) and Cu2(btmpa). L=H2O in an aqueous solution, and probably rapidly exchanging with phosphate in a phosphate buffer.
2. Results and Discussion
2.1. The (electronic) structure of Cu2(btmpa)
The dinuclear complex Cu2(btmpa) was synthesized from the btmpa ligand and Cu(OTf)2, while the ligand was synthesized according to an earlier reported synthesis that was slightly adjusted to increase the purity and yield (see supporting info). [19b] EPR and SQUID measurements did not show a large coupling interaction between the two Cu2+ centers of the complex (Figures S1 and S2). We found the copper centers of Cu2(btmpa) are reduced simultaneously in an 0.1 m phosphate buffer solution of pH 7 (Figure S3), similar to earlier studies in organic solvents. [19c] In addition, the CuII/I redox potential shifted 0.3 V positively with respect to the mononuclear Cu(tmpa) towards 0.51 V versus the Reversible Hydrogen Electrode (RHE). A previously published crystal structure of a [(btmpa)Cu2(CH3CN)2(ClO4)2]2+ complex showed that the Cu−N bond of the bipyridine moiety has longer distances (2.4 Å) than the other pyridines (2.0 Å). [19c] As a result, the CuII site is likely less electron dense than Cu(tmpa) which explains the positive shift of the CuII/I redox couple. The CuII/I redox couple of Cu2(bmpta) has a relatively large peak separation, which increases with increasing scan rate (Figure S3B). In line with Marcus theory, in which a higher reorganization energy is linked to slower electron transfer, [20] this points to a relative slow electron transfer process. In contrast to Cu2(btmpa), the reduction of the mononuclear Cu(tmpa) complex is a very fast process [17] due to the easy transition of a trigonal bipyramidal geometry of the CuII complex to the preferred tetragonal geometry for the CuI state by the elongation of Cu−N distance of the tertiary amine from 2.10 to 2.43 Å. [21] In contrast, the CuII geometry of Cu2(btmpa) leans towards a more pseudo‐octahedral geometry, [19c] and it seems unlikely that Cu2(btmpa) can easily obtain the preferred tetragonal geometry for the CuI state during redox state changes.
2.2. Electrocatalysis in presence of Cu2(btmpa)
During reduction from the +II to the +I oxidation state Cu2(btmpa) has a tendency to adsorb on the electrode. This behavior was studied in detail with electrochemical quartz crystal microbalance (EQCM) studies (Figure 1 and Figure S4), which showed that the reduction of Cu2(btmpa) from a total 4+charge to 2+charge does trigger adsorption on the electrode, whereas electrochemical oxidation triggers desorption. Although the EQCM data show that the potential‐dependent adsorption is reversible on gold electrodes, the carbon‐based GC electrode might have a stronger affinity with Cu2(btmpa).
Figure 1.
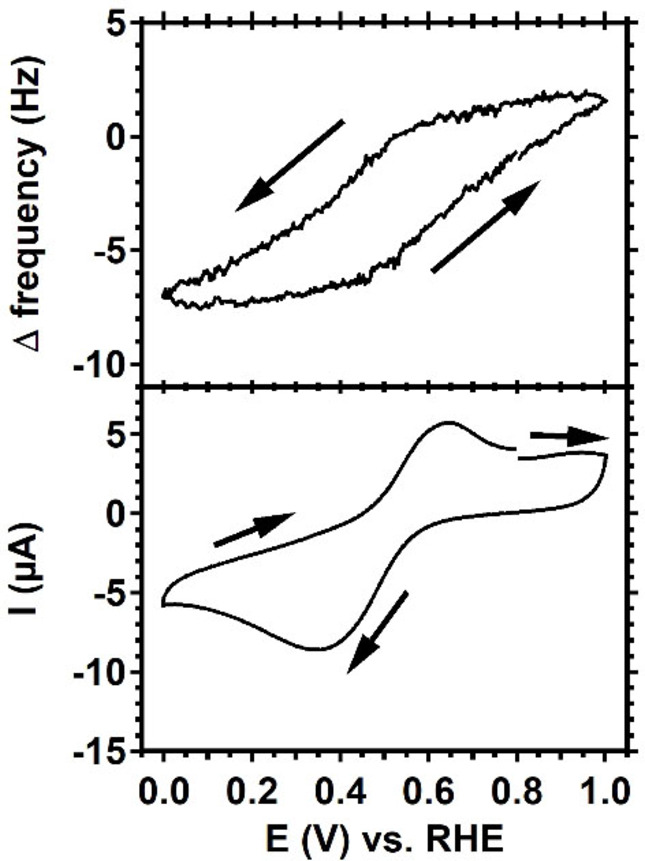
Electrochemical quartz crystal microbalance measurement with a gold work electrode of 0.15 mm Cu2(btmpa) in 0.1 m phosphate buffer of pH 7. The bottom panel shows the second scan of a CV at 50 mV/s scan rate under argon atmosphere. The top panel shows the relative frequency of the quartz crystal and its response with respect to the applied potential.
Studies with a rotating ring disk electrode (RRDE) setup of the O2 reduction reaction (ORR) showed that Cu2(btmpa) reduces O2 to H2O2 with an onset of 0.50 V versus RHE (Figure 2). At potentials lower than 0.35 V, the GC electrode itself reduces O2 to H2O2 as well (Figure S5) and increases the reductive current when performing cyclic voltammetry (CV).Compared to Cu(tmpa) the O2 reduction and in particular the H2O2 reduction reactions mediated by Cu2(btmpa) are slow (see supporting info). This is in line with the electron transfer rates being significantly slower as well in case of Cu2(btmpa). In addition the binding affinity of dioxygen are also lower, which is most likely due to the copper +I oxidation state being relatively stable, illustrated by the higher E1/2 value of Cu2(bmpta) compared to Cu(tmpa). The slow H2O2 reduction in presence of Cu2(btmpa) results in a relatively high selectivity towards hydrogen peroxide. Whereas the rate determining step in case of the oxygen reduction reaction mediated by Cu(tmpa) is binding of dioxygen, [17] the rate determining step of hydrogen peroxide reduction most likely involves cleavage of the O−O bond via a Fenton like mechanism. [22] It is not unlikely that the reaction rate of the more complex reaction step is slowed down the most by more problematic electron transfer. Although we cannot rule out that activation of the btmpa‐ligand initiated by reactive oxygen species (ROS) produced by either the copper species and/or the carbon electrode may play a role as well in these observations.
Figure 2.
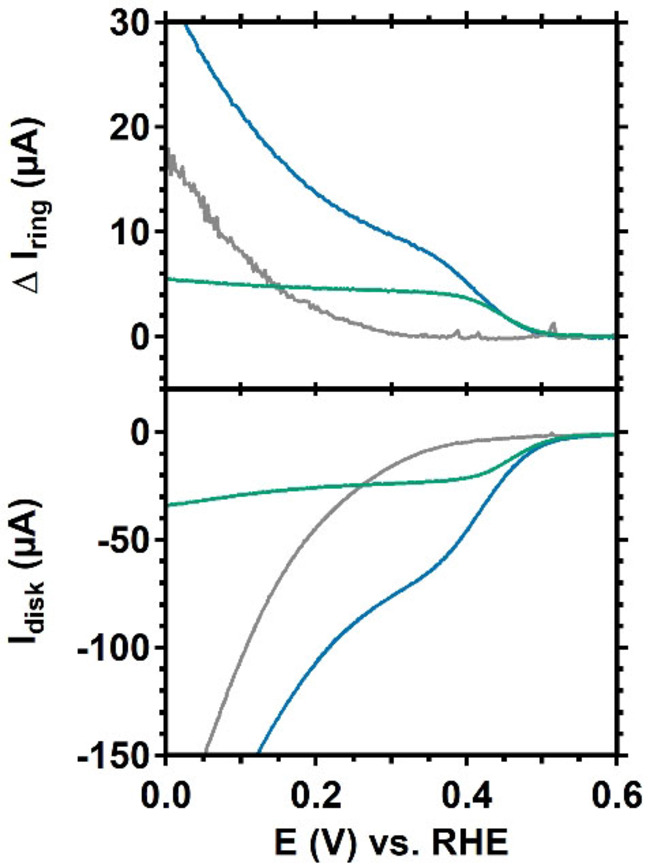
Linear sweep voltammograms with a rotating ring disk electrode setup of 0.15 mm Cu2(btmpa) under argon (green) and O2 atmosphere (blue). The cyclic voltammogram of the GC disk (bottom panel) and the current response of the Pt ring (top panel) are shown. The grey line represents the GC disk in catalyst‐free, O2 purged electrolyte. The voltammograms were recorded at 50 mV/s in a 0.1 m phosphate buffer of pH 7. A rotation rate of 1600 rpm and a Pt ring potential of 1.2 V were applied.
Since Cu2(btmpa) adsorbs at the electrode when negative potentials are applied, this allows one to significantly increase the number of active copper sites at the electrode interface over time and thereby strongly increase the peroxide productivity during amperometry measurements. Chronoamperometry in presence of Cu2(btmpa) was performed using a rotating ring disk setup (Figure S6B). A (collection efficiency of the ring electrode) of 17.5 % was determined and used to calculate the % H2O2 for this measurement (see Figures S7 and S8 for the method). The selectivity for H2O2 production by Cu2(btmpa) initially starts at 90 %. Over the course of 15 minutes, the selectivity lowers to 70 %. A selectivity below 100 % suggests that over‐reduction of H2O2 takes place. For that purpose, H2O2 reduction by Cu2(btmpa) under argon atmosphere was studied with non‐rotating and rotating electrodes (Figure S9). H2O2 is reduced by Cu2(btmpa) indeed and the reducing current increases with the H2O2 concentration. However, the H2O2 reduction by Cu2(btmpa) is very sluggish which explains the high selectivity for H2O2 when performing O2 reduction. When chronoamperometry measurements were performed for a longer period, a significant drop in selectivity was observed. This is most likely linked to formation of deposited copper (depCu) at the cathode because a brown‐colored, metallic deposit could be observed on the surface of the electrode (Figure S10). Formation of this copper deposit inherently changes the selectivity from H2O2 to H2O over the course of time. To counter formation of depCu we applied stripping intervals, wherein the potential at the working electrode is periodically increased to 0.8 V vs RHE, which is sufficient to strip depCu from the electrode, yet insufficient to oxidize H2O2 itself (Figure S9).
2.3. Employing stripping intervals to produce hydrogen peroxide over several hours
O2 reduction with Cu2(btmpa) was monitored over a 2 hour period in O2 saturated phosphate buffer. To do so, a rotating disk setup was used for constant diffusion of O2 saturated electrolyte at 1600 rpm rotation rate. We chose 0.0 V as the most ideal potential because a background hydrogen evolution reaction is not to be expected here, and background O2 reduction reactions on GC are still minimal at this potential, while significant currents were generated at this potential in presence of Cu2(btmpa) in amperometry experiments. Three different types of measurements were performed (Figure 3). First, a GC electrode in absence of Cu2(btmpa) was tested as blank measurement (grey line). Second, a GC electrode in 0.15 mM catalyst solution was tested while continuously applying 0.0 V (orange). Last, a GC electrode in catalyst solution was tested with intervals (blue): after 20 minutes of 0.0 V, the potential at the disk was briefly set at 0.8 V for 4 minutes (see the scheme in the top panel of Figure 3. The results of the continuous measurements with and without catalyst show that the current is significantly higher in the presence of Cu2(btmpa) (−0.25 mA versus −0.05 mA in the first minutes) and increases gradually over the course of 2 h. In the first half hour, there is a large increase in current from −0.25 to −0.37 mA. This feature of quick increase within the first 30 minutes of the measurement is observed in all cases when catalyst is present but not in absence of the catalyst. At 0.0 V, O2 reduction by Cu2(btmpa) is still kinetically limited in a cyclic voltammetry experiment (Figure S5). As a result, the large increase in reductive current can be explained by an increase in active sites due to accumulation of the catalyst on the electrode. The EQCM measurements points towards this behavior as well (Figure 1). While the electrochemical production of hydrogen peroxide appears to be catalyzed by catalytic material adsorbed on the electrode interface, it seems likely that it is the adsorbed Cu2(btmpa) complex, or a reaction product thereof that still retains some form of an organic ligand, that is responsible for the two‐electron reduction of dioxygen. Heterogeneous copper species, either with Cu in the +0 oxidation state, or in a partly oxidized form are not expected to produce hydrogen peroxide in significant concentrations. [22]
Figure 3.
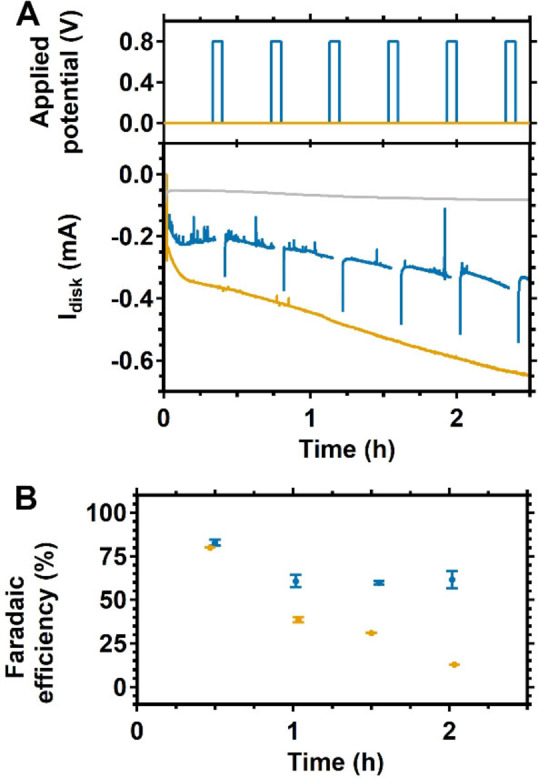
Rotating disk chronoamperometry of a GC disk at 0.0 V in a O2 saturated Cu2(btmpa) solution in a continuous measurement (orange) or a 20 minute interval measurement (blue). For the latter, a 0.8 V potential was applied for 4 minutes to re‐oxidize adsorbed Cu0 deposition every 20 minutes according to the sequence shown in panel (A). The Faradaic efficiency for H2O2 is given in (B). The black dots represent the Faradaic efficiency of the time window since the last H2O2 measurement. The disk was rotated at 1600 rpm in a 0.1 m phosphate buffer of pH 7 with 0.15 mm catalyst.
Interestingly, the magnitude of the current at 0.0 V after a 4‐minute 0.8 V interval is equal to the magnitude of the final part of the preceding 20 minutes amperogram. This indicates that the adsorbed Cu2(btmpa) largely retains on the GC electrode even when a potential of 0.8 V is briefly applied. Only thoroughly rinsing the electrode could remove most of the adsorbed catalyst and lower the O2 reduction current to the same level of a bare GC electrode in a catalyst‐free electrolyte (see Figure S11). After 1.5 hours, the reducing current of the continuous measurement became close to the estimated diffusion limited current (−0.49 mA) that one would expect for reduction of O2 to H2O2 under these conditions. The estimation is based on the diffusion limited current that a Pt disk of the same size (0.196 cm2) reaches under the same conditions for the 4‐electron reduction of O2 to H2O, for which −0.98 mA was obtained. [17] However, the current continues to rise even further in longer measurements (Figure S13), pointing to a significant overreduction of H2O2.
When O2 reduction at 0.0 V was alternated with short periods of depCu stripping at 0.8 V (blue line of Figure 3) the magnitude of the reducing current at 0.0 V is lower as compared to the continuous measurement suggesting that less over‐reduction of H2O2 takes place. Visibly, this interval procedure prevents the over‐reduction by the Cu deposition to a certain extent with respect to a continuous measurement.
The Faradaic efficiency for H2O2 at 0.0 V was monitored to study the effect of over‐reduction of H2O2 by either Cu2(btmpa) or depCu. Two different methods were considered. The first method used the same RRDE set‐up as used for Figure S6 that utilizes the Pt ring as electrochemical H2O2 sensor. Here, we found that the ring is not suited as quantitative peroxide sensor during long‐term electrolysis (see Figure S12). The formation of high amounts of H2O2 results in oxidation of Pt to produce PtOx resulting in deactivation of the activity of the Pt ring. (Figures S7 and S8). [23] However, the data did suggest that there was a slow build‐up of H2O2 within the reaction mixture. Therefore, we applied a second method: bulk electrolysis with an RDE setup for which the bulk concentration of H2O2 was periodically determined with an enzyme based photometric analysis using a reflectometer. The Faradaic efficiency was determined for measurements with 4 minute intervals and no intervals of 0.8 V. The results of the 4‐minute interval and continuous measurement are shown in Figure 3B. Within the first 30 minutes, a Faradaic efficiency of 83 % was obtained which is in good agreement with the selectivity that was found with the 15 minute RRDE measurement at 0.2 V (Figure S6). Likewise, in the continuous measurement without intervals, an efficiency of 80 % was found after the first 30 minutes (Figure 3). The continuous measurement showed a drastic drop in efficiency to 40 % 1 hour after the start and stagnated around 10 % after 2 h. The Faradaic efficiencies remained at 60 to 70 % during the interval experiments in the same time window clearly indicating that the interval procedure greatly enhances the Faradaic efficiency. Typically, peroxide concentrations between 0.15 mM (after 2 h) and 0.5 mM (after 8 hours) are obtained, which are of sufficient concentration of a substantial number of direct applications as anti‐bacteria and anti‐algae reagent. Measurements for longer than 2 h showed that the formation of depCu starts to decrease the Faradaic efficiency also in case of experiments with intervals (Figure S13 and S14). Here, it appears that the rising H2O2 concentration leads to a faster formation of depCu. Most likely, the ligand is (partially) oxidized that leads to degradation of the complex (see supporting information). XPS measurements of the electrode post catalysis confirm the presence of a copper species different from Cu2(btmpa) on the electrode (Figure S17). Experiments wherein the electrolyte solution was spiked deliberately with hydrogen peroxide showed significant levels of overreduction, while treatment of Cu2(btmpa) with H2O2 resulted to visual color changes that can be attributed to ligand oxidation.
2.4. Pinpointing the activity to an active species
Cu2(btmpa) was found to adsorb reversibly at the electrode, and as more material deposits the activity of the ORR increases. Simultaneously the Faradaic efficiency towards formation of H2O2 decreases from >80 to >60 % during this stage. After several hours the catalytic currents increase, mostly due to more efficient reduction reaction of hydrogen peroxide, resulting in a decrease in Faradaic efficiency. Selectivity of the deposited material can be restored by stripping depCu from the electrode. These observations point to a gradual degradation process of adsorbed Cu2(btmpa) to depCu. Reactive oxidation species generated from O2 and in particular H2O2 are likely to play a role herein. We recently have shown that the reduction of peroxide mediated by Cu(tmpa) most likely proceeds via a Fenton like mechanism wherein ROS are involved. [22a] It is unclear which species along the path from Cu2(btmpa) to depCu is responsible for the high ORR rates leading to selective formation of H2O2. However, it is clear that this is not the final species in the sequence. Control experiments with Cu2+ salts do not lead to significant amounts of peroxide being formed, [23] suggesting that the active species at least contains some form of an organic ligand. The degradation of the active species, however, can be largely prevented by interception of H2O2. Studies with an RRDE setup (Figure S12) wherein the H2O2 is continuously reduced to O2 by the platinum ring displayed a high selectivity and stable catalytic rates throughout an entire 8 hour measurement, which would make these systems very suitable for flow cell chemistry.
3. Conclusion
We have shown that significant amounts of hydrogen peroxide can be produced during long term amperometry experiments employing a copper catalyst. Due to accumulation of the reduced catalysts on the electrode surface, sufficiently high currents could still be obtained which are very close to the mass transport limited currents that one in principle could reach for a two electron reduction reaction involving dioxygen with the RRDE setup of study. Periodic stripping of the cathode is important for long term selectivity, as it removes depCu formation at the electrode interface. We anticipate that our results will allow for the next step, which is the incorporating the catalyst in electrochemical flow cell devices for the direct electrochemical production of H2O2 from O2.
Conflict of interest
The authors declare no conflict of interest.
4.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Dr. Andrey Konovalov and the Competence Center in Magnetometry at Institut Jean Lamour in Nancy are kindly thanked for the measurement, simulation and interpretation of the SQUID data. Max Makurat is kindly thanked for the measurement of the Raman spectra. This work has been financially supported by the European Research Council (ERC starting grant 637556 Cu4Energy to Dr. D. G. H. Hetterscheid).
B. van Dijk, R. Kinders, T. H. Ferber, J. P. Hofmann, D. G. H. Hetterscheid, ChemElectroChem 2022, 9, e202101692.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1.R. Ciriminna, L. Albanese, F. Meneguzzo, D. Pagliaro, ChemSusChem 2016, 9, 3374-3381. . [DOI] [PubMed]
- 2. Eul W., Moeller A., Steiner N., in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., 2001. DOI: 10.1002/0471238961.0825041808051919.a01.pub2. [Google Scholar]
- 3.
- 3a. Hage R., Lienke A., Angew. Chem. Int. Ed. 2006, 45, 206–222; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 212–229; [Google Scholar]
- 3b. Süss H. U., in Ullmann's Encyclopedia of Industrial Chemistry, 2006. DOI: 10.1002/14356007.a04_191.pub2. [Google Scholar]
- 4.
- 4a. Legrini O., Oliveros E., Braun A. M., Chem. Rev. 1993, 93, 671–698; [Google Scholar]
- 4b. Tchobanoglous G., Stensel H. D., Tsuchihashi R., Burton F., Abu-Orf M., Bowden G., Pfrang W., in Wastewater Engineering: Treatment and Resource Recovery, 5th edition, McGraw-Hill Education, New York, US, 2013, pp. 510-521. [Google Scholar]
- 5. Goor G., Catalytic Oxidations with Hydrogen Peroxide as Oxidant (Ed: Strukul G.), Kluwer Academic Publishers, The Netherlands, 1992, pp. 13–43. [Google Scholar]
- 6. Campos-Martin J. M., Blanco-Brieva G., Fierro J. L. G., Angew. Chem. Int. Ed. 2006, 45, 6962–6984; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 7116–7139. [Google Scholar]
- 7. Berl E., Trans. Electrochem. Soc. 1939, 76, 359. [Google Scholar]
- 8. Xia C., Xia Y., Zhu P., Fan L., Wang H., Science 2019, 366, 226. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Jiang Y., Ni P., Chen C., Lu Y., Yang P., Kong B., Fisher A., Wang X., Adv. Energy Mater. 2018, 8, 1801909; [Google Scholar]
- 9b. Siahrostami S., Verdaguer-Casadevall A., Karamad M., Deiana D., Malacrida P., Wickman B., Escudero-Escribano M., Paoli E. A., Frydendal R., Hansen T. W., Chorkendorff I., Stephens I. E. L., Rossmeisl J., Nat. Mater. 2013, 12, 1137–1143; [DOI] [PubMed] [Google Scholar]
- 9c. Verdaguer-Casadevall A., Deiana D., Karamad M., Siahrostami S., Malacrida P., Hansen T. W., Rossmeisl J., Chorkendorff I., Stephens I. E. L., Nano Lett. 2014, 14, 1603–1608; [DOI] [PubMed] [Google Scholar]
- 9d. Jirkovský J. S., Panas I., Ahlberg E., Halasa M., Romani S., Schiffrin D. J., J. Am. Chem. Soc. 2011, 133, 19432–19441. [DOI] [PubMed] [Google Scholar]
- 10. Song C., Zhang J., in PEM Fuel Cell Electrocatalysts and Catalyst Layers: Fundamentals and Applications (Ed.: Zhang J.), Springer London, London, UK, 2008, pp. 89–134. [Google Scholar]
- 11.
- 11a. Chen S., Chen Z., Siahrostami S., Kim T. R., Nordlund D., Sokaras D., Nowak S., To J. W. F., Higgins D., Sinclair R., Nørskov J. K., Jaramillo T. F., Bao Z., ACS Sustainable Chem. Eng. 2018, 6, 311–317; [Google Scholar]
- 11b. Liu Y., Quan X., Fan X., Wang H., Chen S., Angew. Chem. Int. Ed. 2015, 54, 6837–6841; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6941–6945. [Google Scholar]
- 12.
- 12a. Kim H. W., Ross M. B., Kornienko N., Zhang L., Guo J., Yang P., McCloskey B. D., Nat. Catal. 2018, 1, 282–290; [Google Scholar]
- 12b. Lu Z., Chen G., Siahrostami S., Chen Z., Liu K., Xie J., Liao L., Wu T., Lin D., Liu Y., Jaramillo T. F., Nørskov J. K., Cui Y., Nat. Catal. 2018, 1, 156–162. [Google Scholar]
- 13.
- 13a. Iglesias D., Giuliani A., Melchionna M., Marchesan S., Criado A., Nasi L., Bevilacqua M., Tavagnacco C., Vizza F., Prato M., Fornasiero P., Chem. 2018, 4, 106–123; [Google Scholar]
- 13b. Chen S., Chen Z., Siahrostami S., Higgins D., Nordlund D., Sokaras D., Kim T. R., Liu Y., Yan X., Nilsson E., Sinclair R., Nørskov J. K., Jaramillo T. F., Bao Z., J. Am. Chem. Soc. 2018, 140, 7851–7859; [DOI] [PubMed] [Google Scholar]
- 13c. Han L., Sun Y., Li S., Cheng C., Halbig C. E., Feicht P., Hübner J. L., Strasser P., Eigler S., ACS Catal. 2019, 9, 1283–1288; [Google Scholar]
- 13d. Fellinger T.-P., Hasché F., Strasser P., Antonietti M., J. Am. Chem. Soc. 2012, 134, 4072–4075. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Fukuzumi S., Yamada Y., Karlin K. D., Electrochim. Acta 2012, 82, 493–511; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Fukuzumi S., Lee Y.-M., Nam W., ChemCatChem 2018, 10, 9–28; [Google Scholar]
- 14c. Pegis M. L., Wise C. F., Martin D. J., Mayer J. M., Chem. Rev. 2018, 118, 2340–2391. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Nagao K., Hiroshi S., Tetsuo O., Chem. Lett. 1985, 14, 1917–1920; [Google Scholar]
- 15b. He Q., Mugadza T., Hwang G., Nyokong T., Int. J. Electrochem. Sci. 2012, 7, 7045–7064. [Google Scholar]
- 16.
- 16a. Kuwana T., Fujihira M., Sunakawa K., Osa T., J. Electroanal. Chem. Interfacial Electrochem. 1978, 88, 299–303; [Google Scholar]
- 16b. Bettelheim A., Kuwana T., Anal. Chem. 1979, 51, 2257–2260; [Google Scholar]
- 16c. Costentin C., Dridi H., Savéant J.-M., J. Am. Chem. Soc. 2015, 137, 13535–13544; [DOI] [PubMed] [Google Scholar]
- 16d. Kobayashi N., Nishiyama Y., J. Phys. Chem. 1985, 89, 1167–1170. [Google Scholar]
- 17. Langerman M., Hetterscheid D. G. H., Angew. Chem. Int. Ed. 2019, 58, 12974–12978; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 13108–13112. [Google Scholar]
- 18.
- 18a. Chan R. J. H., Su Y. O., Kuwana T., Inorg. Chem. 1985, 24, 3777–3784; [Google Scholar]
- 18b. D′Souza F., Deviprasad R. G., Hsieh Y.-Y., J. Electroanal. Chem. 1996, 411, 167–171; [Google Scholar]
- 18c. D′Souza F., Hsieh Y.-Y., Deviprasad G. R., J. Electroanal. Chem. 1997, 426, 17–21; [Google Scholar]
- 18d. Geiger T., Anson F. C., J. Am. Chem. Soc. 1981, 103, 7489–7496; [Google Scholar]
- 18e. Kang C., Anson F. C., Inorg. Chem. 1995, 34, 2771–2780; [Google Scholar]
- 18f. Smith P. T., Kim Y., Benke B. P., Kim K., Chang C. J., Angew. Chem. Int. Ed. 2020, 59, 4902–4907; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 4932–4937. [Google Scholar]
- 19.
- 19a. Døssing A., Hazell A., Toftlund H., Acta Chem. Scand. 1996, 50, 95–101; [Google Scholar]
- 19b. Dürr H., Zengerle K., Trierweiler H.-P., Z. Naturforsch. B 1988, 43, 361–368; [Google Scholar]
- 19c. Lee D.-H., Murthy N. N., Karlin K. D., Inorg. Chem. 1997, 36, 5785–5792. [DOI] [PubMed] [Google Scholar]
- 20. Marcus R. A., Rev. Mod. Phys. 1993, 65, 599–610. [Google Scholar]
- 21. Lim B. S., Holm R. H., Inorg. Chem. 1998, 37, 4898–4908. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Benzbiria N., Zertoubi M., Azzi M., SN App. Sci. 2020, 2, 2101; [Google Scholar]
- 22b. van Dijk B., Hofmann J. P., Hetterscheid D. G. H., Phys. Chem. Chem. Phys. 2018, 20, 19625–19634; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22c. Smits N. W. G., Rademaker D., Konovalov A. I., Siegler M. A., Hetterscheid D. G. H., Dalton Trans. 2022, 51, 1206–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hall S. B., Khudaish E. A., Hart A. L., Electrochim. Acta 1998, 43, 579–588. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.