

Abstract
Oxidative stress is closely related to the pathogenesis of Parkinson’s disease (PD), a typical neurodegenerative disease. NADPH oxidase 2 (NOX2) is involved in hydrogen peroxide (H2O2) generation. Recently, we have reported that treatment with H2O2 and PD toxins, including 6-hydroxydopamine (6-OHDA), 1-Methyl-4-phenylpyridin-1-ium (MPP+) and rotenone, induces neuronal apoptosis by inhibiting the mTOR pathway. Here, we show that treatment with 6-OHDA, MPP+ or rotenone induced H2O2 generation by upregulating the levels of NOX2 and its regulatory proteins (p22phox, p40phox, p47phox, p67phox, and Rac1), leading to apoptotic cell death in PC12 cells and primary neurons. Inhibition of NOX2 with apocynin or diphenyleneiodonium, or knockdown of NOX2 powerfully attenuated PD toxins-evoked NOX2 and H2O2, thereby hindering activation of AMPK, inhibition of Akt/mTOR, and induction of apoptosis in neuronal cells. Pretreatment with catalase, a H2O2-scavenging enzyme, blocked the effects of PD toxins on NOX2-dependent H2O2 production, AMPK/Akt/mTOR signaling and apoptosis in the cells. Similar effects were also seen in the cells pretreated with Mito-TEMPO, a mitochondria-selective superoxide scavenger, implying a mitochondrial H2O2-dependent mechanism involved. Further research revealed that ectopic expression of constitutively active Akt or dominant negative AMPKα, or inhibition of AMPK with compound C suppressed PD toxins-induced expression of NOX2 and its regulatory proteins, as well as consequential H2O2 production and apoptosis in the cells. Taken together, these results indicate that certain PD toxins can impede the AMPK/Akt-mTOR signaling pathway leading to neuronal apoptosis by eliciting NOX2-derived H2O2 production. Our findings suggest that neuronal loss in PD may be prevented by regulating the NOX2, AMPK/Akt-mTOR signaling and/or applying antioxidants to ameliorate oxidative stress.
Keywords: NADPH oxidase 2, Hydrogen peroxide, Neuronal cells, AMPK, Akt, mTOR
1. Introduction
Parkinson’s disease (PD) is a progressive neurodegenerative disease characterized by loss of dopamine-producing neurons in the ventral midbrain substantia nigra (SN) [1–3]. As the disease progresses, typical motor symptoms may occur, such as bradykinesia, rigidity, impaired postural balance, a characteristic resting tremor, and consequential dementia in PD patients [4,5]. Overwhelming evidence has demonstrated that neuronal loss is linked to protein aggregation and oxidative stress [6], and especially oxidative stress is increasingly recognized as a central event contributing to the degeneration of dopaminergic neurons in the pathogenesis of PD [7]. It is well-known that oxidative stress is due to the disequilibrium between the production of reactive oxygen species (ROS) and the availability of antioxidants or radical scavengers, which creates a perilous state contributing to cellular damage [5,8,9]. ROS are conventionally considered cytotoxic byproducts of abnormal metabolism [10]. Excessive or sustained ROS can attack all macromolecules including lipids, proteins and nucleic acids, leading to defects in their physiological functions and more ROS production and ultimately SN neuronal damage and death in PD [5,9,10]. Thus, understanding the underlying mechanisms by which oxidative stress contributes to the loss of dopaminergic neurons may help develop effective approaches in the prevention and treatment of PD.
Many studies have reported that the primary ROS sources start with the formation of a superoxide (O2−•) and subsequently produces hydrogen peroxide (H2O2), which is closely associated with NADPH-oxidases (NOXs) family, including NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1 and DUOX2 enzymes, in phagocytes and numerous non-phagocytic cells, including neurons [11–15]. NOX2, also known as gp91phox, is abundant in phagocytes [11–15]. Membrane-bound catalytic subunit NOX2 and noncatalytic subunit p22phox interact with the cytosolic subunits p40phox, p47phox, p67phox, and the small GTPase Rac1, and thus NOX2 is activated to evoke ROS generation [13,15]. There exists high expression of NOX2 in the central nervous system (CNS), and especially abnormal activity of NOX2 causes excess amounts of ROS production in the pathophysiology of neurodegenerative disorders, such as PD, Alzheimer’s disease (AD), Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS) and multiple sclerosis (MS) [13,15–17]. Several studies have demonstrated that ROS produced by NOX2 may alter the phosphorylation of diverse signaling molecules through activating kinases and/or inactivating protein phosphatases [12,14,16].
The AMP-activated protein kinase (AMPK), a master energy sensor, maintains cellular energy homeostasis during metabolic stress and controls cell survival status in response to oxidative stress [18,19]. It has been shown that overactivation or deactivation of AMPK due to oxidative stress is implicated in various signaling pathways that are involved in neurodegenerative diseases’ progression [18–22]. The mammalian target of rapamycin (mTOR) senses and integrates a variety of environmental cues (growth factors, nutrients, and energy/oxidative stress) to control protein/lipid/nucleic acid synthesis and cell growth/proliferation/survival, and is negatively regulated by AMPK [23]. Akt/protein kinase B (PKB) lies upstream of mTOR, so activated Akt may positively regulate mTOR, leading to increased phosphorylation of ribosomal p70 S6 kinase 1 (S6K1) and eukaryotic initiation factor 4E binding protein 1 (4E-BP1), two best characterized downstream effector molecules of mTOR [23]. Our group has observed that H2O2 inhibits the mTOR signaling by activation of AMPK leading to apoptosis of neuronal cells [24]. PD toxins, such as 6-hydroxydopamine (6-OHDA), 1-Methyl-4-phenylpyridin-1-ium (MPP+) and rotenone, can activate AMPK and inactivate Akt, causing neuronal cell death via inhibiting mTOR-mediated phosphorylation of S6K1 and 4E-BP1 [25]. We have also demonstrated that rotenone triggers H2O2-dependent inhibition of mTOR pathway leading to neuronal apoptosis [26]. Here we show that upregulation of NOX2 and its regulatory proteins (p22phox, p40phox, p47phox, p67phox, and Rac1) contributes to excessive generation of H2O2 in neuronal cells, which impedes the AMPK/Akt-mTOR signaling pathway leading to apoptotic cell death in the cells. Our findings suggest that proper co-manipulation of the NOX2, AMPK/Akt-mTOR signaling and/or administration of antioxidants to ameliorate oxidative stress may be a potential strategy for prevention and treatment of PD.
2. Materials and methods
2.1. Materials
Rotenone, 6-hydroxydopamine (6-OHDA), poly-d-lysine (PDL), 2′7′-dichlorodihydrofluorescein diacetate (H2DCFDA), 4′,6-diamidino-2-phenylindole (DAPI), catalase (CAT), apocynin, and protease inhibitor cocktail were purchased from Sigma (St Louis, MO, USA). Mito-TEMPO was acquired from ALEXIS Biochemicals Corporation (San Diego, CA, USA). Diphenyleneiodonium (DPI) was from MedChemExpress (Shanghai, China). Compound C and 1-methyl-4-phenylpyridin-1-ium (MPP+) were provided by Calbiochem (San Diego, CA, USA). Dulbecco’s Modified Eagle’s Medium (DMEM), 0.05% Trypsin–EDTA, NEU-ROBASAL™ Media, and B27 Supplement were from Invitrogen (Grand Island, NY, USA). Horse serum and fetal bovine serum (FBS) were supplied by Hyclone (Logan, UT, USA). Enhanced chemiluminescence solution was from Sciben Biotech Company (Nanjing, China). Other chemicals were purchased from local commercial sources and were of analytical grade, unless stated elsewhere.
2.2. Cell culture
Rat pheochromocytoma (PC12) cell line (RRID: CVCL_0481) was obtained from American Type Culture Collection (ATCC) (Manassas, VA, USA). Because of the replicative nature and cost effectiveness, the cell line is widely used as a neuronal cell model, so it was employed in this study. PC12 cells, seeded in a 6-well plate (5 × 105 cells/well) or 96-well plate (1 × 104 cells/well) pre-coated with 0.2 μg/ml PDL, were cultured in antibiotic-free DMEM supplemented with 10% horse serum and 5% FBS in a humidified incubator of 5% CO2 at 37 °C. To verify the data obtained from PC12 cells, primary neurons were also used in this study. For this, primary murine neurons were isolated from fetal mouse cerebral cortexes of 16–18 days of gestation in female ICR mice (being pregnant) as described [24], and seeded in a 6-well plate (5 × 105 cells/well) or 96-well plate (1 × 104 cells/well) coated with 10 μg/ml PDL for experiments after 6 days of culture. The experiments involving animals in this study were approved by the Institutional Animal Care and Use Committee of Nanjing Normal University (Certificate NO. 200408), and were conducted in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
2.3. Recombinant adenoviral constructs and infection of cells
The recombinant adenoviral vectors encoding HA-tagged myristoylated, constitutively active Akt (Ad-myr-Akt), HA-tagged dominant negative AMPKα (Ad-dn-AMPKα), and the control adenovirus expressing β-galactosidase (Ad-LacZ) were described previously [24,27,28]. The viruses were amplified, titrated and used as described [24,29]. For experiments, PC12 cells were cultured in the growth medium, and infected with the individual adenovirus for 24 h at 5 of multiplicity of infection (MOI = 5). Subsequently, cells were used for experiments. Cells infected with Ad-LacZ alone served as a control. Expression of HA-tagged myr-Akt or dn-AMPKα was determined by Western blotting with antibodies to HA.
2.4. Lentiviral shRNA cloning, production, and infection
Lentiviral shRNAs to NOX2 and GFP (for control) were generated as described [27,30]. For use, a monolayer of PC12 cells, when grown to about 70% confluence, were infected with the corresponding lentivirus-containing medium in the presence of 8 μg/ml polybrene for 12 h twice at an interval of 6 h. Uninfected cells were eliminated by exposure to 2 μg/ml puromycin for 48 h before use. After 5 days of culture, cells were used for experiments.
2.5. Assays for cell caspase-3/7 activity
PC12 cells and primary neurons were seeded in a PDL-coated 96-well plate (1 × 104 cells/well). The next day, cells were treated with/without 6-OHDA (30, 60 and/or 120 μM), MPP+ (0.5, 1 and/or 1.5 mM) or rotenone (0.5, 1 and/or 2 μM) for 6, 12 and/or 24 h with five replicates of each treatment. Subsequently, caspase-3/7 activity was detected using Caspase-Glo®3/7 Assay Kit (Promega, Madison, WI, USA), according to the manufacturer’s protocol.
2.6. DAPI and TUNEL staining
PC12 cells and primary neurons, or PC12 cells infected with lentiviral shRNA to NOX2 or GFP, or PC12 cells infected with Ad-myr-Akt, Ad-dn-AMPKα or Ad-LacZ, respectively, were seeded at a density of 5 × 105 cells/well in a 6-well plate containing a PDL-coated glass coverslip per well. The next day, cells were treated with/without 6-OHDA (30, 60 and/or 120 μM), MPP+ (0.5, 1 and/or 1.5 mM) or rotenone (0.5, 1 and/or 2 μM) for 6, 12 and/or 24 h, or treated with/without 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h following pre-incubation with/without a NOX inhibitor apocynin (50 μM), a canonical NOX2 inhibitor DPI (10 μM), a H2O2-scavenging enzyme CAT (350 U/ml), or a mitochondria-targeted antioxidant Mito-TEMPO (10 μM), for 1 h, or an AMPK inhibitor compound C (20 μM) for 2 h with 5 replicates of each treatment. Afterwards, the cells with fragmented and condensed nuclei were determined using DAPI staining as described [31]. For TUNEL staining, TUNEL reaction mixture (TdT enzyme solution and labeling solution) was added according to the manufacturer’s protocols of In Situ Cell Death Detection Kit® (Roche, Mannheim, Germany). Finally, slides were mounted in glycerol/phosphate buffered saline (PBS) (1:1, v/v) containing 2.5% 1,4-diazabiclo-(2,2,2) octane. Photographs were taken under a fluorescence microscope (200×) (Leica DMi8, Wetzlar, Germany) equipped with a digital camera. For quantitative analysis of the fluorescence intensity using TUNEL staining, the integral optical density (IOD) was determined by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
2.7. Apoptotic cell detection by flow cytometry
PC12 cells were seeded in a 6-well plate (5 × 105 cells/well). The next day, cells were treated with/without 6-OHDA (30, 60 and/or 120 μM), MPP+ (0.5, 1 and/or 1.5 mM) or rotenone (0.5, 1 and/or 2 μM) for 6, 12 and/or 24 h with five replicates of each treatment. Then, the ratios of live, necrotic, early and late apoptotic cells were monitored by a fluorescence-activated cell sorter (FACS) flow cytometer (CytoFLEX S, Beckman, USA) using Annexin V-FITC/PI Apoptosis Detection Kit (Beyotime, Hangzhou, China).
2.8. Intracellular H2O2 imaging
Intracellular H2O2 level was imaged by using a nonfluorescent probe, H2DCFDA. This peroxide-selective dye can penetrate into the intracellular matrix of cells, where it is cleaved by intracellular esterases and oxidized by H2O2 to form fluorescent DCF [32]. In brief, the indicated cells, after treatment as described above, were loaded with H2DCFDA (20 μM) for 1 h. Subsequently, all stained specimens were rinsed three times with PBS, followed by imaging under a fluorescence microscope, and quantitatively measuring integral optical density (IOD) of the fluorescence intensity by Image-Pro Plus 6.0 software (Media Cybernetics Inc., Newburyport, MA, USA).
2.9. Western blot analysis
The indicated cells, after treatments, were briefly washed with cold PBS, and then on ice, lysed in the radioimmunoprecipitation assay buffer. Lysates were sonicated for 10 s and centrifuged at 14,000 rpm for 10 min at 4 °C, followed by collecting the supernatants. Protein con-centration was determined by bicinchoninic acid assay with bovine serum albumin as a standard (Pierce, Rockford, IL, USA). Afterwards, Western blotting was performed as described previously [24]. In brief, lysates containing equivalent amounts of protein were separated on 7–12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA, USA). Membranes were incubated with PBS containing 0.05% Tween 20 and 5% nonfat dry milk to block nonspecific binding, and then with primary antibodies against phosphorylated Akt (p-Akt) (Ser473), p-Akt (Thr308), p-S6K1 (Thr389), p-4E-BP1 (Thr70), 4E-BP1, cleaved caspase-3 (all from Cell Signaling Technology, Danvers, MA, USA); β-tubulin, p-mTOR (Ser2448), mTOR, p-AMPKα (Thr172), AMPKα, p40phox, p47phox, HA (all from Sigma); Akt, S6K1, p22phox (all from Santa Cruz Biotechnology, Dallas, TX, USA); NOX2, p67phox (all from Epitomics, Burlingame, CA, USA), or Rac1 (Cytoskeleton, Denver, CO, USA) overnight at 4 °C, respectively, followed by incubation with appropriate secondary antibodies including goat anti-rabbit IgG-horseradish peroxidase (HRP), goat anti-mouse IgG-HRP, and rabbit anti-goat IgG-HRP (Pierce, Rockford, IL, USA) overnight at 4 °C. Immunoreactive bands were visualized by using enhanced chemiluminescence solution.
2.10. Statistical analysis
All data were expressed as means ± standard error of the mean (means ± SEM). Student’s t-test for non-paired replicates was used to identify statistically significant differences between treatment means. Group variability and interaction were compared using either one-way or two-way ANOVA followed by Bonferroni’s post-tests to compare replicate means. A level of P < 0.05 was considered to be significant.
3. Results
3.1. PD toxins induce H2O2 production contributing to apoptosis by upregulating the levels of NOX2 and its regulatory proteins in neuronal cells
It is well-known that NOX2 and its regulatory proteins (p22phox, p40phox, p47phox, p67phox, and Rac1) are involved in H2O2 generation [13,33]. Our group has demonstrated that rotenone induces H2O2 production leading to neuronal apoptosis [26]. Here, PD toxins (6-OHDA, MPP+ and rotenone) were used to treat PC12 cells and primary neurons for 0–24 h or 24 h, followed by Western blot analysis. We found that treatment with 6-OHDA, MPP+ or rotenone upregulated the expression of NOX2, p22phox, p40phox, p47phox, p67phox and Rac1 in a time- and dose-dependent manner in the cells (Fig. 1A, B). We also observed time- and dose-dependent induction of intracellular H2O2 production by 6-OHDA, MPP+ or rotenone (Fig. 1C–E), as evidenced by imaging (in green) (Fig. 1C) and quantifying (Fig. 1D, E) using a peroxide-selective probe, H2DCFDA [32]. Sequentially, we investigated the cells with nuclear fragmentation and condensation, a hallmark of apoptosis [34], using DAPI staining, live, early apoptotic, late apoptotic and necrotic cells, using annexin-V-FITC/PI staining, and the cells with fragmented DNA, using TUNEL staining, in PC12 cells and/or primary neurons. Imaged and/or quantified results revealed that 6-OHDA, MPP+ or rotenone markedly increased the percentage of the cells with nuclear fragmentation and condensation (arrows) in PC12 cells and primary neurons (Fig. 2A–C), the ratios of apoptotic cells in PC12 cells (Fig. 2D–F), and the number of TUNEL-positive cells with fragmented DNA (in red) in primary neurons (Fig. 2G–I). In line with the above observations, using caspase3/7 activity assay, treatment with these PD toxins profoundly induced activation of caspases 3/7 in the cells as well (Fig. 2J, K). Taken together, the results imply that treatment with PD toxins (6-OHDA, MPP+ or rotenone) might induce H2O2 production contributing to apoptosis by upregulating the levels of NOX2 and its regulatory proteins in neuronal cells.
Fig. 1.
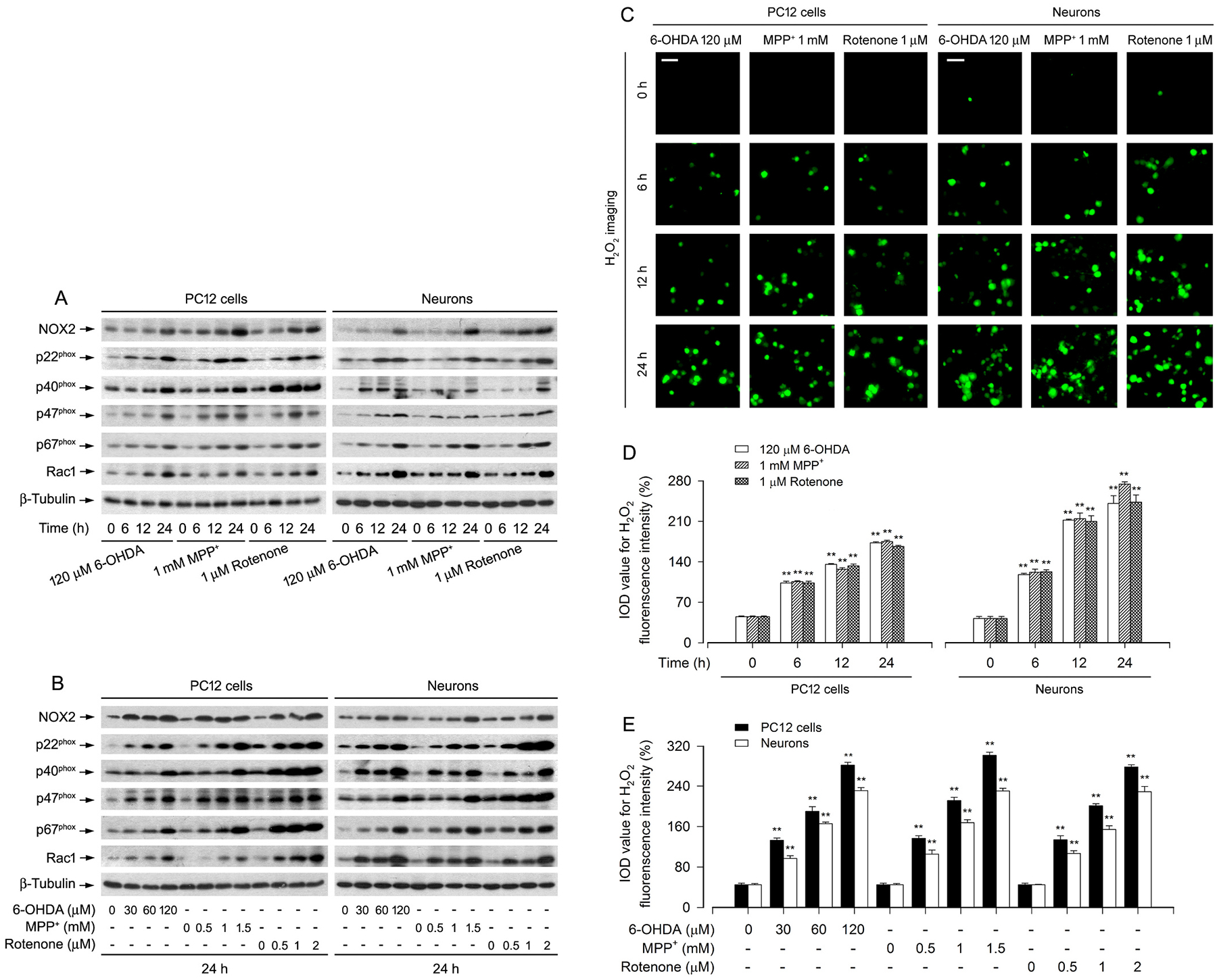
Upregulation of NOX2 and its regulatory proteins is associated with excessive H2O2 generation in PD toxins-induced neuronal cells. PC12 cells and primary neurons were treated with 6-OHDA (30, 60 and/or 120 μM), MPP+ (0.5, 1 and/or 1.5 mM) or rotenone (0.5, 1 and/or 2 μM) for 6, 12 and/or 24 h, as described in Materials and Methods. A and B Total cell lysates were subjected to western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. C Intracellular H2O2 was imaged (in green) using a peroxide-selective probe H2DCFDA. Scale bar: 20 μm. D and E Fluorescent intensity of intracellular H2O2 imaging was quantified. Results were presented as mean ± SEM, n = 5. **P < 0.01, difference with control group.
Fig. 2.
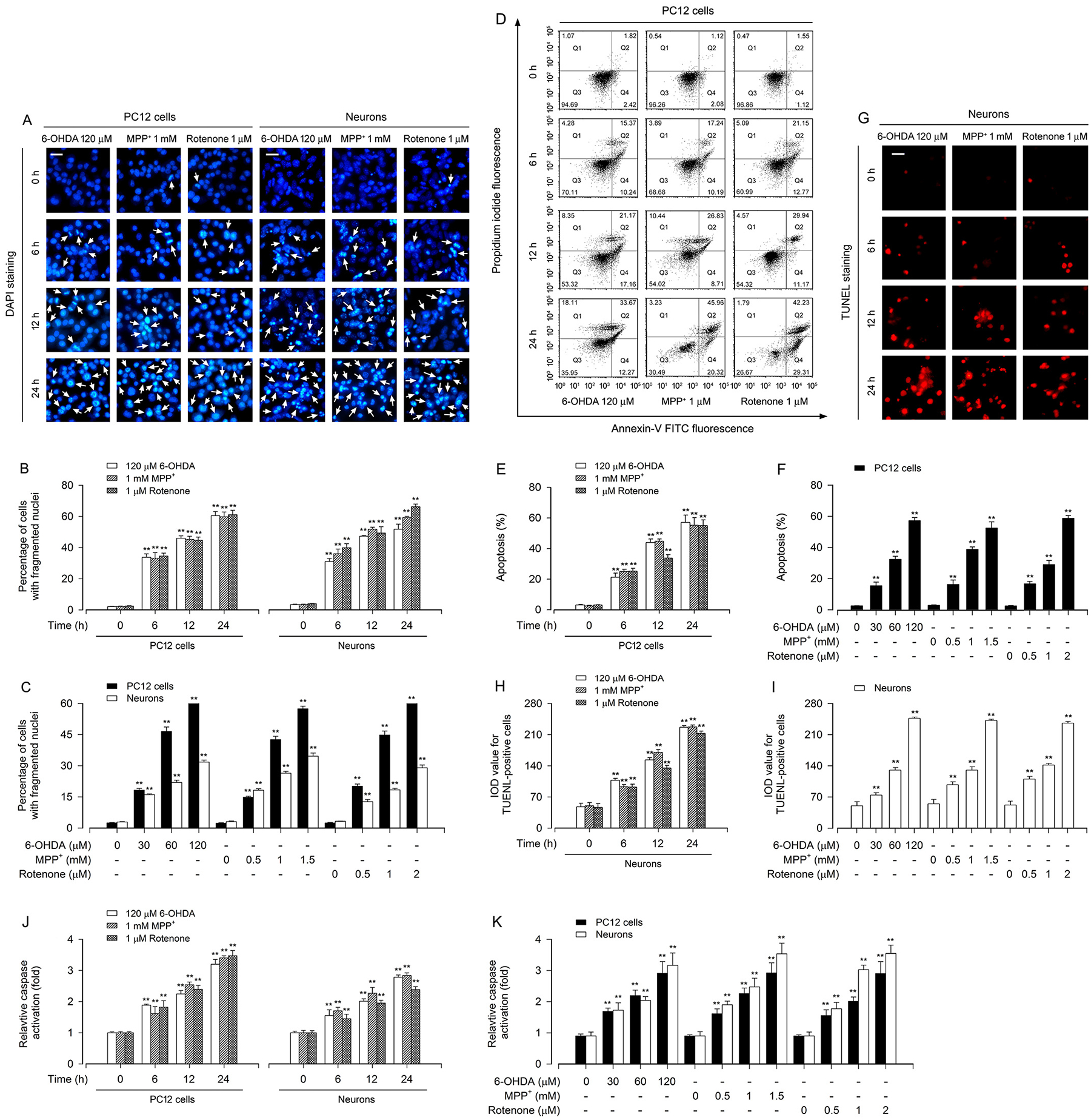
Apoptosis is triggered in PD toxins-induced neuronal cells. PC12 cells and primary neurons were treated with 6-OHDA (30, 60 and/or 120 μM), MPP+ (0.5, 1 and/or 1.5 mM) or rotenone (0.5, 1 and/or 2 μM) for 6, 12 and/or 24 h. A Apoptotic cells were evaluated by nuclear fragmentation and condensation (arrows) using DAPI staining. Scale bar: 20 μm. B and C The percentage of cells with fragmented nuclei was quantified. D The percentages of necrotic (Q1), late apoptotic (Q2), live (Q3) and early apoptotic (Q4) cells were determined by FACS using annexin-V-FITC/PI staining. The results from a representative experiment in PC12 cells are shown. E and F Quantitative analysis of apoptotic cells by FACS assay. G Apoptotic cells were evaluated by in situ detection of fragmented DNA (in red) using TUNEL staining. Scale bar: 20 μm. H and I IOD values of TUNEL-positive cells with the fluorescence staining were quantified. J and K Caspase-3/7 activities were detected using Caspase-3/7 Assay Kit. Results were presented as mean ± SEM, n = 5. **P < 0.01, difference with control group.
3.2. PD toxins activate AMPK and inactivate Akt, resulting in inhibition of the mTOR signaling, as well as apoptosis by a NOX2-dependent mechanism in neuronal cells
Our previous studies have found that PD toxins activate AMPK and inactivate Akt, causing neuronal apoptosis via inhibiting the mTOR signaling pathway [25]. Therefore, we sought to validate whether the toxins impair the AMPK/Akt-mTOR signaling pathway leading to neuronal apoptosis in a NOX2-dependent manner. For this, apocynin, an inhibitor of NOXs [35] and DPI, a long-acting inhibitor of NOX2 [36], were employed. The results showed that apocynin (50 μM) or DPI (10 μM) obviously suppressed 6-OHDA-, MPP+- or rotenone-induced upregulation of NOX2 and generation of H2O2 in PC12 cells and primary neurons (Fig. 3A, B, D, E). Of importance, apocynin or DPI rescued the cells from PD toxins-elicited increase in p-AMPKα and decrease in p-Akt, p-mTOR, p-S6K1 and p-4E-BP1 (Fig. 3A, D). In line with this, apocynin or DPI also potently attenuated PD toxins-induced cleavage of caspase-3 and apoptosis in the cells (Fig. 3A, C, D, F).
Fig. 3.
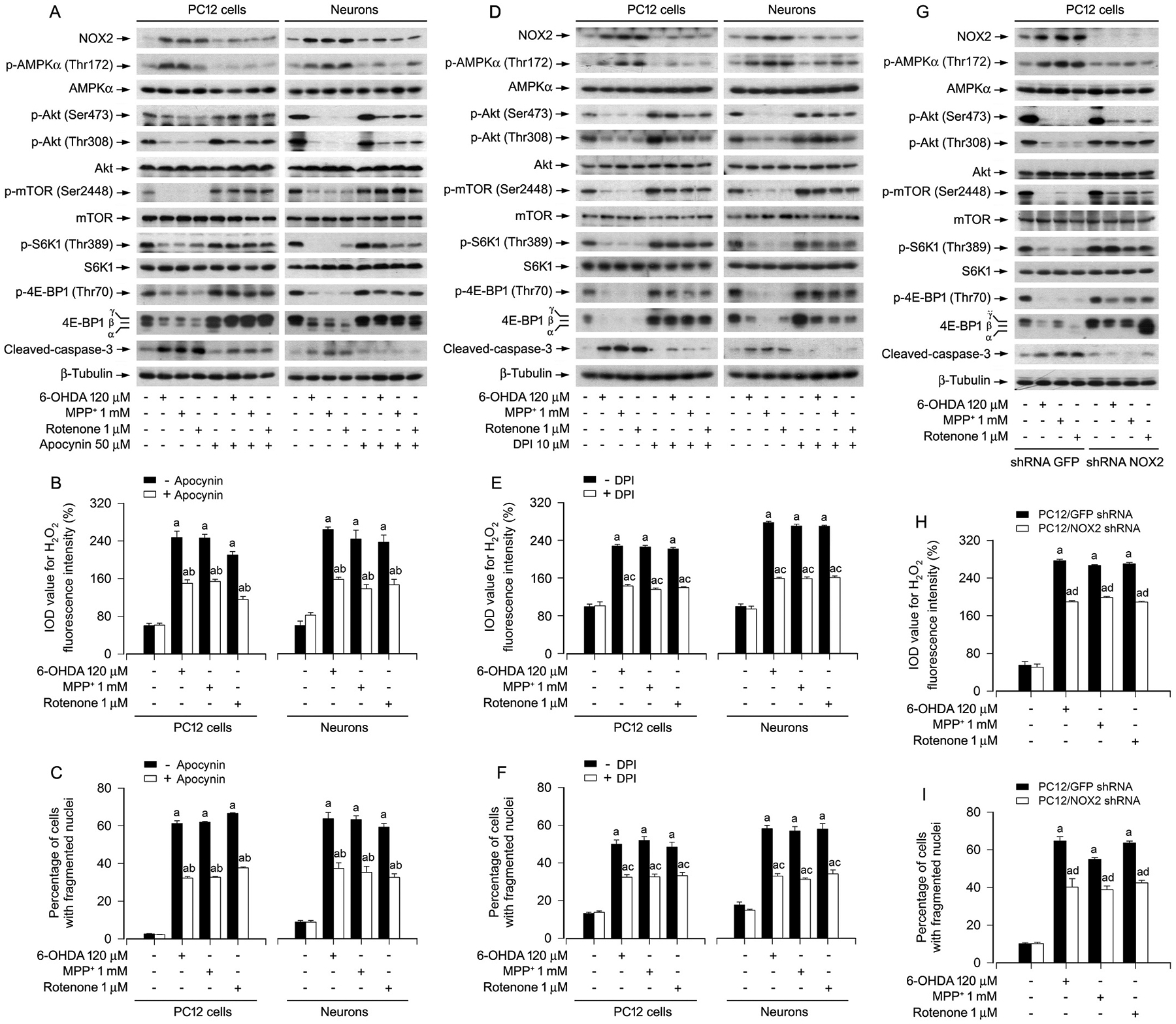
Pharmacological inhibition of NOX2 with apocynin or DPI, or knockdown of NOX2 prevents activation of AMPK, inhibition of Akt/mTOR, generation of H2O2, and apoptosis in PD toxins-induced neuronal cells. PC12 cells and primary neurons, or PC12 cells infected with lentiviral shRNA to NOX2 or GFP (as control), respectively, were treated with/without 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h following pre-incubation with/without apocynin (50 μM) or DPI (10 μM) for 1 h, or treated with/without 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. A, D and G Total cell lysates were subjected to western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B, E and H Intracellular H2O2 was imaged and quantified using a peroxide-selective probe H2DCFDA. C, F and I Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results were presented as mean ± SEM, n = 5. aP < 0.05, difference with control group; bP < 0.05, + Apocynin group vs - Apocynin group; cP < 0.05, + DPI group vs - DPI group; dP < 0.05, NOX2 shRNA group vs GFP shRNA group.
To further corroborate the role of NOX2 in the PD toxins-induced activation of AMPK, inhibition of Akt-mTOR signaling pathway and neuronal apoptosis, PC12 cells, infected with lentiviral shRNA to NOX2 or GFP, were exposed to 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. As shown in Fig. 3G, NOX2 expression was down-regulated by ~90% in shRNA NOX2-infected cells compared to shRNA GFP-infected cells. Knockdown of NOX2 did not affect the basal phosphorylation of AMPKα, Akt, mTOR, S6K1 and 4E-BP1, and cleavage of caspase-3. However, knockdown of NOX2 remarkably suppressed 6-OHDA-, MPP+- or rotenone-induced increase of p-AMPKα and decreases of p-Akt, p-mTOR, p-S6K1 and p-4E-BP1 (Fig. 3G). Consistently, depletion of NOX2 also potently attenuated PD toxins-elicited cleavage of caspase-3 (Fig. 3G), H2O2 production (Fig. 3H), and apoptosis (Fig. 3I) in the cells. Taken together, these data verify that the PD toxins activate AMPK and inactivate Akt, resulting in inhibition of the mTOR signaling, as well as apoptosis through a NOX2-dependent mechanism in neuronal cells.
3.3. PD toxins’ induction of intracellular and mitochondrial H2O2 hinders the AMPK/Akt-mTOR signaling pathway leading to apoptosis in neuronal cells
To pinpoint whether the effects of PD toxins on AMPK/Akt-mTOR and apoptosis are attributed to induction of intracellular H2O2, catalase (CAT), a H2O2-scavenging enzyme, was employed. The results showed that pretreatment with CAT strikingly blocked 6-OHDA-, MPP+- or rotenone-induced upregulation of NOX2 and its regulatory proteins, as well as generation of H2O2 in PC12 cells and primary neurons (Fig. 4A, B). PD toxins-induced activation of AMPK and inactivation of Akt, as well as inhibition of mTOR-mediated phosphorylation of S6K1 and 4E-BP1 were reversed by pretreatment with CAT in the cells (Fig. 4C). CAT also profoundly diminished the activation of caspases-3 and the number of fragmented nuclear cells in the cells exposed to the PD toxins (Fig. 4C, D). The findings support that the PD toxins induce intracellular H2O2, which mediates the AMPK/Akt-mTOR signaling pathway leading to apoptosis by upregulating the expression of NOX2 and its regulatory proteins in neuronal cells.
Fig. 4.
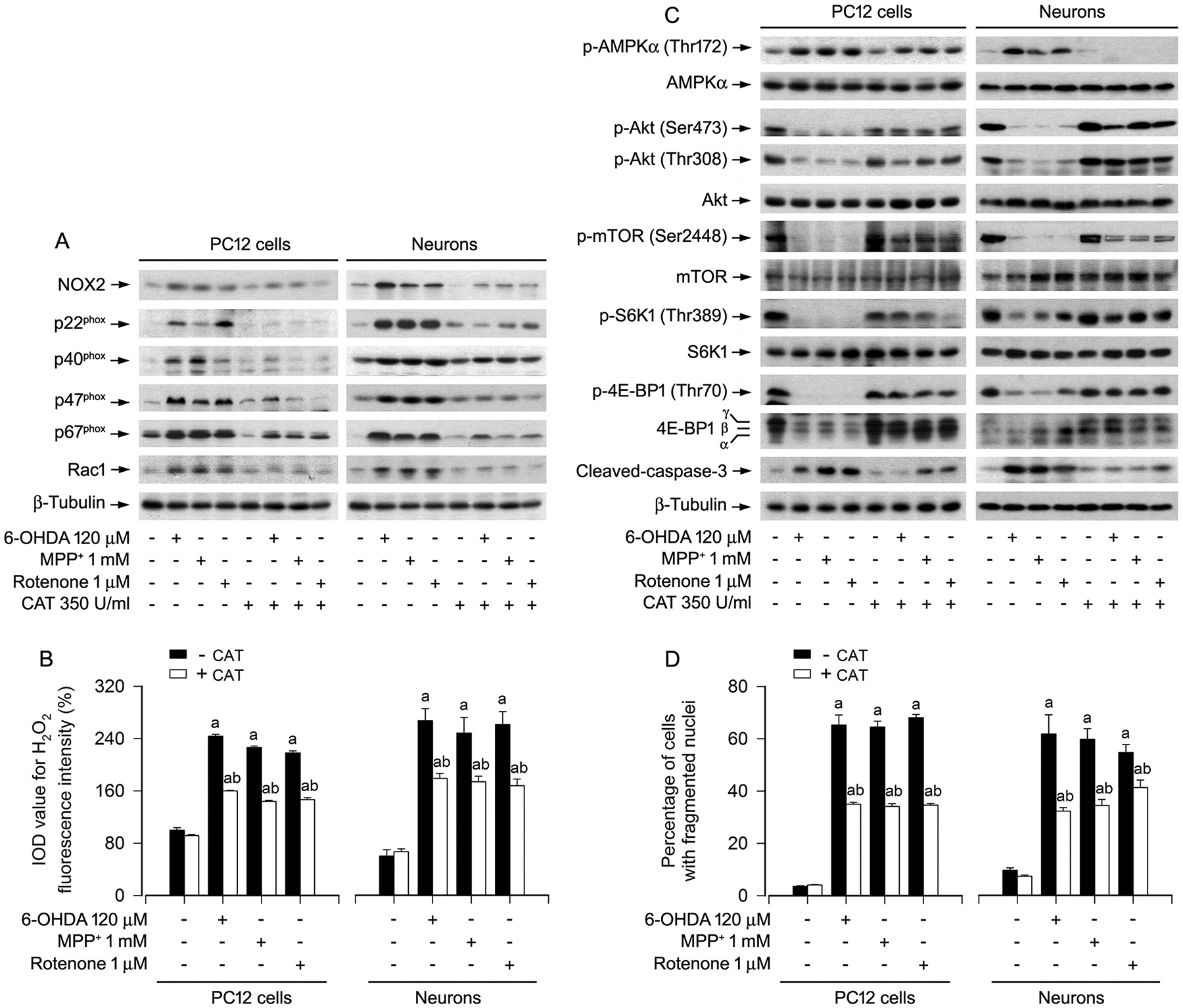
Scavenging intracellular H2O2 with catalase ameliorates abnormal NOX2 and its regulatory proteins, as well as AMPK/Akt-mTOR signaling pathway and apoptosis in PD toxins-induced neuronal cells. PC12 cells and primary neurons were pretreated with/without CAT (350 U/ml) for 1 h and then exposed to 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. A and C Total cell lysates were subjected to western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B Intracellular H2O2 was imaged and quantified using a peroxide-selective probe H2DCFDA. D Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results were presented as mean ± SEM, n = 5. aP < 0.05, difference with control group; bP < 0.05, + CAT group vs - CAT group.
Next, we studied whether the effects of PD toxins on AMPK/Akt-mTOR and apoptosis are related to excessive H2O2 production in the mitochondria of neuronal cells. To this end, PC12 cells and primary neurons were pretreated with/without a mitochondria-targeted antioxidant Mito-TEMPO (10 μM) for 1 h, and then exposed to 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. The results showed that pretreatment with Mito-TEMPO powerfully mitigated exorbitant expression of NOX2 and its regulatory proteins, as well as excessive H2O2 production in the cells induced by the PD toxins (Fig. 5A, B). Consistently, Mito-TEMPO substantially reversed 6-OHDA-, MPP+- or rotenone-triggered increase of p-AMPKα and decreases of p-Akt, p-mTOR, p-S6K1 and p-4E-BP1 (Fig. 5C), as well as cleavage of caspases-3 and apoptosis in the cells (Fig. 5C, D). The results indicate that the PD toxins indeed evoke mitochondrial H2O2, which impairs the AMPK/Akt-mTOR signaling pathway leading to apoptosis by upregulating the expression of NOX2 and its regulatory proteins in neuronal cells.
Fig. 5.
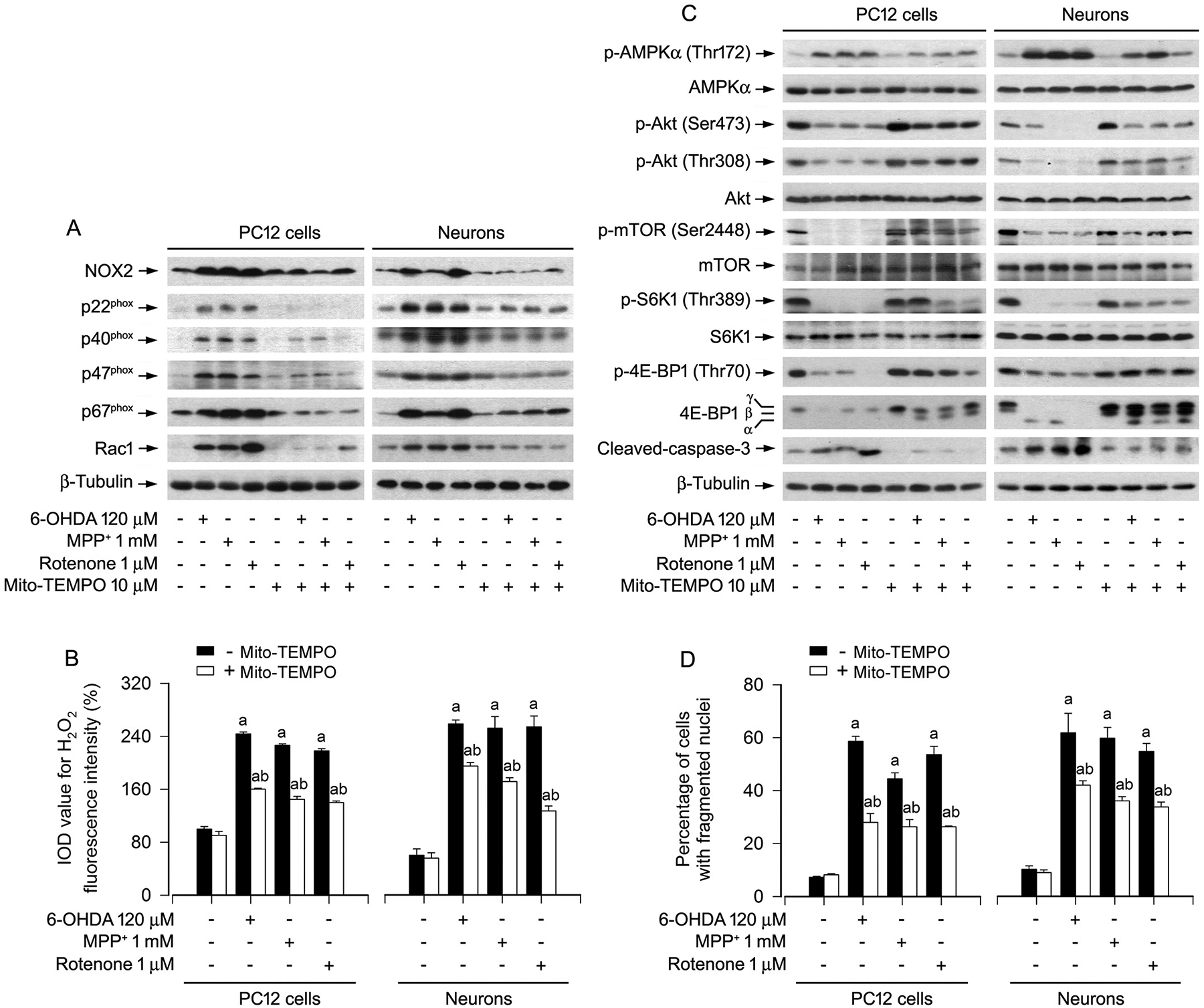
Scavenging of mitochondrial H2O2 with Mito-TEMPO ameliorates abnormal NOX2 and its regulatory proteins, as well as AMPK/Akt-mTOR signaling pathway and apoptosis in PD toxins-induced neuronal cells. PC12 cells and primary neurons were pretreated with/without Mito-TEMPO (10 μM) for 1 h and then exposed to 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. A and C Total cell lysates were subjected to western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B Cell H2O2 was imaged and quantified using a peroxide-selective probe H2DCFDA. D Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results were presented as mean ± SEM, n = 5. aP < 0.05, difference with control group; bP < 0.05, + Mito-TEMPO group vs - Mito-TEMPO group.
3.4. Modulation of Akt and AMPK activity interferes with PD toxins’ induction of NOX2-derived H2O2 and apoptosis in neuronal cells
To investigate whether modulation of Akt activity impacts on NOX2-derived H2O2 and apoptosis in neuronal cells in response to PD toxins, a recombinant adenovirus expressing HA-tagged myristoylated constitutively active Akt (Ad-myr-Akt) was employed. We observed that ectopic expression of myr-Akt partially prevented PC12 cells from upregulation of NOX2, p22phox, p40phox, p47phox, p67phox and Rac1, as well as excessive H2O2 production induced by 6-OHDA, MPP+ or rotenone (Fig. 6A, B). Of importance, overexpression of myr-Akt potently rendered resistance to the PD toxins-induced apoptosis (Fig. 6C) in the cells.
Fig. 6.
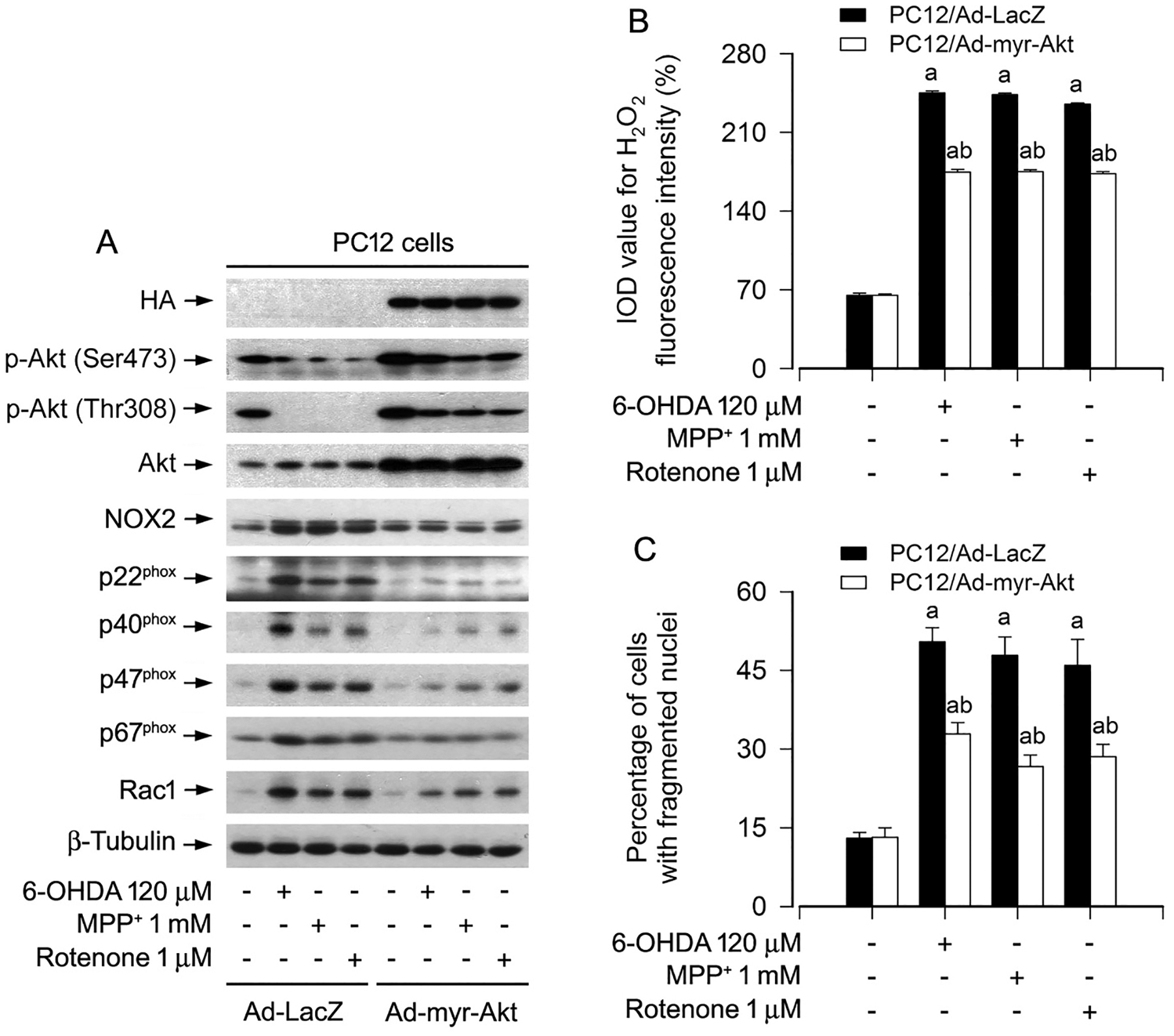
Ectopic expression of constitutively active Akt attenuates upregulation of NOX2 and its regulatory proteins, generation of H2O2, as well as apoptosis in PD toxins-induced neuronal cells. PC12 cells, infected with Ad-myr-Akt or Ad-LacZ (as control), respectively, were treated with/without 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. A Total cell lysates were subjected to western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B Intracellular H2O2 was imaged and quantified using a peroxide-selective probe H2DCFDA. C Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results were presented as mean ± SEM, n = 5. aP < 0.05, difference with control group; bP < 0.05, Ad-myr-Akt group vs Ad-LacZ group.
To gain more insights into the role and significance of AMPK in inducing NOX2-derived H2O2 and neuronal apoptosis by PD toxins, PC12 cells and primary neurons were pretreated with/without AMPK inhibitor compound C (20 μM) for 2 h and then exposed to 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. As demonstrated in Fig. 7A, B, 6-OHDA, MPP+ or rotenone markedly evoked the elevation of p-AMPKα, and the upregulation of NOX2, p22phox, p40phox, p47phox, p67phox and Rac1, as well as the increase of H2O2 production. The effects were substantially attenuated in the cells pretreated with compound C (Fig. 7A, B). Sequentially, we also observed that addition of compound C conferred high resistance to the PD toxins-induced neuronal apoptosis (Fig. 7C). These results support the idea that PD toxins induce NOX2-derived H2O2 and apoptosis in part by activating AMPK in neuronal cells.
Fig. 7.
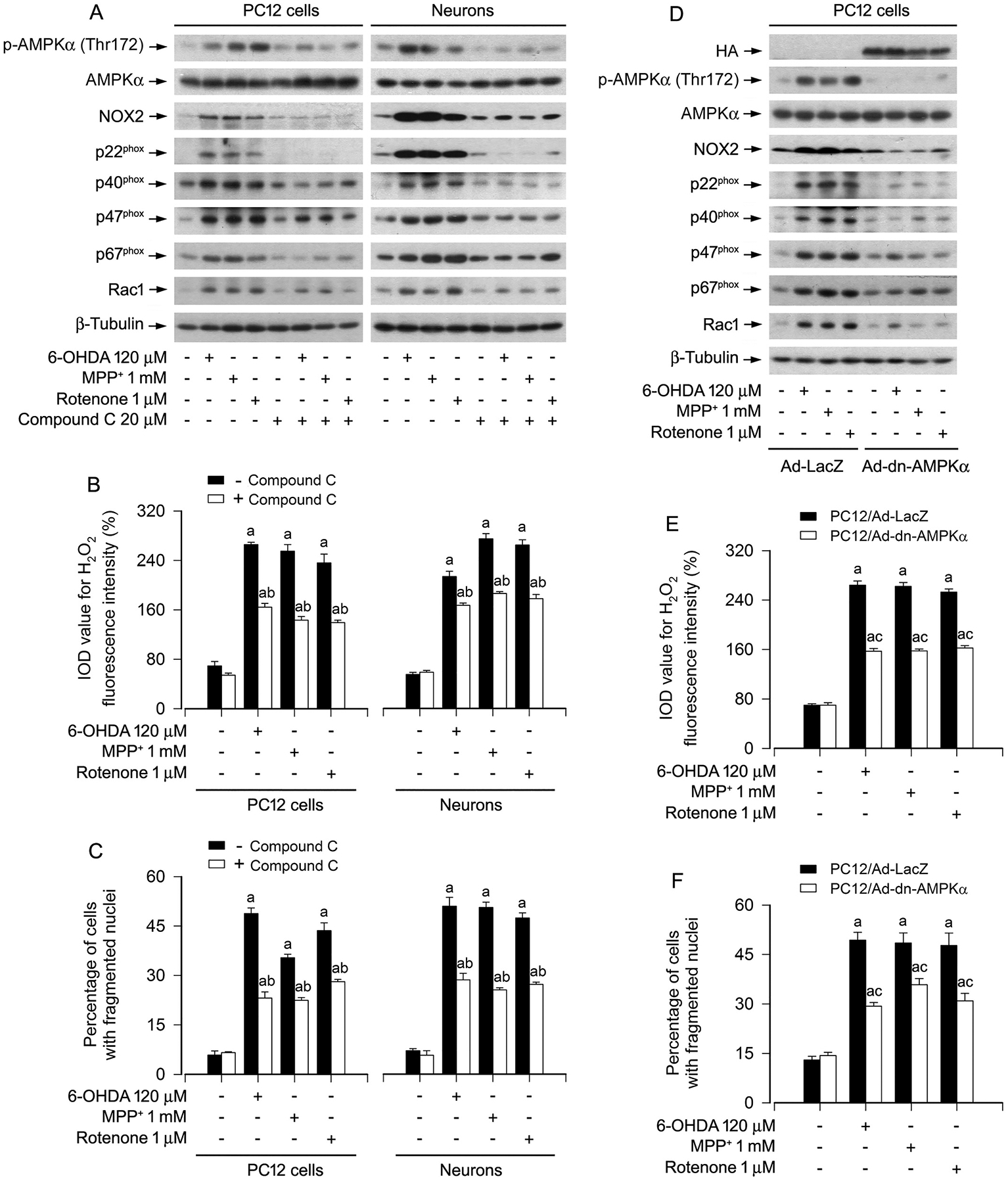
Pharmacological inhibition of AMPKα with compound C or expression of dominant negative AMPKα attenuates upregulation of NOX2 and its regulatory proteins, generation of H2O2, as well as apoptosis in PD toxins-induced neuronal cells. PC12 cells and primary neurons, or PC12 cells infected with Ad-dn-AMPKα or Ad-LacZ (as control), respectively, were treated with/without 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h following pre-incubation with/without compound C (20 μM) for 2 h, or treated with/without 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. A and D Total cell lysates were subjected to western blotting using indicated antibodies. The blots were probed for β-tubulin as a loading control. Similar results were observed in at least three independent experiments. B and E Intracellular H2O2 was imaged and quantified using a peroxide-selective probe H2DCFDA. C and F Apoptotic cells were evaluated by nuclear fragmentation and condensation using DAPI staining. Results were presented as mean ± SEM, n = 5. aP < 0.05, difference with control group; bP < 0.05, + Compound C group vs - Compound C group; cP < 0.05, Ad-dn-AMPKα group vs Ad-LacZ group.
To confirm the above findings, PC12 cells, infected with recombinant adenoviruses expressing HA-tagged dominant negative AMPKα1 (Ad-dn-AMPKα) and control adenovirus expressing galactosidase alone (Ad-LacZ), respectively, were exposed to 6-OHDA (120 μM), MPP+ (1 mM) or rotenone (1 μM) for 24 h. As expected, a high level of HA-tagged dn-AMPKα was seen in Ad-dn-AMPKα-infected cells, but not in Ad-LacZ-infected cells (control) (Fig. 7D). Ectopic expression of dn-AMPKα almost completely blocked the AMPK activity, since the basal or the PD toxins-inhibited p-AMPK was substantially suppressed (Fig. 7D). Of importance, expression of dn-AMPKα markedly reversed 6-OHDA-, MPP+- or rotenone-triggered increase in NOX2, p22phox, p40phox, p47phox, p67phox, Rac1, H2O2 production and apoptosis in the cells (Fig. 7D–F). Taken together, our data demonstrate the importance of PD toxins-activated AMPKα in their induction of NOX2-derived H2O2 and neuronal apoptosis.
4. Discussion
PD is a neurodegenerative disease characterized by a major loss of dopaminergic nigrostriatal neurons [1–3]. A large number of reports have demonstrated a close relationship of oxidative stress, e.g., ROS, as a critical etiological factor of neuronal loss, to pathogenesis of PD [6,7,37]. Excessive intracellular ROS can directly oxidize lipids, proteins, and nucleic acids, and thus activate or inhibit related signaling pathways, thereby leading to damage of the basic cell structures and consequential CNS dysfunction [5,9,24,37,38]. Our previous studies have shown that PD toxins, including 6-OHDA, MPP+ and rotenone, inhibit mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death through activating AMPK and inactivating Akt [25]. However, the underlying mechanism remains enigmatic. Here we present evidence that the PD toxins induced upregulation of NOX2, p22phox, p40phox, p47phox, p67phox, and Rac1, as well as excessive generation of H2O2, resulting in apoptosis in PC12 cells and primary neurons. Further, we found that the PD toxins-induced H2O2 inhibited the mTOR pathway, at least in part by activating the negative regulator AMPKα [23] and concurrently by inhibiting the positive regulator Akt [23], leading to apoptosis of the neuronal cells.
There are many sources of ROS, and one of the major sources is attributed to the generation by the NOXs family, including NOX1, NOX2, NOX3, NOX4, NOX5, DUOX1 and DUOX2 enzymes [10,13]. Among them, NOX2 is the most abundant and widely expressed in phagocytes [11–15]. NOX2, as a catalytic subunit, interacts with a noncatalytic subunit p22phox transmembrane protein along with the cytosolic subunits p40phox, p47phox, p67phox and Rac 1 [13,15,17]. It is known that NOX2 is highly expressed in cells throughout the central nervous system, and ROS generation by NOX2 is implicated in a variety of neurological disorders, including PD, AD, HD, ALS and MS [13,15–17]. Rotenone induces ROS/H2O2 contributing to apoptosis in neuronal cells [26], yet the underlying mechanism is poorly defined. Here, for the first time, we show that, on the one hand, induction of H2O2 generation by 6-OHDA, MPP+ or rotenone was closely related to upregulated expression of the H2O2-generating enzyme NOX2 and its regulatory proteins, as well as neuronal apoptosis (Figs. 1 and 2); on the other hand, PD toxins-induced H2O2 stimulated the expression of NOX2 family members, because treatment with CAT, a H2O2-scavenging enzyme, abolished PD toxins-induced H2O2 and also negated PD toxins-induced expression of NOX2, p22phox, p40phox, p47phox, p67phox, and Rac1, as well as apoptosis in the cells (Fig. 4). The results imply a positive feedback loop involved in the regulation of the H2O2 production and NOX2 activity.
In this study, we asked whether 6-OHDA-, MPP+- or rotenone-impaired AMPK/Akt-mTOR signaling pathway in neuronal cells was due to abnormal manifestation of NOX2-derived H2O2 induced by the PD toxins. For this, apocynin, a pharmacological inhibitor for ROS-generating enzyme NOX activity by blocking the assembly of NOX complex [35,42], and DPI, a long-acting NOX2 inhibitor [36], respectively, were used. The results showed that apocynin (50 μM) or DPI (10 μM) effectively suppressed 6-OHDA-, MPP+- or rotenone-stimulated NOX2 activation and H2O2 generation (Fig. 3A, B, D, E). Of importance, apocynin or DPI substantially suppressed 6-OHDA-, MPP+- or rotenone-induced activation of AMPK, inactivation of Akt, inhibition of mTOR-mediated phosphorylation of S6K1 and 4E-BP1, increase of cleaved caspase-3 and apoptosis in PC12 cells and primary neurons (Fig. 3A, C, D, F). This was further supported by the observations in NOX2-knockdown cells (Fig. 3G–I). Taking all these findings into account, we deduce that PD toxins trigger NOX2-derived H2O2 generation impeding the AMPK/Akt-mTOR signaling pathway and consequentially resulting in apoptosis in neuronal cells.
Mitochondria are significant contributors to ROS generation [5]. Any dysfunction of this cell organelle can be harmful to cell function and viability, as seen in PD [5,7,9]. For example, mitochondrial dysfunction leads to excessive ROS production that is detrimental for dopaminergic SN cells [5,7]. 6-OHDA, MPP+ or rotenone evokes ROS generation by inhibiting mitochondrial complex I [9,26,39]. There exists a much enhanced H2O2 formation in mitochondria in situ in isolated nerve terminals when mitochondrial complex I is inhibited at a small degree [40]. In the present study, to determine the association of PD toxins-induced mitochondrial H2O2 generation with neuronal apoptosis, mitochondria-targeted antioxidant Mito-TEMPO [41] was employed. We found that treatment with Mito-TEMPO dramatically diminished H2O2 production and apoptosis in PC12 cells and primary neurons triggered by 6-OHDA, MPP+ or rotenone (Fig. 5). Meanwhile, treatment with Mito-TEMPO also powerfully blocked the expression of NOX2, p22phox, p40phox, p47phox, p67phox, and Rac1 in the cells in response to 6-OHDA, MPP+ or rotenone (Fig. 5). The findings suggest a mitochondrial H2O2-dependent mechanism involved. In this study, we also found that treatment with CAT and Mito-TEMPO conferred high resistance to 6-OHDA-, MPP+- or rotenone-induced activation of AMPK, inactivation of Akt, inhibition of mTOR-mediated phosphorylation of S6K1 and 4E-BP1, and increase of cleaved caspase-3 (Figs. 4C, 5C), respectively. Collectively, our data support that excessive mitochondrial H2O2 generation can impede the AMPK/Akt-mTOR signaling, resulting in neuronal apoptosis in the context of PD.
AMPK and Akt are respectively negative and positive regulators of mTOR, and especially their regulation of mTOR plays a key role in cell survival [18,23,43,44]. The observations from our group and others have demonstrated that 6-OHDA-, MPP+- and/or rotenone-induced neuronal cell death is associated with or through AMPK activation and/or Akt inactivation [2,25,45]. In the current study, we further confirmed these, as ectopic expression of constitutively active Akt (myr-Akt) or dominant negative AMPKα (dn-AMPK), or inhibition of AMPKα with compound C markedly rescued from 6-OHDA-, MPP+- or rotenone-induced apoptosis in PC12 cells and/or primary neurons (Figs. 6 and 7). However, interestingly, we noticed that modulation of Akt and AMPK activity by RNA interference or pharmacological inhibition rendered a high resistance to 6-OHDA-, MPP+- or rotenone-upregulated NOX2, p22phox, p40phox, p47phox, p67phox, and Rac1, as well as H2O2 generation in the cells (Figs. 6 and 7), suggesting that activated AMPK or deactivated Akt may feedback upregulate the PD toxins-stimulated NOX2 and its regulatory proteins, as well as H2O2 production. The findings enhance our understanding of PD toxins-stimulated H2O2-generating enzyme NOX2 and its regulatory proteins, which is vital for PD toxins-induced activation of AMPK, deactivation of Akt, and neuronal apoptosis, and provide an expanded conceptual view of blocking NOX2-derived H2O2 in the prevention and treatment of PD. Since the specific assembly process for NOX2 activation is a particularly complex mechanism, currently, we do not know whether there is causal regulation between NOX2 assembly and the AMPK/Akt-mTOR signaling pathway in the development of PD. Undoubtedly, more studies are needed to address these issues.
In conclusion, we have identified that PD toxins upregulate the expression of H2O2-generating NOX2 and its regulatory proteins, and thus evoke intracellular H2O2 and concomitant mitochondrial H2O2. This results in activation of AMPK and inhibition of the Akt-mTOR pathway, leading to apoptosis in neuronal cells (Fig. 8). Our results highlight that PD stresses impede the AMPK/Akt-mTOR signaling pathway causing neuronal apoptosis by eliciting NOX2-derived H2O2. The findings suggest that proper co-manipulation of NOX2, AMPK/Akt-mTOR signaling and/or administration of antioxidants to ameliorate oxidative stress may be a potential strategy for prevention and treatment of PD.
Fig. 8.
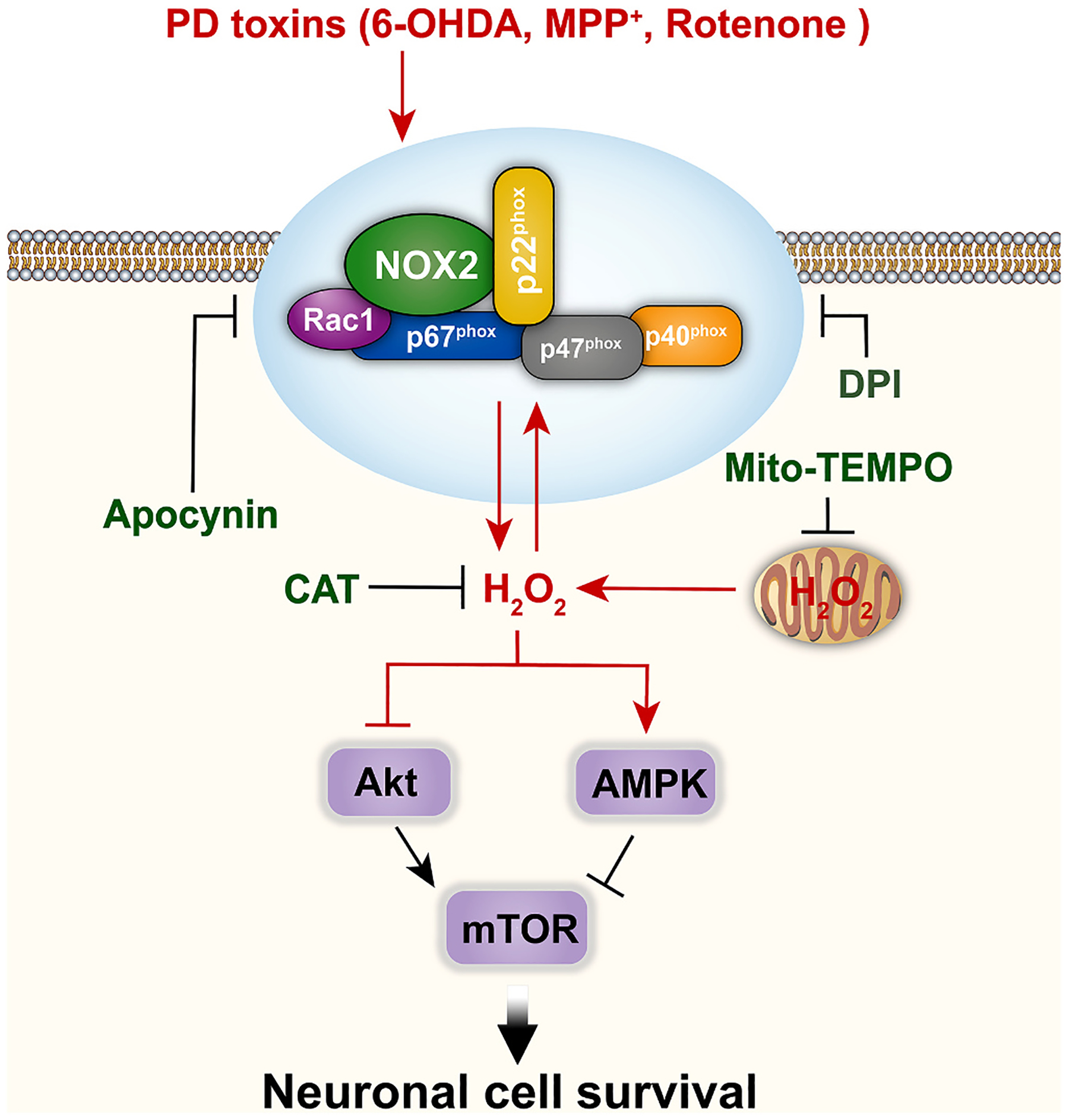
A schematic model of how PD toxins (6-OHDA, MPP+ or rotenone) induce NOX2-derived H2O2 leading to neuronal apoptosis. PD toxins upregulate the expression of NOX2 and its regulatory proteins, and thus evoke intracellular H2O2 and concomitant mitochondrial H2O2. This results in activation of AMPK and inactivation of Akt, convergently inhibiting the mTOR pathway contributing to apoptosis in neuronal cells.
Funding information
This work was supported in part by the grants from National Natural Science Foundation of China (No. 81873781, 81271416, 82101337), National Institutes of Health (CA115414), Project for the Priority Academic Program Development of Jiangsu Higher Education Institutions of China (PAPD-14KJB180010), BSKY Scientific Research from Anhui Medical University (XJ201813), and American Cancer Society (RSG-08-135-01-CNE).
Abbreviations:
- 4E-BP1
eukaryotic initiation factor 4E binding protein 1
- 6-OHDA
6-hydroxydopamine
- AD
Alzheimer’s disease
- ALS
amyotrophic lateral sclerosis
- AMPK
AMP-activated protein kinase
- CAT
catalase
- DAPI
4′,6-diamidino-2-phenylindole
- DMEM
Dulbecco’s Modified Eagle’s Medium
- DPI
diphenyleneiodonium
- FBS
fetal bovine serum
- HA
hemagglutinin
- HD
Huntington’s disease
- H2DCFDA
2′7′-dichlorodihydrofluorescein diacetate
- H2O2
hydrogen peroxide
- MPP+
1-methyl-4-phenylpyridin-1-ium
- mTOR
mammalian target of rapamycin
- MS
multiple sclerosis
- PDL
poly-d-lysine
- NOX
NADPH oxidase
- PBS
phosphate buffered saline
- PD
Parkinson disease
- PI3K
phosphatidylinositol 3′-kinase
- Akt (PKB)
protein kinase B
- ROS
reactive oxygen species
- S6K1
ribosomal p70 S6 kinase 1
- O2−•
superoxide
- TUNEL
the terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick-end labeling
Footnotes
Ethics declarations
The experiments involving animals in this study were approved by the Institutional Animal Care and Use Committee of Nanjing Normal University (Certificate NO. 200408), and were conducted in compliance with the guidelines set forth by the Guide for the Care and Use of Laboratory Animals.
Declaration of Competing Interest
The authors declared that there are no conflicts of interests.
References
- [1].Choi DH, Cristovao AC, Guhathakurta S, Lee J, Joh TH, Beal MF, Kim YS, NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease, Antioxid. Redox Signal 16 (10) (2012) 1033–1045, 10.1089/ars.2011.3960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Malagelada C, Jin ZH, Greene LA, RTP801 is induced in Parkinson’s disease and mediates neuron death by inhibiting Akt phosphorylation/activation, J. Neurosci 28 (53) (2008) 14363–14371, 10.1523/JNEUROSCI.3928-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Maiti P, Manna J, Dunbar GL, Current understanding of the molecular mechanisms in Parkinson’s disease: targets for potential treatments, Transl. Neurodegener 6 (2017) 28, 10.1186/s40035-017-0099-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Moustafa AA, Chakravarthy S, Phillips JR, Crouse JJ, Gupta A, Frank MJ, Hall JM, Jahanshahi M, Interrelations between cognitive dysfunction and motor symptoms of Parkinson’s disease: behavioral and neural studies, Rev. Neurosci 27 (5) (2016) 535–548. [DOI] [PubMed] [Google Scholar]
- [5].Zaman V, Shields DC, Shams R, Drasites KP, Matzelle D, Haque A, Banik NL, Cellular and molecular pathophysiology in the progression of Parkinson’s disease, Metab. Brain Dis 36 (5) (2021) 815–827, 10.1007/s11011-021-00689-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Trist BG, Davies KM, Cottam V, Genoud S, Ortega R, Roudeau S, Carmona A, De Silva K, Wasinger V, Lewis SJG, Sachdev P, Smith B, Troakes C, Vance C, Shaw C, Al-Sarraj S, Ball HJ, Halliday GM, Hare DJ, Double KL, Amyotrophic lateral sclerosis-like superoxide dismutase 1 proteinopathy is associated with neuronal loss in Parkinson’s disease brain, Acta Neuropathol. 134 (1) (2017) 113–127, 10.1007/s00401-017-1726-6. [DOI] [PubMed] [Google Scholar]
- [7].Guo JD, Zhao X, Li Y, Li GR, Liu XL, Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (review), Int. J. Mol. Med 41 (4) (2018) 1817–1825, 10.3892/ijmm.2018.3406. [DOI] [PubMed] [Google Scholar]
- [8].Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK, Reactive oxygen species in metabolic and inflammatory signaling, Circ. Res 122 (6) (2018) 877–902, 10.1161/CIRCRESAHA.117.311401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Dias V, Junn E, Mouradian MM, The role of oxidative stress in Parkinson’s disease, J. Parkinsons Dis 3 (4) (2013) 461–491, 10.3233/JPD-130230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lin CS, Lee SH, Huang HS, Chen YS, Ma MC, H2O2 generated by NADPH oxidase 4 contributes to transient receptor potential vanilloid 1 channel-mediated mechanosensation in the rat kidney, Am. J. Physiol. Renal 309 (4) (2015) F369–F376, 10.1152/ajprenal.00462.2014. [DOI] [PubMed] [Google Scholar]
- [11].Block K, Gorin Y, Aiding and abetting roles of NOX oxidases in cellular transformation, Nat. Rev. Cancer 12 (9) (2012) 627–637, 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Olguin-Albuerne M, Moran J, ROS produced by NOX2 control in vitro development of cerebellar granule neurons development, ASN Neuro 7 (2) (2015), 1759091415578712, 10.1177/1759091415578712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Bedard K, Krause KH, The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology, Physiol. Rev 87 (1) (2007) 245–313, 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- [14].Frey RS, Ushio-Fukai M, Malik AB, NADPH oxidase-dependent signaling in endothelial cells: role in physiology and pathophysiology, Antioxid. Redox Signal 11 (4) (2009) 791–810, 10.1089/ars.2008.2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Brown DI, Griendling KK, Nox proteins in signal transduction, Free Radic. Biol. Med 47 (9) (2009) 1239–1253, 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Koppula S, Kumar H, Kim IS, Choi DK, Reactive oxygen species and inhibitors of inflammatory enzymes, NADPH oxidase, and iNOS in experimental models of Parkinson’s disease, Mediat. Inflamm 2012 (2012), 823902, 10.1155/2012/823902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tarafdar A, Pula G, The role of NADPH oxidases and oxidative stress in neurodegenerative disorders, Int. J. Mol. Sci 19 (12) (2018) 3824, 10.3390/ijms19123824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chiang MC, Nicol CJ, Cheng YC, Resveratrol activation of AMPK-dependent pathways is neuroprotective in human neural stem cells against amyloid-beta-induced inflammation and oxidative stress, Neurochem. Int 115 (2018) 1–10, 10.1016/j.neuint.2017.10.002. [DOI] [PubMed] [Google Scholar]
- [19].Garcia D, Shaw RJ, AMPK: mechanisms of cellular energy sensing and restoration of metabolic balance, Mol. Cell 66 (6) (2017) 789–800, 10.1016/j.molcel.2017.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ding R, Wu W, Sun Z, Li Z, AMP-activated protein kinase: an attractive therapeutic target for ischemia-reperfusion injury, Eur. J. Pharmacol 888 (2020), 173484, 10.1016/j.ejphar.2020.173484. [DOI] [PubMed] [Google Scholar]
- [21].Domise M, Vingtdeux V, AMPK in neurodegenerative diseases, Exp. Suppl 107 (2016) 153–177, 10.1007/978-3-319-43589-3_7. [DOI] [PubMed] [Google Scholar]
- [22].Qin S, Tang H, Li W, Gong Y, Li S, Huang J, Fang Y, Yuan W, Liu Y, Wang S, Guo Y, Guo Y, Xu Z, AMPK and its activator berberine in the treatment of neurodegenerative diseases, Curr. Pharm. Des 26 (39) (2020) 5054–5066, 10.2174/1381612826666200523172334. [DOI] [PubMed] [Google Scholar]
- [23].Liu GY, Sabatini DM, mTOR at the nexus of nutrition, growth, ageing and disease, Nat. Rev. Mol. Cell Biol 21 (4) (2020) 183–203, 10.1038/s41580-019-0199-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen L, Xu B, Liu L, Luo Y, Yin J, Zhou H, Chen W, Shen T, Han X, Huang S, Hydrogen peroxide inhibits mTOR signaling by activation of AMPKalpha leading to apoptosis of neuronal cells, Lab. Investig 90 (5) (2010) 762–773, 10.1038/labinvest.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu Y, Liu C, Chen S, Ye Y, Guo M, Ren Q, Liu L, Zhang H, Xu C, Zhou Q, Huang S, Chen L, Activation of AMPK and inactivation of Akt result in suppression of mTOR-mediated S6K1 and 4E-BP1 pathways leading to neuronal cell death in in vitro models of Parkinson’s disease, Cell. Signal 26 (8) (2014) 1680–1689, 10.1016/j.cellsig.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou Q, Liu C, Liu W, Zhang H, Zhang R, Liu J, Zhang J, Xu C, Liu L, Huang S, Chen L, Rotenone induction of hydrogen peroxide inhibits mTOR-mediated S6K1 and 4E-BP1/eIF4E pathways, leading to neuronal apoptosis, Toxicol. Sci 143 (1) (2015) 81–96, 10.1093/toxsci/kfu211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S, Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways, Oncogene 25 (53) (2006) 7029–7040, 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- [28].Fujio Y, Guo K, Mano T, Mitsuuchi Y, Testa JR, Walsh K, Cell cycle withdrawal promotes myogenic induction of Akt, a positive modulator of myocyte survival, Mol. Cell. Biol 19 (7) (1999) 5073–5082, 10.1128/MCB.19.7.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Liu L, Chen L, Chung J, Huang S, Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins, Oncogene 27 (37) (2008) 4998–5010, 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Xu C, Wang X, Gu C, Zhang H, Zhang R, Dong X, Liu C, Hu X, Ji X, Huang S, Chen L, Celastrol ameliorates cd-induced neuronal apoptosis by targeting NOX2-derived ROS-dependent PP5-JNK signaling pathway, J. Neurochem 141 (1) (2017) 48–62, 10.1111/jnc.13966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chen L, Liu L, Luo Y, Huang S, MAPK and mTOR pathways are involved in cadmium-induced neuronal apoptosis, J. Neurochem 105 (1) (2008) 251–261, 10.1111/j.1471-4159.2007.05133.x. [DOI] [PubMed] [Google Scholar]
- [32].Bao L, Avshalumov MV, Rice ME, Partial mitochondrial inhibition causes striatal dopamine release suppression and medium spiny neuron depolarization via H2O2 elevation, not ATP depletion, J. Neurosci 25 (43) (2005) 10029–10040, 10.1523/JNEUROSCI.2652-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bertolotti M, Farinelli G, Galli M, Aiuti A, Sitia R, AQP8 transports NOX2-generated H2O2 across the plasma membrane to promote signaling in B cells, J. Leukoc. Biol 100 (5) (2016) 1071–1079, 10.1189/jlb.2AB0116-045R. [DOI] [PubMed] [Google Scholar]
- [34].Hao B, Cheng S, Clancy CJ, Nguyen MH, Caspofungin kills Candida albicans by causing both cellular apoptosis and necrosis, Antimicrob. Agents Ch 57 (1) (2013) 326–332, 10.1128/AAC.01366-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Barbieri SS, Sandrini L, Musazzi L, Popoli M, Ieraci A, Apocynin prevents anxiety-like behavior and histone deacetylases overexpression induced by sub-chronic stress in mice, Biomolecules 11 (6) (2021) 885, 10.3390/biom11060885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Wang Q, Chu CH, Oyarzabal E, Jiang L, Chen SH, Wilson B, Qian L, Hong JS, Subpicomolar diphenyleneiodonium inhibits microglial NADPH oxidase with high specificity and shows great potential as a therapeutic agent for neurodegenerative diseases, Glia 62 (12) (2014) 2034–2043, 10.1002/glia.22724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yeung AWK, Tzvetkov NT, Georgieva MG, Ognyanov IV, Kordos K, Jozwik A, Kuhl T, Perry G, Petralia MC, Mazzon E, Atanasov AG, Reactive oxygen species and their impact in neurodegenerative diseases: literature landscape analysis, Antioxid. Redox Signal 34 (5) (2021) 402–420, 10.1089/ars.2019.7952. [DOI] [PubMed] [Google Scholar]
- [38].Chen L, Xu B, Liu L, Luo Y, Zhou H, Chen W, Shen T, Han X, Kontos CD, Huang S, Cadmium induction of reactive oxygen species activates the mTOR pathway, leading to neuronal cell death, Free Radic. Biol. Med 50 (5) (2011) 624–632, 10.1016/j.freeradbiomed.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dabbeni-Sala F, Di Santo S, Franceschini D, Skaper SD, Giusti P, Melatonin protects against 6-OHDA-induced neurotoxicity in rats: a role for mitochondrial complex I activity, FASEB J. 15 (1) (2001) 164–170, 10.1096/fj.00-0129com. [DOI] [PubMed] [Google Scholar]
- [40].Adam-Vizi V, Production of reactive oxygen species in brain mitochondria: contribution by electron transport chain and non-electron transport chain sources, Antioxid. Redox Signal 7 (9–10) (2005) 1140–1149, 10.1089/ars.2005.7.1140. [DOI] [PubMed] [Google Scholar]
- [41].Yeh YT, Yeh H, Su SH, Lin JS, Lee KJ, Shyu HW, Chen ZF, Huang SY, Su SJ, Phenethyl isothiocyanate induces DNA damage-associated G2/M arrest and subsequent apoptosis in oral cancer cells with varying p53 mutations, Free Radic. Biol. Med 74 (2014) 1–13, 10.1016/j.freeradbiomed.2014.06.008. [DOI] [PubMed] [Google Scholar]
- [42].Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D, Classical inhibitors of NOX NAD(P)H oxidases are not specific, Curr. Drug Metab 9 (8) (2008) 686–696, 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- [43].Cornu M, Albert V, Hall MN, mTOR in aging, metabolism, and cancer, Curr. Opin. Genet. Dev 23 (1) (2013) 53–62, 10.1016/j.gde.2012.12.005. [DOI] [PubMed] [Google Scholar]
- [44].Hardie DG, Ross FA, Hawley SA, AMPK: a nutrient and energy sensor that maintains energy homeostasis, Nat. Rev. Mol. Cell Biol 13 (4) (2012) 251–262, 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chung JY, Lee SJ, Lee SH, Jung YS, Ha NC, Seol W, Park BJ, Direct interaction of alpha-synuclein and AKT regulates IGF-1 signaling: implication of Parkinson disease, Neurosignals 19 (2) (2011) 86–96, 10.1159/000325028. [DOI] [PubMed] [Google Scholar]