

Abstract
Biocatalysis in organic solvents (OSs) enables more efficient routes to the synthesis of various valuable chemicals. However, OSs often reduce enzymatic activity, which limits the use of enzymes in OSs. Herein, we report a comprehensive understanding of interactions between surface polar substitutions and DMSO by integrating molecular dynamics (MD) simulations of 45 variants from Bacillus subtilis lipase A (BSLA) and substitution landscape into a “BSLA‐SSM” library. By systematically analyzing 39 structural‐, solvation‐, and interaction energy‐based observables, we discovered that hydration shell maintenance, DMSO reduction, and decreased local flexibility simultaneously govern the stability of polar variants in OS. Moreover, the fingerprints of 1631 polar‐related variants in three OSs demonstrated that substituting aromatic to polar amino acid(s) hold great potential to highly improve OSs resistance. Hence, surface polar engineering is a powerful strategy to generate OS‐tolerant lipases and other enzymes, thereby adapting the catalyst to the desired reaction and process with OSs.
Keywords: biocatalysis, molecular dynamics simulation, surface polar engineering, organic solvent resistance, directed evolution
Behold, the surface: Investigating the molecular dynamics (MD) simulations of 45 variants from Bacillus subtilis lipase A (BSLA) and 1631 substitutions’ landscape in “BSLA‐SSM” library obtained a comprehensive molecular understanding of interactions between surface polar substitutions and DMSO. The results suggest that surface polar engineering, driven by three determinants, is a promising rational strategy to tailor OS‐tolerant enzymes.

Introduction
Biocatalysis in organic (co‐)solvents (OSs) provides numerous industrially attractive advantages, for example, the favorable shift of reaction equilibria, increased solubility of substrate/product, alternation of substrate specificity and enantioselectivity, suppression of water‐dependent side reactions, and easy product recovery. [1] Enzymatic selectivity (e. g., substrate, stereo‐, regio‐, and chemoselectivity) can be markedly affected by OSs. [2] As such, enzymatic reactions conducted in OSs would enable to combine the synthetic power of enzymes with chemical synthesis efficiently in industrial and pharmaceutical fields.[ 1b , 3 ] However, OSs frequently lead to a dramatic drop in enzymes’ catalytic activity and even deactivation. [4]
A plethora of approaches have been applied to improve enzyme activity and stability in the presence of OSs, for example, immobilization and encapsulation of enzymes, [5] chemical/physical modification,[ 5d , 6 ] single enzyme nanoparticles, [5d] and protein engineering. [7] Especially, the evolved OS tolerant enzymes by directed evolution are highly promising as they have changed their inherent functions and can be combined with other immobilization/modification techniques to reach the icing on the cake. [8] For instance, Tian et al. reported the semi‐rational method for directed Thermomyces lanuginosus lipase evolution to improve methanol tolerance by targeting high B‐factor residues for iterative saturation mutagenesis (ISM), and obtain the best double substitution TLL‐S105C/D27R showing 30 % greater methanol tolerance than TLL wild‐type (WT). [8a] In the directed evolution campaign of metalloprotease PT121 with random mutagenesis method, eleven variants with the noticeable difference in 25 % (v/v) acetonitrile resistance were obtained. [9] And three variants (T46Y, H224F, and H224Y) of PT121 showed excellent OS stability in the presence of acetonitrile and acetone, which increased their half‐lives 1.2–3.5‐fold as compared to WT. [9] Yedavalli and Rao combined six mutations, obtained from loop engineering, into Bacillus subtilis lipase A (BSLA) gene (6SR), and presented that 6SR has eight times higher catalytic turnover in 60 % (v/v) dimethyl sulfoxide (DMSO). [10] Recently, 2GenReP and InSiRep recombination strategies yielded a supercharged BSLA variant M4 (I12R/Y49R/E65H/N98R/K122E/L124K), having up to 14.6‐fold 1,4‐dioxane (DOX) resistance improvement after the screening of about 270 clones. [7c] Interestingly, 58–93 % of beneficial BSLA variants towards OS resistance led to chemically different amino acids. [7d] Much work on the potential of charged substitutions to tailor the OS resistant enzymes has been carried out in our previous studies,[ 7b , 7c , 11 ] but other amino acid changes are still unexplored.
A clear molecular understanding of the protein‐dynamics‐function relationship enables to facilitate protein engineering emerging as a better tool to optimize enzymes with achieving the desired properties (e. g., (co)solvent resistance,[ 7b , 7c , 11 ] selectivity,[ 8b , 12 ] thermostability,[ 11 , 13 ] and electrocatalysis [14] ). Although the OS type profoundly affects the kinetics of biocatalysis, [15] the general effect of OS on enzymatic activity is mainly through “disturbing” the structure and dynamics within enzymes. [16] For example, polar OSs showed a deeper penetration into the enzyme than non‐polar OSs, thereby inducing destructive secondary/tertiary structural changes. [17] Meanwhile, the occupation of OSs also stripped off the essential water molecules from the protein surface. [7b] While the water activity (a w) can connect enzyme activity with the water in nonaqueous OS reaction conditions.[ 7b , 18 ] Protein hydration shells have essential roles in keeping the enzyme activity and improving 2,2,2‐trifluoroethanol (TFE) and DOX resistance of enzymes.[ 7b , 7c , 16 ] Besides, OS introduced inhibition and the substrate/intermediate transition state change indeed affect the enzymatic catalysis performance.[ 7b , 19 ] Interestingly, the effects of OS on enzymes are also dependent on the types of enzyme. [20] And the specific residue‐OS interaction pattern can provide a particular modification clue to engineer the favorable amino acid positions. Collectively, there is still a need for dealing with the complicated and puzzled OS‐enzyme interaction in depth. Besides the experimental techniques (e. g., circular dichroism (CD), [21] NMR, [7a] and IR spectroscopy [22] ), the all‐atom molecular dynamics (MD) simulations open the gate to characterize the correlation between protein structure/dynamics and their stability in OSs, which has been confirmed to hold remarkable consistency with their experimental studies.[ 20 , 23 ]
Generally, design principle(s) are challenging to be summarized in a directed evolution campaign with random mutagenesis since only a small fraction of amino acid substitutions is explored when compared to the diversity that offers. [24] A main conclusion by comparing a standard epPCR library of BSLA with a BSLA‐SSM library (site saturation mutagenesis is denoted as SSM) that offered the full natural diversity of BSLA at each amino acid position, revealed that general design principles can be discovered in SSM‐libraries in contrast to the biased epPCR based directed evolution campaigns.[ 24b , 25 ] An overview study of the BSLA‐SSM library revealed that resistance/tolerance of enzyme in three OS media could be predominantly achieved through polar residue substitutions, especially in DMSO with up to 34 % beneficial rate.[ 7d , 16 ] Based on the gained knowledge on the BSLA‐libraries, we report here the comprehensive results of MD simulations of 45 BSLA variants in DMSO and the experimental OS resistance pattern of 1631 polar‐related variants in the BSLA‐SSM library. Forty‐five BSLA variants included 30 beneficial polar substitutions (the substitution of any type of residues for a polar amino acid) and 15 non‐beneficial substitutions. DMSO was chosen as the model OS due to its favorable properties (amphiphilicity, dissolving ability, low chemical reactivity) and has been extensively used in organic synthesis and biocatalysis fields. [26] After the evaluation of in total 39 structural‐, solvation‐, energy‐based observables and aligning them with experimental results (Table S1), we uncover the ubiquitous importance of the main driving forces within enhanced polar variants resistance in OSs, and address the question of how in the future to rationally recover/improve activity and resistance through engineering the lipase surface by substitutions to polar residues.
Results and Discussion
Overall structure, dynamics, and solvation behaviors in BSLA variants
Considering our previous study and the reasonable computational calculation,[ 7d , 16 ] in total, forty‐five BSLA variants on 15 amino acid positions (30 beneficial polar substitutions and 15 non‐beneficial substitutions) obtained from the BSLA‐SSM library (Figure 1a and Tables S2,S3) were used for MD simulations in DMSO (Figures S1–S3). To mostly avoid the experimental bias, the selection criteria were as follows: a) two beneficial polar and one non‐beneficial variant at each position was selected for better comparison; b) the positions distributed on the whole BSLA amino acid sequence evenly and belonged to diverse secondary structure; c) the positions should be on the enzyme surface; d) the amino acid type of non‐beneficial variant is random. Figure 1b shows a statistical overview by comparing the beneficial variants and their corresponding non‐beneficial ones regarding overall structural, solvation, and interaction energy changes within 17 observables. In terms of overall geometrical properties, seven observables were investigated, including time‐averaged root‐mean‐square deviation (RMSD), the radius of gyration (R g), internal H‐bond, thermodynamic stability, and SASA. Briefly, the results of comparison mentioned above suggested there is no universal or predominant trend towards the overall conformational change in BSLA (see more details in Supporting Information, Figures 1b and S1–S8). The latter can be expected since all mutations are single point variants, which cannot provide considerable variations in the overall dimensions among different variants.[ 7b , 14a ]
Figure 1.

Overall structural and dynamics observables of BSLA variants in 60 % (v/v) DMSO. (a) Visualization of the 15 beneficial/non‐beneficial amino acid positions in the 3D structure of the BSLA wild type (PDB: 1i6w, [28] Chain A). Catalytic triad residues (S77, D133, H156) are presented as the cyan sphere. Selected amino acid positions are given as orange sticks. (b) Structural properties (7 observables), solvation phenomenon (4 observables), and interaction energy (6 observables) investigation towards overall BSLA for 15 non‐beneficial and 30 beneficial variants in 60 % (v/v) DMSO. The percentage indicates the beneficial variants exhibiting higher (purple), within deviation (orange), lower (green) properties compared to a non‐beneficial variant in the same position. (c) Contact frequency between the overall BSLA protein and DMSO towards beneficial/non‐beneficial variant in 60 % (v/v) DMSO. (d) Contact frequency between the overall BSLA protein and water towards beneficial/non‐beneficial variant in 60 % (v/v) DMSO. Twenty beneficial variants were divided into beneficial group 1 (denoted as beneficial 1) and beneficial group 2 (denoted as beneficial 2) for better visualization. The ramp was colored from white to red/blue to indicate BSLA–DMSO/water contact frequency change from low to high. The minimum distance of 2.5 Å or less was chosen as the residue–water/DMSO contact cutoff to identify the strong interactions. Contact frequency was averaged over the last 40 ns from three independent MD runs.
Solvation universally plays an essential role in maintaining the functional enzyme structure and participating in various noncovalent interactions such as electrostatic interactions, van der Waals interactions, π‐effects, and hydrophobic effect. [17b] As shown in Figures 2, S9 and S10, DMSO interacted with the BSLA surface to form the DMSO layer. The solvation phenomenon called “water stripping” occurred in all 45 BSLA variants in DMSO, as presented for enzymes in polar OSs.[ 15c , 16 , 27 ] Notably, 100 % beneficial variants kept consistent or even enhanced (∼30.0 % (9 : 30), e. g., K95 N, G145Q, and L168 N) hydration shell around the whole BSLA variants compared to their corresponding non‐beneficial variants (Figures 1b and S9). In contrast, up to 53.3 % (16 : 30) and 33.3 % (10 : 30) beneficial variants showed decreased and comparable DMSO layer. Only 13.3 % (4 : 30) displayed an increased DMSO layer around the whole BSLA beneficial variants. As expected, similar observations were confirmed by investigating the water/DMSO‐BSLA contact frequency (Figure 1b, c, and d) and 13 energy‐based observables (Figure 1b and Tables S4, S5) as well. Remarkably, the DMSO‐BSLA contact frequency of non‐beneficial L168I decreased 4.5‐5.5 % when introducing the polar residues (L168T and L168 N, Figure 1c). And the contact frequency of water molecules increased from 55.8 % in non‐beneficial N4I to 61.0 % in beneficial N4S (Figure 1d). Consequently, these results suggested the hydration shell and DMSO layer might be the pivotal factor(s) to govern the BSLA resistance in DMSO.
Figure 2.
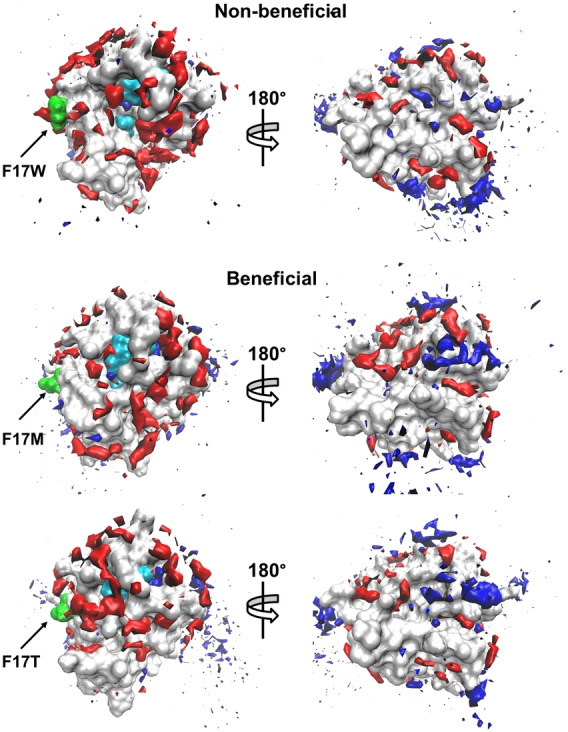
Spatial distribution of water and DMSO occupancy at the surface of the BSLA variants on position F17 in 60 % (v/v) DMSO. The BSLA surface is shown in grey, S77, D133, H156 (the catalytic triad) in cyan, the OS molecules in red with a solid surface, the water molecules in blue with a solid surface. The substituted sites are labeled and shown in green. A 180° rotation view around the vertical axis is offered to give a complete view of the surface. The contours are shown with the isovalue 16, 9 for DMSO, water, respectively. Each view of the BSLA has the same orientation.
Local structure dynamics and solvation behavior around the substitution sites
In order to validate the introduced substitutions are the source of the global change within BSLA, we looked into local structure dynamics and solvation behaviors around the substituted sites (15 observables, Figure 3a). It emerged that almost all beneficial variants (21 variants, 70 %) gain a distinctly reduced number of DMSO molecules around the substituted site (70 %, Figure 3a and b), but the non‐reduced number of water molecules (22 variants, 73 %, Figures 3a and S11). The spatial distribution function (SDF) study of substitutions on amino acid position F17 as examples were shown in Figure 2 and 3b to give a better visualization. These results agree well with the global solvation change. It is generally accepted that the amino acid type enables to influence the protein‐OS interaction modes and the behaviors of solvent molecules like their orientation, distribution, and assembly.[ 7a , 7b , 7c , 29 ] Besides, as another site‐resolved property, the local contact frequency was investigated towards the DMSO/water‐residues interaction. Surprisingly, the lowest contact frequency of DMSO molecules decreased to 7 % on the substituted residue positions of non‐beneficial variants (e. g., N94P; Figures 4a, S12a, and S13). And water‐substituted residue contact frequency showed up to 71‐times improvement in K95S compared to non‐beneficial (Figures 4b, S12b, and S14). A similar hydration phenomenon was observed in charged variants studies.[ 7b , 7c ] Furthermore, the results of six energy‐based factors in Figure 3a were well correlated to the above solvation foundlings and demonstrated that DMSO molecules in beneficial polar variants interact with the enzyme less tightly than non‐beneficial ones. Oppositely, water molecules are bound more tightly (more descriptions are in Supporting Information, Figure 3a and Tables S6, S7).
Figure 3.

Local structural and dynamics observables for 45 BSLA variants (including 15 non‐beneficial and 30 beneficial variants) in 60 % (v/v) DMSO. (a) Structural properties (5 observables), solvation phenomenon (4 observables), and interaction energy (6 observables) investigation on the substituted sites of BSLA non‐beneficial and beneficial variants in 60 % (v/v) DMSO. (b) The number of DMSO molecules around the BSLA substituted residue in 60 % (v/v) DMSO. The calculated number of DMSO was averaged over the last 40 ns from three independent MD runs. Left panel: position 1–94; right panel: position 95–181. The cut‐off distance was determined from the radial distribution function (RDF) of DMSO around BSLA residues when the “central” atom S2 showed first minima approximately at this distance, [32] thereby a 6.8 Å cut‐off was employed for DMSO. (c) The RMSF of the substituted residue in BSLA non‐beneficial/beneficial (group 1) variants in 60 % (v/v) DMSO during MD simulations. The RMSF was averaged over the last 40 ns from three independent MD runs.
Figure 4.

Contact frequency of substituted sites between the BSLA beneficial/non‐beneficial variants and (a) DMSO, (b) water molecule in 12 % (v/v) TFE, respectively. The definition of residue‐DMSO/water contact frequency was similar to BSLA–DMSO/water. Specific substituted sites are labeled with amino acid position numbers. Except for the substituted sites, the rest surface is colored yellow. Back‐side (rotated by 180°) views are shown to give a complete view of the BSLA variants with the same orientation.
Towards local conformational change, RMSF values for the individual residue of BSLA variants are shown in Figures S17 and 18, mainly varying between 0.3 Å and 5.0 Å. The results of the RMSF of the specific substitution site are given in Figure 3a and c. There was a significantly decreased RMSF value change in 73 % (22 : 30) beneficial variants compared to non‐beneficial ones, indicating the substituted site has worse flexibility. The latter is opposite from the previous report that the enzyme flexibility increased with increasing hydration level. [30] There is no doubt that an appropriate balance between structural flexibility and rigidity is favorable for better enzymatic functions. [31] Interestingly, 70 % (21 : 30) beneficial variants displayed reduced residue SASA than non‐beneficial, which might be caused by their short side chain in the polar residue (Figures 3a and S19). However, the latter local SASA change does not map to the total SASA possibly due to the complex residue network in structure.
The active site is the most important enzyme region since it directly catalyzes the chemical reaction. Therefore, we investigated the conformational and solvation change in the substrate binding cleft as well. Except for DMSO penetration into substrate binding site of all variants as shown in previous studies,[ 16 , 33 ] there is no universal or predominant trend observed in terms of the distance change between substitution positions and catalytic triad (Figures 3a and S20), and the structure, solvation change in the active site (7 observables; Figures S21–S23). And more details are described in Supporting Information.
The experimental resistance landscape of polar‐related variants in OSs revealed the surface polar engineering strategy
Collectively, the aforementioned computational studies revealed the three potential factors for governing the improved DMSO resistance in 45 BSLA variants, including less DMSO layer and at least consistent hydration and reduced flexibility. To expand the application scope of our molecular findings to more variants and OSs and therefore conclude the potential general engineering strategy, we statistically analyzed all 1631 polar‐related variants in the experimental BSLA‐SSM library towards DMSO, DOX, and TFF resistance (Figures 5 and S24, S25). The polar‐related variants are defined as the single substitutions that all types of amino acids are substituted to polar residue (In total, 1267 BSLA variants, Table S8), or polar amino acids are substituted to non‐polar residue (364, Table S9). The trend of beneficial rate in 1267 variants is as follows: DMSO (12.2 %)>TFE (6.6 %)>DOX (3.7 %), indicating the polar variant works much better in DMSO than the rest. Of course, as previously reported,[ 7d , 16 ] non‐polar variants (i. e., aromatic, aliphatic, and charged) can still obtain the beneficial variants (Table S8).
Figure 5.

Resistance landscape of surface polar relevant BSLA variants towards DMSO. Residual activity, activity, variants, and the empty vector are denoted as R, A, V, and EV, respectively. The OS resistance was measured in the absence or presence of 60 % (v/v) DMSO cosolvents after 2 h incubation with crude culture supernatant. The beneficial, unchanged, decreased, and inactive variants are colored green, yellow, red, and purple, respectively.
In addition, 896 surface polar variants on 128 positions were identified (Table S10) and presented a bit higher beneficial rate in DOX and TFE, but a much lower factuality rate (9.6–14.6 %) in all OSs when compared to the pattern in the whole 1267 polar variants (factuality rate: 15.2–21.5 %). To answer the question of which kind of surface amino acid substituting to polar residue holds the “best” promise to improve the OS resistance efficiently, we investigated their detailed substitution landscape on the BSLA surface. As shown in Tables 1 and S11, the substitution of aromatic residues for polar amino acid behaves superior in boosting the BSLA stability in OSs (4.8–14.3 %) and keeping acceptable fatality rates (11.9–14.3 %), when compared to other changes. Interestingly, we noticed that most cases (60 %, 9/15) of non‐beneficial substitutions selected for MD studies are the introduction of an aromatic residue, which further confirms the aforementioned statistical trend. Furthermore, these observations were also proved by the specific MD simulation studies, such that F17 M and F17T showed much better solvation phenomena (Figures 2, 3b, S11) and favorable flexibility (Figures 3c and S18). It is particularly interesting to note that reducing the area of water‐accessible hydrophobic surface prefer to increase the stability of proteins. [34] When comparing aromatic residues, the polar amino acids orient the water neighbors in a quantitatively stronger manner. [35] These observations hold true for other technically important enzymes (e. g., PETase, cytochrome P450, and organophosphorus hydrolase). [35]
Table 1.
Analysis of the polar variants in which different amino acids exchanged to polar residue towards three OSs resistance in BSLA–SSM library.[a]
|
Exchange |
Fraction of substitution [%] |
|||
|---|---|---|---|---|
|
|
beneficial |
unchanged |
decreased |
inactive |
|
polar to polar |
4.0–11.1 |
74.4.2–83.1 |
3.0–7.3 |
7.3–12.6 |
|
aromatic to polar |
4.8–14.3 |
66.7–77.4 |
4.6–8.3 |
11.9–14.3 |
|
aliphatic to polar |
2.9–14.0 |
60.4–72.1 |
7.1–17.2 |
8.4–18.5 |
|
charged to polar |
2.0–7.4 |
62.6–78.8 |
2.0–16.7 |
13.8–16.3 |
[a] 22 % (v/v) DOX, 60 % (v/v) DMSO, and 12 % (v/v) TFE were used; amino acid is classified as follows: aromatic: F, Y, W; aliphatic: A, V, L, I, G; polar: C, M, P, S, T, N, Q; charged: D, E, K, H, R.
The generally high activity and stability in OSs could result from precise control of protein surface region, [36] thereby maximizing the other outstanding performances such as the stereo‐, regio‐, and chemoselectivity in organic synthesis. Since there is a relationship between OS resistance and thermostability,[ 7c , 11 ] and numerous polar variants had an enhanced thermodynamic stability, [37] we speculate the surface polar engineering might be applied to yield more thermotolerant enzymes. Interestingly, not only the computational and experimental results of polar variants in DMSO can be well reflected in DOX and TFE (our case), but also other OSs (e. g., cyclohexane, octane, n‐decane, methanol, acetone) have good transferability with DMSO in biocatalysis.[ 7b , 38 ] Furthermore, the representative OS DMSO worked well in offering a general explanation and universal picture of the different behavior of protein structure‐function observed in the presence of cosolvents. [39] These findings confirm surface polar engineering, promoting favorable flexibility and solvation in enzymes, hold the promise to be a common tool to tailor the stability of the enzyme in a wide range of OSs.
Conclusions
Molecular dynamics (MD) simulation studies on a model enzyme Bacillus subtilis lipase A (BSLA) provided a comprehensive picture of organic solvent (OS)–enzyme interactions and revealed molecular principles governing OS resistance improvement in polar BSLA variants. And three drivers were explored on a level pegging, including the hydration shell maintenance, DMSO reduction, and decreased local flexibility around the substituted site. The functional enzymes require packed three‐dimensional structures and “balanced” dynamics with a favorable solvation environment. Experimental investigation of 1631 polar BSLA variants aligned well with our molecular understanding and demonstrated that substituting aromatic to polar residue wins the “crown” for enhancing the stability of enzymes in OSs by keeping a lower inactive ratio. These results proved that surface polar engineering (especially aromatic substitute to polar) is a promising rational design strategy to tailor the high OS‐tolerant lipases and most likely transferable to other enzymes. Furthermore, integrating the surface polar engineering with different computational and/or experimental approaches (e. g., CompassR, [40] KnowVolution, [24a] NMR, [7a] CASTing, [41] InSiPeP, and 2GenReP [7c] ) would minimize the screening efforts to achieve the targeted properties in non‐conventional solvents efficiently. Also, the decryption of the molecular principles which lead to the enzyme resistance in OSs brings substantial knowledge to guide protein engineering campaigns and promote biocatalysis in OSs.
Experimental and Computational Section
In silico generation of variants and stability analysis
The starting coordinates of BSLA WT were taken from the crystal structure of BSLA (PDB ID: 1i6w, [28] chain A, resolution 1.5 Å). The selected beneficial polar and non‐beneficial variants in Table S2–S3 were obtained from previous studies. [7d] All variant structures were generated based on BSLA WT using the FoldX method and through the FoldX plugin implemented in YASARA Structure version 17.8.19. [42] Default FoldX parameters were applied (Temperature 298 K; ionic strength 0.05 M; pH 7). The structure of the BSLA WT was energy minimized using the “RepairObject” command and rotamerized with optimizing the residues by removing Van der Waals clashes and bad contacts (<6 Å).[ 40 , 42a ] “Mutate residue” command was applied to generate the 3D structure of BSLA substitution and calculate the ΔΔG fold (ΔΔG fold=ΔG fold,sub−ΔG fold,WT) of substitutions. Five FoldX runs were carried out for each variant to ensure the minimum energy conformation of substitution is identified.
Molecular dynamics simulations
MD simulations and analysis were performed with GROMACS v5.1.2 simulation package. [43] The DMSO‐BSLA system was set up regarding our previous studies under the GROMOS96 (54a7) force field.[ 16 , 44 ] The validated DMSO model was taken from the literature.[ 16 , 45 ] The BSLA structure was solvated into a cubic box of SPCE water [46] with a minimal distance of 12 Å from the box edge to the centered protein. The simulation systems were filled with ∼7109 water molecules and ∼956 DMSO molecules, to simulate the experimental conditions (60 % (v/v) DMSO). Na+ and Cl−, as counter ions, were used to neutralize the total net charge of the systems to achieve a net charge of zero. The electrostatic interactions were calculated by using the particle mesh Ewald (PME) method as previously reported.[ 16 , 47 ]
To avoid the most unfavorable interactions, energy minimization using the steepest descent method was performed before MD simulations. As our previous report, [16] 100 ps NVT ensemble was performed by temperatures kept close to 298 K. Then 100 ps NPT ensemble with position restraints on the BSLA was chosen for equilibration, followed by the production simulation runs (100 ns at 298 K, 1 bar, and time step 1 fs). Three independent MD runs with different starting atomic velocities were presented to circumvent a dependence of the results on the starting conditions of the simulation. Coordinates, energies, and velocities were stored every 0.5 ns in MD runs and used for trajectories analysis. All the observables (Table S1) were calculated by GROMACS analysis tools. The software Pymol and VMD 1.9.2 were used for visualization. [48]
OSs resistance of variants in BSLA‐SSM library
The screening experiments with standard p‐nitrophenyl butyrate (pNPB) assay in 60 % (v/v) DMSO, 22 % (v/v) DOX, and 12 % (v/v) TFE was performed as our previous studies. [7d] This DMSO, DOX, TFE concentration for “BSLA‐SSM” library screening was chosen to ensure an RWT of ∼30 %, a suitable condition to estimate the performance change of the BSLA variant.[ 7b , 29 ] Residual activities of WT/variants in OSs were calculated as the following equation 1:
| (1) |
Residual activity is abbreviated R, variant as V, wild‐type as WT, empty vector as EV. OS resistance of BSLA (WT or variant) was evaluated as activity in the presence of OS divided by activity in the absence of OS, [7d] and shown in equation 2:
| (2) |
Beneficial variants are variants with improved resistance (R V)≥resistance of BSLA WT (R WT)+3σ. Non‐beneficial variants are variants with three type: unchanged resistance (R WT−3σ<R V<R WT+3σ), decreased resistance (R EV+3σ<R V <R WT−3σ) and inactive (A V<A EV+3σ). σ is the standard deviation of the screening system in the presence of OS.[ 7d , 16 ]
We loosely define the polar variant is the substitution that introducing the polar amino acid into BSLA, regardless of the original type of amino acid in wild‐type BSLA. Amino acid is classified as follows: Aromatic: F, Y, W; aliphatic: A, V, L, I, G; polar: C, M, P, S, T, N, Q (termed neutral in the cited papers); charged: D, E, K, H, R. [7d]
Notes
The authors declare no competing financial interest.
Supporting Information
Detailed structural analysis, interaction energy investigation, properties of variants, and experimental results.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Simulations were performed with computing resources granted by JARA‐HPC from RWTH Aachen University under projects JARA0169 and JARA0189. Open Access funding enabled and organized by Projekt DEAL.
H. Cui, M. Vedder, L. Zhang, K.-E. Jaeger, U. Schwaneberg, M. D. Davari, ChemSusChem 2022, 15, e202102551.
Contributor Information
Prof. Dr. Ulrich Schwaneberg, Email: u.schwaneberg@rwth-aachen.de, Email: u.schwaneberg@biotec.rwth-aachen.de.
Dr. Mehdi D. Davari, Email: mehdi.davari@ipb-halle.de.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Gupta M. N., Eur. J. Biochem. 1992, 203, 25–32; [DOI] [PubMed] [Google Scholar]
- 1b. Klibanov A. M., Nature 2001, 409, 241; [DOI] [PubMed] [Google Scholar]
- 1c. Carrea G., Riva S., Angew. Chem. Int. Ed. 2000, 39, 2226–2254; [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 2312–2341; [Google Scholar]
- 1d. Kumar A., Dhar K., Kanwar S. S., Arora P. K., Biol. Proced. Online 2016, 18, 1–11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1e. Kajiwara S., Komatsu K., Yamada R., Matsumoto T., Yasuda M., Ogino H., Biochem. Eng. J. 2019, 142, 1–6; [DOI] [PubMed] [Google Scholar]
- 1f. Wang Y., Zhang N., Tan D., Qi Z., Wu C., Front. Bioeng. Biotechnol. 2020, 8, 714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Brink L. E. S., Tramper J., Luyben K. C. A. M., Van′t Riet K., Enzyme Microb. Technol. 1988, 10, 736–743; [Google Scholar]
- 2b. Davison B. H., Barton J. W., Petersen G. R., Biotechnol. Prog. 1997, 13, 512–518. [Google Scholar]
- 3. Secundo F., Carrea G., J. Mol. Catal. B 2002, 19, 93–102. [Google Scholar]
- 4.
- 4a. Wang S., Meng X., Zhou H., Liu Y., Secundo F., Liu Y., Catalysts 2016, 6, 32; [Google Scholar]
- 4b. Lombard C., Saulnier J., Wallach J., Protein Pept. Lett. 2005, 12, 621–629; [DOI] [PubMed] [Google Scholar]
- 4c. Sheldon R. A., Brady D., Chem. Commun. 2018, 54, 6088–6104; [DOI] [PubMed] [Google Scholar]
- 4d. O′Reilly E., Köhler V., Flitsch S. L., Turner N. J., Chem. Commun. 2011, 47, 2490–2501. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Takahashi H., Li B., Sasaki T., Miyazaki C., Kajino T., Inagaki S., Microporous Mesoporous Mater. 2001, 44, 755–762; [Google Scholar]
- 5b. Fernandez-Lopez L., Pedrero S. G., Lopez-Carrobles N., Gorines B. C., Virgen-Ortíz J. J., Fernandez-Lafuente R., Enzyme Microb. Technol. 2017, 98, 18–25; [DOI] [PubMed] [Google Scholar]
- 5c. Thangaraj B., Solomon P. R., ChemBioEng Rev. 2019, 6, 157–166; [Google Scholar]
- 5d. Chapman R., Stenzel M. H., J. Am. Chem. Soc. 2019, 141, 2754–2769. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Li X., Zhang C., Li S., Huang H., Hu Y., Ind. Eng. Chem. Res. 2015, 54, 8072–8079; [Google Scholar]
- 6b. Rueda N., Dos Santos J. C., Ortiz C., Torres R., Barbosa O., Rodrigues R. C., Berenguer-Murcia Á., Fernandez-Lafuente R., Chem. Rec. 2016, 16, 1436–1455. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Nordwald E. M., Armstrong G. S., Kaar J. L., ACS Catal. 2014, 4, 4057–4064; [Google Scholar]
- 7b. Cui H., Zhang L., Eltoukhy L., Jiang Q., Korkunç S. K., Jaeger K.-E., Schwaneberg U., Davari M. D., ACS Catal. 2020, 10, 14847–14856; [Google Scholar]
- 7c. Cui H., Jaeger K. E., Davari M. D., Schwaneberg U., Chem. Eur. J. 2021, 27, 2789–2797; [DOI] [PubMed] [Google Scholar]
- 7d. Frauenkron-Machedjou V. J., Fulton A., Zhao J., Weber L., Jaeger K.-E., Schwaneberg U., Zhu L., Bioresour. Bioprocess. 2018, 5, 2. [Google Scholar]
- 8.
- 8a. Tian K., Tai K., Chua B. J. W., Li Z., Bioresour. Technol. 2017, 245, 1491–1497; [DOI] [PubMed] [Google Scholar]
- 8b. Arnold F. H., Angew. Chem. Int. Ed. 2018, 57, 4143–4148; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 4212–4218. [Google Scholar]
- 9. Zhu F., He B., Gu F., Deng H., Chen C., Wang W., Chen N., J. Biotechnol. 2020, 309, 68–74. [DOI] [PubMed] [Google Scholar]
- 10. Yedavalli P., Rao N. M., Protein Eng. Des. Sel. 2013, 26, 317–324. [DOI] [PubMed] [Google Scholar]
- 11. Cui H., Eltoukhy L., Zhang L., Markel U., Jaeger K.-E., Davari M. D., Schwaneberg U., Angew. Chem. Int. Ed. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.
- 12a. Turner N. J., Nat. Chem. Biol. 2009, 5, 567–573; [DOI] [PubMed] [Google Scholar]
- 12b. Wang J.-b., Li G., Reetz M. T., Chem. Commun. 2017, 53, 3916–3928; [DOI] [PubMed] [Google Scholar]
- 12c. Ji Y., Islam S., Cui H., Dhoke G. V., Davari M. D., Mertens A. M., Schwaneberg U., Catal. Sci. Technol. 2020, 10, 2369–2377. [Google Scholar]
- 13. Fasan R., Jennifer Kan S. B., Zhao H., ACS Catal. 2019, 9, 9775–9788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.
- 14a. Zhang L., Cui H., Dhoke G. V., Zou Z., Sauer D. F., Davari M. D., Schwaneberg U., Chem. Eur. J. 2020, 26, 4974–4979; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14b. Zhang L., Cui H., Zou Z., Garakani T. M., Novoa-Henriquez C., Jooyeh B., Schwaneberg U., Angew. Chem. Int. Ed. 2019, 58, 4562–4565; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 4610–4613; [Google Scholar]
- 14c. Cui H., Zhang L., Söder D., Tang X., Davari M. D., Schwaneberg U., ACS Catal. 2021, 11, 2445–2453. [Google Scholar]
- 15.
- 15a.A. A. Pollardo, H.-s. Lee, D. Lee, S. Kim, J. Kim, J. Cleaner Prod. 2018, 185, 382–388;
- 15b.N. Yaacob, N. H. Ahmad Kamarudin, A. T. C. Leow, A. B. Salleh, R. N. Z. Raja Abd Rahman, M. S. Mohamad Ali, Molecules 2017, 22; [DOI] [PMC free article] [PubMed]
- 15c.S. Dutta Banik, M. Nordblad, J. M. Woodley, G. n. H. Peters, ACS Catal. 2016, 6, 6350–6361;
- 15d.K. Watanabe, T. Yoshida, S. Ueji, Bioorg. Chem. 2004, 32, 504–515. [DOI] [PubMed]
- 16. Cui H., Stadtmüller T. H. J., Jiang Q., Jaeger K. E., Schwaneberg U., Davari M. D., ChemCatChem 2020, 12, 4073. [Google Scholar]
- 17.
- 17a. Mohtashami M., Fooladi J., Haddad-Mashadrizeh A., Housaindokht M. R., Monhemi H., Int. J. Biol. Macromol. 2019, 122, 914–923; [DOI] [PubMed] [Google Scholar]
- 17b. Gorman L. A. S., Dordick J. S., Biotechnol. Bioeng. 1992, 39, 392–397. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Rezaei K., Jenab E., Temelli F., Crit. Rev. Biotechnol. 2007, 27, 183–195; [DOI] [PubMed] [Google Scholar]
- 18b. Yang L., Dordick J. S., Garde S., Biophys. J. 2004, 87, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Kim A. S., Kakalis L. T., Abdul-Manan N., Liu G. A., Rosen M. K., Nature 2000, 404, 151–158; [DOI] [PubMed] [Google Scholar]
- 19b. Xu Z., Affleck R., Wangikar P., Suzawa V., Dordick J. S., Clark D. S., Biotechnol. Bioeng. 1994, 43, 515–520. [DOI] [PubMed] [Google Scholar]
- 20. Pramanik S., Dhoke G. V., Jaeger K.-E., Schwaneberg U., Davari M. D., ACS Sustainable Chem. Eng. 2019. [Google Scholar]
- 21. Greenfield N. J., Nat. Protoc. 2006, 1, 2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Maeda Y., Langmuir 2001, 17, 1737–1742. [Google Scholar]
- 23.
- 23a. Klähn M., Lim G. S., Wu P., Phys. Chem. Chem. Phys. 2011, 13, 18647–18660; [DOI] [PubMed] [Google Scholar]
- 23b. Burney P. R., Nordwald E. M., Hickman K., Kaar J. L., Pfaendtner J., Recent Pat. Mater. Sci. 2015, 83, 670–680. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Cheng F., Zhu L., Schwaneberg U., Chem. Commun. 2015, 51, 9760–9772; [DOI] [PubMed] [Google Scholar]
- 24b. Markel U., Zhu L., Frauenkron-Machedjou V. J., Zhao J., Bocola M., Davari M. D., Jaeger K.-E., Schwaneberg U., Catalysts 2017, 7, 142. [Google Scholar]
- 25. Zhao J., Kardashliev T., Joëlle Ruff A., Bocola M., Schwaneberg U., Biotechnol. Bioeng. 2014, 111, 2380–2389. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Hill J., Donald K., Griffiths D. E., Donald G., Nucleic Acids Res. 1991, 19, 5791; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Tjernberg A., Markova N., Griffiths W. J., Hallén D., J. Biomol. Screening 2006, 11, 131–137. [DOI] [PubMed] [Google Scholar]
- 27. Khmelnitsky Y. L., Belova A. B., Levashov A. V., Mozhaev V. V., FEBS Lett. 1991, 284, 267–269. [DOI] [PubMed] [Google Scholar]
- 28. Van Pouderoyen G., Eggert T., Jaeger K. E., Dijkstra B. W., J. Mol. Biol. 2001, 309, 215–226. [DOI] [PubMed] [Google Scholar]
- 29. Cui H., Pramanik S., Jaeger K.-E., Davari M. D., Schwaneberg U., Green Chem. 2021. [Google Scholar]
- 30. Broos J., Visser A. J., Engbersen J. F., Verboom W., Van Hoek A., Reinhoudt D. N., J. Am. Chem. Soc. 1995, 117, 12657–12663. [Google Scholar]
- 31.
- 31a. Teilum K., Olsen J. G., Kragelund B. B., Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 969–976; [DOI] [PubMed] [Google Scholar]
- 31b. Bhalla J., Storchan G. B., MacCarthy C. M., Uversky V. N., Tcherkasskaya O., Mol. Cell. Proteomics 2006, 5, 1212–1223. [DOI] [PubMed] [Google Scholar]
- 32. Wedberg R., Abildskov J., Peters G. H., J. Phys. Chem. B 2012, 116, 2575–2585. [DOI] [PubMed] [Google Scholar]
- 33. Lousa D., Baptista A. M., Soares C. M., Phys. Chem. Chem. Phys. 2013, 15, 13723–13736. [DOI] [PubMed] [Google Scholar]
- 34. Strub C., Alies C., Lougarre A., Ladurantie C., Czaplicki J., Fournier D., BMC Biochem. 2004, 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qiao B., Jiménez-Ángeles F., Nguyen T. D., De La Cruz M. O., Proc. Natl. Acad. Sci. USA 2019, 116, 19274–19281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.
- 36a. Shivange A. V., Hoeffken H. W., Haefner S., Schwaneberg U., BioTechniques 2016, 61, 305–314; [DOI] [PubMed] [Google Scholar]
- 36b. Winkler C. K., Schrittwieser J. H., Kroutil W., ACS Cent. Sci. 2021, 7, 55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pokkuluri P. R., Raffen R., Dieckman L., Boogaard C., Stevens F. J., Schiffer M., Biophys. J. 2002, 82, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Kawata T., Ogino H., Biotechnol. Prog. 2009, 25, 1605–1611; [DOI] [PubMed] [Google Scholar]
- 38b. Zumárraga M., Bulter T., Shleev S., Polaina J., Martínez-Arias A., Plou F. J., Ballesteros A., Alcalde M., Chem. Biol. 2007, 14, 1052–1064; [DOI] [PubMed] [Google Scholar]
- 38c. Ogino H., Uchiho T., Doukyu N., Yasuda M., Ishimi K., Ishikawa H., Biochem. Biophys. Res. Commun. 2007, 358, 1028–1033. [DOI] [PubMed] [Google Scholar]
- 39. Roy S., Jana B., Bagchi B., J. Chem. Phys. 2012, 136, 03B608. [DOI] [PubMed] [Google Scholar]
- 40. Cui H., Cao H., Cai H., Jaeger K.-E., Davari M. D., Schwaneberg U., Chem. Eur. J. 2020, 26, 643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reetz M. T., Wang L. W., Bocola M., Angew. Chem. Int. Ed. 2006, 45, 1236–1241; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 1258–1263. [Google Scholar]
- 42.
- 42a. Van Durme J., Delgado J., Stricher F., Serrano L., Schymkowitz J., Rousseau F., Bioinformatics 2011, 27, 1711–1712; [DOI] [PubMed] [Google Scholar]
- 42b. Schymkowitz J., Borg J., Stricher F., Nys R., Rousseau F., Serrano L., Nucleic Acids Res. 2005, 33, W382–388; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42c. Krieger E., Joo K., Lee J., Lee J., Raman S., Thompson J., Tyka M., Baker D., Karplus K., Proteins Struct. Funct. Bioinf. 2009, 77, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., SoftwareX 2015, 1, 19–25. [Google Scholar]
- 44. Schmid N., Eichenberger A. P., Choutko A., Riniker S., Winger M., Mark A. E., van Gunsteren W. F., Eur. Biophys. 2011, 40, 843. [DOI] [PubMed] [Google Scholar]
- 45. Malde A. K., Zuo L., Breeze M., Stroet M., Poger D., Nair P. C., Oostenbrink C., Mark A. E., J. Chem. Theory Comput. 2011, 7, 4026–4037. [DOI] [PubMed] [Google Scholar]
- 46. Mark P., Nilsson L., J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar]
- 47.
- 47a. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G., J. Chem. Phys. 1995, 103, 8577–8593; [Google Scholar]
- 47b. Pramanik S., Dhoke G. V., Jaeger K.-E., Schwaneberg U., Davari M. D., ACS Sustainable Chem. Eng. 2019, 7, 11293–11302. [Google Scholar]
- 48.
- 48a.W. L. DeLano, http://www.pymol.org 2002;
- 48b. Humphrey W., Dalke A., Schulten K., J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.