

Abstract
An energy transfer‐based signal amplification relay concept enabling transmission of bioorthogonally activatable fluorogenicity of blue‐excitable coumarins to yellow/red emitting cyanine frames is presented. Such relay mechanism resulted in improved cyanine fluorogenicities together with increased photostabilities and large apparent Stokes‐shifts allowing lower background fluorescence even in no‐wash bioorthogonal fluorogenic labeling schemes of intracellular structures in live cells. These energy transfer dyads sharing the same donor moiety together with their parent donor molecule allowed three‐color imaging of intracellular targets using one single excitation source with separate emission windows. Sub‐diffraction imaging of intracellular structures using the bioorthogonally activatable FRET dyads by STED microscopy is also presented.
Keywords: bioorthogonal, fluorogenic, FRET dyad, multicolor, single excitation
A concept for the transformation of the excellent tetrazine‐ligation dependent fluorogenicity of blue‐excitable cores to biologically preferable emission ranges is presented herein. Such relay mechanism resulted in improved yellow/red fluorogenicities together with increased photostabilities and large apparent Stokes‐shifts. The presented fluorogenic dyads were utilized in multicolor single excitation imaging schemes and STED imaging as well.
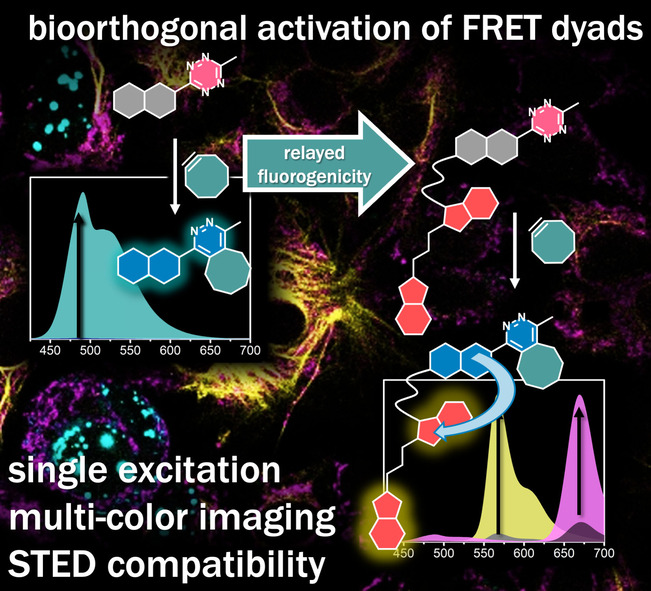
Introduction
Advances in super‐resolution microscopy (SRM) techniques enabled the investigation of biological structures and functions in live cells in the sub‐diffraction range. [1] Due to these hardware developments it is rather the limited number of membrane permeable synthetic probes with proper optical characteristics for multi‐color imaging and potential for site‐specific labeling inside living cells that is considered as major constraint in SRM techniques. [2] Even with an ideal probe[ 3 , 4 , 5 ] in hand that possesses optimal photophysical features, the achievable signal‐to‐noise ratio is often impaired by auto‐fluorescence of endogenous fluorophores and/or the background fluorescence of non‐specifically bound labels especially under no‐wash conditions. Autofluorescence‐related issues are efficiently treated by applying probes with excitation bands in the red, far‐red or near infra‐red region or possessing large Stokes‐shifts. [6] Problems arising from background fluorescence on the other hand, are best addressed by the use of fluorogenic probes,[ 7 , 8 , 9 , 10 ] which exist in a quenched state until they are transformed to an emissive form in a specific reaction. Amongst fluorogenic probes, tetrazine modulated fluorophores are remarkable examples due to the 2‐in‐1 feature of the tetrazine moiety, such as being the quencher of fluorescence and the bioorthogonal handle at the same time.[ 11 , 12 ] Indeed, we and others have demonstrated that tetrazines efficiently quench [13] the fluorescence of various blue/green excitable cores, e.g., coumarins and BODIPYs, via energy (e.g., FRET,[ 14 , 15 ] TBET[ 10 , 11 ]) or electron transfer (PET [10] ) processes. Direct conjugation of tetrazines[ 16 , 17 ] to a fluorescent frame may also result in a dark lowest lying excited state. These examples highlighted the strengths and also the limitations of the tetrazine‐based modulation strategy in terms of ligation schemes and fluorogenic behavior. In combination with genetic code expansion (GCE), [18] bioorthogonal labeling schemes provide an elegant biocompatible approach even for the direct labeling of hardly accessible proteins[ 19 , 20 ] in live cells with minimal perturbation of the biomolecule of interest with practically zero linkage error also holding great promise for single molecule applications. [21] Therefore, tetrazine modulated fluorogenic probes are highly desirable. There are several tetrazine‐quenched blue‐ and green‐light excitable probes, which show orders of magnitude increase in fluorescence intensity upon biorthogonal ligation to a target structure.[ 11 , 12 ] However, this excellent modulation efficiency of the tetrazine moiety drastically declines towards the red end of the spectrum. [9] Our continuing efforts to address this problem resulted in somewhat improved fluorogenicities of cyanine [7] or siliconrhodamine [8] frames, however, these were still behind the expectations. Very recently, the Wombacher group [22] presented a new design concept that was applied to various xanthene frames giving rise to improved fluorogenicities in the red range. By now, it has become evident that alternative, possibly more generalizable approaches need to be sought in order to improve the fluorogenicity of tetrazine modulated probes emitting in the red edge of the spectrum.
Multichromophoric donor‐acceptor systems often possess better overall photophysical features than the individual components. The large apparent Stokes‐shift resulting from the non‐radiative relay of excitation energy from the donor to the acceptor allows complete separation of excitation and emission bands eliminating self‐quenching or errors during detection due to backscattering.
Recent examples show that fast transfer of the excitation energy protects the donor from photobleaching, [23] thus enabling longer exposure of such constructs to excitation light. [24] Laursen and co‐workers have just demonstrated that FRET‐based transfer of energy from a highly absorbing donor to an emitter with long fluorescent lifetime leads to significantly brighter probes. [25] Besides the classical examples taking advantage of the strong distance dependency of the FRET efficiency,[ 26 , 27 , 28 , 29 , 30 ] numerous examples exploit the modulation of the energy transfer process [27] in the sensing of various analytes e.g., H+,[ 31 , 32 , 33 ] O2−[34] or Hg2+,[ 27 , 35 ] however, to the best of our knowledge no bioorthogonal ligation‐assisted modulation of FRET dyads has been presented.
The challenges in tetrazine‐modulated fluorogenic probe design and the considerations on energy transfer dyads prompted us to design constructs where the bioorthogonally controlled fluorogenicity of blue‐light excitable coumarins is relayed to yellow/ red emitters via non‐radiative long range dipole‐dipole interactions (Figure 1). The broad emission band of the activated, de‐quenched form of the coumarin shows appreciable spectral overlap with the absorption spectra of several fluorescent frames emitting in the yellow—red range. By transmitting the excellent bioorthogonal ligation activated turn‐on features of the blue/ green chromophores to yellow/ red acceptors with good quantum yields, such dyads are expected to have improved fluorogenicities and remarkably large apparent Stokes‐shifts, a combination that facilitates easy reduction of both background and autofluorescence. Multicolor imaging of subcellular targets is commonly challenged by the considerable spectral overlap of probes leading to cross‐talk and bleed‐through of signals. Such challenges are addressed by complicated optical setups or sophisticated data processing. [36] Sharing the very same donor moiety, our proposed energy transfer constructs in combination with their parent donor as a third component are also expected to be used in 3‐color imaging of distinct biological targets using a single excitation source. Such a feature is highly desirable in order to avoid excitation‐derived chromatic aberration without the need for complicated optical setups. To date, the number of such large Stokes‐shift dyes that enable single source excitation is quite limited. Although quantum dots with their broad absorption spectra [37] are suitable for single excitation—different color detection imaging, these nanoparticles are not suitable for single molecule tracking or organelle imaging. [38]
Figure 1.

A) Representation of the bioorthogonally controlled fluorogenicity relay concept, B) Structures of bioorthogonal fluorogenic dyads 1–4 and further probes (5–7) used in this study.
Results and Discussion
To evaluate our hypothesis, we have designed a set of bioorthogonally activatable dyads (1–4, Figure 1) comprised of tetrazine modulated coumarins as energy donor units and Cy3 or a Cy5 as energy acceptors linked together via a flexible spacer (Figure 1). In these constructs we either took advantage of our recently described, [17] directly conjugated tetrazine‐chromophore design, where quenching is effected by internal conversion (1, 2) or the classic TBET setup, [12] where the tetrazine is linked to the coumarin via a conjugatable, but otherwise twisted linker (3, 4). Probes 1–4 were accessed through a convergent synthetic route as outlined in the Supporting Information. With the probes in hand, we have acquired the absorption, excitation and emission spectra of probes 1–4 and compared it with their cyclooctyne (BCN) clicked products 1.BCN–4.BCN. As expected, absorption spectra of all probes and their BCN products showed the characteristic bands of both chromophores (Figure 2 and Figure S4). Excitation spectra, monitored at the emission maxima of the respective cyanine, however, showed essential differences. While each BCN‐clicked product had enhanced excitation band at around the absorption of the coumarin compared to the respective parent tetrazine, in case of directly conjugated tetrazine coumarins, 1 and 2, this difference was more profound as they possess virtually no excitation in the coumarin regime (i.e., ≈405 nm). Such an essential difference is explained by the much more efficient quenching in case of vinylene linked coumarin‐tetrazines, 1 and 2. The residual fluorescence and thus the resulting excitation of the cyanines at 405 nm in case of 3 and 4 is probably due to the non‐aligning transition moments of the coumarin and the tetrazine, necessary for efficient energy transfer. The more efficient quenching of 1 and 2 was also translated in larger fluorogenicities as can be seen in the emission spectra (Figure 2). We have also calculated FRET efficiencies between the respective donor‐acceptor pairs, in probes 1 and 2. As expected, the better spectral overlap between the emission spectrum of the coumarin unit and the absorption spectrum of Cy3 in probe 1 resulted in a ca. 25 % more efficient energy transfer compared to 2 as calculated from photobleaching half‐times (see Supporting Information, Table S3).
Figure 2.
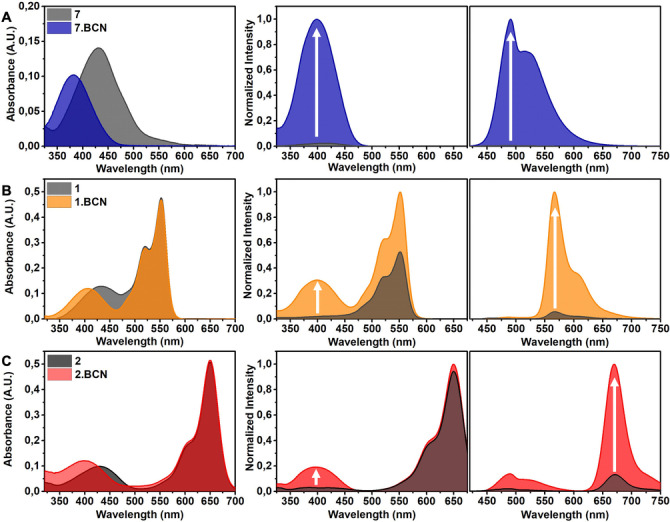
Absorption (5 μM), excitation and emission spectra (1 μM) of (A) 7, λem: 560 nm, λex: 405 nm; B) 1, λem: 620 nm, λex: 405 nm; C) 2, λem: 690 nm, λex: 405 nm and their BCN conjugates in PBS‐SDS; fluorescence spectra were normalized for each BCN conjugates.
Tetrazine‐cyclooctyne reaction activated fluorogenic behavior of probes 1 and 2 were also compared to the respective, tetrazine modulated Cy3 and Cy5 derivatives, 5 and 6 (Figure 1). While 5 gave half of the fluorogenicity of probe 1, 6 gave a confusingly high increase upon reaction with BCN (Table 1) presumably due to the high aggregation potential of Cy5 cores in aqueous media, resulting in the formation of non‐fluorescent aggregates of the tetrazine form. To explore this possibility, we have compared the fluorescence enhancement values in different media with increasing water content (see Supporting Information, Figure S9). These studies revealed that in a pure organic solvent such as MeCN, dyads 1 and 2 outperform cyanines 5 and 6, although the FE values are considerably lower than in PBS(0.1 % SDS). Increasing the water content of the solvents resulted in dramatic changes especially in case of probe 6. It is known that aggregation of indocyanines is efficiently prevented in the presence of proteins like FBS.[ 39 , 40 ] We therefore measured the fluorescence of the probes and their BCN conjugates in complete culture medium (DMEM, without phenol red with 10 % FBS). To our delight, under these conditions the FE pattern of the probes was similar to those measured in pure MeCN, with the exception that probe 1 showed a remarkable 27‐fold increase.
Table 1.
Main photophysical data of probes 1–4 and their BCN conjugates.
|
|
λabs,max [nm] |
λem,max [nm] |
ϵ [a] /×104 M−1 cm−1 |
Stokes‐shift[b] [nm] |
ΦD [c] [%] |
ΦBCN/ΦTet |
IBCN/ITet [d] |
|---|---|---|---|---|---|---|---|
|
1 |
434/552 |
567 |
2.5/9.5 |
146 |
1.0 |
14.5 |
15.9 |
|
1.BCN |
406/552 |
566 |
2.2/9.3 |
14.5 |
|||
|
2 |
428/650 |
670 |
2.1/10.4 |
247 |
2.0 |
6.2 |
7.2 |
|
2.BCN |
403/650 |
669 |
2.3/10.4 |
12.4 |
|||
|
3 |
371/552 |
566 |
2.0/9.8 |
199 |
2.0 |
7.1 |
5.2 |
|
3.BCN |
367/552 |
566 |
2.0/9.7 |
14.2 |
|||
|
4 |
372/650 |
672 |
2.1/14.6 |
280 |
3.3 |
5.1 |
2.5 |
|
4.BCN |
370/650 |
670 |
2.0/14.3 |
16.8 |
[a] determined at λabs,max. [b] apparent Stokes‐shifts. [c] excited at the absorption maxima of the donors (relative to the respective standard (see Supporting Information)). [d] calculated at λem,max of the acceptor when excited at the λabs, max of the donor.
We also wished to extend this fluorogenicity relay concept to other dyads. Following spectral evaluation of possible chromophore pairs, we have selected a tetrazine‐quenched rhodamine donor in combination with a Cy5.5 acceptor (see Supporting Information). Unexpectedly, reaction with BCN did not lead to enhanced Cy5.5 emission. In fact, it led to increased rhodamine (donor) and virtually unchanged Cy5.5 (acceptor) emission. Evaluation of absorption, excitation and emission spectra suggests, that the FRET efficiency between the rhodamine donor and the Cy5.5 acceptor is unchanged irrespective of the actual form of the donor indicating that the energy transfer rate has already reached its limit in the quenched form. These results highlight the importance of the fluorogenicity mechanism suggesting that directly conjugated tetrazines such as in 1 and 2, are quenched much faster via internal conversion [17] restricting the competing processes such as fluorescence or energy transfer.
Before applying the probes to live cells, we have tested the cytotoxicity of probes 1, 2, 5, 6 and their BCN conjugates (see Supporting Information). MTT assay indicated that neither of the fluorogenic dyads nor their BCN adducts were toxic within the 0.3–10 μM concentration range.
On the contrary, tetrazine quenched cyanines 5 and 6 were found to cause a slight decrease of viability, which effect diminished upon BCN conjugation (see Supporting Information, Figure S11).
Following these gratifying results, we moved on to test the applicability of these probes in intracellular protein labeling studies in live cells. To this end, we chose inner nuclear membrane‐anchored protein, Lamin, which was either fused to HaloTag or to a fluorescent protein (miRFP or GFP). In these latter constructs an Amber stop codon was also involved either in the linker between Lamin and the fluorescent protein (LTAG‐miRFP) or within the fluorescent protein (GFP39TAG, see Supporting Information). While Lamin‐HaloTag [41] enables incorporation of a BCN moiety via treatment with cyclooctynylated Halo‐Tag substrate (Halo‐BCN [42] ), Lamin‐LTAG‐miRFP and Lamin‐GFPTAG offer BCN‐bearing non‐canonical amino acid implementation through Amber‐codon suppression. First, HEK293T cells transfected with Lamin‐HaloTag were treated with Halo‐BCN, washed then the respective probes were added. Following a 90 min incubation time, cells were washed, fixed then subjected to confocal microscopy (Figure 3). This experiment revealed that the bioorthogonally activatable fluorogenic dyads 1 and 2, are i) membrane permeable, ii) can stand the challenging environment of live cell labeling and subsequent fixation and iii) are suitable for specific labeling. Figure 3 D on the contrary indicates that 6 is not suitable for specific labeling of intracellular proteins, due to its high background fluorescence. The fluorogenicity of both dyads (1 and 2) turned out to be sufficiently high to tell specifically bound activated and non‐specifically adsorbed probes apart allowing us to carry out these experiments under no‐wash conditions (Figure S12).
Figure 3.
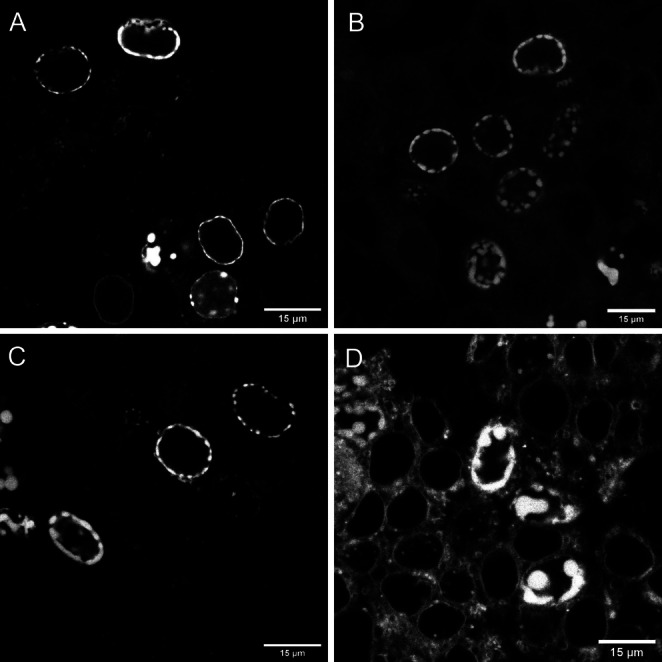
Confocal microscopy images of Lamin‐HaloTag expressing HEK293T cells treated with Halo‐BCN (3 μM) and probes 1 (A), 2 (B) and 5 (C), 6 (D) (1 μM) following washing. Spectral detection: A) (dye 1): λexc: 405 nm/ λem: 560–800 nm; B) (dye 2): λexc: 405 nm/ λem: 560–800 nm; C) (5): λexc: 543 nm/ λem: 560–800 nm; D) (6): λexc: 633 nm/ λem: 650–800 nm.
We also aimed to demonstrate the use of our fluorogenic dyads in combination with genetically encoded bioorthogonal non‐canonical amino acid Lys(BCN). To this end we have treated Lamin‐LTAG‐miRFP or Lamin‐GFPTAG transfected cells with Lys(BCN). Following removal of unincorporated Lys(BCN), the live cells were treated with 1 and 2 or 5 and 6, washed, fixed then imaged. The confocal images (Figure 4) showed similar results to the HaloTag fused proteins, further confirming the potential of the fluorogenic cassettes in specific live cell labeling schemes (note the considerably higher background in case of 5 and 6; cf. more details in the Supporting Information).
Figure 4.

Confocal microscopy images of Lamin‐LTAG‐miRFP (A–F) or Lamin‐GFPTAG (G–L) expressing HEK293T cells treated with Lys(BCN) and probes 1 (A–C), 2 (G–I) or 5 (D–F), 6 (J–L). Scale bar: 20 μm. Spectral detection: A,D) (miRFP/reporter channel): λexc: 633 nm/ λem: 645–800 nm; G,J) (GFP/reporter channel): λexc: 488 nm/ λem: 495–600 nm; B) (1): λexc: 405 nm/ λem: 560–600 nm; E) (5): λexc: 543 nm/ λem: 560–600 nm; H) (2): λexc: 405 nm/ λem: 620–800 nm; K) (6): λexc: 633 nm/ λem: 645–800 nm. Overlay pictures demonstrate the merged fluorescent signals of the reporter and dye channels.
Due to the salient feature of the large apparent Stokes‐shifts, excitation of the dyads at the very same wavelength (i.e., 405 nm) results in the appearance of two distinct emission windows (i.e., 540–600 nm and 650–800 nm; Figure 5 A). Adding the tetrazine modulated donor itself (7, emission window: 420–500 nm) to this set, we foresaw a 3‐color imaging Scheme, where all probes are excited at 405 nm, while detected at three different channels. For such multicolor staining experiment, we have combined immunostaining with HaloTag‐based labeling. More precisely, we have stained HaloTagged Lamin (Lamin‐HaloTag) or lysosomal protein Lamp1‐HaloTag with Halo‐BCN.7, while structural protein, keratin‐19 (CK‐19), and mitochondrial protein, TOMM20 were stained with appropriate (secondary) antibodies prelabeled with 2 and 1, respectively. The as‐treated cells were subjected to confocal imaging using 405 nm laser for excitation and 420–500 nm, 570–600 nm, and 660–800 nm as detection windows for Lamin‐7 (or Lamp1‐7 in Supporting Information, Figure S13), TOMM20‐1 and CK19‐2, respectively.
Figure 5.

Multicolor imaging of HepG2 cells. A) Lamin‐HaloTag treated with 7 tagged Halo‐substrate (Halo‐7), λexc: 405 nm/ λem: 420–500 nm); B) TOMM20 stained with 1‐tagged antibody, λexc: 405 nm/ λem: 570–600 nm); C) CK‐19 stained with 2‐tagged antibody, λexc: 405 nm/ λem: 660–800 nm; D) Overlay of A, B and C; E) magnification of D, Scale bar: 20 μm; F) Emission spectra of dyes 7 (cyan), 1 (yellow) and 2 (magenta) together with excitation wavelength (405 nm) and detection windows.
Gratifyingly, the images showed selective, spectrally distinct labeling of the intracellular structures (Figure 5) without cross‐talk after subtracting the cyan channel from yellow and magenta as well as yellow from magenta. Importantly, our approach could enable simultaneous detection of complex biological events even in real time, using a single excitation source and three detectors without the need for spectral unmixing.
A further advantage of relaying the excitation energy to acceptors is the increased photostability of the donor moiety. [24] To compare the photostabilities of probes 1, 2 and 7, HEK293T Lamin‐HaloTag expressing HEK293T cells were treated with Halo‐BCN.7 or Halo‐BCN. In this latter case the cells were subsequently stained with 1 and 2 and fixed. Determination of bleaching half time values using 405 nm for excitation and 415–520 nm (for 7) or 540–800 nm (for 1 and 2) as detection windows revealed 62.5±1.9 s (n=9), 546.7±11.6 s (n=14) and 218.3±2.7 s (n=13) for 7, 1 and 2, respectively (Supporting Information Figure S14). This suggests that besides relayed fluorogenicity, the dyads possess improved photostabilities as well.
The spectral properties, the fluorogenic behavior and the increased photostability together prompted us to test our fluorogenic dyads in super‐resolution—STED—imaging experiments. Fixed COS‐7 cells were immunostained with anti‐TOMM20 combined with secondary antibody labeled with 1 or 2 and imaged under confocal microscopy using 405 nm for excitation. For super‐resolution images a pulsed, 775 nm depletion laser was applied (Figure 6 and Figure S15). To our delight, both dyads were found suitable for STED imaging and provided super‐resolved images of the outer membrane of mitochondria.
Figure 6.

Confocal (A,B,E,F) and STED (C,D,G,H) images of COS‐7 cells immunostained for TOMM20 with secondary antibodies labeled with probe 1 (A–D) and probe 2 (E–H) (λexc: 405 nm/ λem: 560–755 nm; λSTED=775 nm (pulsed)). Scale bar: 5 μm (A,C,E,G) and in magnified pictures (B,D,F,H): 1 μm. FWHM: full width at half maxima.
Conclusion
In conclusion, we have demonstrated that the bioorthogonally activatable fluorogenicity of blue‐excitable coumarins can be efficiently relayed to yellow/red emitting cyanine cores via long‐range dipole‐dipole interaction‐based energy transfer. The concept enables transformation of the excellent tetrazine‐ligation dependent fluorogenicity of blue‐excitable cores to biologically more preferable emission ranges. Such relay mechanism resulted in improved yellow/ red fluorogenicities together with increased photostabilities and large apparent Stokes‐shifts. To the best of our knowledge this is the first time when bioorthogonally modulated fluorogenicity is relayed to other chromophores via energy transfer. Toxicity and intracellular protein labeling studies with subsequent confocal fluorescence microscopy imaging served evidence for the applicability of these bioorthogonally activatable fluorogenic dyads in live cell labeling experiments even under no‐wash conditions. Furthermore, the large apparent Stokes‐shifts of the dyads in combination with the parent coumarin donor enabled multicolor imaging of distinct intracellular structures using a single excitation wavelength with different emission windows. Moreover, the increased photostability enabled the use of these fluorogenic dyads in STED imaging studies using 405 nm laser for excitation and 775 nm for depletion. The modularity of the design allows a more generalizable approach that can be applied to various fluorogenic scaffolds in combination with suitable acceptors. Thus, we believe that such an energy transfer‐based relay of bioorthogonal reaction aided fluorogenicity may address the wavelength limitation of tetrazine modulated fluorogenic probes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work has been implemented with the support provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the NKFIH‐K‐131439, NKFIH‐FK‐137589 and NKFIH‐PD‐135121 funding Schemes. We are also grateful for the generous support of Eötvös Loránd Research Network (KEP‐6, FIKU). DK is grateful for the support of the ÚNKP‐21‐3 (New National Excellence Program of the Ministry for Innovation and Technology).
E. Albitz, D. Kern, A. Kormos, M. Bojtár, G. Török, A. Biró, Á. Szatmári, K. Németh, P. Kele, Angew. Chem. Int. Ed. 2022, 61, e202111855.
Dedicated to Professor Otto S. Wolfbeis on the occasion of his 75th birthday
Contributor Information
Dr. Márton Bojtár, Email: bojtar.marton@ttk.hu.
Dr. Krisztina Németh, Email: nemeth.krisztina@ttk.hu.
Dr. Péter Kele, Email: kele.peter@ttk.hu.
References
- 1. Sigal Y. M., Zhou R., Zhuang X., Science 2018, 361, 880–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wang L., Frei M. S., Salim A., Johnsson K., J. Am. Chem. Soc. 2019, 141, 2770–2781. [DOI] [PubMed] [Google Scholar]
- 3. Grimm J. B., English B. P., Choi H., Muthusamy A. K., Mehl B. P., Dong P., Brown T. A., Lippincott-Schwartz J., Liu Z., Lionett T., Lavis L. D., Nat. Methods 2016, 13, 985–988. [DOI] [PubMed] [Google Scholar]
- 4. Grimm J. B., English B. P., Chen J., Slaughter J. P., Zhang Z., Revyakin A., Patel R., Macklin J. J., Normanno D., Singer R. H., Lionnet T., Lavis L. D., Nat. Methods 2015, 12, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang L., Tran M., D′Este E., Roberti J., Koch B., Xue L., Johnsson K., Nat. Chem. 2020, 12, 165–172. [DOI] [PubMed] [Google Scholar]
- 6. Sednev M. V., Belov V. N., Hell S. W., Methods Appl. Fluoresc. 2015, 3, 042004. [DOI] [PubMed] [Google Scholar]
- 7. Knorr G., Kozma E., Schaart J. M., Németh K., Török G., Kele P., Bioconjugate Chem. 2018, 29, 1312–1318. [DOI] [PubMed] [Google Scholar]
- 8. Kozma E., Estrada Girona G., Paci G., Lemke E. A., Kele P., Chem. Commun. 2017, 53, 6696–6699. [DOI] [PubMed] [Google Scholar]
- 9. Németh E., Knorr G., Németh K., Kele P., Biomolecules 2020, 10, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wieczorek A., Werther P., Euchner J., Wombacher R., Chem. Sci. 2017, 8, 1506–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carlson J. C. T., Meimetis L. G., Hilderbrand S. A., Weissleder R., Angew. Chem. Int. Ed. 2013, 52, 6917–6920; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 7055–7058. [Google Scholar]
- 12. Meimetis L. G., Carlson J. C. T., Giedt R. J., Kohler R. H., Weissleder R., Angew. Chem. Int. Ed. 2014, 53, 7531–7534; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7661–7664. [Google Scholar]
- 13. Pinto-Pacheco B., Carbery W. P., Khan S., Turner D. B., Buccella D., Angew. Chem. Int. Ed. 2020, 59, 22140–22149; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 22324–22333. [Google Scholar]
- 14. Devaraj N. K., Hilderbrand S., Upadhyay R., Mazitschek R., Weissleder R., Angew. Chem. Int. Ed. 2010, 49, 2869–2872; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2931–2934. [Google Scholar]
- 15. Yang J., Šečkutė J., Cole C. M., Devaraj N. K., Angew. Chem. Int. Ed. 2012, 51, 7476–7479; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 7594–7597. [Google Scholar]
- 16. Lee Y., Cho W., Sung J., Kim E., Park S. B., J. Am. Chem. Soc. 2018, 140, 974–983. [DOI] [PubMed] [Google Scholar]
- 17. Bojtár M., Németh K., Domahidy F., Knorr G., Verkman A., Kállay M., Kele P., J. Am. Chem. Soc. 2020, 142, 15164–15171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chin J. W., Annu. Rev. Biochem. 2014, 83, 379–408. [DOI] [PubMed] [Google Scholar]
- 19. Bessa-Neto D., Kuhlemann A., Beliu G., Pecoraro V., Doose S., Retailleau N., Chevrier N., Perrais D., Sauer M., Choquet D., bioRxiv 2021, 10.1101/2021.02.27.433189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Arsić A., Hagemann C., Stajković N., Schubert T., Nikić-Spiegel I., bioRxiv 2021, 10.1101/2021.01.14.426692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. König A. I., Sorkin R., Alon A., Nachmias D., Dhara K., Brand G., Yifrach O., Arbely E., Roichman Y., Elia N., Nanoscale 2020, 12, 3236–3248. [DOI] [PubMed] [Google Scholar]
- 22. Werther P., Yserentant K., Braun F., Grußmayer K., Navikas V., Yu M., Zhang Z., Ziegler M. J., Mayer C., Gralak A. J., Busch M., Chi W., Rominger F., Radenovic A., Liu X., Lemke E. A., Buckup T., Herten D.-P., Wombacher R., ACS Cent. Sci. 2021, 7, 1561–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Petrovics R., Söveges B., Egyed A., Knorr G., Kormos A., Imre T., Török G., Zeke A., Kocsmár É., Lotz G., Kele P., Németh K., Org. Biomol. Chem. 2018, 16, 2997–3005. [DOI] [PubMed] [Google Scholar]
- 24. Basu S., Needham L., Lando D., Taylor E. J. R., Wohlfahrt K. J., Shah D., Boucher W., Tan Y. L., Bates L. E., Tkachenko O., Cramard J., Lagerholm B. C., Eggeling C., Hendrich B., Klenerman D., Lee S. F., Laue E. D., Nat. Commun. 2018, 9, 2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kacenauskaite L., Bisballe N., Mucci R., Santella M., Pullerits T., Chen J., Vosch T., Laursen B. W., J. Am. Chem. Soc. 2021, 143, 1377–1385. [DOI] [PubMed] [Google Scholar]
- 26. Wu L., Huang C., Emery B. P., Sedgwick A. C., Bull S. D., He X. P., Tian H., Yoon J., Sessler J. L., James T. D., Chem. Soc. Rev. 2020, 49, 5110–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fan J., Hu M., Zhan P., Peng X., Chem. Soc. Rev. 2013, 42, 29–43. [DOI] [PubMed] [Google Scholar]
- 28. Jia X., Chen Q., Yang Y., Tang Y., Wang R., Xu Y., Zhu W., Qian X., J. Am. Chem. Soc. 2016, 138, 10778–10781. [DOI] [PubMed] [Google Scholar]
- 29. Yuan L., Lin W., Chen B., Xie Y., Org. Lett. 2012, 14, 432–435. [DOI] [PubMed] [Google Scholar]
- 30. Yu H., Fu M., Xiao Y., Phys. Chem. Chem. Phys. 2010, 12, 7386–7391. [DOI] [PubMed] [Google Scholar]
- 31. Wu Y.-X., Zhang X.-B., Li J.-B., Zhang C.-C., Liang H., Mao G.-J., Zhou L.-Y., Tan W., Yu R.-Q., Anal. Chem. 2014, 86, 10389–10396. [DOI] [PubMed] [Google Scholar]
- 32. Fang M., Xia S., Bi J., Wigstrom T. P., Valenzano L., Wang J., Mazi W., Tanasova M., Luo F. T., Liu H., Chem. Commun. 2018, 54, 1133–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yuan L., Lin W., Cao Z., Wang J., Chen B., Chem. Eur. J. 2012, 18, 1247–1255. [DOI] [PubMed] [Google Scholar]
- 34. Albers A. E., Okreglak V. S., Chang C. J., J. Am. Chem. Soc. 2006, 128, 9640–9641. [DOI] [PubMed] [Google Scholar]
- 35. Yu H., Xiao Y., Guo H., Qian X., Chem. Eur. J. 2011, 17, 3179–3191. [DOI] [PubMed] [Google Scholar]
- 36. Zimmermann T., Marrison J., Hogg K., O'Toole P. in Confocal Microscopy: Methods and Protocols (Ed.: Paddock S. W.), Springer, New York, NY, 2014, pp. 129–148. [Google Scholar]
- 37. Wu X., Liu H., Liu J., Haley K. N., Treadway J. A., Larson J. P., Ge N., Peale F., Bruchez M. P., Nat. Biotechnol. 2003, 21, 41–46. [DOI] [PubMed] [Google Scholar]
- 38. Wegner K. D., Hildebrandt N., Chem. Soc. Rev. 2015, 44, 4792–4834. [DOI] [PubMed] [Google Scholar]
- 39. Jung B., Vullev V. I., Anvari B., IEEE J. Sel. Top. Quantum Electron. 2014, 20, 149–157. [Google Scholar]
- 40. Onoe S., Temma T., Shimizu Y., Ono M., Saji H., Cancer Med. 2014, 3, 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Szatmári Á., Cserép G. B., Molnár T. Á., Söveges B., Biró A., Várady G., Szabó E., Németh K., Kele P., Molecules 2021, 26, 4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Murrey H. E., Judkins J. C., am Ende C. W., Ballard T. E., Fang Y., Riccardi K., Di L., Guilmette E. R., Schwartz J. W., Fox J. M., Johnson D. S., J. Am. Chem. Soc. 2015, 137, 11461–11475. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information