

Abstract
Background
Bintrafusp alfa (BA) is a bifunctional fusion protein designed for colocalized, simultaneous inhibition of two immunosuppressive pathways, transforming growth factor-β (TGF-β) and programmed death-ligand 1 (PD-L1), within the tumor microenvironment (TME). We hypothesized that targeting PD-L1 to the tumor by BA colocalizes the TGF-β trap (TGF-βRII) to the TME, enabling it to sequester TGF-β in the tumor more effectively than systemic TGF-β blockade, thereby enhancing antitumor activity.
Methods
Multiple technologies were used to characterize the TGF-β trap binding avidity. BA versus combinations of anti-PD-L1 and TGF-β trap or the pan-TGF-β antibody fresolimumab were compared in proliferation and two-way mixed lymphocyte reaction assays. Immunophenotyping of tumor-infiltrating lymphocytes (TILs) and RNA sequencing (RNAseq) analysis assessing stromal and immune landscape following BA or the combination therapy were performed in MC38 tumors. TGF-β and PD-L1 co-expression and their associated gene signatures in MC38 tumors and human lung carcinoma tissue were studied with single-cell RNAseq (scRNAseq) and immunostaining. BA-induced internalization, degradation, and depletion of TGF-β were investigated in vitro.
Results
BA and fresolimumab had comparable intrinsic binding to TGF-β1, but there was an ~80× avidity-based increase in binding affinity with BA. BA inhibited cell proliferation in TGF-β-dependent and PD-L1-expressing cells more potently than TGF-β trap or fresolimumab. Compared with the combination of anti-PD-L1 and TGF-β trap or fresolimumab, BA enhanced T cell activation in vitro and increased TILs in MC38 tumors, which correlated with efficacy. BA induced distinct gene expression in the TME compared with the combination therapy, including upregulation of immune-related gene signatures and reduced activities in TGF-β-regulated pathways, such as epithelial-mesenchymal transition, extracellular matrix deposition, and fibrosis. Regulatory T cells, macrophages, immune cells of myeloid lineage, and fibroblasts were key PD-L1/TGF-β1 co-expressing cells in the TME. scRNAseq analysis suggested BA modulation of the macrophage phenotype, which was confirmed by histological assessment. PD-L1/TGF-β1 co-expression was also seen in human tumors. Finally, BA induced TGF-β1 internalization and degradation in the lysosomes.
Conclusion
BA more effectively blocks TGF-β by targeting TGF-β trap to the tumor via PD-L1 binding. Such colocalized targeting elicits distinct and superior antitumor responses relative to single agent combination therapy.
Keywords: Immunotherapy; Lymphocytes, Tumor-Infiltrating; Tumor Microenvironment; Gene Expression Profiling; Immunoassay
WHAT IS ALREADY KNOWN ON THIS TOPIC
Simultaneous targeting of programmed death-ligand 1 (PD-(L)1) and transforming growth factor-β (TGF-β) showed additive preclinical antitumor activity. Bintrafusp alfa (BA) demonstrated enhanced efficacy compared with the combination therapy, but the mechanism of action (MoA) of targeting PD-L1 to colocalize the TGF-β trap to the tumor microenvironment (TME) has only been hypothesized.
WHAT THIS STUDY ADDS
We provide evidence that the bifunctional design of BA more effectively blocks TGF-β by targeting TGF-β trap to the tumor via PD-L1 binding, thereby eliciting distinct and superior antitumor responses relative to the combination therapy.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE, AND/OR POLICY
Colocalization as a unique MoA to neutralize other immunosuppressive cytokines in the TME may be a general approach to improve the efficacy of the combination therapy.
Background
Immune checkpoint inhibitors (ICIs) are effective cancer therapies that may benefit from concurrently targeting additional immunosuppressive pathways. Transforming growth factor-β (TGF-β) has been identified as a potential resistance mechanism of ICIs.1 Bintrafusp alfa (BA) is a first-in-class bifunctional fusion protein composed of human TGF-β receptor II extracellular domain (TGF-βRII ECD or TGF-β trap) fused to the C-terminus of each heavy chain of a human anti-programmed death-ligand 1 (PD-L1) immunoglobulin G1 (IgG1) antibody.2 BA elicits superior antitumor activity relative to anti-PD-L1(mut)/TGF-β trap control (a TGF-β trap control that is mutated to abrogate PD-L1 binding) and anti-PD-L1 monotherapies in preclinical models,2 and shows early evidence of clinical activity in heavily pretreated patients with advanced solid tumors in phase 1 studies.3–5
The concept of BA is to use one bifunctional fusion protein to simultaneously target PD-L1 and TGF-β. During its design, several factors were taken into consideration. First, targeting of the anti-PD-L1 moiety to the tumor is anticipated to localize the TGF-β trap moiety to sequester TGF-β in the tumor microenvironment (TME). This is especially important because TGF-β is secreted at high levels by tumor cells and tumor-infiltrating immune cells but acts locally as an autocrine or paracrine in the TME. TGF-β induces extracellular matrix (ECM) remodeling, such as increased collagen deposition, creating a physical barrier that blocks the infiltration of immune cells1 and the penetration of anticancer drugs into tumors,6 while blockade of TGF-β reduces tumor stroma and improves drug distribution.7 This role of TGF-β may be particularly suited for intratumoral blockade through BA-mediated colocalization of TGF-β trap and anti-PD-L1.
Supporting this colocalization hypothesis, we and others have shown that anti-PD-L1/TGF-β trap fusion proteins are more efficacious than the combination of anti-PD-L1 and TGF-β trap monotherapies in syngeneic and humanized models.2 8 In fact, BA increases tumor biodistribution and tumor to blood ratio compared with TGF-β trap,9 whereas the pan-TGF-β antibody fresolimumab, which also lacks the ability to bind PD-L1, accumulates in primary tumors and metastases in a manner similar to the IgG control.10 We recently found that the superior effects of BA in combination with radiotherapy could be attributed to its ability to trap TGF-β in relevant PD-L1+ compartments, corroborating the importance of colocalization.11
Second, the bifunctional fusion protein may provide a potential mechanism for the clearance of BA-bound TGF-β in the TME, in contrast to an anti-TGF-β antibody, which can build up the concentration of the antibody/TGF-β complex in circulation.12 This clearance may occur when the anti-PD-L1 moiety internalizes upon binding to PD-L1+ cells, including tumor cells, lymphocytes, tumor-associated myeloid cells,13 and liver sinusoidal endothelial cells14 that are responsible for clearing circulating small immune complexes from the blood.15
Third, the use of the natural receptor TGF-βRII as a ligand trap ensures neutralization with high affinity for TGF-β1 and TGF-β3,16 whereas an antibody may not completely neutralize TGF-β and may even act as a carrier protein to potentiate cytokine activity.17 Regardless of this difference, the intrinsic binding affinities of TGF-βRII for TGF-β1 and TGF-β316 are comparable to those reported for fresolimumab.18 Importantly, a structural TGF-β model shows that the obligatory dimeric configuration of the TGF-βRII at the C terminus of an antibody allows bivalent binding of the TGF-β, which occurs naturally as a homodimer.2 This avidity-based gain of affinity may have significant functional relevance as BA can compete more effectively with cellular surface TGF-β receptors.
The present study evaluated the biophysical and functional properties of BA that may contribute to its enhanced efficacy and identified gene expression profiles and signaling pathways specifically modulated by BA compared with the combination of anti-PD-L1 and TGF-β trap in MC38 tumors.
Methods
Binding affinity and avidity of BA for TGF-β were investigated using surface plasmon resonance (SPR), isothermal titration calorimetry (ITC), mass photometry, micro-scale thermophoresis (MST), dynamic light scattering (DLS), and electron microscopy (EM). TGF-β targeting and sequestration were measured via cell proliferation in the Detroit 562 cell line overexpressing PD-L1 and via interferon gamma (IFN-γ) production in two-way mixed lymphocyte reaction (MLR) assays. Immune profiling was measured in MC38 tumor-bearing mice using flow cytometry. Gene expression changes were explored in MC38 tumor-bearing mice via bulk RNA sequencing (RNAseq) and further analyzed using MSigDB and gene set enrichment analysis (GSEA). Single-cell RNAseq (scRNAseq) was used to investigate TME reprogramming and co-expression of TGF-β and PD-L1 in MC38 tumor samples. Immunostaining was also performed to investigate TGF-β and PD-L1 co-expression on human non-small cell lung cancer (NSCLC) tumor samples. PD-L1-dependent internalization of BA-bound TGF-β was measured in cell-based assays by flow cytometry. Internalization of TGF-β into lysosomes was determined by the live-cell imaging systems IncuCyte Zoom and CellDiscoverer7, and by confocal microscopy.
For all other materials and methods, please see online supplemental material 1.
jitc-2021-004122supp003.pdf (2.6MB, pdf)
Results
Simultaneous binding of both TGF-βRII moieties results in an avidity-based increase in affinity for BA
The kinetics of interactions between BA or fresolimumab with TGF-β were measured by SPR with immobilized ligands for comparison with published data. Binding affinity of BA to immobilized TGF-β1 (KD=3.6 nM) and TGF-β3 (KD=0.4 nM) was similar to that of fresolimumab (TGF-β1=1.7 nM; TGF-β3=0.7 nM). Binding affinity of fresolimumab for TGF-β2 was 1.8 nM, consistent with previously published values,18 while that for BA could not be detected (online supplemental figure S1A, table S1). This is presumably because TGF-β2 was immobilized on the surface of the chip and BA binds with avidity to only soluble TGF-β2.2
ITC was used to analyze binding interactions in solution (online supplemental figure S1B). Similar to fresolimumab, BA bound TGF-β1 with an estimated KD ≤2 nM and followed a near 1:1 stoichiometry. To explore potential avidity within the BA/TGF-β1 complex, we generated a monovalent BA one-leg variant, bearing a single TGF-βRII moiety. ITC revealed a significantly weaker affinity (KD=8.3 nM) for this variant, and the stoichiometry implied a 2:1 binding of BA one-leg variant with the bivalent TGF-β1 dimer. Similarly, ITC titration of TGF-β1 with monovalent fresolimumab one-arm or with the fresolimumab Fab revealed a consistent stoichiometry of N~2, matching published structural data.18
The affinity in some of the complexes was too tight for exact differentiation via ITC so we employed MST as a complementary method. The MST assay setup was validated by titrating fluorescently labeled TGF-β1 with fresolimumab Fab, resulting in an average KD of ~1.6 nM (online supplemental figure S1C), consistent with ITC results. MST binding isotherms of labeled TGF-β1 with BA were fitted using a Hill function, showing KD=55 pM (figure 1A). Finally, BA one-leg variant reported a KD of 4.3 nM (online supplemental figure S1D), again consistent with ITC values and reflecting the intrinsic affinity at one binding site. Overall, the MST results demonstrate that BA (two TGF-βRII moieties) gains significant affinity (~80-fold) toward the TGF-β1 dimer compared with bintrafusp one-leg variant (one TGF-βRII moiety). Such avidity-induced affinity gain was not observed for fresolimumab, with the SPR KD (1.7 nM) being nearly identical to the intrinsic MST KD (1.6 nM) measured for fresolimumab Fab (online supplemental table S1).
Figure 1.
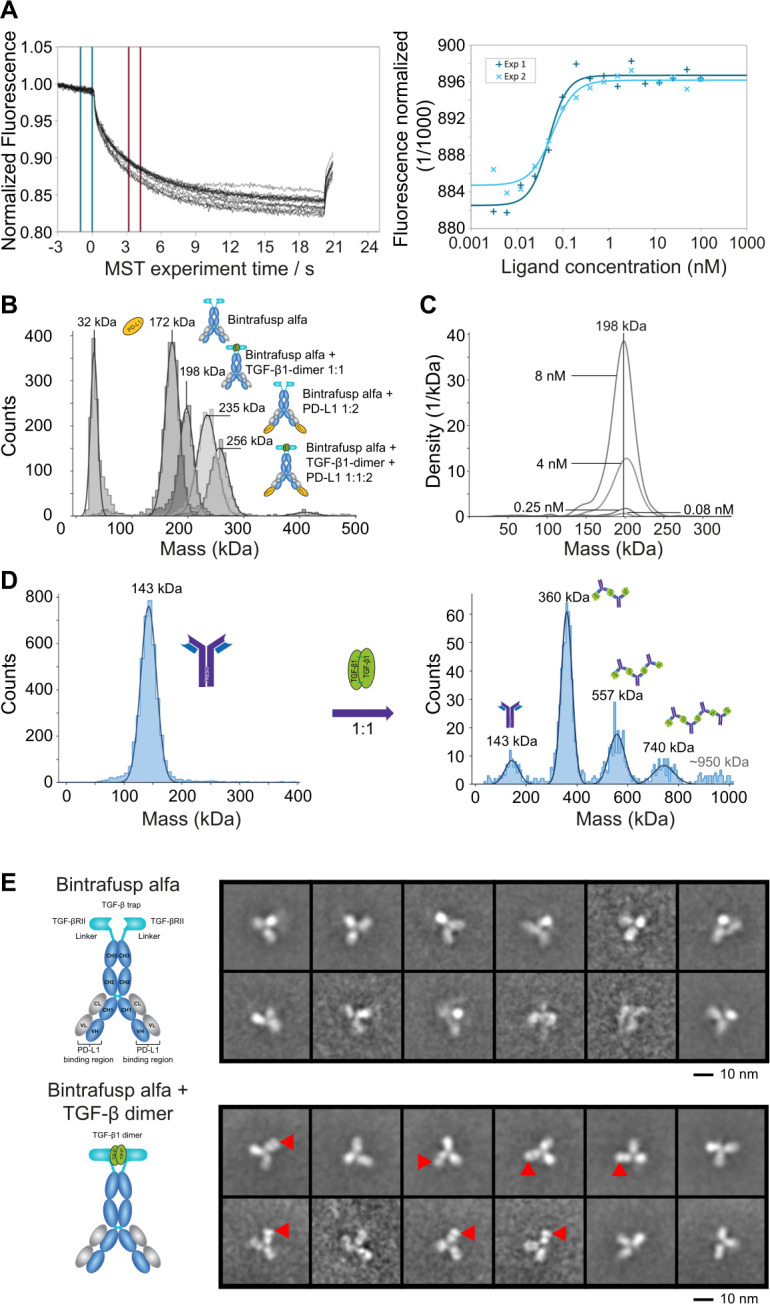
Avidity-based binding affinity of BA to TGF-β1 is greater than that of fresolimumab. (A) MST dose response for TGF-β1 dimer and BA. Left panel: representative MST traces. Blue lines indicate the area for averaging the baseline fluorescence and red lines indicate the area for averaging the fluorescence after thermal induction. Right panel: combined dose response curve for fluorescence change from two experiments. (B) Mass photometry of binary and ternary complexes of BA /TGF-β1 dimer/PD-L1. Overlay of Gaussian distribution plots with indicated center masses for the individual native biomolecules and complexes shown to the right. (C) Detection limits (as low as 80 pM) for BA /TGF-β1 dimer complex. The complex is detected as a uniform distribution plot with a center mass of 198 kDa matching a 1:1 ratio. (D) Oligomeric complex formation of fresolimumab with TGF-β1 dimer. Left panel: detection of free fresolimumab. Right panel: multiple Gaussian peaks correspond to higher order associations of fresolimumab with TGF-β1 dimer. (E) Negative-stain EM analysis. Top panel: typical reference-free 2D class averages corresponding to BA (selected from 40 classes). Bottom panel: typical reference-free 2D class averages corresponding to BA+TGF-β1 dimer (selected from 60 classes). The red arrowheads indicate the position of the extra density attributed to the TGF-β1 dimer. 2D, two-dimensional; BA, bintrafusp alfa; EM, electron microscopy; MST. micro-scale thermophoresis; PD-L1, programmed death-ligand 1; TGF-β, transforming growth factor-β.
Although both BA and fresolimumab revealed a 1:1 binding stoichiometry in ITC experiments (online supplemental figure S1B), the binding mode was different. Equimolar mixtures of BA with TGF-β1 analyzed by DLS showed a small increase of the hydrodynamic radius (online supplemental figure S1E), which fits with the formation of a 1+1 complex but not with higher order associations (eg, 2:2). Fresolimumab and TGF-β1 do not form 1+1 complexes but rather hetero-oligomeric assemblies. For stoichiometric mixtures, the hydrodynamic radius increased to 47.7 nm compared with only 5.25 nm for free fresolimumab, indicating the presence of large polydisperse oligomers.
The complex formation in solution was further analyzed by mass photometry19 (figure 1B). The uniform BA/TGF-β1 complex was detected at a center mass of 198 kDa matching a 1+1 ratio (figure 1C). Interestingly, further uniform complexes of BA with PD-L1 (1+2 complex) as well as the ternary complex of BA/TGF-β1/PD-L1 with a 1+1+2 ratio were generated. All species were detected with center masses matching the theoretical mass of the complexes (online supplemental figure S1F). In contrast, a 1:1 mixture of fresolimumab and TGF-β1 (nM concentrations) revealed heterogeneous higher order complex formation, where a fresolimumab/TGF-β1 2+3 complex was most abundant but even species in agreement with 3+4 and 4+5 associations were observed (figure 1D). Adding a six-fold excess of TGF-β1 shifted the equilibrium toward smaller complexes and preferentially a 1+2 complex was formed (online supplemental figure S1G).
The complex between BA and TGF-β1 was detected in mass photometry at a uniform center mass of 198 kDa (figure 1C), even after dilution to 80 pM. Negative-stain EM revealed the expected IgG-like domain structural organization for BA alone (online supplemental figure S1H) and in complex with TGF-β1 (figure 1E). Different two-dimensional classes were obtained due to the inherent flexibility of the Fab domains with respect to the Fc domain. An extra density was visualized close to the BA molecules when TGF-β1 was present (figure 1E). Considering the size of the density and molecular weight of TGF-β1, only one bound TGF-β1 was visualized per BA. This extra density was only bound to one out of the three main IgG domains, consistent with the TGF-β1 binding at the Fc domain side of BA, where the TGF-βRII receptor moieties are connected.
BA enhances TGF-β sequestration by targeting the cell surface through PD-L1 binding
The Detroit 562 human pharyngeal carcinoma cell line20 is dependent on TGF-β for growth but expresses low levels of endogenous PD-L1. In vitro treatment with either BA or TGF-β trap dose-dependently inhibited proliferation of Detroit 562 cells (Detroit-parental) (figure 2A) and Detroit 562 cells transfected to overexpress PD-L1 (Detroit–PD-L1, figure 2B, online supplemental figure S2). At the lowest concentrations, BA, but not TGF-β trap, significantly inhibited proliferation of Detroit–PD-L1 but not Detroit-parental cells. Similarly, BA was more potent than fresolimumab at inhibiting the proliferation of Detroit–PD-L1, but not Detroit-parental cells (figure 2C, D). Furthermore, pre-incubating Detroit–PD-L1 cells with anti-PD-L1 almost completely reversed the potency of BA, whereas the effect of TGF-β trap was relatively unchanged (figure 2E). The data together support that targeting TGF-β trap to the cell surface enhanced TGF-β sequestration.
Figure 2.

BA enhances TGF-β blockade by colocalizing the TGF-β trap moiety to the cell surface via binding to PD-L1. (A–E) CellTiter-Glo 2.0 proliferation assays using luminescence (RLU) to measure cell numbers 2 weeks after the start of treatment. Mean and SD are shown with nonlinear best fit curves. (A) Detroit parental or (B) Detroit–PD-L1 cells were plated and treated with varying concentrations of TGF-β trap or BA for 2 weeks. (C) Detroit parental or (D) Detroit–PD-L1 cells were plated and treated with varying concentrations of fresolimumab or BA. (E) Detroit–PD-L1 cells were plated and treated with anti-PD-L1 or an isotype control antibody 30 min prior to TGF-β trap or BA treatment. (F–H) Two-way MLR assays, using ELISA to measure IFN-γ production after the coculture of PBMCs from one individual (responder) and PBMCs from another individual (stimulator). Fold changes with mean±SE of the mean are shown. (F) Results from 6 assays with 6 different donor pairs, cocultured at 1:1 ratio for 3 days, are plotted as fold changes over isotype control (set to 1). Cells were treated (10 µg/mL) with isotype, BA, anti-PD-L1, TGF-β trap or anti-PD-L1+TGF-β trap. P values were calculated using a one-way ANOVA with Dunnett’s multiple comparisons test, where *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. (G–H) Results at day 3 from 4 assays with 4 different donor pairs treated with varying concentrations of BA and (G) anti-PD-L1+TGF-β trap, or (H) anti-PD-L1+fresolimumab. ANOVA, analysis of variance; BA, bintrafusp alfa; IFN-γ, interferon gamma; MLR, mixed lymphocyte reaction; PBMCs, peripheral blood mononuclear cells; PD-L1, programmed death-ligand 1; RLU, relative light unit; TGF-β, transforming growth factor-β.
In a two-way MLR, anti-PD-L1 or TGF-β trap alone led to slightly enhanced IFN-γ production, which was further enhanced with their combination. However, BA was more potent than the combination of anti-PD-L1 and TGF-β trap or fresolimumab, suggesting that targeting TGF-β blockade to the cell surface of immune cells further potentiates TGF-β inhibition and may ameliorate TGF-β-induced immunosuppression (figure 2F–H).
BA increases median survival, tumor regressions, and TILs compared with the combination therapy in the MC38 model
BA treatment extended median survival and resulted in more complete tumor regressions compared with the combination of anti-PD-L1 and TGF-β trap in a dose-dependent manner in MC38 tumor-bearing mice (figure 3A). Furthermore, BA increased total and proliferating CD4+ tumor-infiltrating lymphocytes (TILs) (figure 3B, C, online supplemental figure S3), CD8+ TILs (figure 3D, E), CD8+/P15E TILs (figure 3F, G), as well as CD8+ effector and central memory TILs (figure 3H, I), relative to anti-PD-L1, TGF-β trap or fresolimumab monotherapies, and the combination of anti-PD-L1 with TGF-β trap or fresolimumab.
Figure 3.

BA extended survival and tumor regressions in MC38 tumors and increased various TIL populations compared with the combination therapy. (A) µMt− mice (n=14 per group) bearing MC38 tumors were treated iv with isotype control (400 µg), anti-PD-L1 (133 µg)+TGF-β trap (164 µg) (low dose), BA (164 µg) (low dose), anti-PD-L1 (400 µg)+TGF-β trap (492 µg) (high dose), or BA (492 µg) (high dose) on day 0, day 3, and day 6. Percent survival, median survival days, and the percentage of complete tumor regressions are shown. (B–I) C57BL/6 mice (n=8 per group) bearing MC38 tumors were treated intravenously on day 0, day 1, and day 2 with isotype control (400 µg), TGF-β trap (492 µg), anti-PD-L1 (400 µg), BA (492 µg), fresolimumab (200 µg), or a combination of anti-PD-L1 with TGF-β trap or fresolimumab. Tumors were harvested on day 6 and dissociated for flow cytometry analysis. Expression of (B) CD4+ TILs, (C) CD4+ Ki67+ TILs, (D) CD8+ TILs, (E) CD8+ Ki67+ TILs, (F) CD8+ P15E Pentamer+ TILs, (G) CD8+ P15E Pentamer+ Ki67+ TILs, (H) CD8+ EM TILs, and (I) CD8+ CM TILs. Expression of markers per 100 mg tumor are shown, and p values were calculated using a one-way ANOVA with Dunnett’s multiple comparisons test, where *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. ANOVA, analysis of variance; BA, bintrafusp alfa; CM, central memory; EM, effector memory; PD-L1, programmed death-ligand 1; TIL, tumor-infiltrating lymphocyte; TGF-β, transforming growth factor-β.
BA elicits distinct gene expression changes, induces reprogramming of stromal and immune landscape, and suppresses genes associated with metastasis, ECM, and SMAD signaling in MC38 TME
To identify the effects on biological functions and signaling pathways that differentiate BA treatment from the combination therapy, MC38 tumors were collected for RNAseq analysis. Differentially expressed genes (DEGs) were identified for the following comparisons: (1) BA versus isotype control, (2) BA versus the combination therapy, and (3) the combination therapy versus isotype control. Their distribution was visualized (figure 4A, B).
Figure 4.

BA treatment induces distinct gene expression changes and signaling pathway activation when compared with the combination therapy. C57BL/6 mice (n=15 per group) bearing MC38 tumors were treated intravenously with isotype control (400 µg), anti-PD-L1 (400 µg), TGF-β trap (492 µg), BA (492 µg), or the combination of anti-PD-L1 and TGF-β trap on day 0, day 1, and day 2. Mice were sacrificed on day 6 and RNAseq analysis was performed on tumor samples. (A) Venn diagram showing the distribution of DEGs in the following comparisons: (1) BA versus isotype control, (2) BA versus the combination therapy, and (3) the combination therapy versus isotype control. DEGs were defined by genes with expression fold change equal to or greater than 1.5 and an FDR corrected p value of 0.05 or less based in their log2TPM values. 72 genes were regulated by BA but not by the combination therapy. (B) Heatmap of top DEGs regulated by both BA and combination therapy versus isotype control. Red and blue colors represent positive and negative z-score, respectively. A positive z-score indicates gene expression higher than the mean expression across all treatment groups, while a negative z-score indicates expression lower than the mean value. (C) Enrichment plots and gene signature plots of gene sets enriched in MC38 tumor-bearing mice treated with BA versus those treated with the combination therapy are presented. GSEA identified 297 gene sets that were significantly enriched at FDR <25% and 218 gene sets that were significantly enriched at a nominal p value of <0.01 with BA. The positive NES indicates pathways that were enriched in BA-treated mice. (D) Enrichment plots and gene signature plots of gene sets suppressed in MC38 tumor-bearing mice treated with BA versus those treated with the combination therapy are presented. GSEA identified 143 gene sets were significant at FDR <25% and 140 gene sets were significantly enriched at a nominal p value of <0.01 with combination therapy. The negative NES indicates pathways that were suppressed by BA versus combination therapy. Statistical significance of gene signature scores was examined by one-way ANOVA and multiple comparison correction. ANOVA, analysis of variance; BA, bintrafusp alfa; DEGs, differentially expressed genes; FDR, False Discovery Rate; GSEA, gene set enrichment analysis; NES, normalized enrichment score; PD-L1, programmed death-ligand 1; TGF-β, transforming growth factor-β; TPM, transcripts per million.
Among the 2543 DEGs identified in mice treated with BA versus isotype control, 1258 of them overlapped with DEGs in mice treated with the combination therapy versus isotype control. Top DEGs regulated by both BA and the combination therapy were further investigated. While these shared DEGs suggested that dual blockade of PD-L1 and TGF-β signaling by either approach could modulate similar biological responses in the TME, greater magnitude of regulation was observed for BA treatment versus the combination therapy. For example, stronger induction of perforin 1, a cytolytic protein essential to cytotoxic T and natural killer (NK) cell-mediated antitumor immunity, and greater suppression of metastasis-related and invasion-related matrix metalloproteinase-10 were demonstrated by BA versus the combination therapy (online supplemental figure S4A). Furthermore, 1285 DEGs were only modulated by BA versus isotype control but not by the combination therapy versus isotype control, including 72 BA-specific DEGs using both isotype control and the combination therapy as baselines, suggesting that the colocalization mechanism of BA also contributed to distinct biological responses in the TME.
To identify potential biological functions that are modulated specifically by BA, the list of 72 BA-specific DEGs, along with a list of the 530 significantly upregulated and 603 significantly downregulated genes that were induced or suppressed only by BA versus isotype control, but not by the combination therapy versus isotype control, were evaluated against the hallmark (H) and curated (C2) gene sets in MSigDB (V.7.3). The 72 BA-specific DEGs significantly overlapped with the gene signatures representing epithelial-mesenchymal transistion (EMT) and ECM biology (online supplemental table S2). The 530 upregulated genes significantly overlapped with B cell maturation, NKT cell differentiation, NK cell reprogramming, and immune-related cytokines (online supplemental table S3), while the 603 downregulated genes significantly overlapped with gene signatures representing metastasis, ECM, and SMAD signaling (online supplemental table S4).
In addition to hypergeometric analysis, GSEA was performed to identify enriched phenotypes21 (online supplemental table S5) and signature scores were computed to visualize the effects of TGF-β and PD-L1 signaling blockade, as well as the differentiation between BA and the combination therapy. Consistent with the overlapping gene signature analysis, GSEA demonstrated significant increases in immune responses, as indicated by enrichment of a gene signature specific to IL-12 signaling, of which significant induction was only observed in mice treated with BA, as well as enrichment of T cell receptor signaling and IFN-γ response in BA-treated mice (figure 4C, online supplemental figure S4B). GSEA also revealed the suppression of gene signatures specific to metastasis, ECM organization, collagen deposition, and TGF-β signaling activity by BA (figure 4D, online supplemental figure S4C). Based on the gene signature scores, BA was the only treatment that significantly reduced the expression of matrisome and ECM glycoprotein signatures, suggesting enhanced targeting of the tumor stroma by BA.
scRNAseq shows treatment-induced immune modulation, identified TGF-β and PD-L1 co-expressing cell populations, and reveals treatment-specific macrophage and fibroblast subclusters in the MC38 TME
scRNAseq was performed on MC38 tumors collected from mice treated with isotype control, anti-PD-L1, TGF-β trap, BA, or the combination therapy. After quality check (online supplemental S5A, B) and unsupervised clustering analysis, 36 distinct cell clusters were identified (figure 5A) and cell clusters were annotated by evaluating the expression levels of cell type-specific markers (online supplemental table S6). Treatment-based differences were identified and visualized in the uniform manifold approximation and projection representation (figure 5B) and the contribution of individual clusters by different treatment groups was computed to identify treatment-specific clusters. Cluster 25, which emerged only in BA-treated mice, was enriched for immune activation markers and effector molecules, including Tnfrsf9, Ifng, and Gzmb. Compared with other treatment groups, BA also had higher contribution to clusters 11 (Cd3d+, Cd3e+, and Cd3g+ T cells) and 34 (Cd79a+ and Cd79b+ B cells). Conversely, BA treatment had a lower contribution to the Cd68+ myeloid cluster 33 (figure 5C). Subsequent DEG and enrichment analysis showed that in regulatory T cell (Treg) and fibroblasts, immunity-related signatures like IFN-α response and IFN-γ response pathways, and inflammation-related signatures like complement pathway and inflammatory response pathway had lower activities for mice treated with the combination therapy relative to BA treatment (online supplemental table S7).
Figure 5.

Evaluation of TME reprogramming and identification of cell populations co-expressing TGF-β and PD-L1. C57BL/6 mice (n=3 per group) bearing MC38 were treated as described in figure 4 legend. Mice were sacrificed on day 6 and tumor samples were dissociated for scRNAseq. (A) UMAP representation and graph-based clustering of merged scRNAseq data from all treatment groups. Cell clusters were annotated by evaluation of canonical markers for different cell lineages (PanglaoDB: single cell sequencing resource for gene expression data; https://panglaodb.se) and the DEGs computed between two groups cells using the MAST algorithm. Cell clusters were annotated that the average expression of the group of markers is greater than 1 unit and the percentage of expression is greater than 30. (B) UMAP representation of treatment-based differences. (C) Contribution to individual clusters by different treatment groups and treatment-specific clusters. (D) Cell clusters demonstrating high expression of TGF-β and PD-L1 were identified based on the average expression of both these genes above the selected cut-off of 0.5. The majority of co-expressing cells were found in clusters 0, 22, 24, 26, 33, and 35. (E) Expression levels of TGF-β and PD-L1 in annotated cell clusters. (F) Characterization of macrophages by subclustering revealed 11 distinct macrophage populations in the MC38 TME. (G) Treatment-specific subclusters were identified among macrophage populations. (H) TGF-β and PD-L1 expression levels in different macrophage subclusters. Newly emerged BA-specific clusters express low levels of TGF-β, PD-L1, or both. (I) Global assessment of the single-cell transcriptome profiles of macrophages showed that BA-induced macrophage populations possessed unique gene expression pattern. (J) Evaluation of key macrophage markers. BA-induced subclusters 4, 9, and 10 express lower levels of M2 markers and higher levels of M1 markers compared with common macrophage subclusters 0, 1, and 2. BA, bintrafusp alfa; DEGs, differentially expressed genes; MAST, model-based analysis of single-cell transcriptomics; PD-L1, programmed death-ligand 1; TGF-β, transforming growth factor-β; TME, tumor microenvironment; scRNAseq, single-cell RNA sequencing; UMAP, uniform manifold approximation and projection.
Since cell populations that co-express high levels of TGF-β and PD-L1 may be preferentially targeted by BA to contribute to the TME modulation observed in the bulk RNAseq data, cell clusters that demonstrated high expression of both genes were identified. The majority of the co-expressing cells were observed in clusters 0, 22, 24, 26, 33, and 35 (figure 5D). These clusters were identified as Cd4+/Foxp3+/Tigit+/Cd274+ Treg cells (clusters 22 and 35), Cd83+ dendritic cells (DCs) (cluster 26), Cd68+/S100a4+/Cd164+ myeloid cells (cluster 33), and Mrc1+/Itgam+/Cd86+ macrophages (clusters 0 and 24). TGF-β1 and PD-L1 expression in all annotated cell types was visualized (figure 5E, online supplemental figure S5C).
As one of the major TGF-β and PD-L1 co-expressing cell types, single cell transcriptome of macrophages was further evaluated to assess treatment impact. Expression profile-based subclustering of macrophages revealed 11 distinct macrophage subclusters (figure 5F). While most macrophages (subclusters 0, 1 and 2) could be identified from all treatment groups, emergence of treatment-specific subcluster was observed (figure 5G, H). Analysis of TGF-β and PD-L1 expression levels in these subclusters showed that macrophage clusters 0, 1 and 2 express high levels of both genes, while newly emerged BA-specific clusters (4, 5, 9 and 10) express low levels of either TGF-β or PD-L1, or both. For combination therapy-specific macrophages, subclusters 3 and 8 expressed low levels of TGF-β and PD-L1, and subcluster 6 retained high expression of both genes (figure 5H). Global assessment of the single-cell transcriptome profiles of macrophages revealed that, compared with isotype control and combination therapy, BA-induced macrophage populations possessed unique gene expression pattern, demonstrating the impact of co-localized targeting of TGF-β and PD-L1 signaling in double positive cell populations (figure 5I). Furthermore, BA-induced subclusters 4, 9, and 10 expressed lower levels of M2 markers, including Arg1, Tgm2 and Il1r2, and higher levels of M1 markers, including Cd40 and Cd80, compared with subcluster 0, 1 and 2, suggesting a potential expansion of the M1 population and suppression of M1 to M2 polarization by BA (figure 5J).
To investigate the impact on tumor stroma by BA, as demonstrated by bulk RNAseq, similar subclustering on fibroblasts revealed 12 distinct subclusters (online supplemental figure S5D) most shared by all treatment groups (online supplemental figure S5E). Subcluster 9 was anti-PD-L1-specific, while subcluster 10 was observed only in BA-treated mice (online supplemental figure S5F). Interestingly, while expression of TGF-β or PD-L1 was detected in most fibroblast subpopulations, BA-specific subcluster 10 was the only fibroblast population that expressed low levels of both genes (online supplemental figure S5G). BA-induced fibroblasts expressed significantly higher levels of the IFN-γ-inducible gene Gpb2 (log2 fold change=1.465, FDR adjusted p=2.70E-04) and the immune-related gene Cacybp (log2 fold change=1.410, FDR adjusted p=0.006), which has been shown to negatively correlate to PD-1 levels in hepatocellular carcinoma.22 These fibroblasts expressed lower levels of Col3a1 (log2 fold change=−1.477, FDR adjusted p=0.050) and lncRNA MALAT1 (log2 fold change=−4.194, FDR adjusted p=6.11E-47), which has been shown to promote migration, wound healing, and collagen production.23 Overall, these observations suggest that BA treatment induces the emergence of a fibroblast subpopulation with lower pro-tumorigenic potential in an immunity enhanced TME.
BA suppresses collagen deposition and increases M1/M2 macrophage ratio in MC38 tumors
Bulk and single-cell transcriptome analysis identified that BA treatment modulated macrophage and ECM phenotypes. To confirm these findings, histological assays were performed on MC38 tumor tissue samples. Multiplex immunohistochemistry of CD68 and the M1 macrophage marker inducible nitric oxide synthase, as well as CD68 and the M2 macrophage marker arginase-1 showed that, compared with isotype control and the combination therapy, BA led to the increase of M1 and decrease of M2 cell density in the MC38 TME. An overall increase of M1/M2 ratio in BA-treated animals was also observed (figure 6A), confirming our transcriptome profiling observations.
Figure 6.

Modulation of macrophage phenotype by BA in MC38 tumors and visualization of PD-L1 and TGF-β1 co-expressing macrophages and fibroblasts in human NSCLC samples. (A) Representative micrographs of multiplex chromogenic IHC of CD68/iNos and CD68/arginase-1, and quantitation of M1 and M2 macrophage densities in MC38 tumors. Scale bar=100 µm; brown signal: CD68; red signal: iNos/arginase-1. P values were generated with one-way ANOVA with Dunnett’s multiple comparison. (B) and (C) Micrographs of multiplex IF of PD-L1/TGF-β1/F4-80 and PD-L1/TGF-β1/α-SMA in human NSCLC tissue samples demonstrating the presence of PD-L1/TGF-β1 double positive macrophage and fibroblasts, respectively. Scale bar=10 µm. ANOVA, analysis of variance; BA, bintrafusp alfa; FDR, false discovery rate; IHC, immunohistochemistry; IF, immunofluorescent; iNos, inducible nitric oxide synthase; NSCLC, non-small cell lung cancer; PD-L1, programmed death-ligand 1; SMA, smooth muscle actin; TGF-β, transforming growth factor-β.
Masson’s trichrome staining showed that BA and combination therapy significantly reduced collagen deposition compared with isotype control. A more consistent reduction of collagen staining intensity and positivity was observed for BA compared with combination therapy for the animals evaluated (online supplemental figure S5H).
Immunofluorescent staining reveals TGF-β and PD-L1 co-expressing cells in human lung tumors
Immunofluorescent (IF) staining of PD-L1, TGF-β, and cell type-specific markers on human advanced stage NSCLC tissue sections was performed to visualize the presence of PD-L1 and TGF-β co-expressing cells in a human disease setting. Multiplex IF results revealed the presence of PD-L1+/TGF-β1+/F4-80+ macrophages (figure 6B) and PD-L1+/TGF-β1+/α-SMA+ fibroblasts (figure 6C) in human lung tumors.
BA depletes bound TGF-β through PD-L1 dependent internalization and lysosomal degradation
The binding of BA/TGF-β1 complex on the cell surface was first measured by flow cytometry. On HEK293 cells transfected to overexpress PD-L1 (HEK293-PD-L1), BA bound PD-L1 and captured TGF-β1 in a dose-dependent manner (online supplemental figure S6A, B). On 4T1 cells, TGF-β1 captured by BA was dependent on PD-L1 expression induced by IFN-γ (online supplemental figure S6C-D). In HEK293-PD-L1 cells, TGF-β1 captured by BA was ~30× higher than TGF-β1 captured by endogenous TGF-βR (figure 7A), suggesting BA may be advantageous over endogenous TGF-βR in capturing TGF-β1 on cells with a high ratio of PD-L1/TGF-βR. We next measured, via flow cytometry, whether TGF-β can be internalized by BA. The internalization of BA in HEK293-PD-L1 cells was ~32% at 4 hours and increased to 69% at 20 hours, while the internalization of the TGF-β1/BA complex was about 44% at 4 hours and increased to 83% at 20 hours (figure 7B, online supplemental figure S7A).
Figure 7.

TGF-β1 is internalized by BA in HEK293-PD-L1 cells and degraded in the lysosomes. (A) Biotinylated TGF-β1 binding to BA/PD-L1 complex or binding to TGF-βR on the cells surface was measured via MFI of SA-APC by flow cytometry. (A) Left panel: biotinylated TGF-β1 binding to TGF-βR was blocked by pre-incubation of cells with unlabeled TGF-β1 (130 ng/mL). Right panel: the binding of biotinylated TGF-β1 to TGF-βR was measured without pre-blocking TGF-βR. The box highlights the biotinylated TGF-β1 binding to the endogenous TGF-βR in the control groups, in which neither isotype control (inactive anti-PD-L1) nor TGF-β trap antibody binds to PD-L1. (B) Internalization of TGF-β1 by BA measured by flow cytometry at 20 hours (n=2) compared with 4 hours (n=3). The internalization of BA serves as a positive control. (C) Live-cell imaging for internalization/colocalization of biotinylated TGF-β1/SA-AF488 by BA in LE and lysosomes measured at time 0 hour and time 10 hours. Cells treated with the combination of anti-PD-L1 and TGF-β trap served as a control. The BA and anti-PD-L1+TGF-β trap combination were labeled with pHrodo conjugated goat anti-human IgG to track the internalization of BA or anti-PD-L1 to low pH organelles such as LE and lysosomes. (D) Imaging for internalization/colocalization of TGF-β1 by BA in lysosomes measured at 8 hours post treatment. AF488 conjugated anti-TGF-β1 antibody and AF594 conjugated anti-LAMP2 antibody were used to recognize TGF-β1 and LAMP2, respectively. Individual channel and merged image are shown. (E) MFI of Bio-TGF-β1/SA-AF488 with or without chloroquine (a lysosomal inhibitor) and sodium azide (an inhibitor for receptor internalization) was measured every 12 hours for 3 days. Data represent 3 independent assays (n=3). (F) The extracellular biotinylated TGF-β1 in the culture supernatant at 48 hours was measured by a sandwich ELISA. The HEK293-PD-L1 cells were pre-treated with or without anti-PD-L1. Treatment with BA or TGF-β trap was compared. (G) Model of simultaneous binding/internalization/degradation of active TGF-β by BA. Controls for figure 7 are located in online supplemental figure S7. APC, antigen-presenting cell; BA, bintrafusp alfa; IgG, immunoglobulin G; LE, late endosomes; MFI, median fluorescence intensity; PD-L1, programmed death-ligand 1; SA, streptavidin; TGF-β, transforming growth factor-β.
The subcellular location of TGF-β1 internalized by BA was first determined using CellDiscoverer 7 (figure 7C, online supplemental figure S7B (control groups), online supplemental video S1 and S2). pHrodo-labeled BA or anti-PD-L1 antibody was undetectable on the cell surface but became visible in late endosomes (LE) and even more so in lysosomes, where the pH turned more acidic. The SA-AF488 labeled biotinylated TGF-β1 was weakly visible on the cell surface and concentrated in intracellular vesicles only in cells treated with BA. Confocal microscopy of HEK293-PD-L1 cells at 8 hours post treatment confirmed that TGF-β1 colocalized with BA in lysosomes, as TGF-β1 overlapped with the lysosome marker LAMP2 only in cells treated with BA (figure 7D, online supplemental figure S7C (control groups)).
jitc-2021-004122supp001.wmv (387.8KB, wmv)
jitc-2021-004122supp002.wmv (387.8KB, wmv)
Lysosomal degradation of TGF-β1 internalized by BA in HEK293-PD-L1 cells was next quantified using IncuCyte Zoom (figure 7E, online supplemental figure S7D (images), online supplemental figure S7E (antibody control)). TGF-β1 bound to the cell surface only in BA-treated cells. The relative integrated intensity was gradually reduced when TGF-β1/BA internalized and localized to the lysosome over 72 hours. Treatment with either the receptor internalization inhibitor sodium azide or the lysosome inhibitor chloroquine significantly inhibited the decay of either TGF-β1 or BA, indicating that BA-bound TGF-β1 is internalized and degraded in the lysosome.
TGF-β1 in cell culture medium was depleted after 2 days in BA-treated, but not TGF-β trap-treated HEK293-PD-L1 cells. Furthermore, the depletion was completely inhibited by blocking the cells with anti-PD-L1 (figure 7F), which confirmed that depletion of extracellular TGF-β1 by BA is dependent on binding to PD-L1 (figure 7G).
Discussion
We hypothesized that the anti-PD-L1 moiety of BA may facilitate the targeting of TGF-β signaling in PD-L1-expressing tumor cells and tumor infiltrating immune cells, leading to a more profound inhibition in tissue compartments that are relevant to tumor progression compared with a non-targeting TGF-β inhibitor.
We first demonstrated that the bivalent BA exhibits a strong gain of functional affinity for TGF-β1 to 55 pM from the 4.3 nM affinity of the monovalent BA one-leg variant. We regard this ~80-fold affinity increase to be due to an avidity effect, by simultaneous binding of both TGF-βRII moieties from the same BA molecule, each with one of the protomers of the TGF-β1 dimer. This is further supported by the 1:1 stoichiometry in ITC and the 1+1 complex species between BA and TGF-β1 in mass photometry even at sub-nM concentrations as well as in negative-stain EM. This avidity-based gain of affinity might have significant functional relevance as BA can compete more effectively with cellular surface TGF-β receptors for trapping TGF-β1. As a result of avidity binding and the finding that active TGF-β captured by BA can be further internalized and degraded in lysosomes, there should be little risk of TGF-β captured by BA being released into the TME.
We also showed that by targeting TGF-β blockade to the cell surface through binding to PD-L1, BA greatly enhanced the potency of TGF-β sequestration. This effect was further confirmed in a two-way MLR assay, in which BA is more potent than the combination of anti-PD-L1 and a non-targeting TGF-β inhibitor. In a tumor model, BA enhanced antitumor activity significantly more than the combination of anti-PD-L1 with TGF-β trap or fresolimumab. This bifunctional approach, which has been corroborated by others using a number of TGF-β trap fusion proteins targeting other immune cell markers such as CTLA-48 and CD4,24 demonstrates that the bifunctional molecule, besides sequestering free TGF-β in circulation,2 can uniquely neutralize locally activated TGF-β on the surface of the target cells.
Gene expression analysis identified that BA-modulated DEGs significantly overlapped with the gene signatures representing EMT and ECM biology, including activated cancer-associated fibroblast (CAF)-specific proteins MFAP525 26 and FAP,27 as well as collagen protein COL12A1.28 These genes, which are upregulated in CAFs on activation by TGF-β in the TME,29 were significantly downregulated by BA but not by the combination therapy, suggesting more effective targeting of reactive stroma and CAF activation by BA. CAF enrichment is associated with aggressive phenotypes and poorer survival outcomes in multiple tumor types.30–32 While heterogeneity of CAFs may play diverse roles in the TME,33 34 high TGF-β fibroblast signature and collagen rich stroma are associated with poor responses to ICI,1 and neutralization of TGF-β in the TME leads to a reduction of myofibroblast subsets and the formation of fibroblast populations characterized by increased immunomodulatory properties.35
TGF-β plays central roles in immune suppression within the TME36 and inhibits interleukin 12 (IL-12) induced IFN-γ production by T and NK cells.37 38 TGF-β also inhibits IL-12 responsiveness in T cells by inhibiting IL-12-induced phosphorylation of the STAT4 transcription factor.39 We showed that BA, but not the combination therapy, enriched the gene signature specific to IL-12 signaling. Indeed, the combination of an HPV vaccine with NHS-IL12 and BA has been evaluated preclinically40 and is being investigated in an ongoing phase 2 trial (NCT04287868).
Enrichment of immune-related gene signatures and suppression of tumor metastasis-related, ECM-related, and collagen deposition-related genes in our RNAseq data suggest that colocalized inhibition of TGF-β and PD-L1 in the TME by BA may be more effective in restoring antitumor immunity and inhibiting aggressive cancer phenotypes compared with systemic TGF-β blockade in combination with anti-PD-L1. We hypothesized that BA trapping of locally activated TGF-β in PD-L1high/TGF-βhigh TME could lead to enhanced TGF-β neutralization. scRNAseq analysis indicated that Tregs, macrophages, and immune cells of myeloid linage, all of which secrete TGF-β, also co-express PD-L1. Targeting TGF-β in these cell types may contribute to a more immunogenic TME and lead to enhanced antitumor immunity. In preclinical models, selective blockade of TGF-β1 production by Tregs with antibodies against GARP-TGF-β1 complexes sensitizes resistant tumors to anti-PD-1 immunotherapy.41
Interestingly, Cd4+/Foxp3+ Tregs that co-express PD-L1/TGF-β1 also express high levels of the coinhibitory molecule TIGIT. Tregs expressing TIGIT inhibit the proinflammatory T helper 1 (Th1) and Th17 cell responses.42 In preclinical studies, TIGIT blockade has limited antitumor efficacy, but the combination of anti-TIGIT and anti-PD-(L)1 leads to tumor rejection.43 However, the combination of tiragolumab and atezolizumab did not meet a primary endpoint of progression-free survival.44 In our study, assessment of the pathway level differences between BA-treated and the combination-treated mice suggested higher IFN and inflammatory response signatures in Tregs of BA-treated mice. This, taken together with the evidence that Treg-mediated TGF-β1 production contributes to immunotherapy resistance, suggests that combination of anti-TIGIT and BA warrants further preclinical and clinical investigation.
Phenotypes of macrophages and fibroblasts are closely regulated by TGF-β and associated with antitumor immunity.45–47 Through subclustering, we identified populations with distinct gene expression patterns consistent with current knowledge of macrophage and CAF heterogeneity.48 49 Interestingly, we also uncovered several treatment-specific subclusters. Unlike macrophages that were found in all treatment groups, BA-induced macrophages were low expressors of TGF-β or PD-L1. They also had lower M2 and higher M1 marker expression. TGF-β plays important roles in M2 polarization of macrophages50 and TGF-β1 secreted by M2 macrophages enhances stemness and migratory potential of glioma cells via the SMAD2/3 signaling pathway.51 Therefore, reducing M2 populations and targeting TGF-β1-expressing macrophages may inhibit aggressive tumor phenotypes. Indeed, using a TGFBR2 knockout mouse model, researchers demonstrated that TGF-β signaling in myeloid cells is required for tumor metastasis, suggesting a pro-tumorigenic role for TGF-β that is dependent on myeloid-specific signaling.52
BA treatment also led to the emergence of a fibroblast subpopulation that expresses low levels of collagen type III, TGF-β and PD-L1. Collagen-producing reactive fibroblasts play important roles in tumor progression via remodeling of the interstitial matrix.53 Following treatment, BA-induced fibroblasts accounted for 2% of all fibroblasts and further investigation of this potentially less immunosuppressive and tumorigenic subpopulation could elucidate their biological functions. In fact, antitumorigenic CAFs, including antigen-presenting CAFs and tumor-restraining CAFs, were recently identified in both human and mouse tumors.54
IF staining on NSCLC tissue revealed PD-L1/TGF-β1 co-expressing fibroblasts. Localized targeting of double positive, activated fibroblasts by BA may contribute to the suppression of matrix remodeling observed with GSEA. In fact, our recent investigation of fibroblastic lung also showed that M2-like lipofibroblasts co-express PD-L1 and TGF-β.11 In lung fibrosis, lipofibroblasts have been shown to be precursors of activated myofibroblasts.55
In conclusion, our study demonstrates that BA, as a bifunctional molecule, more effectively blocks TGF-β on the surface of cells expressing high levels of PD-L1 through colocalization, avidity binding of the TGF-β trap, and PD-L1-dependent degradation of the BA-bound TGF-β. Through colocalization, BA enhanced T cell activation in vitro, and increased TILs in MC38 tumors, relative to the single-agent combination therapy. scRNAseq analysis suggests that the superior antitumor responses elicited by BA is via immunomodulation of the TME, where Tregs, macrophages, immune cells of myeloid linage and fibroblasts are key PD-L1/TGF-β1 co-expressing cells and hence the relevant cell compartment for BA colocalization. Importantly, co-expression of TGF-β and PD-L1 was also seen in human tumors.
Acknowledgments
The authors thank Eva-Maria Leibrock & Michelle Seifert for protein sample preparations and assistance with biophysical assays as ITC and DLS. Maximilian Plach (2Bind, Regensburg) for executing the MST studies; Nicolas Bocquet (LeadXPro, Basel) for supporting the negative stain EM analysis.
Footnotes
Twitter: @vanitasood2
Contributors: YL, T-LY, and HH: contributed equally to this work. YL, T-LY, HH, AW, SS, MT-A, AL, GL, AWG, MB, AS, HM, VDS, DZ, FJ, and K-ML: conceptualization and methodology. T-LY, HH, AW, SS, MT-A, AL, OT, HW, JQ, GL, DK, GQ, BM, HY, L-YC, MS, MB, and FJ: investigation. YL, T-LY, HH, AW, SS, MT-A, AL, OT, HW, JQ, GL, DK, GQ, BM, HY, L-YC, AWG, MS, MB, HM, VS, DZ, FJ, and K-ML: formal analysis. YL, T-LY, HH, AW, SS, MT-A, MHJ, AWG, MGD, FJ, and K-ML: writing—original draft and visualization. All authors contributed to writing—review and editing. YL and K-ML are responsible for the overall content as guarantors.
Funding: This work was supported by EMD Serono Research & Development Institute, Inc., Billerica, USA; an affiliate of Merck KGaA, Darmstadt, Germany.
Competing interests: YL, T-LY, HH, SS, MHJ, HW, JQ, GL, GQ, BM, HY, L-YC, AWG, MGD, MS, AS, HM, VS, FJ and K-ML are all employees of EMD Serono Research and Development Institute, Billerica, Massachusetts, USA, an affiliate of Merck KGaA, Darmstadt, Germany. AW is an employee of Merck KGaA, Darmstadt, Germany, and MT-A and DK are employees of Inter-lab, a subsidiary of Merck KGaA, Yavne, Israel. GL is an inventor on the US20210196822A1 patent held by Merck Patent GmbH, 'Treatment of triple negative breast cancer with targeted TGF-β inhibition.' K-ML is the inventor on the US Patent 9,676,863 B2, 'Targeted TGF-β inhibition,' issued on June 13, 2017, and held by the Merck Patent GmbH, covering M7824 (bintrafusp alfa), its methods of making, and its methods of use. All other authors disclose no competing interests.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not applicable.
Ethics approval
All in vivo protocols were approved by the Institutional Animal Care and Use Committee of EMD Serono Research and Development Institute.
References
- 1.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. 10.1038/nature25501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lan Y, Zhang D, Xu C, et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med 2018;10. 10.1126/scitranslmed.aan5488. [Epub ahead of print: 17 01 2018]. [DOI] [PubMed] [Google Scholar]
- 3.Strauss J, Heery CR, Schlom J, et al. Phase I trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGFβ, in advanced solid tumors. Clin Cancer Res 2018;24:1287–95. 10.1158/1078-0432.CCR-17-2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paz-Ares L, Kim TM, Vicente D, et al. Bintrafusp alfa, a bifunctional fusion protein targeting TGF-β and PD-L1, in second-line treatment of patients with NSCLC: results from an expansion cohort of a phase 1 trial. J Thorac Oncol 2020;15:1210–22. 10.1016/j.jtho.2020.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gulley JL, Heery CR, Schlom J, et al. Preliminary results from a phase 1 trial of M7824 (MSB0011359C), a bifunctional fusion protein targeting PD-L1 and TGF-β, in advanced solid tumors. Journal of Clinical Oncology 2017;35:3006–06. 10.1200/JCO.2017.35.15_suppl.3006 [DOI] [Google Scholar]
- 6.Furler RL, Nixon DF, Brantner CA, et al. TGF-β Sustains Tumor Progression through Biochemical and Mechanical Signal Transduction. Cancers 2018;10. 10.3390/cancers10060199. [Epub ahead of print: 14 06 2018]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Liao S, Diop-Frimpong B, et al. TGF-β blockade improves the distribution and efficacy of therapeutics in breast carcinoma by normalizing the tumor stroma. Proc Natl Acad Sci U S A 2012;109:16618–23. 10.1073/pnas.1117610109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ravi R, Noonan KA, Pham V, et al. Bifunctional immune checkpoint-targeted antibody-ligand traps that simultaneously disable TGFβ enhance the efficacy of cancer immunotherapy. Nat Commun 2018;9:741. 10.1038/s41467-017-02696-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burvenich IJG, Goh YW, Guo N, et al. Radiolabelling and preclinical characterization of 89Zr-Df-radiolabelled bispecific anti-PD-L1/TGF-βRII fusion protein bintrafusp alfa. Eur J Nucl Med Mol Imaging 2021;48:3075-3088. 10.1007/s00259-021-05251-0 [DOI] [PubMed] [Google Scholar]
- 10.Oude Munnink TH, Arjaans MEA, Timmer-Bosscha H, et al. PET with the 89Zr-labeled transforming growth factor-β antibody fresolimumab in tumor models. J Nucl Med 2011;52:2001–8. 10.2967/jnumed.111.092809 [DOI] [PubMed] [Google Scholar]
- 11.Lan Y, Moustafa M, Knoll M, et al. Simultaneous targeting of TGF-β/PD-L1 synergizes with radiotherapy by reprogramming the tumor microenvironment to overcome immune evasion. Cancer Cell 2021;39:1388-1403.e10. 10.1016/j.ccell.2021.08.008 [DOI] [PubMed] [Google Scholar]
- 12.Finkelman FD, Madden KB, Morris SC, et al. Anti-cytokine antibodies as carrier proteins. Prolongation of in vivo effects of exogenous cytokines by injection of cytokine-anti-cytokine antibody complexes. J Immunol 1993;151:1235–44. [PubMed] [Google Scholar]
- 13.Jin H, D'Urso V, Neuteboom B, et al. Avelumab internalization by human circulating immune cells is mediated by both Fc gamma receptor and PD-L1 binding. Oncoimmunology 2021;10:1958590. 10.1080/2162402X.2021.1958590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hutchins NA, Wang F, Wang Y, et al. Kupffer cells potentiate liver sinusoidal endothelial cell injury in sepsis by ligating programmed cell death ligand-1. J Leukoc Biol 2013;94:963–70. 10.1189/jlb.0113051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anderson CL. The liver sinusoidal endothelium reappears after being eclipsed by the Kupffer cell: a 20th century biological delusion corrected. J Leukoc Biol 2015;98:875–6. 10.1189/jlb.4VMLT0215-054R [DOI] [PubMed] [Google Scholar]
- 16.Lin HY, Moustakas A, Knaus P, et al. The soluble exoplasmic domain of the type II transforming growth factor (TGF)-beta receptor. A heterogeneously glycosylated protein with high affinity and selectivity for TGF-beta ligands. J Biol Chem 1995;270:2747–54. 10.1074/jbc.270.6.2747 [DOI] [PubMed] [Google Scholar]
- 17.Martin CE, van Leeuwen EMM, Im SJ, et al. IL-7/anti-IL-7 mAb complexes augment cytokine potency in mice through association with IgG-Fc and by competition with IL-7R. Blood 2013;121:4484–92. 10.1182/blood-2012-08-449215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moulin A, Mathieu M, Lawrence C, et al. Structures of a pan-specific antagonist antibody complexed to different isoforms of TGFβ reveal structural plasticity of antibody-antigen interactions. Protein Sci 2014;23:1698–707. 10.1002/pro.2548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young G, Hundt N, Cole D, et al. Quantitative mass imaging of single biological macromolecules. Science 2018;360:423–7. 10.1126/science.aar5839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Aarsen LAK, Leone DR, Ho S, et al. Antibody-Mediated blockade of integrin alpha v beta 6inhibits tumor progression in vivo by a transforming growth factor-beta-regulated mechanism. Cancer Res 2008;68:561–70. 10.1158/0008-5472.CAN-07-2307 [DOI] [PubMed] [Google Scholar]
- 21.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Peng Y, Liu C, Li M, et al. Identification of a prognostic and therapeutic immune signature associated with hepatocellular carcinoma. Cancer Cell Int 2021;21:98. 10.1186/s12935-021-01792-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liang Z-H, Pan Y-C, Lin S-S, et al. LncRNA MALAT1 promotes wound healing via regulating miR-141-3p/ZNF217 axis. Regen Ther 2020;15:202–9. 10.1016/j.reth.2020.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li S, Liu M, Do MH, et al. Cancer immunotherapy via targeted TGF-β signalling blockade in TH cells. Nature 2020;587:121–5. 10.1038/s41586-020-2850-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leung CS, Yeung T-L, Yip K-P, et al. Calcium-dependent FAK/CREB/TNNC1 signalling mediates the effect of stromal MFAP5 on ovarian cancer metastatic potential. Nat Commun 2014;5:5092. 10.1038/ncomms6092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou Z, Cui D, Sun M-H, et al. CAFs-derived MFAP5 promotes bladder cancer malignant behavior through NOTCH2/HEY1 signaling. Faseb J 2020;34:7970–88. 10.1096/fj.201902659R [DOI] [PubMed] [Google Scholar]
- 27.Teichgräber V, Monasterio C, Chaitanya K, et al. Specific inhibition of fibroblast activation protein (FAP)-alpha prevents tumor progression in vitro. Adv Med Sci 2015;60:264–72. 10.1016/j.advms.2015.04.006 [DOI] [PubMed] [Google Scholar]
- 28.Jiang X, Wu M, Xu X, et al. COL12A1, a novel potential prognostic factor and therapeutic target in gastric cancer. Mol Med Rep 2019;20:3103–12. 10.3892/mmr.2019.10548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yeung T-L, Leung CS, Wong K-K, et al. TGF-β modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res 2013;73:5016–28. 10.1158/0008-5472.CAN-13-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elmusrati AA, Pilborough AE, Khurram SA, et al. Cancer-associated fibroblasts promote bone invasion in oral squamous cell carcinoma. Br J Cancer 2017;117:867–75. 10.1038/bjc.2017.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanley CJ, Mellone M, Ford K, et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J Natl Cancer Inst 2018;110:109–20. 10.1093/jnci/djx121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sandberg TP, Stuart MPME, Oosting J, et al. Increased expression of cancer-associated fibroblast markers at the invasive front and its association with tumor-stroma ratio in colorectal cancer. BMC Cancer 2019;19:284. 10.1186/s12885-019-5462-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Friedman G, Levi-Galibov O, David E, et al. Cancer-associated fibroblast compositions change with breast cancer progression linking the ratio of S100A4+ and PDPN+ CAFs to clinical outcome. Nat Cancer 2020;1:692–708. 10.1038/s43018-020-0082-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kieffer Y, Hocine HR, Gentric G, et al. Single-Cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov 2020;10:1330–51. 10.1158/2159-8290.CD-19-1384 [DOI] [PubMed] [Google Scholar]
- 35.Grauel AL, Nguyen B, Ruddy D, et al. TGFβ-blockade uncovers stromal plasticity in tumors by revealing the existence of a subset of interferon-licensed fibroblasts. Nat Commun 2020;11:6315. 10.1038/s41467-020-19920-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Batlle E, Massagué J. Transforming growth factor-β signaling in immunity and cancer. Immunity 2019;50:924–40. 10.1016/j.immuni.2019.03.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter CA, Bermudez L, Beernink H, et al. Transforming growth factor-beta inhibits interleukin-12-induced production of interferon-gamma by natural killer cells: a role for transforming growth factor-beta in the regulation of T cell-independent resistance to Toxoplasma gondii. Eur J Immunol 1995;25:994–1000. 10.1002/eji.1830250420 [DOI] [PubMed] [Google Scholar]
- 38.Sudarshan C, Galon J, Zhou Y, et al. Tgf-Beta does not inhibit IL-12- and IL-2-induced activation of Janus kinases and STATs. J Immunol 1999;162:2974–81. [PubMed] [Google Scholar]
- 39.Pardoux Cécile, Ma X, Gobert Stéphanie. Downregulation of interleukin-12 (IL-12) responsiveness in human T cells by transforming growth factor-β: relationship with IL-12 signaling. Blood, The Journal of the American Society of Hematology 1999;93:1448–55. [PubMed] [Google Scholar]
- 40.Smalley Rumfield C, Pellom ST, Morillon Ii YM, et al. Immunomodulation to enhance the efficacy of an HPV therapeutic vaccine. J Immunother Cancer 2020;8. 10.1136/jitc-2020-000612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.de Streel G, Bertrand C, Chalon N, et al. Selective inhibition of TGF-β1 produced by GARP-expressing Tregs overcomes resistance to PD-1/PD-L1 blockade in cancer. Nat Commun 2020;11:4545. 10.1038/s41467-020-17811-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Joller N, Lozano E, Burkett PR, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 2014;40:569–81. 10.1016/j.immuni.2014.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ge Z, Peppelenbosch MP, Sprengers D, et al. TIGIT, the Next Step Towards Successful Combination Immune Checkpoint Therapy in Cancer. Front Immunol 2021;12:699895. 10.3389/fimmu.2021.699895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mullard A. Roche’s anti-TIGIT drug suffers a phase III cancer setback. Nat Rev Drug Discov 2022;21:327. 10.1038/d41573-022-00068-4 [DOI] [PubMed] [Google Scholar]
- 45.Gratchev A. TGF-β signalling in tumour associated macrophages. Immunobiology 2017;222:75–81. 10.1016/j.imbio.2015.11.016 [DOI] [PubMed] [Google Scholar]
- 46.Pan Y, Yu Y, Wang X, et al. Tumor-Associated macrophages in tumor immunity. Front Immunol 2020;11:583084. 10.3389/fimmu.2020.583084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chung JY-F, Chan MK-K, Li JS-F, et al. TGF-β Signaling: From Tissue Fibrosis to Tumor Microenvironment. Int J Mol Sci 2021;22:7575. 10.3390/ijms22147575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Y-K, Wang M, Sun Y, et al. Macrophage spatial heterogeneity in gastric cancer defined by multiplex immunohistochemistry. Nat Commun 2019;10:3928. 10.1038/s41467-019-11788-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanzaki R, Pietras K. Heterogeneity of cancer-associated fibroblasts: Opportunities for precision medicine. Cancer Sci 2020;111:2708–17. 10.1111/cas.14537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gong D, Shi W, Yi S-ju, et al. Tgfβ signaling plays a critical role in promoting alternative macrophage activation. BMC Immunol 2012;13:31. 10.1186/1471-2172-13-31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu Z, Kuang W, Zhou Q, et al. TGF-β1 secreted by M2 phenotype macrophages enhances the stemness and migration of glioma cells via the SMAD2/3 signalling pathway. Int J Mol Med 2018;42:3395–403. 10.3892/ijmm.2018.3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pang Y, Gara SK, Achyut BR, et al. TGF-β signaling in myeloid cells is required for tumor metastasis. Cancer Discov 2013;3:936–51. 10.1158/2159-8290.CD-12-0527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nissen NI, Karsdal M, Willumsen N. Collagens and cancer associated fibroblasts in the reactive stroma and its relation to cancer biology. J Exp Clin Cancer Res 2019;38:115. 10.1186/s13046-019-1110-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ganguly D, Chandra R, Karalis J, et al. Cancer-associated fibroblasts: versatile players in the tumor microenvironment. Cancers 2020;12. 10.3390/cancers12092652. [Epub ahead of print: 17 09 2020]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.El Agha E, Moiseenko A, Kheirollahi V, et al. Two-way conversion between lipogenic and myogenic fibroblastic phenotypes marks the progression and resolution of lung fibrosis. Cell Stem Cell 2017;20:571. 10.1016/j.stem.2017.03.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
jitc-2021-004122supp003.pdf (2.6MB, pdf)
jitc-2021-004122supp001.wmv (387.8KB, wmv)
jitc-2021-004122supp002.wmv (387.8KB, wmv)
Data Availability Statement
Data are available upon reasonable request.